 Naoyuki Sato
Naoyuki Sato Ryuichi Morishita
Ryuichi Morishita- 1Department of Clinical Gene Therapy, Graduate School of Medicine, Osaka University, Osaka, Japan
- 2Department of Geriatric Medicine, Graduate School of Medicine, Osaka University, Osaka, Japan
Emerging evidence suggests that diabetes affects cognitive function and increases the incidence of dementia. However, the mechanisms by which diabetes modifies cognitive function still remains unclear. Morphologically, diabetes is associated with neuronal loss in the frontal and temporal lobes including the hippocampus, and aberrant functional connectivity of the posterior cingulate cortex and medial frontal/temporal gyrus. Clinically, diabetic patients show decreased executive function, information processing, planning, visuospatial construction, and visual memory. Therefore, in comparison with the characteristics of AD brain structure and cognition, diabetes seems to affect cognitive function through not only simple AD pathological feature-dependent mechanisms but also independent mechanisms. As an Aβ/tau-independent mechanism, diabetes compromises cerebrovascular function, increases subcortical infarction, and might alter the blood–brain barrier. Diabetes also affects glucose metabolism, insulin signaling, and mitochondrial function in the brain. Diabetes also modifies metabolism of Aβ and tau and causes Aβ/tau-dependent pathological changes. Moreover, there is evidence that suggests an interaction between Aβ/tau-dependent and independent mechanisms. Therefore, diabetes modifies cognitive function through Aβ/tau-dependent and independent mechanisms. Interaction between these two mechanisms forms a vicious cycle.
Introduction
More than 30 million patients suffer from dementia (1), while 285 million battle diabetes in this aging society (2). Emerging evidence suggests that diabetes increases the incidence of dementia (3–8). Indeed, diabetes in mid-life is associated with mild cognitive impairment (MCI) (3), and impaired glycemia increases disease progression to dementia in patients with MCI (4). Moreover, numerous epidemiological studies have also demonstrated that patients with diabetes have a significantly higher risk of developing AD (5–8). While genetic and non-genetic risk factors contribute to sporadic AD (9), APOEε4 is the strongest genetic risk factor for sporadic AD and is believed to promote the development of senile plaques. However, the mechanisms by which diabetes modifies cognitive function still remain unclear (10). Here, we review recent advances in brain alterations and clinical symptoms in dementia associated with diabetes and its Aβ/tau-dependent and independent mechanisms.
Brain Alterations in Dementia Associated with Diabetes
Diabetes causes functional and structural deficits in the brain (Table 1). Diabetes is associated with reduced volume of the hippocampus (3, 11), whole brain (3), gray (12), and white matter (11). Gray matter loss is distributed in the medial temporal, anterior cingulate, and medial frontal lobes (3, 11, 12), and white matter loss occurs in the frontal and temporal regions (11), whereas, in AD, gray matter loss is in temporal lobe, hippocampus, entorhinal, and parietal lobes (13–15), and white matter loss is in the temporal region (16). In addition to neuronal loss, diabetes also affects functional connectivity. Resting-state functional connectivity, measured by functional MRI, is used to access brain function. Disruption of resting-state functional connectivity in the default mode network, which is most active during rest, is recently believed to be a predictor of current and future cognitive dysfunction, especially in AD (17, 18). Diabetic patients develop aberrant functional connectivity of the posterior cingulate cortex with the medial temporal gyrus (19) and medial frontal gyrus (20), reflecting white matter abnormalities in diabetes. Diabetic patients also have decreased spontaneous brain activity in the occipital lobe and postcentral gyrus (21). Moreover, functional MRI during task performance demonstrates that diabetic patients show reduced activation of the dorsolateral prefrontal cortex during encoding (22). These studies suggest that diabetes is associated with neuronal loss in the frontal and temporal lobes including the hippocampus, and aberrant functional connectivity between the posterior cingulate cortex and medial frontal/temporal gyrus.
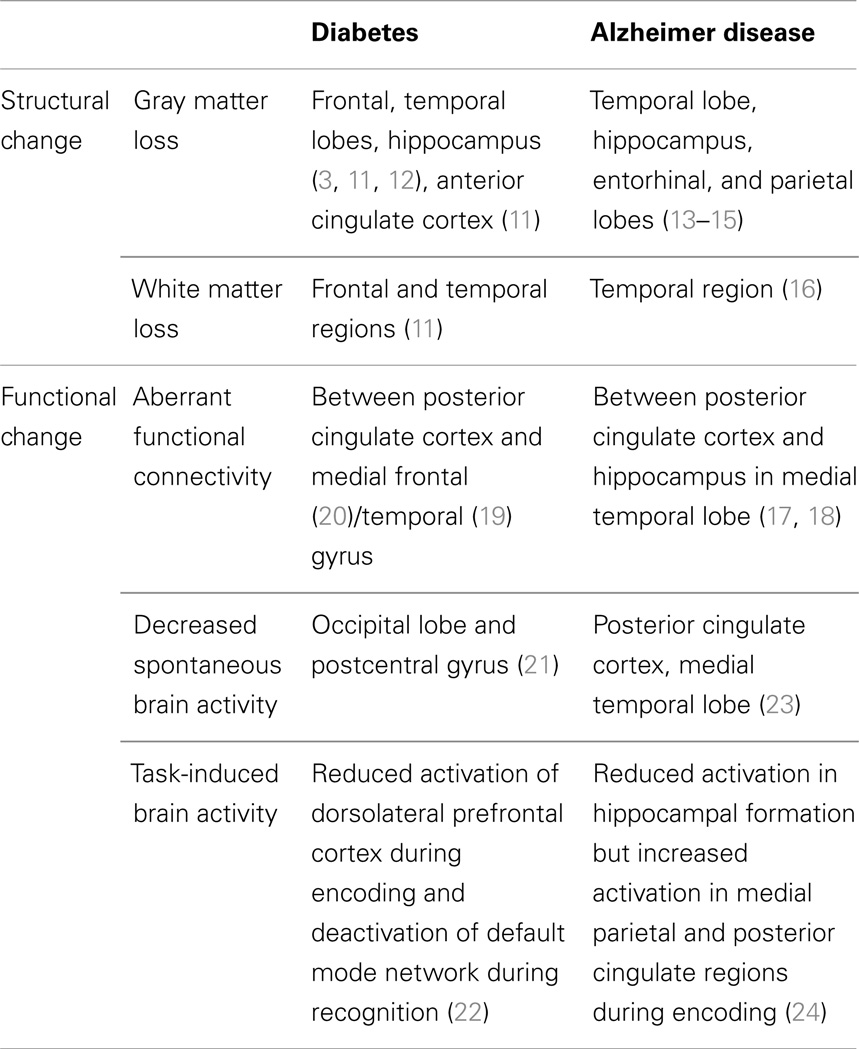
Table 1. Comparison of brain structural and functional alteration in diabetes and Alzheimer disease.
Clinical Symptoms in Dementia Associated with Diabetes
Diabetic patients show impaired cognitive function, increased behavioral symptoms, and decreased activity of daily living (Table 2). Diabetic patients show impairment of memory (25), attention (26, 27), executive function (28, 29), information processing (28, 29), planning (11), visuospatial construction (11), and visual memory (11, 21). Diabetic patients are reported to have impaired memory retrieval rather than encoding (25). Patients with higher HbA1c have increased behavioral and psychological symptoms (30) such as apathy, overeating, and excessive daytime sleeping, and also have impaired activities of daily living (30). Therefore, the knowledge about brain alterations and clinical symptoms suggests that diabetes affects cognitive function through not only simply AD pathological feature-dependent mechanisms but also independent mechanisms (Figure 1).
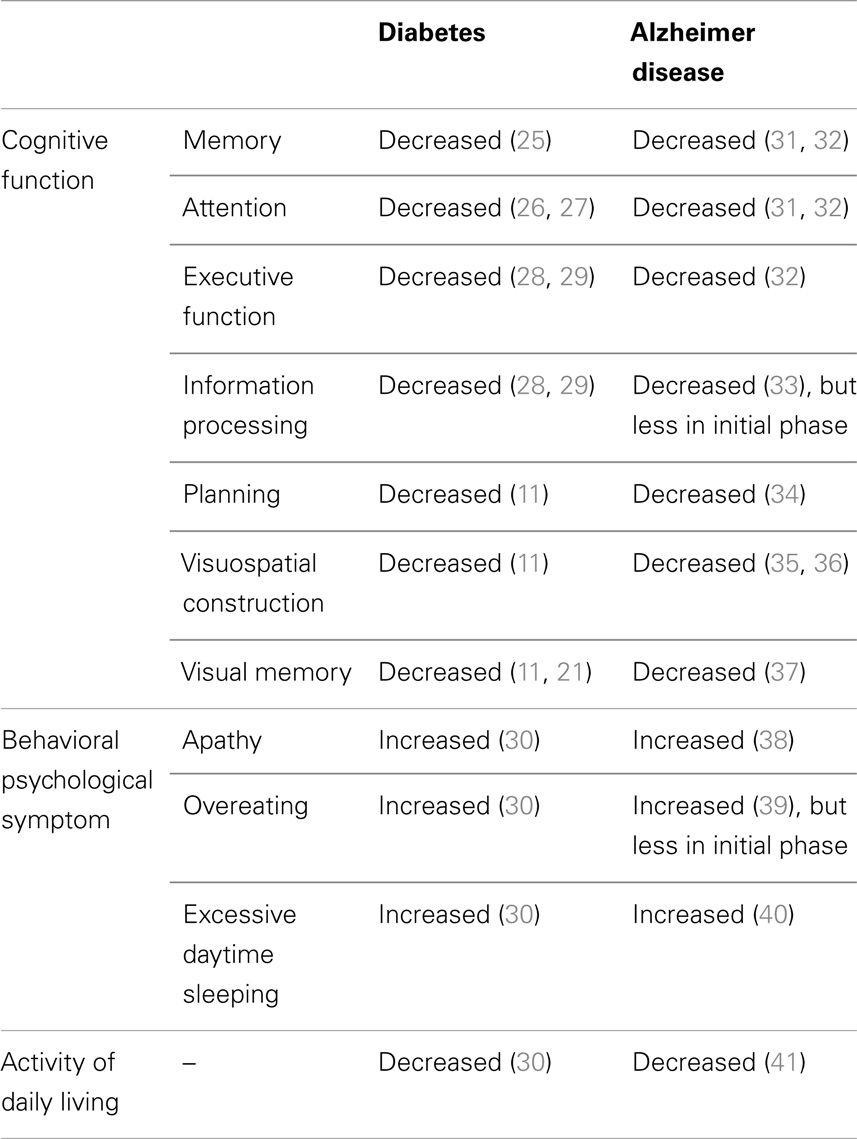
Table 2. Comparison of cognitive and behavioral alterations in diabetes and Alzheimer disease.
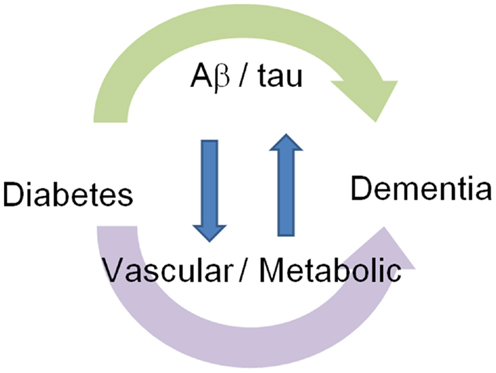
Figure 1. Association of dementia with diabetes: Aβ/tau-dependent and independent mechanisms. Diabetes modifies cognitive function through Aβ/tau-dependent and independent mechanisms. Interaction between these two mechanisms forms a vicious cycle.
Alzheimer Pathological Feature-Independent Mechanisms
Vascular Mechanism
Cerebrovascular damage is related to cognitive function (42–45) and brain atrophy (46). Diabetes is associated with vascular reactivity impairment (47, 48), microangiopathy (49), and cerebrovascular lesions (50), including subcortical infarcts (3). Indeed, microvascular network alteration in the retina, which is believed to be a predictor of vascular changes inside the brain, is associated with increased risk of cognitive dysfunction (51) and AD (52) in diabetes. Vascular permeability is reported to be increased in diabetic retinopathy (53), suggesting that diabetes disrupts the blood–brain barrier (BBB) inside the brain. These data suggest that diabetes compromises cerebrovascular function, increases subcortical infarction, and might cause BBB dysfunction.
Metabolic Mechanism
Hyperglycemia and hypoglycemia – brain glucose metabolism
Hyperglycemia and hypoglycemia have impacts on cognitive function and activity of daily life. Interestingly, Holmes et al. demonstrated that attention and fine motor skills were slowed at altered glucose levels, assessed in diabetic patients during hypoglycemia and hyperglycemia induced by an artificial insulin/glucose infusion system (27). In the long term, the duration of diabetes is associated with impaired cognition in patients with higher HbA1c levels (54). Glucose metabolism declines with age in many brain regions (55), and glucose hypometabolism and brain atrophy are associated with concurrent cognitive dysfunction (56).
Hypoglycemia is associated with cognitive impairment in elderly diabetic patients (57). Because the brain uses mainly glucose as an energy source, hypoglycemia causes defects of neuronal function, though lactate can also be used in such situations (58, 59). In addition to dysfunction of individual cells, failure of neuronal networking also contributes to cognitive impairment in a hypoglycemic state (60). In the long term, repeated episodes of severe hypoglycemia are reported to also be a risk for the development of dementia (61). Hyperglycemia affects cognitive function, and is associated with brain hypometabolism (62), impaired deactivation of the default mode network (22), poorer memory, and reduced hippocampal microstructures (63). Behavioral and psychological symptoms, including apathy, overeating, and excessive daytime sleeping, appear to be increased in patients with HbA1c ≥7.0% (30). In an animal model, hyperglycemia induced by a high-fat diet causes chronic energy imbalance with resulting loss of neurons and reduces olfactory learning (64). These studies suggest that hyperglycemia has an impact on cognition and behavior through glucose-energy imbalance. Although the memory in diabetes (MIND) sub-study of action to control cardiovascular risk in diabetes (ACCORD) suggests that intensive glycemic control has no effect on cognitive function (65), the incidence of hypoglycemia should be considered in interpreting the data (66).
Hyperinsulinemia – brain insulin signaling
Insulin signaling is believed to be decreased in the diabetic brain. Insulin receptors are expressed in the cortex and hippocampus (67–69), and peripheral insulin accesses the brain by crossing the BBB (70). Insulin is also produced in the CNS. Indeed, single-cell PCR reveals that insulin is strongly expressed in GABAergic neurogliaform cells in the cerebral cortex (71). Similarly, insulin-like growth factor-1 (IGF-1), IGF-2, and their receptors exist in the CNS (72–75). When insulin binds to the insulin receptor, IRS-1 and -2 (insulin receptor substrate) undergo tyrosine phosphorylation and bind phosphatidylinositol 3-kinase (PI3K) (76), which activates AKT and glycogen-synthase kinase-3β (GSK3β) (77–79). Importantly, insulin receptor knockout mice show no obvious alteration in the brain (80), suggesting compensation of IGF receptor signaling for insulin signaling. Liu also reported that the levels and activities of several components of the insulin–PI3K–AKT signaling pathway were decreased in patients with diabetes (81). Taken together, these findings indicate that insulin/IGF signaling is impaired in the diabetic brain, and this signaling might have an impact on aging-related brain dysfunction (82). Interestingly, aerobic exercise increases some proteins related to the insulin/IGF-1 pathway in the hippocampus and improves spatial memory in diabetic rats (83).
Brain mitochondrial metabolism
Diabetes deregulates mitochondrial function in mouse (84) and rat (85) neurons. Recent work reveals that hyperglycemia mediates a phenotypic change in mitochondrial biology through alteration of AMP-activated protein kinase (AMPK), a key cellular energy sensor that regulates the activity of a number of metabolic enzymes (86). It is known that an anti-diabetic drug, metformin, activates AMPK kinase. These findings might explain the recent clinical observation that use of metformin is associated with increased risk of cognitive impairment in patients with diabetes (87), though this is still controversial (88). Diabetes is also reported to impair neurite outgrowth through JAK/STAT3 modulation of mitochondrial bioenergetics in neurons (89). Thus, mitochondria might be a new therapeutic target not only for diabetic neuropathy (90) but also for dementia associated with diabetes.
Alzheimer Pathological Feature-Dependent Mechanisms
Aβ-Dependent Mechanism
While AD consists of both familial and sporadic forms, familial AD is caused by mutations in the amyloid precursor protein (91) and presenilin (92). Both mutations cause overproduction of Aβ, particularly its longer form, Aβ42, which is more prone to aggregate in vitro (93) and deposits first in the brain (94) to form senile plaques. Insulin resistance in mid-life is associated with the development of senile plaques (8), though retrospective studies suggest that the magnitude of senile plaques and another hallmark, neurofibrillary tangles, is comparable between AD with and without diabetes (95). Several groups report that a high-fat diet causes Aβ accumulation in the brain of wild type rabbits (96) and APP Tg mice (97, 98). Accumulation of autophagosomes to enhance amyloidogenic APP processing (99) or up-regulation of BACE1 (100) have thus far been proposed as the mechanisms of the increase in Aβ by diabetes. APP+-ob/ob mice, produced by crossing of diabetic and obese ob/ob mice, manifest no change in total brain Aβ level, but increase Aβ deposition in the cerebral vasculature (101). APP+-ob/ob mice show up-regulation of RAGE, the receptor for AGE (102), in the vasculature. Because RAGE mediates amplification of inflammatory responses (103), inflammatory cytokines are upregulated around the cerebrovasculature in APP+-ob/ob (101). Soluble Aβ itself is believed to reduce endothelial function in vitro (27) and vascular reactivity in mice (104) and humans (105, 106). Interestingly, crossing obese and diabetic db/db mice with APP/PS1 knock-in mice leads to severe cerebrovascular pathological features, including aneurysms and small strokes, though no further Aβ deposition in the vasculature, indicating an interaction between soluble Aβ and a diabetic vascular factor (107).
Glucose-energy metabolism is altered in AD (108) and various AD models (109–111). Amyloid burden is accompanied by glucometabolic increases in people at risk for AD (108). Brain 18FDG uptake is a sensitive biomarker for early detection of abnormal metabolism in the 5XFAD mouse (109), indicating that glucose metabolism is decreased in the AD model brain. A glucose transporter, Glut-1, is reduced in the brain capillaries of 18-month-old 3xTg-AD mice (110), suggesting that glucose uptake into the brain might be altered. Another AD model, APP/PS1 mice, also shows alterations in energy-sensor AMPK (111). AMPK could mediate the toxic effects of Aβ through tau phosphorylation (112).
Insulin signaling is also altered by Aβ. In the AD brain, the levels of insulin and IGF (113) and the responses to insulin and IGF (114) are reduced. The levels and activities of the insulin signaling pathway are also decreased in AD (81, 115) and diabetic brains (81), as mentioned above. AD animal models show significant reductions in insulin receptor (111), IRS-1 (116), and IRS-2 (111) in the brain. Therefore, diabetes and Aβ could synergistically affect insulin signaling (101, 117, 118). This impaired insulin signaling could lead to an increase of tau phosphorylation.
Tau-Dependent Mechanism
Diabetes could promote tau phosphorylation, and then formation of neurofibrillary tangles, which is one of the major pathological features of AD. Normal tau promotes the assembly and stabilization of microtubules, but abnormally hyperphosphorylated tau sequesters normal tau and disrupts microtubules (119, 120). Several neuropathological studies suggest that the magnitude of neurofibrillary tangles in the brain at autopsy is not different between AD with and without diabetes (95). However, animal studies show that tau phosphorylation is increased in diabetes (121–125). Tau phosphorylation is increased in the cortex and hippocampus in db/db mice (121) and streptozotocin-induced diabetic mice (122–124). Moreover, streptozotocin exacerbates neurofibrillary tangles in a transgenic mouse model over-expressing the P301L mutant human tau (125). Recently, Takalo et al. report that a high-fat diet induces the expression of four repeat tau mRNA and protein in the temporal cortex (126). Importantly, tau phosphorylation sites in AD are shown to be increased in the diabetic human brain (127). These data suggest that diabetes promotes tau phosphorylation, splicing, and the formation of neurofibrillary tangles.
Impaired insulin signaling in the brain could cause tau phosphorylation (128) and insulin signaling is mainly mediated through the PI3K–AKT–GSK3β pathway (77–79). Because GSK3β phosphorylates tau, insulin inhibits tau phosphorylation through negative regulation of GSK3β (129). Therefore, loss of insulin (130), insulin receptor (80), or IRS-2 (131–133) increases tau phosphorylation. While protein phosphorylation is also regulated by kinases and phosphatases, tau is reported to be dephosphorylated by protein phosphatase 2A (PP2A) (134). As disruption of IRS-2 downregulates PP2A (134), impaired insulin signaling might cause tau phosphorylation by influencing not only kinases but also phosphatases. Importantly, depletion of endogenous tau mitigates behavioral impairment and synaptic deficits induced in diabetic mice (135). Taken together, these findings indicate that diabetes could promote tau phosphorylation via impaired insulin signaling in the brain and then, contribute to cognitive impairment.
Conclusion
Morphologically, diabetes is associated with neuronal loss in the frontal and temporal lobes including the hippocampus, and aberrant functional connectivity of the posterior cingulate cortex and medial frontal/temporal gyrus, and decreased spontaneous brain activity in the occipital lobe and postcentral gyrus. As clinical symptoms, diabetic patients show decreased executive function, information processing, planning, visuospatial construction, and visual memory. Therefore, in comparison with the characteristics in AD brain structure and cognition, diabetes seems to affect cognitive function through not only simply AD pathological feature-dependent but also independent mechanisms. As Aβ/tau-independent mechanisms, diabetes compromises cerebrovascular function, increases subcortical infarction, and might alter BBB. Diabetes also compromises glucose metabolism, insulin signaling, and mitochondrial function. Diabetes also modifies metabolism of Aβ and tau and causes Aβ/tau-dependent pathological changes. Interestingly, in cognitively normal diabetic subjects, higher mean HbA1c levels are associated with lower cognitive performance in ApoEε4 carriers (136), indicating an interaction between Aβ/tau-dependent and independent mechanisms. In conclusion, diabetes modifies cognitive function through Aβ/tau-dependent and independent mechanisms. Interaction between these two mechanisms forms a vicious cycle.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We greatly acknowledge grants from Japan Promotion of Science and the Japanese Ministry of Education, Culture, Sports, Science and Technology (Grant-in-Aid for Scientific Research (B) No. 26293167; to Naoyuki Sato). We also thank members of Department of Clinical Gene Therapy and Department of Geriatric Medicine, past and present.
Abbreviations
AMPK, AMP-activated protein kinase; APP, amyloid precursor protein; BBB, blood–brain barrier; IGF-1, insulin-like growth factor-1; IRS-2, insulin receptor substrate-2; MCI, mild cognitive impairment; RAGE, receptor for advanced glycation endproducts.
References
1. Dartigues JF. Alzheimer’s disease: a global challenge for the 21st century. Lancet Neurol (2009) 8(12):1082–3. doi: 10.1016/S1474-4422(09)70298-4
2. Type 2 diabetes epidemic: a global education. Lancet (2009) 374(9702):1654. doi:10.1016/S0140-6736(09)61974-7
3. Roberts RO, Knopman DS, Przybelski SA, Mielke MM, Kantarci K, Preboske GM, et al. Association of type 2 diabetes with brain atrophy and cognitive impairment. Neurology (2014) 82(13):1132–41. doi:10.1212/WNL.0000000000000269
4. Morris JK, Vidoni ED, Honea RA, Burns JM. Impaired glycemia increases disease progression in mild cognitive impairment. Neurobiol Aging (2014) 35(3):585–9. doi:10.1016/j.neurobiolaging.2013.09.033
5. Maher PA, Schubert DR. Metabolic links between diabetes and Alzheimer’s disease. Expert Rev Neurother (2009) 9(5):617–30. doi:10.1586/ern.09.18
6. Kopf D, Frolich L. Risk of incident Alzheimer’s disease in diabetic patients: a systematic review of prospective trials. J Alzheimers Dis (2009) 16(4):677–85. doi:10.3233/JAD-2009-1011
7. Ott A, Stolk RP, van Harskamp F, Pols HA, Hofman A, Breteler MM. Diabetes mellitus and the risk of dementia: the Rotterdam study. Neurology (1999) 53(9):1937–42. doi:10.1212/WNL.53.9.1937
8. Matsuzaki T, Sasaki K, Tanizaki Y, Hata J, Fujimi K, Matsui Y, et al. Insulin resistance is associated with the pathology of Alzheimer disease: the Hisayama study. Neurology (2010) 75(9):764–70. doi:10.1212/WNL.0b013e3181eee25f
9. Fotuhi M, Hachinski V, Whitehouse PJ. Changing perspectives regarding late-life dementia. Nat Rev Neurol (2009) 5(12):649–58. doi:10.1038/nrneurol.2009.175
10. Sato N, Morishita R. Roles of vascular and metabolic components in cognitive dysfunction of Alzheimer disease: short- and long-term modification by non-genetic risk factors. Front Aging Neurosci (2013) 5:64. doi:10.3389/fnagi.2013.00064
11. Moran C, Phan TG, Chen J, Blizzard L, Beare R, Venn A, et al. Brain atrophy in type 2 diabetes: regional distribution and influence on cognition. Diabetes Care (2013) 36(12):4036–42. doi:10.2337/dc13-0143
12. Garcia-Casares N, Berthier ML, Jorge RE, Gonzalez-Alegre P, Gutiérrez Cardo A, Rioja Villodres J, et al. Structural and functional brain changes in middle-aged type 2 diabetic patients: a cross-sectional study. J Alzheimers Dis (2014) 40(2):375–86. doi:10.3233/JAD-131736
13. Braak H, Braak E. Neuropathological stageing of Alzheimer-related changes. Acta Neuropathol (1991) 82(4):239–59. doi:10.1007/BF00308809
14. Thompson PM, Hayashi KM, de Zubicaray G, Janke AL, Rose SE, Semple J, et al. Dynamics of gray matter loss in Alzheimer’s disease. J Neurosci (2003) 23(3):994–1005.
15. Andrade-Moraes CH, Oliveira-Pinto AV, Castro-Fonseca E, da Silva CG, Guimarães DM, Szczupak D, et al. Cell number changes in Alzheimer’s disease relate to dementia, not to plaques and tangles. Brain (2013) 136(Pt 12):3738–52. doi:10.1093/brain/awt273
16. Mann DM. The topographic distribution of brain atrophy in Alzheimer’s disease. Acta Neuropathol (1991) 83(1):81–6. doi:10.1007/BF00294434
17. Buckner RL, Snyder AZ, Shannon BJ, LaRossa G, Sachs R, Fotenos AF, et al. Molecular, structural, and functional characterization of Alzheimer’s disease: evidence for a relationship between default activity, amyloid, and memory. J Neurosci (2005) 25(34):7709–17. doi:10.1523/JNEUROSCI.2177-05.2005
18. Sorg C, Riedl V, Mühlau M, Calhoun VD, Eichele T, Läer L, et al. Selective changes of resting-state networks in individuals at risk for Alzheimer’s disease. Proc Natl Acad Sci U S A (2007) 104(47):18760–5. doi:10.1073/pnas.0708803104
19. Chen YC, Jiao Y, Cui Y, Shang SA, Ding J, Feng Y, et al. Aberrant brain functional connectivity related to insulin resistance in type 2 diabetes: a resting-state FMRI study. Diabetes Care (2014) 37(6):1689–96. doi:10.2337/dc13-2127
20. Hoogenboom WS, Marder TJ, Flores VL, Huisman S, Eaton HP, Schneiderman JS, et al. Cerebral white matter integrity and resting-state functional connectivity in middle-aged patients with type 2 diabetes. Diabetes (2014) 63(2):728–38. doi:10.2337/db13-1219
21. Cui Y, Jiao Y, Chen YC, Wang K, Gao B, Wen S, et al. Altered spontaneous brain activity in type 2 diabetes: a resting-state functional MRI study. Diabetes (2014) 63(2):749–60. doi:10.2337/db13-0519
22. Marder TJ, Flores VL, Bolo NR, Hoogenboom WS, Simonson DC, Jacobson AM, et al. Task-induced brain activity patterns in type 2 diabetes: a potential biomarker for cognitive decline. Diabetes (2014). doi:10.2337/db13-1783
23. Wang Z, Yan C, Zhao C, Qi Z, Zhou W, Lu J, et al. Spatial patterns of intrinsic brain activity in mild cognitive impairment and Alzheimer’s disease: a resting-state functional MRI study. Hum Brain Mapp (2011) 32(10):1720–40. doi:10.1002/hbm.21140
24. Sperling RA, Bates JF, Chua EF, Cocchiarella AJ, Rentz DM, Rosen BR, et al. fMRI studies of associative encoding in young and elderly controls and mild Alzheimer’s disease. J Neurol Neurosurg Psychiatry (2003) 74(1):44–50. doi:10.1136/jnnp.74.1.44
25. Perlmuter LC, Hakami MK, Hodgson-Harrington C, Ginsberg J, Katz J, Singer DE, et al. Decreased cognitive function in aging non-insulin-dependent diabetic patients. Am J Med (1984) 77(6):1043–8. doi:10.1016/0002-9343(84)90186-4
26. Goh DA, Dong Y, Lee WY, Koay WI, Tay SZ, Soon D, et al. A pilot study to examine the correlation between cognition and blood biomarkers in a Singapore Chinese male cohort with type 2 diabetes mellitus. PLoS One (2014) 9(5):e96874. doi:10.1371/journal.pone.0096874
27. Holmes CS, Hayford JT, Gonzalez JL, Weydert JA. A survey of cognitive functioning at difference glucose levels in diabetic persons. Diabetes Care (1983) 6(2):180–5. doi:10.2337/diacare.6.2.180
28. Nazaribadie M, Amini M, Ahmadpanah M, Asgari K, Jamlipaghale S, Nazaribadie S. Executive functions and information processing in patients with type 2 diabetes in comparison to pre-diabetic patients. J Diabetes Metab Disord (2014) 13(1):27. doi:10.1186/2251-6581-13-27
29. Watari K, Letamendi A, Elderkin-Thompson V, Haroon E, Miller J, Darwin C, et al. Cognitive function in adults with type 2 diabetes and major depression. Arch Clin Neuropsychol (2006) 21(8):787–96. doi:10.1016/j.acn.2006.06.014
30. Sakurai T, Kawashima S, Satake S, Miura H, Tokuda H, Toba K. Differential subtypes of diabetic older adults diagnosed with Alzheimer’s disease. Geriatr Gerontol Int (2014) 14(Suppl 2):62–70. doi:10.1111/ggi.12250
31. Monsell SE, Mock C, Hassenstab J, Roe CM, Cairns NJ, Morris JC, et al. Neuropsychological changes in asymptomatic persons with Alzheimer disease neuropathology. Neurology (2014) 83(5):434–40. doi:10.1212/WNL.0000000000000650
32. Reid W, Broe G, Creasey H, Grayson D, McCusker E, Bennett H, et al. Age at onset and pattern of neuropsychological impairment in mild early-stage Alzheimer disease. A study of a community-based population. Arch Neurol (1996) 53(10):1056–61. doi:10.1001/archneur.1996.00550100142023
33. Huber SJ, Shuttleworth EC, Freidenberg DL. Neuropsychological differences between the dementias of Alzheimer’s and Parkinson’s diseases. Arch Neurol (1989) 46(12):1287–91. doi:10.1001/archneur.1989.00520480029015
34. Starkstein SE, Sabe L, Vazquez S, Teson A, Petracca G, Chemerinski E, et al. Neuropsychological, psychiatric, and cerebral blood flow findings in vascular dementia and Alzheimer’s disease. Stroke (1996) 27(3):408–14. doi:10.1161/01.STR.27.3.408
35. Haxby JV, Grady CL, Koss E, Horwitz B, Schapiro M, Friedland RP, et al. Heterogeneous anterior-posterior metabolic patterns in dementia of the Alzheimer type. Neurology (1988) 38(12):1853–63. doi:10.1212/WNL.38.12.1853
36. Fukui T, Lee E, Kitamura M, Hosoda H, Bokui C, Ikusu K, et al. Visuospatial dysfunction may be a key in the differentiation between Alzheimer’s disease and subcortical cognitive impairment in moderate to severe stages. Dement Geriatr Cogn Disord (2009) 28(4):288–94. doi:10.1159/000245157
37. Sahakian BJ, Morris RG, Evenden JL, Heald A, Levy R, Philpot M, et al. A comparative study of visuospatial memory and learning in Alzheimer-type dementia and Parkinson’s disease. Brain (1988) 111(Pt 3):695–718. doi:10.1093/brain/111.3.695
38. Mori T, Shimada H, Shinotoh H, Hirano S, Eguchi Y, Yamada M, et al. Apathy correlates with prefrontal amyloid beta deposition in Alzheimer’s disease. J Neurol Neurosurg Psychiatry (2014) 85(4):449–55. doi:10.1136/jnnp-2013-306110
39. Burns A, Jacoby R, Levy R. Psychiatric phenomena in Alzheimer’s disease. IV: disorders of behaviour. Br J Psychiatry (1990) 157:86–94. doi:10.1192/bjp.157.1.86
40. Rothman SM, Mattson MP. Sleep disturbances in Alzheimer’s and Parkinson’s diseases. Neuromolecular Med (2012) 14(3):194–204. doi:10.1007/s12017-012-8181-2
41. Patterson MB, Mack JL, Neundorfer MM, Martin RJ, Smyth KA, Whitehouse PJ. Assessment of functional ability in Alzheimer disease: a review and a preliminary report on the Cleveland scale for activities of daily living. Alzheimer Dis Assoc Disord (1992) 6(3):145–63. doi:10.1097/00002093-199206030-00003
42. Iadecola C, Park L, Capone C. Threats to the mind: aging, amyloid, and hypertension. Stroke (2009) 40(3 Suppl):S40–4. doi:10.1161/STROKEAHA.108.533638
43. Dickstein DL, Walsh J, Brautigam H, Stockton SD Jr, Gandy S, Hof PR. Role of vascular risk factors and vascular dysfunction in Alzheimer’s disease. Mt Sinai J Med (2010) 77(1):82–102. doi:10.1002/msj.20155
44. Neltner JH, Abner EL, Baker S, Schmitt FA, Kryscio RJ, Jicha GA, et al. Arteriolosclerosis that affects multiple brain regions is linked to hippocampal sclerosis of ageing. Brain (2014) 137(Pt 1):255–67. doi:10.1093/brain/awt318
45. Richard F, Pasquier F. Can the treatment of vascular risk factors slow cognitive decline in Alzheimer’s disease patients? J Alzheimers Dis (2012) 32(3):765–72. doi:10.3233/JAD-2012-121012
46. Barnes J, Carmichael OT, Leung KK, Schwarz C, Ridgway GR, Bartlett JW, et al. Vascular and Alzheimer’s disease markers independently predict brain atrophy rate in Alzheimer’s disease neuroimaging initiative controls. Neurobiol Aging (2013) 34(8):1996–2002. doi:10.1016/j.neurobiolaging.2013.02.003
47. Caballero AE, Arora S, Saouaf R, Lim SC, Smakowski P, Park JY, et al. Microvascular and macrovascular reactivity is reduced in subjects at risk for type 2 diabetes. Diabetes (1999) 48(9):1856–62. doi:10.2337/diabetes.48.9.1856
48. Pasquier F, Boulogne A, Leys D, Fontaine P. Diabetes mellitus and dementia. Diabetes Metab (2006) 32(5 Pt 1):403–14. doi:10.1016/S1262-3636(07)70298-7
49. Muris DM, Houben AJ, Schram MT, Stehouwer CD. Microvascular dysfunction is associated with a higher incidence of type 2 diabetes mellitus: a systematic review and meta-analysis. Arterioscler Thromb Vasc Biol (2012) 32(12):3082–94. doi:10.1161/ATVBAHA.112.300291
50. van Elderen SG, de Roos A, de Craen AJ, Westendorp RG, Blauw GJ, Jukema JW, et al. Progression of brain atrophy and cognitive decline in diabetes mellitus: a 3-year follow-up. Neurology (2010) 75(11):997–1002. doi:10.1212/WNL.0b013e3181f25f06
51. Bruce DG, Davis WA, Starkstein SE, Davis TM. Mid-life predictors of cognitive impairment and dementia in type 2 diabetes mellitus: the Fremantle diabetes study. J Alzheimers Dis (2014).
52. Cheung CY, Ong YT, Ikram MK, Ong SY, Li X, Hilal S, et al. Microvascular network alterations in the retina of patients with Alzheimer’s disease. Alzheimers Dement (2014) 10(2):135–42. doi:10.1016/j.jalz.2013.06.009
53. Cerani A, Tetreault N, Menard C, Lapalme E, Patel C, Sitaras N, et al. Neuron-derived semaphorin 3A is an early inducer of vascular permeability in diabetic retinopathy via neuropilin-1. Cell Metab (2013) 18(4):505–18. doi:10.1016/j.cmet.2013.09.003
54. West RK, Ravona-Springer R, Schmeidler J, Leroith D, Koifman K, Guerrero-Berroa E, et al. The Association of duration of type 2 diabetes with cognitive performance is modulated by long-term glycemic control. Am J Geriatr Psychiatry (2014). doi:10.1016/j.jagp.2014.01.010
55. Knopman DS, Jack CR Jr, Wiste HJ, Lundt ES, Weigand SD, Vemuri P, et al. F-fluorodeoxyglucose positron emission tomography, aging, and apolipoprotein E genotype in cognitively normal persons. Neurobiol Aging (2014) 35(9):2096–106. doi:10.1016/j.neurobiolaging.2014.03.006
56. Ossenkoppele R, van der Flier WM, Verfaillie SC, Vrenken H, Versteeg A, van Schijndel RA, et al. Long-term effects of amyloid, hypometabolism, and atrophy on neuropsychological functions. Neurology (2014) 82(20):1768–75. doi:10.1212/WNL.0000000000000432
57. Pilotto A, Noale M, Maggi S, Addante F, Tiengo A, Perin PC, et al. Hypoglycemia is independently associated with multidimensional impairment in elderly diabetic patients. Biomed Res Int (2014) 2014:906103. doi:10.1155/2014/906103
58. Rasmussen P, Wyss MT, Lundby C. Cerebral glucose and lactate consumption during cerebral activation by physical activity in humans. FASEB J (2011) 25(9):2865–73. doi:10.1096/fj.11-183822
59. Wyss MT, Jolivet R, Buck A, Magistretti PJ, Weber B. In vivo evidence for lactate as a neuronal energy source. J Neurosci (2011) 31(20):7477–85. doi:10.1523/JNEUROSCI.0415-11.2011
60. Sherin A, Anu J, Peeyush KT, Smijin S, Anitha M, Roshni BT, et al. Cholinergic and GABAergic receptor functional deficit in the hippocampus of insulin-induced hypoglycemic and streptozotocin-induced diabetic rats. Neuroscience (2012) 202:69–76. doi:10.1016/j.neuroscience.2011.11.058
61. Whitmer RA, Karter AJ, Yaffe K, Quesenberry CP Jr, Selby JV. Hypoglycemic episodes and risk of dementia in older patients with type 2 diabetes mellitus. JAMA (2009) 301(15):1565–72. doi:10.1001/jama.2009.460
62. Roberts RO, Knopman DS, Cha RH, Mielke MM, Pankratz VS, Boeve BF, et al. Diabetes and elevated hemoglobin a1c levels are associated with brain hypometabolism but not amyloid accumulation. J Nucl Med (2014) 55(5):759–64. doi:10.2967/jnumed.113.132647
63. Kerti L, Witte AV, Winkler A, Grittner U, Rujescu D, Floel A. Higher glucose levels associated with lower memory and reduced hippocampal microstructure. Neurology (2013) 81(20):1746–52. doi:10.1212/01.wnl.0000435561.00234.ee
64. Thiebaud N, Johnson MC, Butler JL, Bell GA, Ferguson KL, Fadool AR, et al. Hyperlipidemic diet causes loss of olfactory sensory neurons, reduces olfactory discrimination, and disrupts odor-reversal learning. J Neurosci (2014) 34(20):6970–84. doi:10.1523/JNEUROSCI.3366-13.2014
65. Launer LJ, Miller ME, Williamson JD, Lazar RM, Gerstein HC, Murray AM, et al. Effects of intensive glucose lowering on brain structure and function in people with type 2 diabetes (ACCORD MIND): a randomised open-label substudy. Lancet Neurol (2011) 10(11):969–77. doi:10.1016/S1474-4422(11)70188-0
66. Strachan MW, Price JF. Diabetes. Cognitive decline and T2DM – a disconnect in the evidence? Nat Rev Endocrinol (2014) 10(5):258–60. doi:10.1038/nrendo.2014.38
67. Havrankova J, Roth J, Brownstein M. Insulin receptors are widely distributed in the central nervous system of the rat. Nature (1978) 272(5656):827–9. doi:10.1038/272827a0
68. Wickelgren I. Tracking insulin to the mind. Science (1998) 280(5363):517–9. doi:10.1126/science.280.5363.517
69. Hill JM, Lesniak MA, Pert CB, Roth J. Autoradiographic localization of insulin receptors in rat brain: prominence in olfactory and limbic areas. Neuroscience (1986) 17(4):1127–38. doi:10.1016/0306-4522(86)90082-5
70. Banks WA. The source of cerebral insulin. Eur J Pharmacol (2004) 490(1–3):5–12. doi:10.1016/j.ejphar.2004.02.040
71. Molnár G, Faragó N, Kocsis ÁK, Rózsa M, Lovas S, Boldog E, et al. GABAergic neurogliaform cells represent local sources of insulin in the cerebral cortex. J Neurosci (2014) 34(4):1133–7. doi:10.1523/JNEUROSCI.4082-13.2014
72. Shemer J, Raizada MK, Masters BA, Ota A, LeRoith D. Insulin-like growth factor I receptors in neuronal and glial cells. Characterization and biological effects in primary culture. J Biol Chem (1987) 262(16):7693–9.
73. Araujo DM, Lapchak PA, Collier B, Chabot JG, Quirion R. Insulin-like growth factor-1 (somatomedin-C) receptors in the rat brain: distribution and interaction with the hippocampal cholinergic system. Brain Res (1989) 484(1–2):130–8.
74. Rotwein P, Burgess SK, Milbrandt JD, Krause JE. Differential expression of insulin-like growth factor genes in rat central nervous system. Proc Natl Acad Sci U S A (1988) 85(1):265–9. doi:10.1073/pnas.85.1.265
75. Chen DY, Stern SA, Garcia-Osta A, Saunier-Rebori B, Pollonini G, Bambah-Mukku D, et al. A critical role for IGF-II in memory consolidation and enhancement. Nature (2011) 469(7331):491–7. doi:10.1038/nature09667
76. Sun XJ, Rothenberg P, Kahn CR, Backer JM, Araki E, Wilden PA, et al. Structure of the insulin receptor substrate IRS-1 defines a unique signal transduction protein. Nature (1991) 352(6330):73–7. doi:10.1038/352073a0
77. Sutherland C, Leighton IA, Cohen P. Inactivation of glycogen synthase kinase-3 beta by phosphorylation: new kinase connections in insulin and growth-factor signalling. Biochem J (1993) 296(Pt 1):15–9.
78. Cross DA, Alessi DR, Cohen P, Andjelkovich M, Hemmings BA. Inhibition of glycogen synthase kinase-3 by insulin mediated by protein kinase B. Nature (1995) 378(6559):785–9. doi:10.1038/378785a0
79. Neumann KF, Rojo L, Navarrete LP, Farias G, Reyes P, Maccioni RB. Insulin resistance and Alzheimer’s disease: molecular links & clinical implications. Curr Alzheimer Res (2008) 5(5):438–47. doi:10.2174/156720508785908919
80. Schubert M, Gautam D, Surjo D, Ueki K, Baudler S, Schubert D, et al. Role for neuronal insulin resistance in neurodegenerative diseases. Proc Natl Acad Sci U S A (2004) 101(9):3100–5. doi:10.1073/pnas.0308724101
81. Liu Y, Liu F, Grundke-Iqbal I, Iqbal K, Gong CX. Deficient brain insulin signalling pathway in Alzheimer’s disease and diabetes. J Pathol (2011) 225(1):54–62. doi:10.1002/path.2912
82. Steculorum SM, Solas M, Bruning JC. The paradox of neuronal insulin action and resistance in the development of aging-associated diseases. Alzheimers Dement (2014) 10(1 Suppl):S3–11. doi:10.1016/j.jalz.2013.12.008
83. Diegues JC, Pauli JR, Luciano E, de AlmeidaLeme JA, de Moura LP, Dalia RA, et al. Spatial memory in sedentary and trained diabetic rats: molecular mechanisms. Hippocampus (2014) 24(6):703–11. doi:10.1002/hipo.22261
84. Edwards JL, Quattrini A, Lentz SI, Figueroa-Romero C, Cerri F, Backus C, et al. Diabetes regulates mitochondrial biogenesis and fission in mouse neurons. Diabetologia (2010) 53(1):160–9. doi:10.1007/s00125-009-1553-y
85. Pipatpiboon N, Pratchayasakul W, Chattipakorn N, Chattipakorn SC. PPARgamma agonist improves neuronal insulin receptor function in hippocampus and brain mitochondria function in rats with insulin resistance induced by long term high-fat diets. Endocrinology (2012) 153(1):329–38. doi:10.1210/en.2011-1502
86. Chowdhury SK, Smith DR, Fernyhough P. The role of aberrant mitochondrial bioenergetics in diabetic neuropathy. Neurobiol Dis (2013) 51:56–65. doi:10.1016/j.nbd.2012.03.016
87. Moore EM, Mander AG, Ames D, Kotowicz MA, Carne RP, Brodaty H, et al. Increased risk of cognitive impairment in patients with diabetes is associated with metformin. Diabetes Care (2013) 36(10):2981–7. doi:10.2337/dc13-0229
88. Goodarzi MO. Increased risk of cognitive impairment in patients with diabetes is associated with metformin. Diabetes Care (2014) 37(6):e150. doi:10.2337/dc13-2473
89. Saleh A, Chowdhury SK, Smith DR, Balakrishnan S, Tessler L, Schartner E, et al. Diabetes impairs an interleukin-1beta-dependent pathway that enhances neurite outgrowth through JAK/STAT3 modulation of mitochondrial bioenergetics in adult sensory neurons. Mol Brain (2013) 6:45. doi:10.1186/1756-6606-6-45
90. Leinninger GM, Edwards JL, Lipshaw MJ, Feldman EL. Mechanisms of disease: mitochondria as new therapeutic targets in diabetic neuropathy. Nat Clin Pract Neurol (2006) 2(11):620–8. doi:10.1038/ncpneuro0320
91. Goate A, Chartier-Harlin MC, Mullan M, Brown J, Crawford F, Fidani L, et al. Segregation of a missense mutation in the amyloid precursor protein gene with familial Alzheimer’s disease. Nature (1991) 349(6311):704–6. doi:10.1038/349704a0
92. Sherrington R, Rogaev EI, Liang Y, Rogaeva EA, Levesque G, Ikeda M, et al. Cloning of a gene bearing missense mutations in early-onset familial Alzheimer’s disease. Nature (1995) 375(6534):754–60. doi:10.1038/375754a0
93. Jarrett JT, Berger EP, Lansbury PT Jr. The carboxy terminus of the beta amyloid protein is critical for the seeding of amyloid formation: implications for the pathogenesis of Alzheimer’s disease. Biochemistry (1993) 32(18):4693–7. doi:10.1021/bi00069a001
94. Iwatsubo T, Odaka A, Suzuki N, Mizusawa H, Nukina N, Ihara Y. Visualization of A beta 42(43) and A beta 40 in senile plaques with end-specific A beta monoclonals: evidence that an initially deposited species is A beta 42(43). Neuron (1994) 13(1):45–53. doi:10.1016/0896-6273(94)90458-8
95. Kalaria RN. Neurodegenerative disease: Diabetes, microvascular pathology and Alzheimer disease. Nat Rev Neurol (2009) 5(6):305–6. doi:10.1038/nrneurol.2009.72
96. Sparks DL, Scheff SW, Hunsaker JC III, Liu H, Landers T, Gross DR. Induction of Alzheimer-like beta-amyloid immunoreactivity in the brains of rabbits with dietary cholesterol. Exp Neurol (1994) 126(1):88–94. doi:10.1006/exnr.1994.1044
97. Refolo LM, Malester B, LaFrancois J, Bryant-Thomas T, Wang R, Tint GS, et al. Hypercholesterolemia accelerates the Alzheimer’s amyloid pathology in a transgenic mouse model. Neurobiol Dis (2000) 7(4):321–31. doi:10.1006/nbdi.2000.0366
98. Ho L, Qin W, Pompl PN, Xiang Z, Wang J, Zhao Z, et al. Diet-induced insulin resistance promotes amyloidosis in a transgenic mouse model of Alzheimer’s disease. FASEB J (2004) 18(7):902–4. doi:10.1096/fj.03-0978fje
99. Son SM, Song H, Byun J, Park KS, Jang HC, Park YJ, et al. Accumulation of autophagosomes contributes to enhanced amyloidogenic APP processing under insulin-resistant conditions. Autophagy (2012) 8:12. doi:10.4161/auto.21861
100. Guglielmotto M, Aragno M, Tamagno E, Vercellinatto I, Visentin S, Medana C, et al. AGEs/RAGE complex upregulates BACE1 via NF-kappaB pathway activation. Neurobiol Aging (2012) 33(1):.e113–27. doi:10.1016/j.neurobiolaging.2010.05.026
101. Takeda S, Sato N, Uchio-Yamada K, Sawada K, Kunieda T, Takeuchi D, et al. Diabetes-accelerated memory dysfunction via cerebrovascular inflammation and Abeta deposition in an Alzheimer mouse model with diabetes. Proc Natl Acad Sci U S A (2010) 107(15):7036–41. doi:10.1073/pnas.1000645107
102. Brownlee M, Cerami A, Vlassara H. Advanced glycosylation end products in tissue and the biochemical basis of diabetic complications. N Engl J Med (1988) 318(20):1315–21. doi:10.1056/NEJM198805193182007
103. Basta G, Lazzerini G, Massaro M, Simoncini T, Tanganelli P, Fu C, et al. Advanced glycation end products activate endothelium through signal-transduction receptor RAGE: a mechanism for amplification of inflammatory responses. Circulation (2002) 105(7):816–22. doi:10.1161/hc0702.104183
104. Niwa K, Younkin L, Ebeling C, Turner SK, Westaway D, Younkin S, et al. Abeta 1-40-related reduction in functional hyperemia in mouse neocortex during somatosensory activation. Proc Natl Acad Sci U S A (2000) 97(17):9735–40. doi:10.1073/pnas.97.17.9735
105. Dumas A, Dierksen GA, Gurol ME, Halpin A, Martinez-Ramirez S, Schwab K, et al. Functional magnetic resonance imaging detection of vascular reactivity in cerebral amyloid angiopathy. Ann Neurol (2012) 72(1):76–81. doi:10.1002/ana.23566
106. den Abeelen AS, Lagro J, van Beek AH, Claassen JA. Impaired cerebral autoregulation and vasomotor reactivity in sporadic Alzheimer’s disease. Curr Alzheimer Res (2014) 11(1):11–7. doi:10.2174/1567205010666131119234845
107. Niedowicz DM, Reeves VL, Platt TL, Kohler K, Beckett TL, Powell DK, et al. Obesity and diabetes cause cognitive dysfunction in the absence of accelerated beta-amyloid deposition in a novel murine model of mixed or vascular dementia. Acta Neuropathol Commun (2014) 2(1):64. doi:10.1186/2051-5960-2-64
108. Johnson SC, Christian BT, Okonkwo OC, Oh JM, Harding S, Xu G, et al. Amyloid burden and neural function in people at risk for Alzheimer’s disease. Neurobiol Aging (2014) 35(3):576–84. doi:10.1016/j.neurobiolaging.2013.09.028
109. Macdonald IR, DeBay DR, Reid GA, O’Leary TP, Jollymore CT, Mawko G, et al. Early detection of cerebral glucose uptake changes in the 5XFAD mouse. Curr Alzheimer Res (2014) 11(5):450–60. doi:10.2174/1567205011666140505111354
110. Do TM, Alata W, Dodacki A, Traversy MT, Chacun H, Pradier L, et al. Altered cerebral vascular volumes and solute transport at the blood-brain barriers of two transgenic mouse models of Alzheimer’s disease. Neuropharmacology (2014) 81:311–7. doi:10.1016/j.neuropharm.2014.02.010
111. Pedrós I, Petrov D, Allgaier M, Sureda F, Barroso E, Beas-Zarate C, et al. Early alterations in energy metabolism in the hippocampus of APPSwe/PS1dE9 mouse model of Alzheimer’s disease. Biochim Biophys Acta (2014) 1842(9):1556–66. doi:10.1016/j.bbadis.2014.05.025
112. Mairet-Coello G, Courchet J, Pieraut S, Courchet V, Maximov A, Polleux F. The CAMKK2-AMPK kinase pathway mediates the synaptotoxic effects of Abeta oligomers through tau phosphorylation. Neuron (2013) 78(1):94–108. doi:10.1016/j.neuron.2013.02.003
113. Rivera EJ, Goldin A, Fulmer N, Tavares R, Wands JR, de la Monte SM. Insulin and insulin-like growth factor expression and function deteriorate with progression of Alzheimer’s disease: link to brain reductions in acetylcholine. J Alzheimers Dis (2005) 8(3):247–68.
114. Talbot K, Wang HY, Kazi H, Han LY, Bakshi KP, Stucky A, et al. Demonstrated brain insulin resistance in Alzheimer’s disease patients is associated with IGF-1 resistance, IRS-1 dysregulation, and cognitive decline. J Clin Invest (2012) 122(4):1316–38. doi:10.1172/JCI59903
115. Hokama M, Oka S, Leon J, Ninomiya T, Honda H, Sasaki K, et al. Altered expression of diabetes-related genes in Alzheimer’s disease brains: the Hisayama study. Cereb Cortex (2013) 24(9):2476–88. doi:10.1093/cercor/bht101
116. Lourenco MV, Clarke JR, Frozza RL, Bomfim TR, Forny-Germano L, Batista AF, et al. TNF-alpha mediates PKR-dependent memory impairment and brain IRS-1 inhibition induced by Alzheimer’s beta-amyloid oligomers in mice and monkeys. Cell Metab (2013) 18(6):831–43. doi:10.1016/j.cmet.2013.11.002
117. Sato N, Takeda S, Uchio-Yamada K, Ueda H, Fujisawa T, Rakugi H, et al. Role of insulin signaling in the interaction between Alzheimer disease and diabetes mellitus: a missing link to therapeutic potential. Curr Aging Sci (2011) 4(2):118–27. doi:10.2174/1874609811104020118
118. Takeda S, Sato N, Rakugi H, Morishita R. Molecular mechanisms linking diabetes mellitus and Alzheimer disease: beta-amyloid peptide, insulin signaling, and neuronal function. Mol Biosyst (2011) 7(6):1822–7. doi:10.1039/c0mb00302f
119. Iqbal K, Liu F, Gong CX, Alonso Adel C, Grundke-Iqbal I. Mechanisms of tau-induced neurodegeneration. Acta Neuropathol (2009) 118(1):53–69. doi:10.1007/s00401-009-0486-3
120. Iqbal K, Alonso AC, Gong CX, Khatoon S, Singh TJ, Grundke-Iqbal I. Mechanism of neurofibrillary degeneration in Alzheimer’s disease. Mol Neurobiol (1994) 9(1–3):119–23. doi:10.1007/BF02816111
121. Kim B, Backus C, Oh S, Hayes JM, Feldman EL. Increased tau phosphorylation and cleavage in mouse models of type 1 and type 2 diabetes. Endocrinology (2009) 150(12):5294–301. doi:10.1210/en.2009-0695
122. Qu Z, Jiao Z, Sun X, Zhao Y, Ren J, Xu G. Effects of streptozotocin-induced diabetes on tau phosphorylation in the rat brain. Brain Res (2011) 1383:300–6. doi:10.1016/j.brainres.2011.01.084
123. Jolivalt CG, Lee CA, Beiswenger KK, Smith JL, Orlov M, Torrance MA, et al. Defective insulin signaling pathway and increased glycogen synthase kinase-3 activity in the brain of diabetic mice: parallels with Alzheimer’s disease and correction by insulin. J Neurosci Res (2008) 86(15):3265–74. doi:10.1002/jnr.21787
124. Clodfelder-Miller BJ, Zmijewska AA, Johnson GV, Jope RS. Tau is hyperphosphorylated at multiple sites in mouse brain in vivo after streptozotocin-induced insulin deficiency. Diabetes (2006) 55(12):3320–5. doi:10.2337/db06-0485
125. Ke YD, Delerue F, Gladbach A, Gotz J, Ittner LM. Experimental diabetes mellitus exacerbates tau pathology in a transgenic mouse model of Alzheimer’s disease. PLoS One (2009) 4(11):e7917. doi:10.1371/journal.pone.0007917
126. Takalo M, Haapasalo A, Martiskainen H, Kurkinen KM, Koivisto H, Miettinen P, et al. High-fat diet increases tau expression in the brain of T2DM and AD mice independently of peripheral metabolic status. J Nutr Biochem (2014) 25(6):634–41. doi:10.1016/j.jnutbio.2014.02.003
127. Liu Y, Liu F, Grundke-Iqbal I, Iqbal K, Gong CX. Brain glucose transporters, O-GlcNAcylation and phosphorylation of tau in diabetes and Alzheimer’s disease. J Neurochem (2009) 111(1):242–9. doi:10.1111/j.1471-4159.2009.06320.x
128. El Khoury NB, Gratuze M, Papon MA, Bretteville A, Planel E. Insulin dysfunction and tau pathology. Front Cell Neurosci (2014) 8:22. doi:10.3389/fncel.2014.00022
129. Hong M, Lee VM. Insulin and insulin-like growth factor-1 regulate tau phosphorylation in cultured human neurons. J Biol Chem (1997) 272(31):19547–53. doi:10.1074/jbc.272.31.19547
130. Schechter R, Beju D, Miller KE. The effect of insulin deficiency on tau and neurofilament in the insulin knockout mouse. Biochem Biophys Res Commun (2005) 334(4):979–86. doi:10.1016/j.bbrc.2005.07.001
131. Freude S, Hettich MM, Schumann C, Stöhr O, Koch L, Köhler C, et al. Neuronal IGF-1 resistance reduces Abeta accumulation and protects against premature death in a model of Alzheimer’s disease. FASEB J (2009) 23(10):3315–24. doi:10.1096/fj.09-132043
132. Killick R, Scales G, Leroy K, Causevic M, Hooper C, Irvine EE, et al. Deletion of Irs2 reduces amyloid deposition and rescues behavioural deficits in APP transgenic mice. Biochem Biophys Res Commun (2009) 386(1):257–62. doi:10.1016/j.bbrc.2009.06.032
133. Schubert M, Brazil DP, Burks DJ, Kushner JA, Ye J, Flint CL, et al. Insulin receptor substrate-2 deficiency impairs brain growth and promotes tau phosphorylation. J Neurosci (2003) 23(18):7084–92.
134. Sontag E, Nunbhakdi-Craig V, Lee G, Bloom GS, Mumby MC. Regulation of the phosphorylation state and microtubule-binding activity of tau by protein phosphatase 2A. Neuron (1996) 17(6):1201–7. doi:10.1016/S0896-6273(00)80250-0
135. Abbondante S, Baglietto-Vargas D, Rodriguez-Ortiz CJ, Estrada-Hernandez T, Medeiros R, Laferla FM. Genetic ablation of tau mitigates cognitive impairment induced by type 1 diabetes. Am J Pathol (2014) 184(3):819–26. doi:10.1016/j.ajpath.2013.11.021
136. Ravona-Springer R, Heymann A, Schmeidler J, Sano M, Preiss R, Koifman K, et al. The ApoE4 genotype modifies the relationship of long-term glycemic control with cognitive functioning in elderly with type 2 diabetes. Eur Neuropsychopharmacol (2014) 24(8):1303–8. doi:10.1016/j.euroneuro.2014.05.001
Keywords: dementia, diabetes mellitus, Alzheimer disease, abeta, tauopathies
Citation: Sato N and Morishita R (2014) Brain alterations and clinical symptoms of dementia in diabetes: Aβ/tau-dependent and independent mechanisms. Front. Endocrinol. 5:143. doi: 10.3389/fendo.2014.00143
Received: 11 July 2014; Paper pending published: 07 August 2014;
Accepted: 15 August 2014; Published online: 05 September 2014.
Edited by:
Subbiah Pugazhenthi, VA Medical Center-Denver, USAReviewed by:
Jack Tang, Yale University, USAMini Rajan Abraham, Overland Park Medical Specialists, USA
Copyright: © 2014 Sato and Morishita. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Naoyuki Sato, Department of Clinical Gene Therapy, Graduate School of Medicine, Osaka University, 2-2 Yamada-oka, Suita, Osaka 565-0871, Japan e-mail:bnNhdG9AY2d0Lm1lZC5vc2FrYS11LmFjLmpw