 Pere Berbel
Pere Berbel Daniela Navarro
Daniela Navarro Gustavo C. Román
Gustavo C. Román- 1Departamento de Histología y Anatomía, Facultad de Medicina, Universidad Miguel Hernández, Alicante, Spain
- 2Department of Neurology, Weill Cornell Medical College, Cornell University, New York, NY, USA
- 3Methodist Neurological Institute, Houston, TX, USA
The morphological alterations of cortical lamination observed in mouse models of developmental hypothyroidism prompted the recognition that these experimental changes resembled the brain lesions of children with autism; this led to recent studies showing that maternal thyroid hormone deficiency increases fourfold the risk of autism spectrum disorders (ASD), offering for the first time the possibility of prevention of some forms of ASD. For ethical reasons, the role of thyroid hormones on brain development is currently studied using animal models, usually mice and rats. Although mammals have in common many basic developmental principles regulating brain development, as well as fundamental basic mechanisms that are controlled by similar metabolic pathway activated genes, there are also important differences. For instance, the rodent cerebral cortex is basically a primary cortex, whereas the primary sensory areas in humans account for a very small surface in the cerebral cortex when compared to the associative and frontal areas that are more extensive. Associative and frontal areas in humans are involved in many neurological disorders, including ASD, attention deficit-hyperactive disorder, and dyslexia, among others. Therefore, an evo-devo approach to neocortical evolution among species is fundamental to understand not only the role of thyroid hormones and environmental thyroid disruptors on evolution, development, and organization of the cerebral cortex in mammals but also their role in neurological diseases associated to thyroid dysfunction.
Introduction
Evolutionary developmental biology (evo-devo) studies the developmental processes of different organisms to determine the ancestral relationships between them and to discover how developmental processes evolved. It addresses the origin and evolution of embryonic development and the modifications of developmental process that produced novel features (Wikipedia, accessed August 2014). Evo-devo teaches us that some fundamental developmental processes are preserved by the evolution among species (1). The evo-devo approach is not only becoming crucial for the modern study of evolution but also it helps in the understanding of morphofunctional alterations in human psychiatric diseases. For instance, autism spectrum disorders (ASD) show abnormal function of cortical areas, such as the frontal or associative neocortices that are minimally present in rodents (2, 3). An approach to the etiologic factors of psychiatric diseases can be inferred by the study of homologous genetic pathways that lead to similar developmental processes in both humans and other mammals. A second issue is that several psychiatric diseases, including ASD, show a wide spectrum of different phenotypes, which are the result of both genetic (nature) and environmental (nurture) factors (4); including among the latter the interaction of comorbid disorders such as hypothyroidism and hypothyroxinemia (5). We begin this review with a summary of thyroid hormone synthesis, transport, and cell actions, which are regulated by a very complex assembly of transporters, deiodinases, receptors, and cofactors. As such, tissues have some control over thyroid hormone action, independent of circulating levels of thyroid hormones. We continue with the analysis of the role of thyroid hormones at different phases of brain development and maturation, focusing our attention on vulnerable periods. These periods occur during gestation and lactation when genetic and environmental factors, which include nutrients and chemical contaminants, interfere with maternal and offspring thyroid health. There is evidence that anatomical characteristics of autistic brains represent defects in processes that occur early in development, in the first half of gestation. Moreover, genomic studies have revealed a catalog of critical genes for these processes that are regulated by thyroid hormones. Finally, recent studies have reported that thyroid hormone deficiency might contribute to increase the number of autism phenotypes, and that disorders associated with hypothyroidism and hypothyroxinemia, such as intellectual impairment, seizures, and anxiety, are comorbid of ASD.
Thyroid Function during Brain Development
Thyroid hormones (T4, thyroxine; and T3, 3,5,3′-triiodo-l-thyronine) are synthesized in the thyroid gland and are transported to different tissues and organs where they regulate growth, maturation, and function in many organs and systems of vertebrates. In particular, the mammalian central nervous system (CNS) is an important target of thyroid hormones from fetus to adult. However, the maximal vulnerability of the CNS to thyroid hormone imbalance occurs during the earliest stages of brain development (6–15).
In target cells, thyroid hormones can exert their action at three levels: nuclear and mitochondrial (genomic) and non-genomic (16). Genomic actions include (1) thyroid hormone cell membrane transport, (2) thyroid hormone metabolism (involving its activation/degradation), and (3) binding to nuclear thyroid hormone receptors (TRs, also known as THRs), which are ligand-regulated transcription factors (17–25).
Thyroid Hormone Cell Membrane Transport
Thyroid hormone cell membrane transport is mediated by four families of transporters: the Na+/taurocholate cotransporting polypeptide (NTCP), the organic anion transporting polypeptide (OATP), monocarboxylate transporter (MCT), and the heterodimeric amino acid transporter (HAT) (26). From these, Oatp14, Mct8, Mct10, Lat1, and Lat2 have been found to be expressed in the brain (20, 26–33).
Thyroid Hormone Metabolism (Activation/Degradation)
Three selenoproteins catalyzing the deiodination of T4 (thyroxine) and T3 (the active form for the genomic action) have been identified: type 1 (D1), type 2 (D2), and type 3 (D3) iodothyronine deiodinases. Only D2 and D3 have been found expressed in the CNS. D2 has been found in the astrocytes and tanycytes [special ependymal cells, Ref. (34)] and mediates the local generation of T3. D3 mediates the degradation of T3 to T2 (diiodothyronine, 3,5-diiodo-l-thyronine) and T4 to rT3 (35–37). In addition to deiodination, iodothyronines are also metabolized by conjugation of the phenolic hydroxyl group with sulfate or glucuronic acid (38).
Thyroid Hormone Nuclear Receptors
In the CNS, there are three nuclear TR isoforms with high-affinity to T3: TRα1 (codified by the THRA gene), TRβ1, and TRβ2 (codified by the THRB gene) (17, 20, 39, 40). TRα1 is the most ubiquitous; it has been detected in the rat brain by embryonic day 12 (E12) and in the human brain by the 10th week of gestation (41–43), regulating the expression of genes involved in the development and maturation of the brain (44), while TRβ1 is mostly expressed in the adult. In addition, N-terminal truncated TRα1 (also known as p43) can serve as a T3-dependent transcription factor that initiates global mitochondrial transcription (16, 45, 46).
Recent studies have shown that thyroid hormone signaling is more diverse and complex than initially concluded. For instance, apart from the canonic role of thyroid hormones mentioned above, novel THRs synthetic ligands might also modulate TRs action, and intra and extracellular signals can affect cell sensitivity to T3 influencing TRs gene expression, TRs translation and its transport into the nucleus, and the recruitment of co-activators/-inhibitors (21, 24, 47). Furthermore, thyroid hormones can show non-genomic actions by binding to cell surface or cytoplasmic receptors and by interacting with other signaling pathways (16, 21, 48).
In rodents and humans, almost all T3 found in the fetal cerebral cortex is generated through local deiodination of circulating maternal T4 (13, 49, 50). The fetal dependence on maternal T4 is due (i) to the late development of the fetal thyroid gland (in rodents thyroid function begins by E17–18 and in humans by the 18–20 gestational week) and (ii) to the increased activity of D2 and D3 deiodinases in placenta and fetal tissues (13, 35, 51, 52). As a consequence of the increased activity of deiodinases in the fetus, serum T3 levels are maintained low and the local generation of cerebral T3 from T4 is enhanced (13, 50). To respond to this requirement, there is an estrogen-dependent increase of maternal thyroid function that transiently induces an increase of (i) circulating thyroxine-binding globulin, affecting the T4 extra-thyroidal pool, and of (ii) human chorionic gonadotropin, transiently stimulating thyrocytes (53). This increased maternal thyroid function consequently needs increased iodine intake.
Nutritional and Environmental Factors Affecting Thyroid Function
Several factors can affect thyroid function during gestation and early postnatal development, including genetic mutations, infections, nutrients, and environmental contaminants. Iodine deficiency from inadequate alimentary habits is the most common cause of maternal and fetal thyroid dysfunction (54–57). In addition, selenium (a component of deiodinases), iron (a component of the prosthetic heme group associated to the thyroperoxidase), and other micronutrients are required for an adequate life-long thyroid function, especially during development and adolescence (58). Moreover, environmental anti-thyroid contaminants are acquiring increased importance (55, 59–67).
Thyroid Function-Disrupting Chemicals from Environmental Contaminants
A thyroid function-disrupting chemical is an exogenous chemical, or mixture of chemicals, that can interfere with any aspect of hormone action (67). The mechanisms of action of disrupting chemicals on thyroid function are not fully understood; some may reduce serum T4 without increasing serum TSH while others may interfere with thyroid hormone action at sites other than the thyroid gland without altering serum TSH levels (21, 67). Howdeshell (59) listed synthetic chemicals that interfere with thyroid hormone synthesis, transport, and metabolism. Some are quite specific such as perchlorate salts that block the sodium/iodide symporter (68), but the majority affects several phases of thyroid hormone action. Some thyroid disruptors are consumed in the diet (5, 63); for instance, plant isoflavonoids such as genistein and daidzein from soy inhibit thyroperoxidase that catalyzes iodination and thyroid hormone biosynthesis; thiocyanate from cassava not only blocks iodine uptake by thyroid and mammary glands but also interferes with thyroid peroxidase. Organochlorides (including mostly DDT and its derivative: p,p′-DDE, dichlorodiphenyl dichloroethylene; HCB, hexachlorobenzene; PBB, polybrominated biphenyls; and PCB, polychlorinated biphenyls) interfere with thyroid function acting upon iodine uptake, thyroid peroxidase action, thyroid hormone binding proteins, and thyroid hormone metabolism, resulting in a wide spectrum of thyroid-related syndromes (59, 69). The increased use of nanoparticles in several industrial, consumer, and medical applications has revealed their unique physico-chemical properties. However, in vitro and in vivo studies have shown that they may have toxic effects on the endocrine system (70). It has been found that Ag-nanoparticles and cadmium telluride-quantum dots alone induced a reduction in the expression TRβ (71).
Iodine Deficiency Disorders and Neurodevelopmental Damage
As mentioned before, during gestation, the mother must produce sufficient amounts of thyroid hormones (fundamentally T4) for herself and her fetus. Iodine intake is the principal source of circulating inorganic iodine; therefore, sufficient iodine is critical for the thyroid gland to produce adequate amounts of thyroid hormones (9, 13, 53, 57, 72–77). The fetus also depends on the mother for its iodine supply, as does the neonatal thyroid during lactation (78). To achieve this, expecting mothers need to double the recommended normal daily intake of iodine for non-pregnant women by 250–300 μg/day (79).
Useful food strategies developed to increase iodine intake in iodine-deficient areas include (i) use of iodinated salt in the household, (ii) incorporation of iodine to industrially elaborated foods (i.e., bread, milk, and cheese), and (iii) dietary diversification (i.e., consuming food from iodine-sufficient areas and seafood). Despite these strategies, inadequate iodine intake actually affects a large number of women during pregnancy and lactation, and this situation currently persists even in countries classified as free of iodine deficiency where iodized salt consumption has been promoted for years (79–84).
Iodine deficiency is one of the most frequent causes worldwide of preventable mental retardation in children (85). A wide spectrum of iodine deficiency disorders has been described during gestation and the early postnatal period (<3 years of age), ranging from abortion, stillbirths, congenital anomalies, deafness, cretinism, neurocognitive delay, epilepsy, schizophrenia, ASD, as well as attention deficit hyperactive disorder (ADHD), among others (3, 8, 63, 72, 86–94). In children, the severity of the neurodevelopmental damage caused by iodine deficiency during gestation depends on several factors: (i) the developmental period affected, (ii) its severity, (iii) the deficiency of other nutrients such as selenium and iron, and (iv) the interaction with thyroid function disruptors (9, 10, 14, 57, 58). Epidemiological studies performed in several countries have shown that hypothyroxinemia due to mild iodine deficiency during gestation causes neurological alterations, including low IQ in children (8, 87, 88, 95–99). As mentioned above, iodine deficiency in conjunction to the deficiency of other nutrients and the interaction with thyroid function disruptors will cause a wide spectrum of syndromes associated to thyroid pathologies. In countries with severe iodine and selenium deficiencies, a high incidence of Kashin–Beck osteoarthropathy associated with cretinism has been observed (100). The incidence of myxedematous cretinism increases in countries where severe iodine and selenium deficiency is associated with high intake of thyroid disruptors found in foodstuffs such as cassava, which contain thiocyanate (5, 101). The study of the alterations resulting from nutritional deficiencies in combination with thyroid function disruptors should contribute to our understanding of the multiple syndromes observed in thyroid diseases.
Critical Issues of Cerebral Cortex Development
For ethical reasons, the role of thyroid hormones on brain development is currently studied using animal models, usually mice and rats. However, although there are basic common developmental principles regulating brain development between mammals, there are also important differences. For instance, the understanding of how different types of neocortex evolved depends on determining not only the numbers and types of cortical areas that exist but also how the internal organization of those areas was modified in the various lines of evolution, including modifications in columnar organization (102). Sexual dimorphism among species also plays an important role, particularly in humans, while in rodents little is known about sex differences between cerebral hemispheres (103). Changes in the organization and size of neocortex also are reflected in the size of cortico-cortical and subcortical projections, in turn affecting target areas (104). Thus, increasing our evo-devo knowledge on neocortical evolution among species (2) will help us to understand not only the role of thyroid hormones and environmental thyroid disruptors on the development, organization, and evolution of the cerebral cortex in mammals (105), but also their role in human associated diseases. The evo-devo considers crucial for the evolution that homologous developmental gene networks are shared among species (1), and it emerges from the relationship between developmental biology and evolution, which in turn are dynamically coupled (106). For instance, basic gene networks involved in symmetric divisions of ventricular neuroblasts during cerebral corticogenesis are common in rodents and humans, while humans evolved by increasing the number of symmetrical divisions, which results in an increased number of cortical columns and therefore an increased cortical surface [see figure 2A by Rakic (2)]. Apart from homology, convergence can also bring solutions for common functional problems. However, little is known on how functional needs have selected different functional networks to generate a similar function between different species, such as the wings of birds and bats (1). Genetic (nature) and environmental (nurture) factors cooperate along time, resulting in differentiation (4). Psychiatric diseases, such as ASD, occur in cerebral areas (e.g., frontal and associative) that are not present in rodents; however, many homologous functional networks, like those involved in radial migration, have been preserved. Furthermore, ASD show a wide spectrum of different phenotypes, resulting in different degrees of morphofunctional alterations and in the concurrence of different comorbid disorders (107). Thyroid hormone deficiency increases comorbidity and the risk of developing ASD (3). For instance, thyroid hormone deficiency during neocorticogenesis results in abnormal development of cortical gamma-aminobutyric acid (GABA)-ergic neurons, which cause altered columnar function in the cerebral cortex and ASD comorbid seizures (108).
The cerebral cortex in all mammalian species, including humans, differs from the development of other organs of the body and even from the rest of the brain. It is a three-dimensional sheet of layers, parallel to the pial surface, mostly composed of projection (or glutamatergic pyramidal) and local neuronal circuits (or glutamatergic and GABAergic interneurons) organized in vertical (or radial) columns that are stereotypically interconnected and share extrinsic connectivity in order to achieve their functions (109). During telencephalic corticogenesis in mammals, including humans, layer I and subplate (deriving from the superficial primordial plexiform layer) are the first cortical layers to appear (110). Subsequently, young cortical neurons begin to migrate radially from the ventricular zone into the superficial cortical plate, adjacent to layer I, following an “inside-out” gradient (111). While in rodents, the neurogenesis of layer I is arrested when radial migration begins, in primates neurogenesis continues during all the periods of corticogenesis (112). Neurons migrate radially to the increasingly distant cortex following the scaffolding of a transient population of radial glial cells (113), in which many signaling pathways – such as reelin, metabolic functions, and gene expression must be involved (114). This phase of corticogenesis is of capital importance because an evo-devo approach of neocortical development and evolution can be explained by the radial unit hypothesis proposed by Rakic (115). As reported by Rakic (2) and mentioned above, increased number of symmetrical divisions will increase the number of functional columns, resulting in increased tangential cortical surface, while that of asymmetrical ones will increase the number of cells per column, resulting in increased cortical thickness [see also figure 2A by Rakic (2)]. The final number of these divisions will depend of apoptotic, anti-apoptotic, or inhibitory factors, and will give rise to either the small lissencephalic cerebrum of rodents or to the larger convoluted cerebrum of humans, as well as to the emergence of new functional areas, such as the prefrontal cortex and associative perisylvian areas (2). The graded expression of transcription factors such as Emx2, Pax6, Coup-Tf1, and Sp8 are implicated in the arealization of the neocortex (116, 117). Deletion or overexpression of these factors results in changes in gene expression, contractions, and expansions in the sizes of cortical fields, and altered patterns of connectivity from the dorsal thalamus (117). Emx2 and Fgf genes share reciprocal functions in regulating cortical patterning; in the frontal cortex, this is accomplished at least in part through controlling the levels of Erm, Er81, Pea3, and Sp8 expression (118, 119). These results support the protomap model (115, 120, 121) because neurons are committed to their areal position at the time of their last cell division (the asymmetrical one) in the proliferative zones in the absence of thalamic afferent inputs, although individual cortical areas may be selectively changed in size during the course of evolution by altered expression signals of their downstream transcription factor signaling mechanisms, as mentioned above (2). In addition, changes in gene expression extrinsic to the neocortex in response to physical stimuli in a particular environmental context might play a crucial role in the formation of domains and areas in the neocortex (117, 122, 123).
In rodents, radially migrating neurons comprise about 80% of the total cortical neurons and will become glutamatergic neurons. The remaining 20% of the cortical neurons migrate tangentially (i.e., parallel to the pial surface) from the ganglionic eminences to their target area and will become local circuit neurons, mostly GABAergic neurons (124–126). In humans, differently from rodents, a subset of neocortical GABAergic neurons [Mash1-positive; a marker for precursors of glutamic acid decarboxylase (GAD)-expressing cells] originates in the ventricular/subventricular zones of the dorsal telencephalon as a distinct neuronal stem cell lineage [Ref. (127, 128); see figure 5 by Rakic (2)]. The identification of the telencephalic origin of local circuit neurons in cerebral cortex of mammals is of capital importance to understand mechanisms operating during primate brain evolution (2, 129, 130) and the pathogenesis of congenital and acquired neurological disorders, such as ASD, related to defects of separate classes of local circuit neurons (131, 132).
In rats, the bulk of neocortical radial migration starts by embryonic day 13 (E13), while the last cohort of cells leaves the ventricular zone by E20 (133). During this process of radial and horizontal migrations, the subplate neurons attract “waiting” afferents from ipsilateral and contralateral cortical areas (including associative and commissural connections), and subcortical connections [including thalamic, nucleus basalis, and monoamine connections (2), see also the figure 2B of this reference]. At the end of this process, neurons and glial cells grow and differentiate, including the loss of juvenile transient connections, to express their mature phenotype, which also contributes to the radial and tangential expansion of the cortex (134). In humans, neocortical development occurs between the 6th and 24th week of gestation (110, 135). The main waves of radial migration in the human neocortex occur during the first half of gestation, with peaks at 11 and 14 weeks of gestational age (110, 135), and mostly before onset of fetal thyroid hormone secretion by the 18th week of gestation (136). This roughly corresponds to waves of cell migration studied in rats (10), which also occur before onset of fetal thyroid hormone secretion, by E17.5–18 (136). Despite the longer development and maturation of the CNS in humans compared with rats, similarities may be established when the onset of fetal thyroid gland secretion is taken as the reference point. However, when comparing the rodent lissencephalic and the primate convoluted mature neocortex, the major differences are found in the tangential rather than in the radial expansion [see figure 1 by Rakic (2)].
Experimental Models to Study Cortical Alterations Caused by Thyroid Hormone Deficiency
Several experimental models have been developed to study alterations in the CNS caused by thyroid hormone deficiency. These models can be grouped into (i) genetic mutants, (ii) surgically induced hypothyroidism, (iii) metabolite deficient diets, and (iv) thyroid function disruptor models.
Several genetic models were developed during the last decades to study different forms of developmental and postnatal hypothyroidism, such as congenital hypothyroidism (137). Genetic models can be classified into two main groups: (1) mutations affecting thyroid gland development and function, and (2) mutations affecting thyroid hormone sensitivity, which includes thyroid hormone cell membrane transport, metabolism, and action (25). The first group includes mutations of the TSH receptor (hyt−/− mice) (138) and agenesis or functional impairment of thyrocytes (TTF1−/−, TTF2−/−, and Pax8−/− mice) (139). The second group includes thyroid hormone transporters mutants such as Mct8−/y (140, 141), Mct8−/− (142), and Lat2−/− (143). These mutant mice have provided new data to understand thyroid hormone transport in the cell membrane and clarified the physiopathology of the Allan–Herndon–Dudley syndrome, which is caused by MCT8 defect (141, 144–146). Thyroid hormone metabolism in the brain has been studied using different mutant mice affecting D2 and D3 expression (Dio2−/−, Dio3−/−, and Dio2−/−Mct8−/y mice) (147–149). Important genes associated to cortical development are affected in Dio mutants. In particular, the neuronal genes Gls2 (glutaminase 2), Nefh and Nefm (heavy and medium neurofilament polypeptide), Sema7a (semaphorin 7A), Shh (sonic hedgehog), Col6a1 and Col6a2 (type VI α1 and α2 collagen), as well as Slc1a3 (glial high-affinity glutamate transporter) and Itga7 (integrin α7), among others, found in glial cells (148). Mutations of the TR gene include TRα−/−, TRβ−/−, and TRα−/−β−/−mice, as well as TRα and TRβ knock-in mutations (23, 150). Mutations of TRβ gene are associated to the Refetoff syndrome (151, 152). A classification of these mutations and their associated syndromes of impaired sensitivity to thyroid hormone has been recently published (25).
The most common models are based on the administration of anti-thyroid drugs interfering either with the thyrocytes iodine uptake by inhibiting the sodium/iodine symporter (e.g., potassium perchlorate and thiocyanate) or with the iodination of thyroglobulin by thionamide and thiourylene drugs such as propylthiouracil (PTU) and methimazole (MMI) (153–155). In addition, PTU (and less MMI) partially inhibits iodothyronine deiodinases affecting the peripheral deiodination of T4 (154, 156). Anti-thyroid treatments result in maternal, fetal, and neonate hypothyroidism of greater or lesser severity (157). MMI treatment was also used experimentally to induce mild and transient maternal hypothyroxinemia at the onset (E12) of fetal neocorticogenesis (158, 159). Models for iodine deficiency during gestation include monkeys (160), sheep (161), and rats (162, 163). These studies have shown changes in the cerebellum with reduction in weight and cell number, and delayed maturation. The influence of iodine deficiency on neocortical development has been studied in rats that are fed a low iodine diet during pregnancy (163–166).
Alternatively, surgical thyroidectomy can be used to induce hypothyroidism (167, 168), when performed in pregnant dams it causes maternal but neither fetal nor neonate hypothyroidism. Recently, late maternal hypothyroidism (LMH) during gestation has been used as a model to study the role of maternal thyroid hormones from the onset of fetal thyroid function (169).
Alterations in Cortical Development Caused by Thyroid Hormone Deficiencies
Genes Regulated by Thyroid Hormones Involved in Brain Development
Fundamental genes involved in brain development are regulated by thyroid hormones. The irreversibility and importance of damage will depend on when, where, and how the alterations of gene expression occur (10, 20). Early studies showed that maternal thyroid hormones regulate gene expression in fetal development modulating the expression of NSP and Oct-1 genes; T4 injections produced rapid, transient, and selective effects on gene expression in the fetal brain (170). Additional genes regulated by maternal thyroid hormones included Nrgn (neurogranin, also known as RC3), found to be significantly decreased (171), as well as reelin, apolipoprotein E receptor 2 (ApoER2; a reelin receptor involved in the migration young neocortical neurons), very-low-density lipoprotein receptor (VLDLR; a reelin receptor that mediates the stop signal), integrin genes, and genes involved in the downstream phosphorylation of Dab1 (very-low-density lipoprotein receptor 1) (172, 173). cDNA microarray studies have shown a number of genes to be transcriptionally or functionally modulated by T3; most of these are involved in cell division, migration, growth, connectivity, and function of neural cells. Using rat pituitary GC cell line, Miller et al. (174) showed that 358 out of 4,400 genes were regulated by T3; and, in a recent study, Morte et al. (44) found 552 out of 14,209 genes regulated by fetal and maternal thyroid hormones at the end of gestation in rats. The function of some of these genes is unknown but most of them are involved in the regulation of key pathways for the development of the cerebral cortex in rodents and humans. Tables 1–6 list some of the most relevant T3-regulated genes at the transcriptional level. Among those of relevant importance for the development of cortical connections are Nefh, Nefl, and Nefm (coding neurofilament proteins); Slit1, Slit2, Nos1, Camk4, and Creb1 (involved in bifurcation and growth of neural processes); Sema3B, Slit1, and Slit2 (guiding axons); and Slc17a7 (coding vesicular glutamate transporter 1; VGluT1). T3 action on the regulation of the Camk4/Creb pathway and downstream targets (175) in neurons of the CNS is highly relevant since Camk4 has not been found expressed in glial cells (169, 176, 177). Camk4 is directly induced by T3 at the transcriptional level (44), and phosphorylates Creb. Many of the genes under thyroid hormone control contain Creb binding sites in their promoter region (149). On the other hand, Camk4 regulates the transcriptional activity of the TR, which might be due to direct phosphorylation of co-activators or by changing the equilibrium between the co-activators and the silencing mediator for retinoid and thyroid hormone receptors (SMRT) (178, 179). Camk4/Creb pathway and downstream targets are involved in processes such as neurogenesis, biosynthesis, and assembly of cytoskeleton, cell movement and migration, neurite development and maturation, synaptic plasticity, and neurotransmission (44, 180). In humans, Camk4/Creb pathway is involved in psychiatric disorders (181–183). There is a strong evidence for the action of Camk4/Creb pathway in the expression of FMR1 gene, encoding fragile X mental retardation protein (FMRP) (184, 185). Lack of FMRP causes fragile X syndrome, which is the most common cause of inherited mental retardation and ASD (186, 187). In addition, brain-derived neurotrophic factor (BDNF)/Erk signaling modulates FMRP function, affecting neuronal proliferation and differentiation in the cerebral cortex [Ref. (188, 189); Table 5].
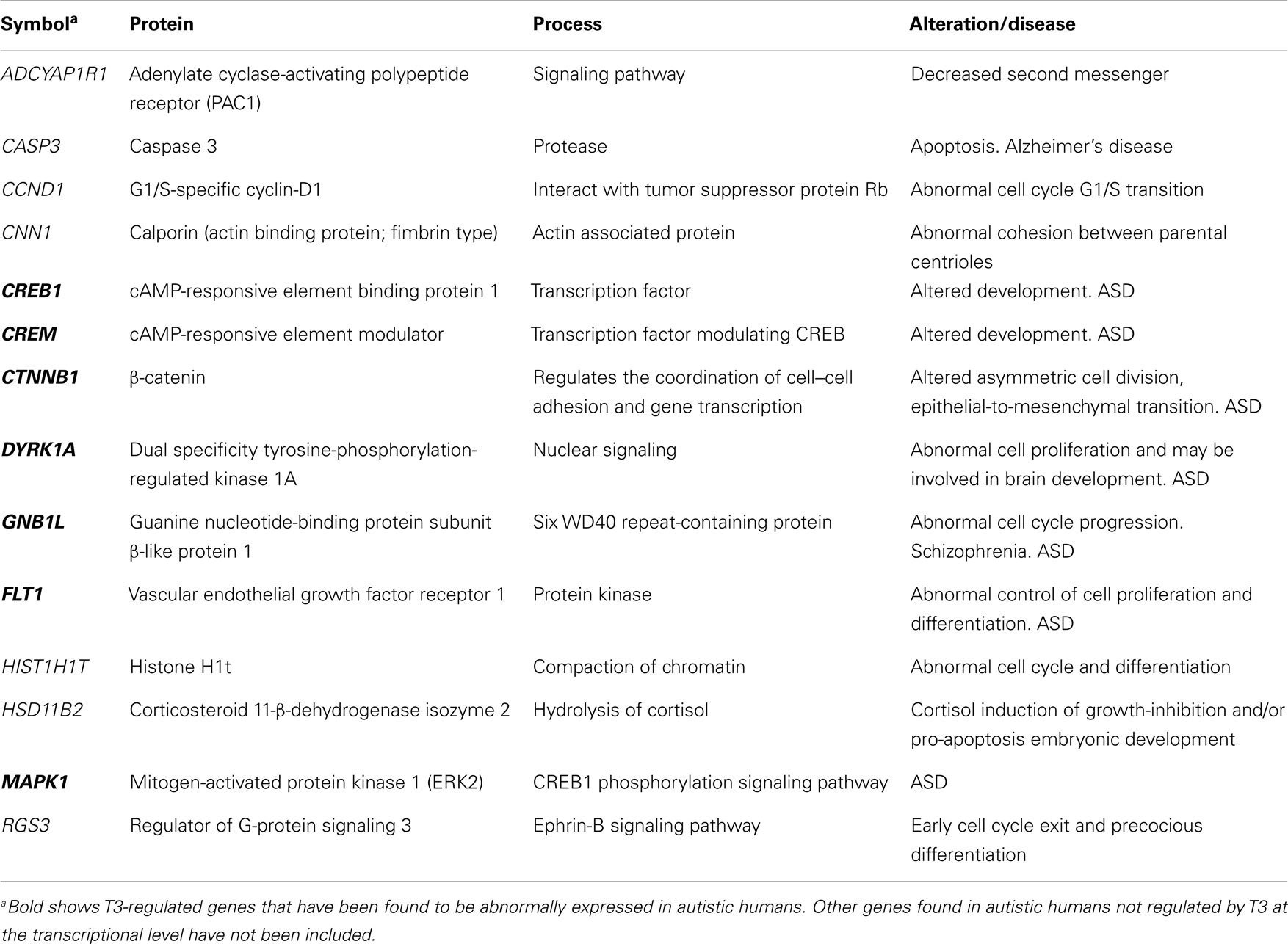
Table 1. Significant T3-regulated genes at the transcriptional level found in the cerebral cortex of rodents, involved in cell division and differentiation: relationship with ASD.
Altered Neurogenesis and Migration during Corticogenesis
Indirect observations based on the cell density estimates and brain size measurements suggested a reduced number of cells in the neocortex of developmentally hypothyroid rats (190). Neural progenitors in the ventricular zone of mouse telencephalon express TRα1, Mct8 transporters, and deiodinases, and maternal hypothyroidism reduces the cell cycle length of these progenitors (191). Since in hypothyroid fetuses the bulk of the neocortical BrdU-labeling occurs between E12 and E19 as in control rats (192), the data by Mohan et al. (191) clearly indicated that the total neuronal progenitor number is reduced in the cerebral cortex. Using 3H-thymidine labeling, a significant reduction was observed in cell acquisition in the granular layer of the hippocampal dentate gyrus in postnatal PTU treated pups (193). These authors also observed that the radial migration of newly generated hippocampal granular cells could be arrested and that the decreased number of labeled cells in the granular layer might result from deficient migration rather that decreased mitotic activity. Several genes involved in cell cycle regulation during neurogenesis have been found to be regulated by T3 [Ref. (44); Table 1]. CCND1 (G1/S-specific cyclin-D1) is downregulated by T3, resulting in an abnormal cell cycle progression (194). In addition, T3-regulated regulator of G-protein signaling 3 (RGS3) plays a key role in ephrin-B signaling, controlling cell cycle exit, and differentiation of neural progenitors (195), and dual specificity tyrosine-phosphorylation-regulated kinase 1A (DYRK1A) is involved in the control of cell proliferation in ASD, causing arrested brain growth (196). Other T3-regulated genes such as CNN1 (calporin), which is an actin associated protein, also exerts a control of cell cycle during neurogenesis (197). Recently, it has been found that developmental mild and severe hypothyroxinemia and MMI-induced hypothyroidism alters Shh signaling pathway in the cerebellar granule cell precursors, resulting in downregulation of D1 and D2 cyclins, of E2F1 expression, and in reduced cell proliferation (198). However, it still remains unclear to what extent thyroid hormones affect symmetrical and asymmetrical divisions of neocortical progenitor cells. Studies on the mouse barrel cortex (192) suggest that both symmetrical and asymmetrical divisions are altered in hypothyroid rats, because the tangential area of the posteromedial barrel subfield stained with cytochrome oxidase (resulting from symmetrical divisions) and the thickness of the barrel cortex (resulting from asymmetrical divisions) are reduced by 27 (Figures 1A–D) and 12.5% (Figure 1E), respectively, in hypothyroid rats. Nevertheless, from these data we could argue that most likely the symmetrical divisions are comparatively more affected in hypothyroid rats. The reduced thickness of the cortex in hypothyroid rats could be explained by a reduction of the columnar neuropile more than by a reduction in the cellular components of the columns. T3-regulated CASP3 (caspase 3) and CTNNB1 (β-catenin) genes are crucial for cerebral cortex expansion (Table 1). Experimental studies using caspase 3 and 9 KO mice (lacking apoptotic signals) (2, 199) and transgenic mice expressing β-catenin (which increases the number of precursor cells) lead to an abnormally convoluted mouse cortex (200). Caspase 3 pathway is downregulated in the cerebral and cerebellar cortices of hypothyroxinemic and hypothyroid rats (201, 202), while β-catenin is T3-downregulated in rat pituitary cultured cells (174). These data show that thyroid hormone deficiency alters the tangential and radial organization of the cortex and might have contributed in the evolutionary elaboration of radial columns, modulating both cortical surface and thickness.
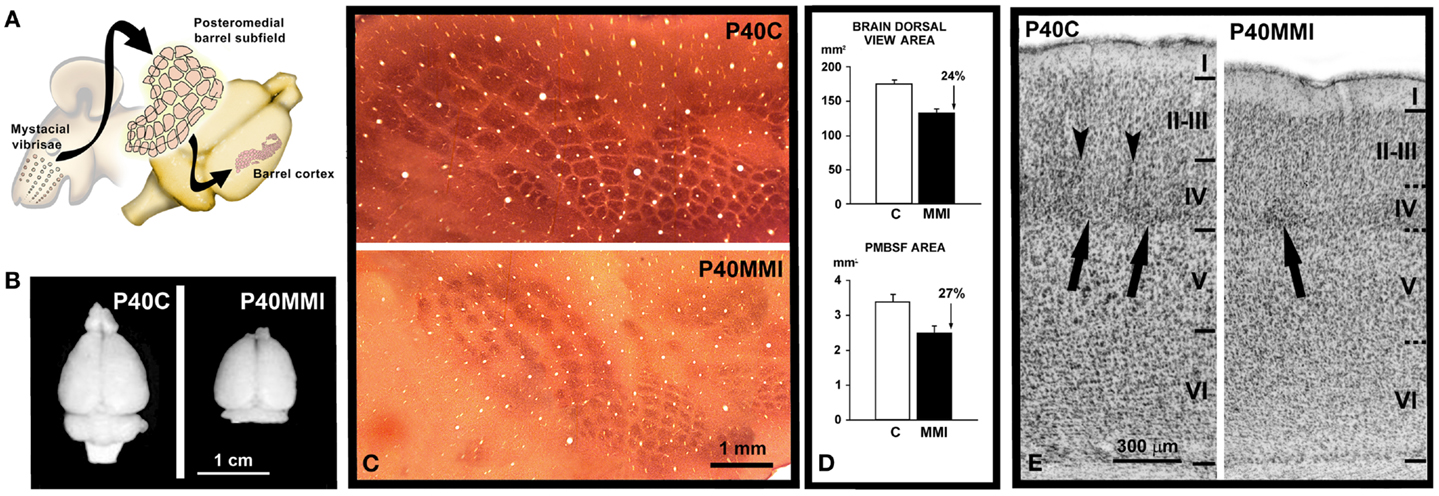
Figure 1. Reduced development of cortical maps in developmental hypothyroidism. (A) Cartoon showing the posteromedial barrel subfield of the primary somatosensory cortex in the brain of a rat. Note the correspondence between mysticial vibrises and the barrels of the posteromedial barrel subfield. (B) Brain dorsal views at P40 of control (C) and MMI pups. (C) Computer reconstruction from photomicrographs of serial tangential sections through layer IV, showing cytochrome oxidase labeling in the barrel cortex of normal and hypothyroid rats. Note the reduced tangential extension of the cytochrome oxidase labeling in hypothyroid with respect to normal rats. (D) Area measurements in normal and hypothyroid rats. The dorsal view brain area was, on average, 24% smaller in hypothyroid rats (upper). A similar reduction (on average, 27%) was observed in the PMBSF tangential area (lower). (E) Photomicrographs of cresyl violet stained coronal sections showing the cytoarchitecture of the barrel cortex of the primary somatosensory cortex at P40 in control (C) and transient MMI treated pups (MMI treatment begun at E12 and finished at E15). Borders between layers (horizontal lines) are clear-cut in C whereas they are more blurred in MMI12 pups. In layer IV of C and dMMI pups, barrels (arrow) are normal and well-defined and demarcated by septae (arrowheads). In contrast, barrels in layer IV of MMI1 pups are not seen. In developmentally hypothyroid pups there is a 10–15% reduction in the cortical thickness of MMI pups compared to controls. (A) Modified from Berbel and Morreale de Escobar (57). (C,D) Modified from Berbel et al. (192). (E) Modified from Ausó et al. (158).
Altered T3-regulated opioid-binding protein/cell adhesion molecule (OPCML; also known as OBCAM) expression affects radial glia function and its transdifferentiation to astrocytes [Ref. (203); Table 2]. In agreement, impaired maturation of radial glia was observed in the hippocampus of pups born to chronic hypothyroxinemic rats (163) and in the neocortex of developmentally hypothyroid pups (204). Abnormal radial migration in the neocortex of developmentally hypothyroid rats was first described in the auditory cortex by combining BrdU and tracer labeling (205). As a result, the radial positioning of migrating neurons was altered, including abnormally located heterotopic neurons in the subcortical white matter (192, 205) (Figure 2C) and corpus callosum (206). Also, altered neuronal migration in the neocortex and hippocampus has been confirmed in hypothyroxinemic rats (158, 159, 164) (Figures 2A–C). Apart from TRs, other nuclear receptors are involved in radial glia maturation and radial migration in the neocortex such as the liver X receptor β (LXRβ) that also regulates the expression of ApoER2 receptor (207). LXRβ−/− mice showed altered cortical migration of later-born neurons (208) and delayed transdifferentiation of radial glial cells into astrocytes (209). Interestingly, LXRs bind to the same response element on DNA as TRs and sometimes regulate the same genes (150, 210). In fact, it has been shown recently that TRα compensates for the lack of LXRβ in cortical development, and a reciprocal compensatory action can also be hypothesized (207).
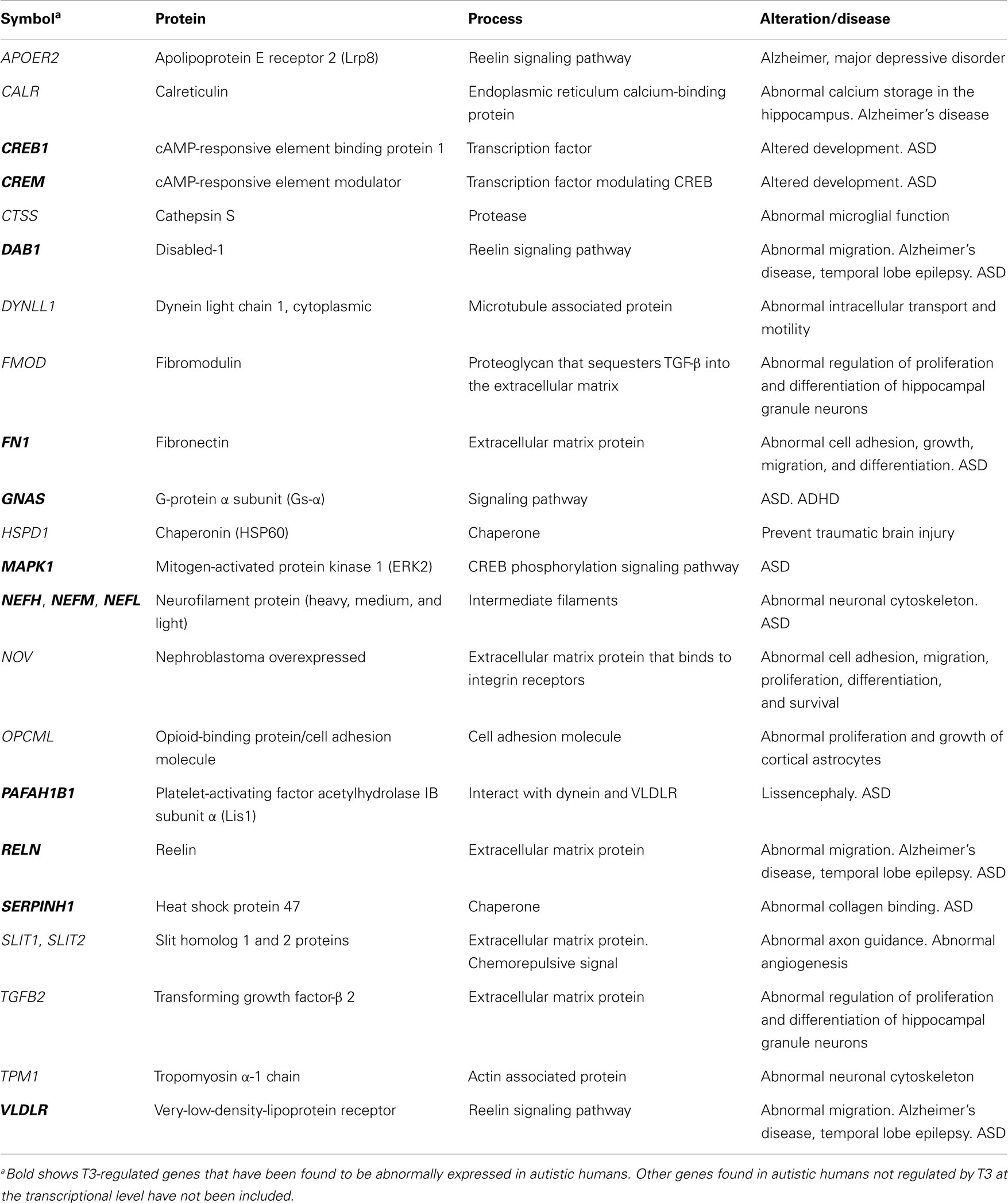
Table 2. Significant T3-regulated genes at the transcriptional level found in the cerebral cortex of rodents, involved in cytoskeleton organization and cell migration: relationship with ASD.
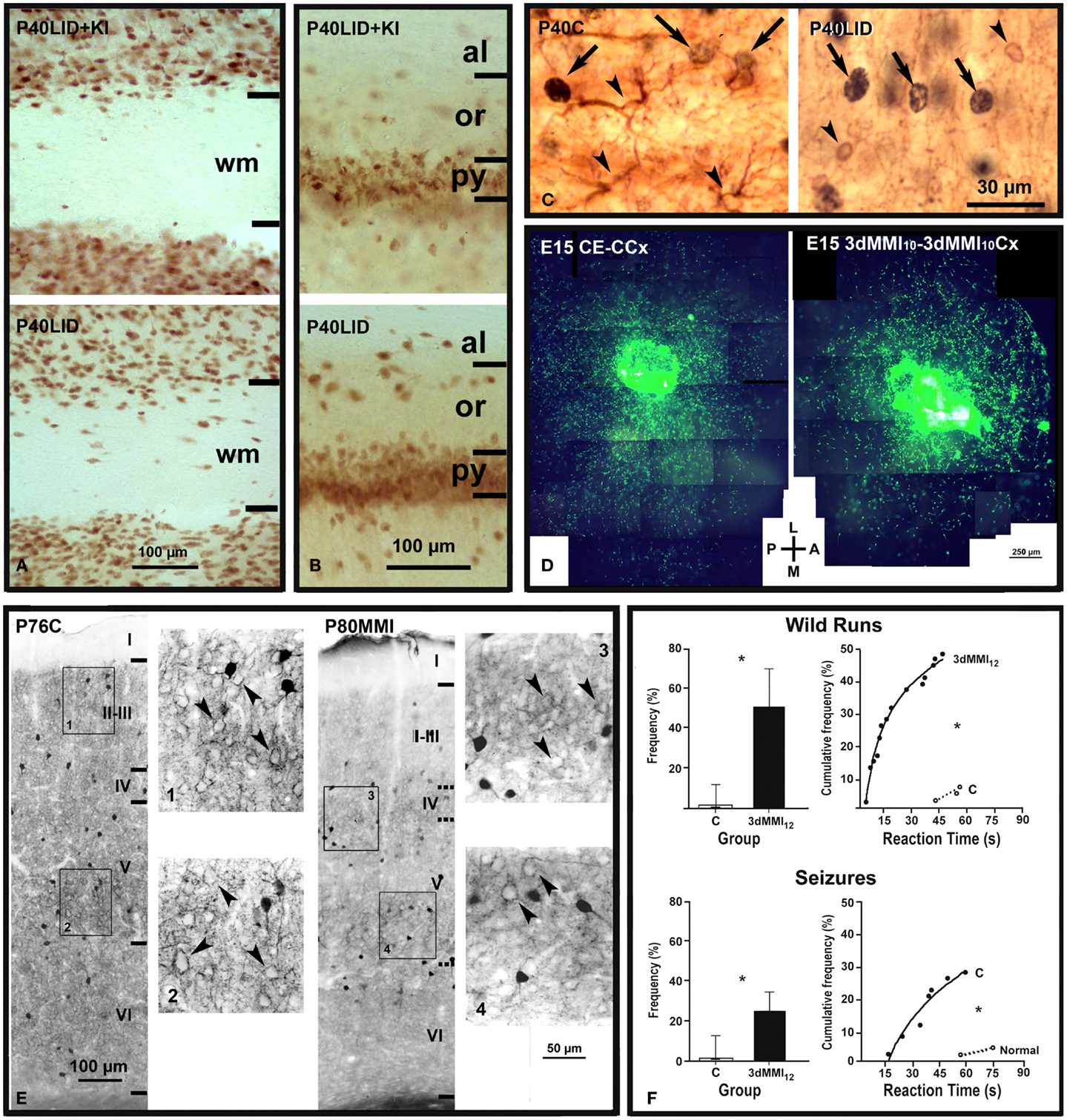
Figure 2. Abnormal neuronal radial and tangential migration, inhibitory local circuits and increased audiogenic seizures in developmental hypothyroidism and hypothyroxinemia. (A,B) Photomicrographs of NeuN-immunostained coronal sections of the primary somatosensory cortex (A) and hippocampal CA1 (B) in LID + KI (rats fed low iodine diet plus approximately 10 μg iodine per day, during gestation and postnatally) and LID (rats fed low iodine diet) progeny at P40. The number of NeuN-labeled neurons increases both in subcortical white matter [wm (A)] and in strata oriens (or) and alveus [al (B)] of hippocampal CA1 of LID pups as compared with LID + KI pups. (C) GFAP- and CNP-positive astrocytes (left panel) and oligodendrocytes (right panel), respectively (arrowheads), and BrdU-positive nuclei (arrows) are shown in layer V of C and LID pups. LID rats received single BrdU injections at E14, E15, and E16. Note that both GFAP-positive astrocytes and CNP-positive oligodendrocytes are BrdU-negative. (D) Low power fluorescent photo- micrograph collage illustrating the tangential distribution of GFP-MGE control migrating neurons (control explant, CE) in wild control flat cortical mounts at E15 (control cortex, CCx; left), and GFP-MGE hypothyroxinemic migrating neurons (3dMMI10) in hypothyroxinemic flat cortical mounts (3dMMI10Cx; right). Note that migrating neurons toward the medial (M) region in the hypothyroxinemic cortical mount (right) expand less than those migrating in the control cortical mount (left). 3dMMI10 rats received MMI treatment from E10 to E12. (E) Photomicrographs through layer V of the auditory cortex immunostained for parvalbumin in normal (C) and hypothyroid (MMI treatment from E14 onward) rats. In normal rats, immunoreactive cells, processes and perisomatic puncta can be seen. In MMI rats, immunoreactive cells, processes and perisomatic puncta can also be seen but they are less prominent than in normal rats. (F) Responses of C and 3dMMI12 (MMI treatment from E12 to E15) pups to an acoustic stimulus. Histograms on the left correspond to the proportion (median with 25th and 75th percentiles) of pups responding with wild runs and with wild runs followed by a seizure, respectively. Graphs on the right represent the cumulative frequency of pups from the same groups that respond with wild runs alone or followed by a seizure, respectively, at the intervals after onset of the stimulus that are shown in the abscissa. (*) Indicates a statistically significant difference compared with control. (A–C) Modified from Lavado-Autric et al. (164). (D) Modified from Cuevas et al. (159). (E) Modified from Berbel et al. (211). (F) Modified from Ausó et al. (158).
Heterotopic cells in the external granular layer of the cerebellar cortex have also been observed (212), as well as in Mct8−/y mice (213). The stunted migration of cerebellar cells found in previous studies (6, 214, 215) and in TRα1 mutant mice (212, 213) suggests that thyroid hormones interfere with different mechanisms involved in the migration of cortical and cerebellar neurons. Cortical neurons retain most of their migratory capacity as can be observed either in studies combining BrdU and tracer labeling (205) or using organotypic cultures (Figure 2D) (159). In the latter, it was found that cells from transient hypothyroid medial ganglionic eminence explants migrate as well as cells from control explants when they were placed on normal host cortex; and reversely, both control and transient hypothyroid median ganglionic eminence cells showed altered latero-medial migration when placed on transient hypothyroid host cortex, which suggests that in the transient hypothyroid cortex the expression of chemo-attractive/-repulsive/-stop signals and/or of their receptors [see review in Ref. (126)] is altered. In fact, some of them, such as Slit1, Slit2, and Sema3B, are regulated by thyroid hormones [Ref. (44); Table 2].
Abnormal Cortical Cytoarchitecture and Connectivity
Blurred neocortical layering can be assessed in the rodent somatosensory barrel cortex owing to the characteristic cytoarchitecture of layer IV (192, 216). The parvalbumin immunostaining pattern in hypothyroid rats is severely altered in the neocortex (211, 217, 218) (Figure 2E) and hippocampus (217, 219). Interestingly, parvalbumin positive neurons (i.e., GABAergic chandelier and basket neurons that migrate tangentially from the medial ganglionic eminence) also exhibit altered tangential migration in the transient hypothyroxinemic cortex (159). The decreased chandelier and basket parvalbumin immunoreactive terminals in the neocortex (211, 217) and hippocampus (217) will affect the inhibitory control of glutamatergic neurons (220) and might explain the high incidence of audiogenic seizures reported in hypothyroid rats (221) and in the pups of mild and transient hypothyroxinemic pregnant rats (158) (Figure 2F).
Early postnatal hypothyroidism affects the growth of dendrites in both the cerebral (193, 222) and cerebellar (223) cortices. Qualitative and quantitative ultrastructural studies of the cerebellar molecular layer in rats show that the retardation in synaptogenesis between Purkinje cell dendritic spines and parallel fibers was associated to hypoplasia of Purkinje cell dendrites and to the retarded development of parallel fibers (223). In the neocortex, it has been found that β-catenin is downexpressed in the dentate gyrus of postnatal hypothyroid rats (165) and the Wnt/β-catenin signaling plays a crucial role for the growth and branching of dendrites (224). Developmental hypothyroidism affects maturation of commissural axons (225–228). In adult hypothyroid rats, the number of myelinated axons was 76 and 66%, respectively, in both the anterior commissure and the corpus callosum compared to controls (228); also, the maturation of cytoskeletal components was altered (226, 229) and the growth of axon caliber was arrested (225, 228). Development and maturation of oligodendrocytes in the forebrain commissures of hypothyroid rats may also be affected. In fact, cortical expression of myelin-associated glycoprotein, proteolipid protein, and myelin basic protein in oligodendrocytes is strongly reduced (230).
Callosal-projecting neurons were found mostly in infra-granular layers of the auditory cortex of developing (205) and adult hypothyroid rats (216). In addition to altered radial distribution, the total number of callosal neurons was increased in auditory (216) and visual (226) cortices, and in cortical projecting neurons such as in the occipito-spinal connections (231), revealing maintenance of exuberant projections in hypothyroid rats. Interestingly, in the hypothyroid MMI model, the heterotopic white matter neurons, in particular, the early BrdU-labeled ones normally destined for the subplate, could provide a target to the transient callosal axons as they might in normal development (134, 232).
Delayed Cortical Maturation
There is a strong evidence that subplate neurons play an important role in thalamocortical axon path finding (233, 234). Subplate neurons may fire action potentials (235) and they are necessary for the establishment of ocular dominance and orientation columns (236) and for the maturation of inhibitory circuits in layer IV (237). The dynamic integration of subplate neurons into the rodent neocortex during postnatal development may play a key role in establishing the cytoarchitectonic pattern in layer IV and to refine layer IV circuitry (238). Recent studies have shown that subplate neurons remain expressing Camk4 in adult hypothyroid rats, while in normal rats, Camk4 is not longer expressed in subplate neurons by P10 [Ref. (239); Figures 3A–C]. Subplate and white matter abnormalities have been related to the pathogenesis of various brain developmental disorders other than ASD, such as periventricular leukomalacia, schizophrenia, and cerebral palsy (240–244). A recent study shows the crucial importance of the identification of subplate cell subpopulations, which may have very different roles in various pathologies such as ASD and schizophrenia (245).
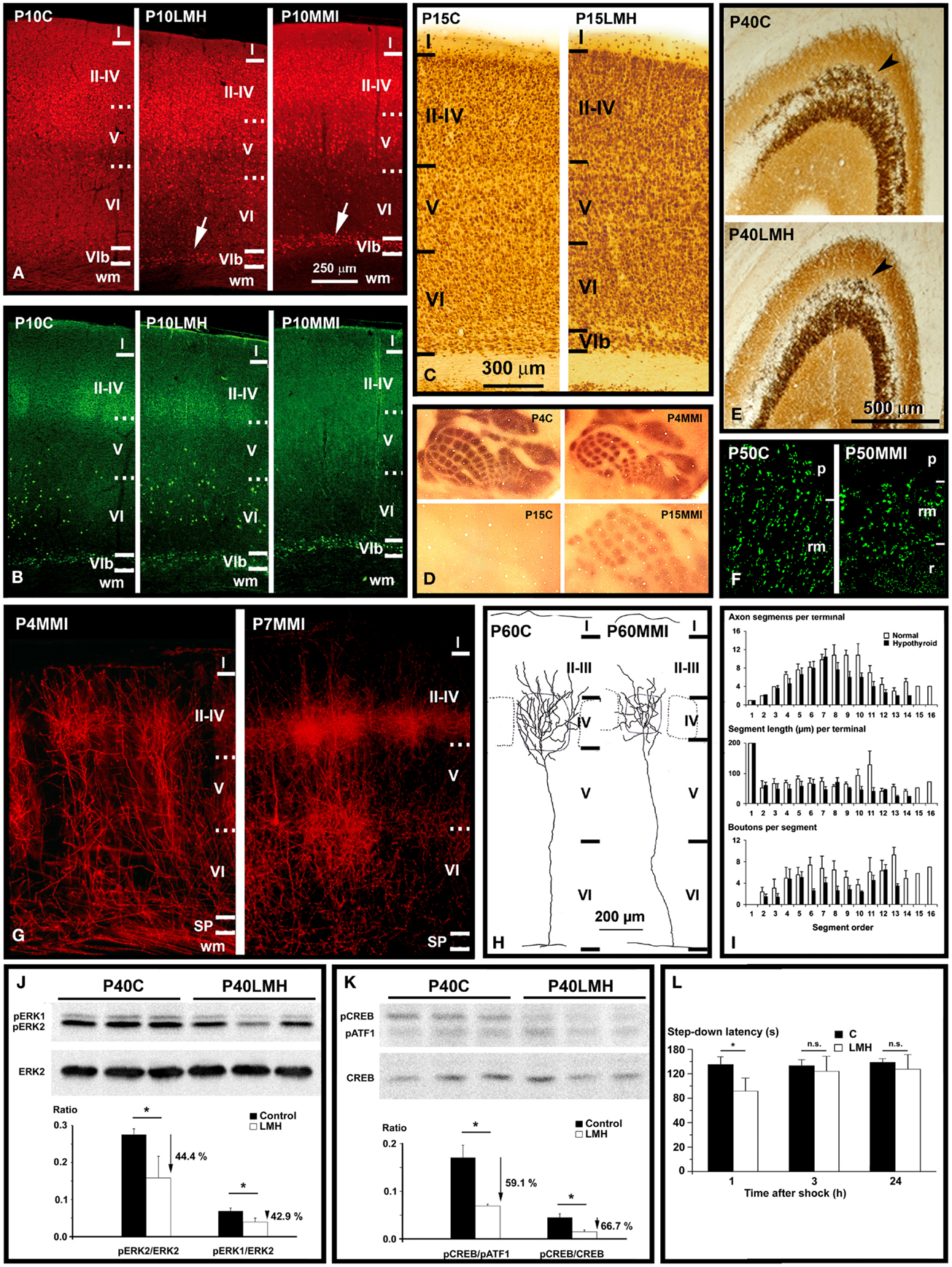
Figure 3. Thyroid hormones affect connectivity, maturation, and function of the cerebral cortex. (A,B) Collages from confocal photo- micrographs (taken with the ×20 objective) showing double immunolabeling for Camk4 [red; (A)] and Nurr1 [green; (B)] in the parietal cortex of P10 control (C), LMH (pups born to dams thyroidectomized at E16), and MMI (MMI treatment starting at E10) pups. Most of Camk4-immunoreactive neurons were located in layers II–IV and upper layer V. Numerous Camk4-immunoreactive neurons can be seen in layer VIb of P10LMH and P10MMI pups (arrows) compared with P10C pups. At P10, about 60% of Camk4-immunoreactive neurons of layer VIb are also Nurr1-immunoreactive. (C) Photomicrographs of coronal sections of the parietal cortex showing NeuN-immunoreactive neurons in C and LMH pups at P15. At P15, the border between the subplate and adjacent layer VI is more clear-cut in LMH than in C pups, showing that a remaining subplate is still present. (D) Photomicrographs of flattened neocortex tangential sections showing 5-HT immunostaining in the posteromedial barrel subfield of the parietal cortex of C and MMI rats at P4 and P15. At P4, heavily immunostained barrels can be seen. Decay of 5-HT labeling occurs by P11 in C and by P16 in MMI rats. Note that in P15C rats, no barrels were immunostained, whereas in P15MMI rats, they are still immunopositive. (E) Photomicrographs of coronal sections of the area CA3 of the hippocampus showing the Zn-labeling of mossy fibers in C and LMH pups at P40. Note the heavier labeling of mossy fibers in the stratum oriens (arrowhead) of CA3 in C compared with LMH pups. (F) Confocal deconvoluted images of vesicular glutamate transporter type 1 (VGluT1, which labels excitatory buttons) of the stratum radiatum (rm) in CA3 in C and MMI rats at P50, labeling mossy fiber buttons. Note the decreased density of immunoreactive buttons in P50MMI pups compared to controls. (G) Photomicrographs of coronal sections from P4 and P7 MMI rats that had the lipophilic carbocyanine DiI tracer (1,1′-dioctadecyl-3,3,3′,3′- tetramethylindocarbocyanine perchlorate) implanted in the ventro-basal thalamic nucleus. At P4, DiI-labeled thalamic afferents enter the somatosensory cortex, and form clusters in layer IV. At P7, collaterals in layer IV form more dense clusters than at P4. These images show that hypothyroid thalamic axons reach their somatosensory target areas as in normal rats. (H) Coronal views of thalamocortical terminal arbors in layer IV in the posteromedial barrel subfield of C and MMI rats at P60. The barrel limit is marked with dashed lines. Note that in MMI rats terminal arbors have shorter and tortuous branches. (I) Histograms representing mean values for the number of axon segments per terminal (upper), segment length (middle), and buttons per segment (bottom) in C (white bars) and MMI (black bars) rats for each segment order. Note that in general MMI mean values are lower. (J,K) Western blots obtained from the hippocampus of C and LMH pups at P40, immunolabeled for ERK2, pERK1, and pERK2 (J) and pATF1, pCREB, and CREB (K). Histograms showing that the pERK1/ERK2 and pERK2/ERK2 (J) and pCREB/pATF1 and pCREB/CREB (K) ratios are reduced by 44.4, 42.9, 59.1, and 66.7%, respectively, in LMH compared with C pups. (L) Histogram showing step-down latencies in seconds at 1, 3, and 24 h after the initial foot shock in C and LMH pups at P39. Pups from LMH dams show 24.9% reduction in the step-down latency at 1 h after the foot shock. (J–L) Error bars represent ± SD; n.s., no significant differences; *P < 0.001 for LMH compared with C group. (A,B) Modified from Navarro et al. (239). (C,E,J,K,L) Modified from Berbel et al. (169). (D,G,H,I) Modified from Ausó et al. (246).
Serotonin (5-HT) immunostaining is a good transient marker for thalamic afferents in the visual, auditory, and somatosensory areas of rats during the first postnatal days. In the barrel cortex of hypothyroid rats, immunolabeling persisted for 5 days until P16–17 [Ref. (246); Figure 3D]. A similar protracted expression of 5-HT transporter (5-HTT) occurred in the ventro-basal thalamic nucleus and cerebral cortex (246). Reduced 5-HT levels during barrel formation delay the differentiation of layers II, III (247, 248) and reduce the tangential extent of thalamocortical arbors within barrels (249). Thus, prolonged 5-HTT expression in the hypothyroid ventro-basal thalamic nucleus should decrease the concentrations of 5-HT in the extracellular space of sensory cortices, affecting their organization and differentiation.
In the barrel cortex of adult hypothyroid rats, the radial distribution of thalamic afferents, anterogradely labeled with dextran–biotin amine and DiI, was reduced compared to normal rats [Ref. (246); Figure 3G]. By single reconstructions of terminal arbors (Figure 3H), these authors showed a reduction of the number of axonal branches reaching layers II–IV, and a 49% reduction in the total length of terminal axon arbors in hypothyroid rats. This arrested growth was also reflected by a 58% reduction in the number of buttons per terminal (Figure 3I). In hypothyroid rats, ramification of the thalamocortical axons would appear to be stalled postnatally, resulting in reduced synaptogenesis as suggested by the reduced number of buttons in thalamocortical axons [Ref. (246); Figure 3I] and in a decreased number of spines along the apical shafts of the hypothyroid pyramidal cells (250). All of the above data show that many target pyramidal cells fail to reach their correct cortical location, not only failing to complete their normal maturation but also that the afferents have arrested growth. In fact, GAP-43 is downregulated, while Sema3A is upregulated in developmentally hypothyroid and hypothyroxinemic pups (251). In agreement, GAP-43−/− mice failed to express 5-HTT in the barrel cortex causing a disrupted segregation of thalamic afferents in the barrel cortex. In addition, recent data show that the density of VGluT1-immunoreactive buttons is decreased in layer IV of the parietal cortex of hypothyroid rats (180). These data show that there is an asynchrony in the maturation of thalamocortical afferents and their cortical targets in hypothyroid rats. Cortical cells could be at a stage of maturation that does not allow them to respond to thalamocortical signals, resulting in abnormal communication between thalamic axons and target cells (e.g., by reduced synaptogenesis). Hypothyroidism seems to dissociate stabilization of juvenile axons from maturation, growth in caliber and myelination, processes, which were previously thought to be necessarily linked (134, 232, 252).
Abnormal patterns of connectivity have been also found in the hippocampus of developmentally hypothyroid rats (193). These authors found in the hippocampus of pups born to hypothyroid dams that CA pyramidal neurons developed atrophic apical (15% shorter) and fewer number of ramifications (about 31 and 36% less in dentate gyrus and CA, respectively). Blurred layering and heterotopic neurons were also found in the hippocampus of pups born to hypothyroxinemic pregnant rats [Ref. (158, 164); Figure 2B]. Decreased mossy fiber zinc density (33–45% reduction) was found in perinatal hypothyroid rats after PTU treatment from E18 to P31 (253) and in postnatal hypothyroid rats (254). P40 pups born to late hypothyroid dams (thyroidectomized by E16; LMH pups), showed a 41.5% decrease in the Zn-positive area in the stratum oriens, in parallel to down expression of the Zn transporter-3 (ZnT-3; Figure 3E) and reduced density of VGluT1-immunoreactive buttons (Figure 3F). In addition, pCreb/pATF1, pCreb/Creb, pErk1/Erk2, and pErk2/Erk2 ratios in the hippocampus decreased in LMH pups (59.1, 66.7, 44.4, and 42.9%, respectively) [Ref. (169); Figures 3J,K]. Recently, the hippocampus of developmentally hypothyroid pups showed altered VGluT1/VGAT immunoreactivity (180). Although Camk4/Creb pathway plays a fundamental role in neurites growth and establishment of synapses, other genes are involved in the development of hippocampal connections. It has been found that the T3-regulated BDNF is involved in the regulation of the translational expression of VGluT1 in cultured hippocampal neurons (255, 256). Interestingly, BDNF was also found involved in the activation of Erk1/2 signaling pathway (188), which affects not only the differentiation of hippocampal neurons but also almost all aspects of corticogenesis (Tables 3–6). However, the inactivation of other pathways such as the Gsk3β/CRMP2 pathway will result in delayed axonal growth (251, 257). Decreased p-Gsk3β labeling and increased labeling of its inactivated p-CRMP2 target protein was seen in the developmentally hypothyroid and hypothyroxinemic rat hippocampus (251).
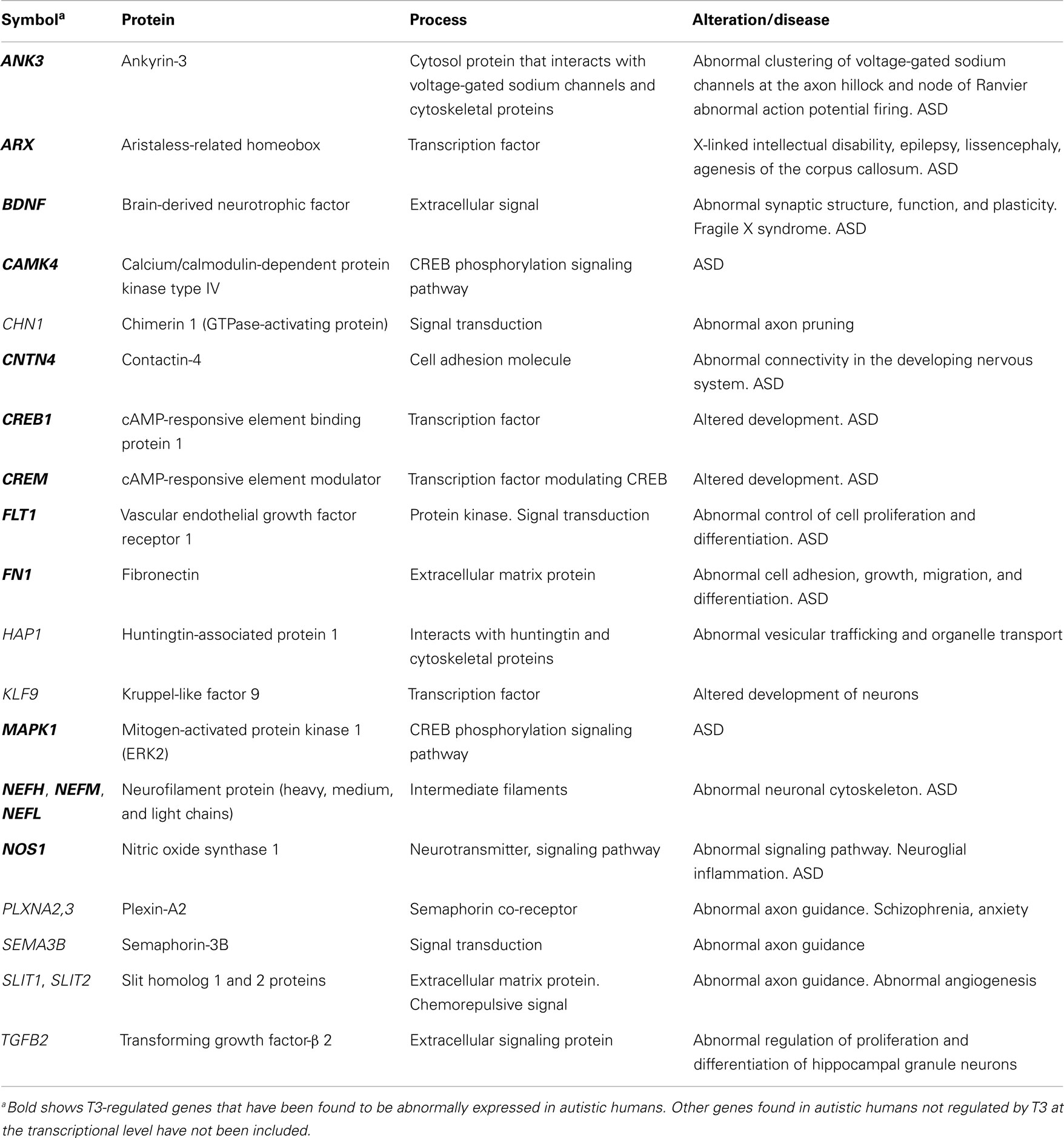
Table 3. Significant T3-regulated genes at the transcriptional level found in the cerebral cortex of rodents, involved in neurite growth, guidance, branching, and maturation: relationship with ASD.
Altered postnatal synaptogenesis has been observed in the molecular layer of the cerebellar cortex of hypo- and hyperthyroid rats (214). Neurotrophins BDNF and NT3 are downexpressed in postnatal PTU treated rats, resulting in atrophy of Purkinje cell dendrites and in a decreased number of synapses (258) and BDNF is also downexpressed in the hippocampus of developing hypothyroid rats (256). However, neurotrophins might have a dual role in developing and adult hypothyroid rats, because, BDNF is highly expressed in layers II, III, and V of the neocortex and in all hippocampal areas of adult hypothyroid rats (259). These authors observed an increased number of apoptotic neurons and astrocytes in the adult hypothyroid cortex, and they suggested that the increase of BDNF in the hypothyroid adult neocortex and hippocampus might have a protective role against cellular stress associated to degenerative processes. Interestingly, in young and adult autistic human cerebellar samples (ages ranging from 7.5 to 34 years) a 40.3% increase in NT3 expression was found (260). Other T3-regulated genes, involved in synaptogenesis, plasticity, and neurotransmission, have been found [Ref. (23, 44); Tables 4 and 5]. The T3-regulated ANXA6 gene codes for the calcium-binding protein annexin 6 that may play a structural role in facilitating membrane association at the cisternal organelle level and/or in stabilizing IP3R1 microdomains in the axonal initial segment of hippocampal neurons (261). NR4A1 codes for the transcription factor Nurr77 that mediates in mechanisms of long-term synaptic plasticity in the hippocampus and consequently, in the consolidation of long-term hippocampus-dependent memory (262). PACSIN2 codes for protein kinase C and casein kinase substrate in neurons protein 2, which interacts with dynamin and synapsin, which affect the recruitment of synaptic vesicles (263), while TGB2 codes for transforming growth factor-beta 2, which is involved in the differentiation of granule neurons of the dentate gyrus (188).
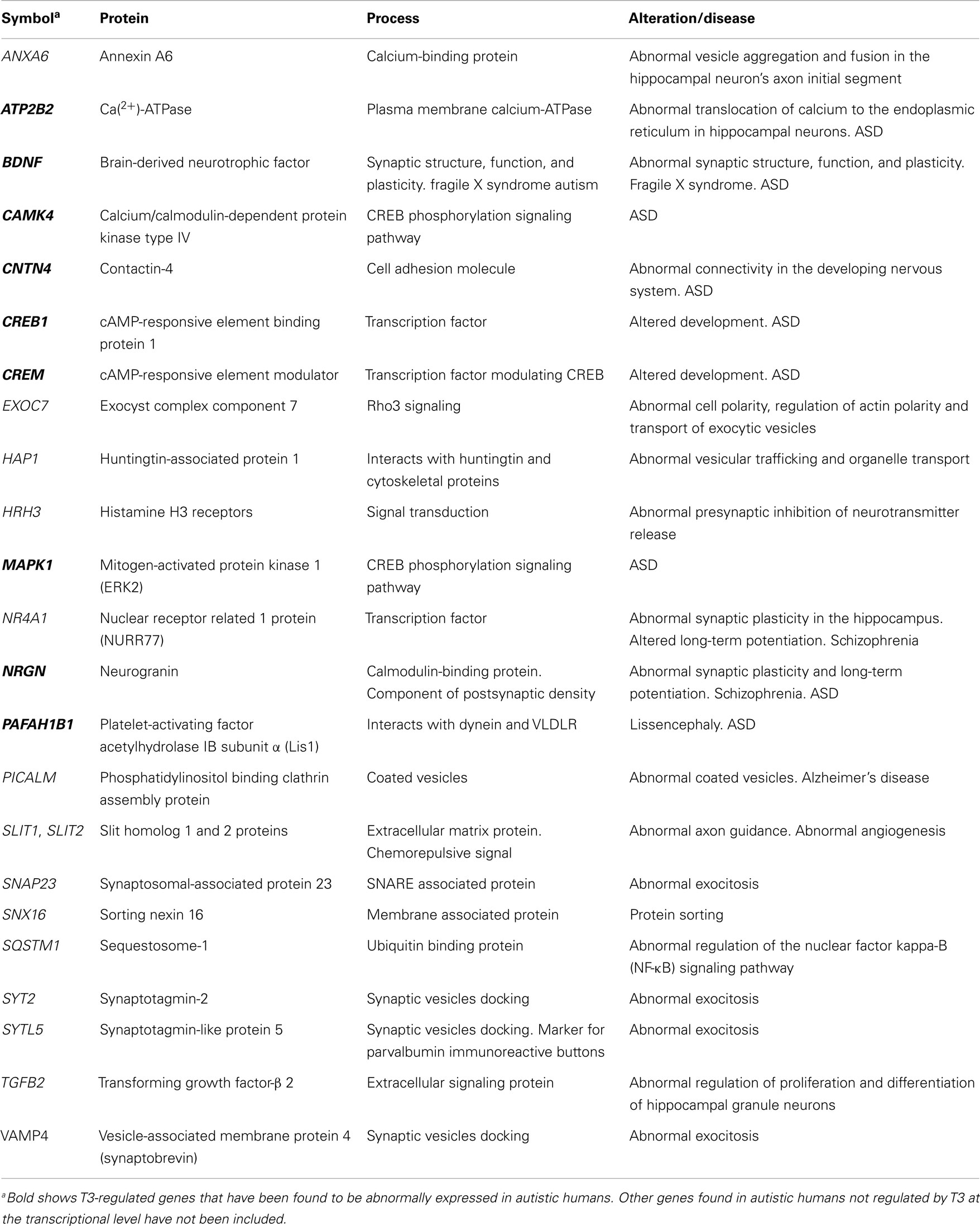
Table 4. Significant T3-regulated genes at the transcriptional level found in the cerebral cortex of rodents, involved in synaptogenesis and plasticity: relationship with ASD.
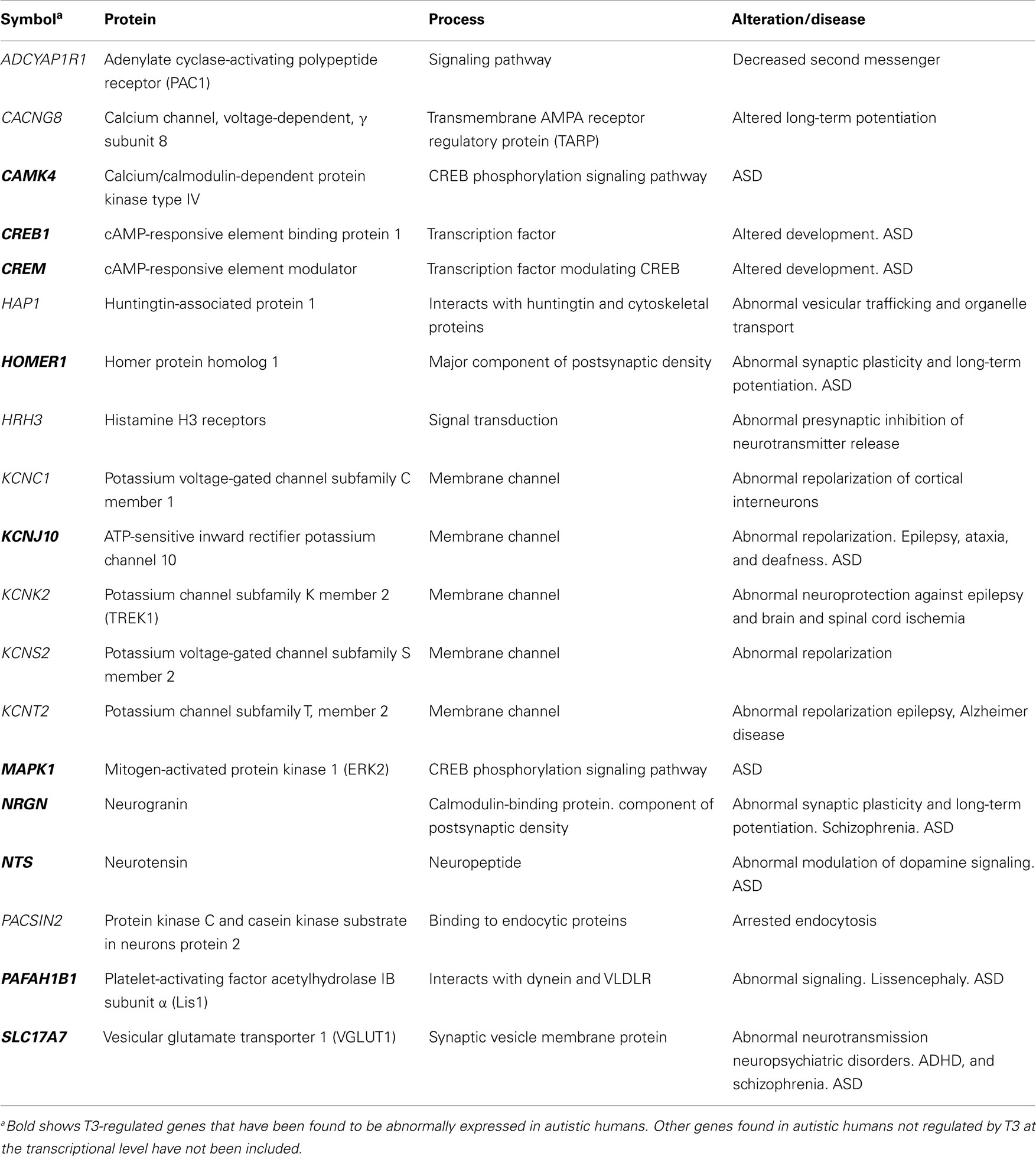
Table 5. Significant T3-regulated genes at the transcriptional level found in the cerebral cortex of rodents, involved in neurotransmission: relationship with ASD.
Structural changes in the cerebral cortex result in altered electrophysiology and behavior of thyroid deficient rats. Most of the electrophysiological studies have been performed in the CA1 hippocampal area. Decreased long-term potentiation (LTP) was shown in developing rats with severe and chronic hypothyroidism (166, 264), as well as in adult hypothyroid rats (265). Altered LTP was also observed in pups born to rats treated with MMI from E12 to E15 (266). Recent studies have shown that developmental hypothyroidism decreases the number of bursting CA1 cells, as well as the number of spikes per burst, resulting from altered low-threshold Ca2+ current (267). As mentioned above, the growth of axon caliber was arrested in the anterior commissure and the corpus callosum of hypothyroid rats (225, 228), which might be relevant for the signal transmission velocity of commissural axons (268). The behavior of hypothyroid rats related to cerebral cortex alterations are mainly based on tests to measure (i) locomotor functional excitability and seizure susceptibility (158, 218, 221, 269), and (ii) learning, attention, and memory deficits (169, 219, 264, 266, 270, 271) (Figure 3L). Despite the differences between rodent and human cerebral cortices, these studies might be useful to find morphofunctional alterations in hypothyroid rodents that might also occur in neurological and mental disorders associated to hypothyroidism in humans, such as ASD and ADHD. Significant T3-regulated genes, involved in memory and behavior have been found [Ref. (23, 44); Table 6]. Among these, ADCY8, ADRBK2, and GRK5 code for proteins associated to G-protein signaling (272–274). The transcription factors DBP, involved in kainite-involved seizures and hippocampal plasticity (275), and NR2A1 were mentioned above.
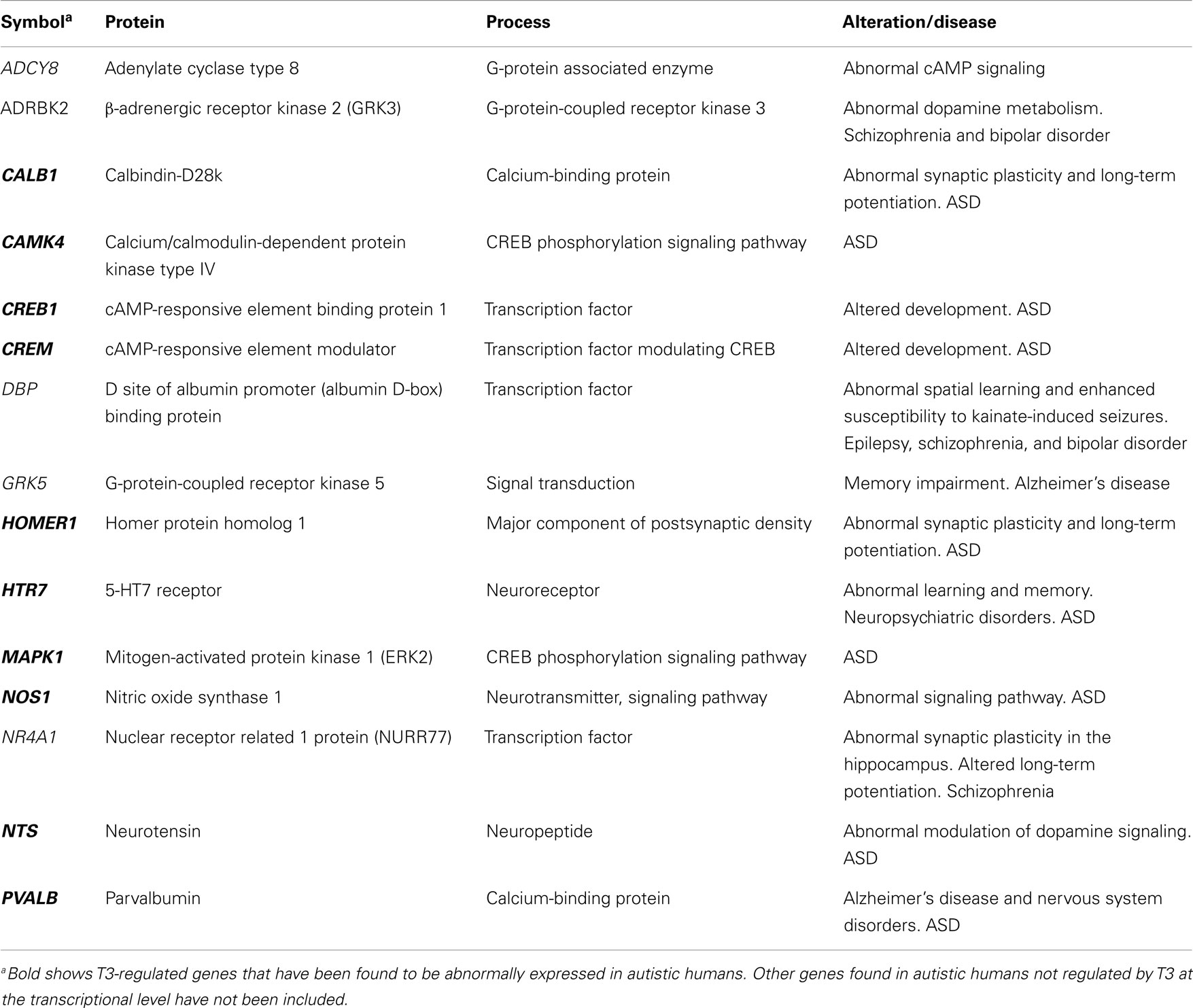
Table 6. Significant T3-regulated genes at the transcriptional level found in the cerebral cortex of rodents, involved in memory and behavior: relationship with ASD.
ASD and Thyroid Hormones during Brain Development
Experimental studies in rodents clearly show that thyroid hormone deficiency results in delayed, temporarily, or permanently suppressed, or abnormal, connections, resulting in behavioral and brain dysfunction (10). Abnormal morphophysiological and behavioral traits established during gestation and early postnatal ages might be maintained throughout life and thereby be a risk factor for the development of behavioral and mental disorders later in life.
Despite differences resulting from age at autopsy and the concomitant effects of seizures and of intellectual disability of variable severity, a substantial body of evidence has accumulated since the 1970s on the fundamental morphological changes affecting the brain of patients with ASD (276, 277). Wegiel et al. (278) reviewed available neuropathological data and concluded that ASD results from dysregulation of the normal mechanisms of neurogenesis and neuronal migration, plus dysplastic changes and defects of neuronal maturation. Thus, the neuropathology of ASD is consistent with a prenatal time of onset. A likely etiological hypothesis posits that ASD may be caused by thyroid hormone deficiencies during cerebral cortex development, either due to a genetic deficiency of the TRIP8 gene (thyroid receptor interacting protein), which codes for a transcriptional regulator associated with nuclear thyroid hormone receptors (279) or associated to maternal hypothyroidism, which increases fourfold the risk of ASD in the child (3, 63).
The morphological brain changes and the genes found to be transcriptionally or functionally involved in ASD will be briefly reviewed here, both from post-mortem data and from in vivo imaging, along with a summary of the neurotransmitters affected. Finally, the relevance of thyroid hormones in accepted animal models of ASD is presented. Thyroid hormone deficiency diseases and ASD share common altered gene pathways and comorbid disorders, and epidemiological studies reported a relationship between thyroid hormone deficiency and ASD, although morphofunctional differences between these two conditions exist.
Brain Alterations
Young children with ASD are megalencephalic, with increased brain size and weight (276, 277). Brain imaging data and head circumference studies have shown two phases of early brain growth in ASD pathology: early brain overgrowth during the first postnatal years and arrest of growth during early childhood (280), which might be overlapped with neuronal degeneration in some brain regions by preadolescence and continued into adulthood (280, 281). Increased number of neurons could contribute to increase brain volume. Macroscopically and on imaging, the cerebral cortex in ASD exhibits an abnormal pattern of convolutions (282) involving the orbitofrontal cortex (283) and the temporal lobes with hyperconvoluted hippocampus (276). Counts performed in Nissl stained sections suggests increased density of cells in the frontal cortex (284). However, other factors, besides of the increase in the number of neurons, can contribute to increase brain volume. Increased cerebrospinal fluid volume, and slight reductions of gray and white matter volume in frontal, temporal, and parietal lobes have been reported (285). In addition, recent studies have shown focal brain inflammation (286, 287) and increased gliosis subjacent to neuronal degeneration (281). Changes do not affect the brain uniformly, i.e., the fusiform face area and the limbic system have increased cell packing density and smaller neuronal size involving hippocampus, subiculum, and amygdala, and to a lesser extent the entorhinal cortex, mammillary bodies, and septal nuclei (277), while other areas are normal; for instance, the posteroinferior occipitotemporal gyrus showed no differences in pyramidal neuron number or size in layers III, V, and VI (288). In Brodmann areas 44–45, Jacot-Descombes et al. (289) demonstrated reduced pyramidal neuron size suggesting impairment of neuronal networks relevant to communication and social behaviors. However, owing to the relatively small number of autistic brains studied up to date and the enormous heterogeneity in ASD phenotypes and comorbid diseases, more neuropathological studies will be need for clarification of neuroanatomy of ASD (107, 290).
Despite changes in brain volume in ASD, some anatomical alterations are common with hypothyroid brains. Microscopic examination reveals dysgenesis of the cerebral cortex (276, 277) with increased cortical thickness, abnormal laminar patterns, high density of hippocampal neurons, presence of neurons in the molecular layer, neuronal disorganization, poor differentiation of the gray–white matter boundary, and neuronal heterotopias. Cortical neurons are small, closely packed, lack dendritic arbors, and appear immature; these changes are consistent with an arrest of cerebral maturation (290, 291). Also, the cortical organization is altered with narrower cortical minicolumns (292, 293). The focal cortical dysplasia of ASD appears to result from loss of synchronized radial and tangential migration of glutamatergic and GABAergic neurons, respectively (294). The CNTNAP2 gene, which codes for contactin associated protein-like 2, is expressed in human frontal areas and has been found to be involved in ASD and language impairment (295, 296). A finding consistent with this view is the demonstration by Kotagiri et al. (297) of cytoarchitectural changes in the ependymal cells of the subventricular zone in ASD, with lower cell density in the septal but not in the striatal zone. A subset of ependymal, astrocyte ribbon, and rostral migratory stream (RMS) cells expressed PCNA, Ki67, PLP, and α-tubulin. In addition, the white matter shows areas of focal increase in the number of heterotopias, reflecting abnormal neuronal migration (278). Using imaging, Gozzi et al. (298) showed that the magnetization transfer ratio of the corpus callosum was significantly higher in children with ASD than in normal controls, indicating abnormal myelination in ASD.
According to a consensus by Fatemi et al. (299), reduction in Purkinje cell and cerebellar granule cell density is consistently observed in ASD (300), along with developmental abnormalities of the inferior olives (301), consistent with abnormal neuronal migration before the 3rd month of gestation. Purkinje cells are decreased in the posterolateral neocerebellar cortex and the archicerebellar cortex (302) with vermis hypoplasia on brain imaging (303, 304). Using MRI tractography in children with ASD, Jeong et al. (305) showed decreased fiber numbers connecting cerebellar cortex to ventral and dorsal dentate nuclei confirming a decrease in connectivity and numbers of Purkinje cells.
Neurotransmitters in ASD
Perry et al. (306) investigated cholinergic biomarkers in the basal forebrain, frontal cortex, and parietal cortex of children with ASD, mental retardation, and epilepsy and found decreased binding of the α4 nicotinic and the muscarinic M1 receptors (α4 nAChR and m1AChR, respectively). In the cerebellum, Lee et al. (307) found decreased α3 and α4 nAChR binding in granule cells, Purkinje cells, and molecular layers along with increased α7 nAChR binding in the granule cell layer. Blatt et al. (308) found that only the GABAergic system was significantly reduced in the hippocampus in ASD; the serotoninergic, cholinergic, and glutamatergic systems were normal. GABAA and GABAB receptor density in the anterior cingulate cortex and fusiform gyrus is decreased (309, 310). The dysregulation of the GABAergic system pathway includes downregulation of GABAA and GABAB receptors (309–311) and reduction of glutamic acid decarboxylase enzymes (312) and metabotropic glutamate receptor type 5 [mGluR5; (313)]. FMRP and mGluR5 are reduced in cerebellar vermis and frontal cortex in ASD (314, 315). In addition, 5-HT neurotransmission has been found to be deficient in ASD; in particular, Oblak et al. (316) showed decrease in 5-HT1A receptor and 5-HT2A receptor-binding density, as well as in 5-HTT in posterior cingulate cortex and fusiform gyrus. Mutations in the GABAA receptor subunit have been associated with ASD and epilepsy (317).
Two relevant genes in the diagnosis of ASD are SHANK3 (318) and GABRB3 (311). SHANK3 is a synaptic scaffolding protein enriched in the postsynaptic density of excitatory synapses, and plays important roles in the formation, maturation, and maintenance of synapses. Several SHANK3 mutations have been identified in a particular phenotypic group of patients with ASD (318). A study of the Danish Newborn Screening Biobank revealed levels of BDNF in the lower 10th percentile during the neonatal period in children later diagnosed with ASD (319). SHANK3 mutations may be involved in ASD, cerebellar development, and cerebellar vermis hypoplasia (320). GABRB3 codes for GABAAβ3 receptor, and is downexpressed in brains of autistic children, particularly in the cerebellum (311).
Thyroid-Related Genes Involved in ASD
Recently, Betancur (321) concluded that despite the more than 100 genetic and genomic disorders associated with ASD, we still lack a clear understanding of its pathogenesis. In 2007, Castermans et al. (279) identified in a subject with ASD a de novo chromosomal anomaly on chromosome 10q21.3 that disrupted the TRIP8 gene and the nearby REEP3 gene that codes for receptor expression-enhancing protein 3, which is a microtubule associated protein sequestering the endoplasmic reticulum away from chromosomes during mitosis. The authors concluded that TRIP8 codes for a protein predicted to be a transcriptional regulator associated with nuclear thyroid hormone receptors but noted that, “no link between thyroid gland and ASD has been reported so far.”
We summarize in Tables 1–6 a list of relevant genes that have been found to be T3-regulated at the transcriptional level in the rodents cerebral cortex (149, 322), and their human homolog genes (marked in bold) that have been found mutated in ASD patients. The list is far to be exhaustive and, most probably, the overlapping between T3-regulated and ASD-mutated (T3/ASD) genes will increase in the near future. Relevant are Creb/Crem transcription factors that are involved in all critical events of corticogenesis, and Camk4 and Erk1/2 kinases that participate in the Camk4/Creb/Crem and Erk1/2/Creb/Crem signaling pathways (44, 183, 323–325).
As mentioned earlier, critical events at the beginning of corticogenesis are cell division and differentiation of neuroblasts to become young migrating neurons, and the migration of young neurons to their final destinations. T3/ASD genes involved in cell division and differentiation (Table 1) are CTNNB1 codes for β-catenin that is involved in the transition of epithelial-to-mesenchymal transition (symmetrical-to-asymmetrical divisions; see above) and in the astrocytes’ differentiation (326); DYRK1A codes for dual specificity tyrosine-phosphorylation-regulated kinase 1A, which is a regulator of brain growth (196); GNB1L code for a 6 WD40 repeats-containing protein most likely involved in cell cycle regulation (327); and FLT1 that codes for vascular endothelial growth factor receptor 1, a tyrosine kinase involved in the control of cell proliferation and differentiation in angiogenesis and neurogenesis; FLT1 has been found reduced in severe autism (328). T3/ASD genes involved in cytoskeleton organization and cell migration (Table 2) are GNAS that codes for G-protein α subunit (Gs-α) (329); FN1 that codes for fibronectin, an extracellular matrix protein involved in cell adhesion and migration, found increased in serum of children with autism (330); SERPINH1 that codes for heat shock protein 47 that binds collagen and was found abnormally expressed in the temporal cortex of ASD patients (331); and NEFH, NEFM, and NEFL code for neurofilament subunits and has been found altered in the frontal cortex neurons in children with autism (332). The genes involved in the reelin signaling pathway include RELN (reelin), DAB1 (disabled-1), VLDLR (very-low-density-lipoprotein receptor) (331–335), and PAFAH1B1 (platelet-activating factor acetylhydrolase IB subunit α; Lis1) that interacts with dimein and VLDLR. Fatemi et al. (312) reported decreased blood levels of reelin in children with ASD. Also, using post-mortem material from superior frontal, parietal, and cerebellar cortex from autistic brains and matched controls, significant reductions in reelin protein, reelin mRNA, and dab1 mRNA along with elevations in VLDLR mRNA in frontal and cerebellar cortex, indicative of impairments in the reelin/dab1 signaling pathway in ASD were observed (336). Genetic susceptibility polymorphisms of the RELN gene have been described in ASD (337–340), although other studies have been negative (341–344). A recent meta-analysis by Wang et al. (345) revealed that the RELN variant rs362691, rather than rs736707 or the GGC repeat variant, might contribute significantly to ASD risk.
The T3/ASD genes involved in neurite development and maturation (Table 3) are ANK3 that codes for ankyrin-3, which participates in the recruitment of voltage-gated sodium channels at the axon hillock and node of Ranvier (346); ARX that codes for the transcription factor Aristaless-Related Homeobox, associated to several neurological and psychiatric disorders, including ASD (347, 348); BDNF has high-affinity for TrkB receptor and is involved in neurite development, neuronal plasticity, LTP, and apoptosis of CNS neurons (348, 349); CNTN4 codes for contactin-4, an Ig-cell adhesion molecule involved in the development and plasticity of neuronal circuits (350); NOS1 codes for nitric oxide synthase 1 that is involved in glutamate-mediated neurotransmission and toxicity (351); FLT1, FN1, and NEFs were mentioned above. T3/ASD genes involved in synaptogenesis and plasticity (Table 4) are ATP2B2 that codes for plasma membrane calcium-ATPase, involved in the translocation of calcium to the endoplasmic reticulum (352); NRGN that codes for neurogranin, involved in synaptic plasticity and LTP (353); BDNF, CNTN4, and PAFAH1B1 mentioned above.
The T3/ASD genes involved in neurotransmission (Table 5) are HOMER1 that codes for homer protein homolog 1, is a major component of postsynaptic density involved in metabotropic glutamate receptor signaling (354); KCNJ10 that codes for ATP-sensitive inward rectifier potassium channel 10, involved in axonal membrane repolarization (355); NTS that codes for neurotensin is involved in modulation of dopamine signaling and focal brain inflammation, and was found increased in serum of ASD children (286); SLC17A7 codes for vesicular glutamate transporter 1 (VGluT1), and is involved in glutamatergic transmission (333); NRGN and PAFAH1B1 were mentioned above.
The T3/ASD genes involved in memory and behavior (Table 6) are CALB1 and PVALB that encode calbindin-D28k and parvalbumin, respectively, are involved in GABAergic transmission (332); HTR7 that codes 5-HT7 receptor is involved in serotonin signal transduction (333, 356); HOMER1, NOS1, and NTS were mentioned above.
Animal Models of ASD
A number of animal models of ASD are the result of insertion/deletion of different ASD-related genes and exposure to environmental factors [reviewed by Gadad et al. and Provenzano et al. (357, 358)]. Sadamatsu et al. (359) proposed the rat with mild and transient neonatal hypothyroidism as a novel model for ASD. Other models include the repetitive behavior observed in C58/J, C57BL/6J, and Grin1 knockdown mice (360). The homeobox-containing transcription factor engrailed-2 (En2) is involved in patterning and neuronal differentiation; Sgadò et al. (361, 362) showed that adult En2−/− mice exhibit reduced brain interneuron expression of GABAergic marker mRNAs, and reduction in parvalbumin, somatostatin, and neuropeptide Y in the cerebellum and cerebral cortex (including hippocampus). The genetically inbred BTBR T+Itpr3tf/J mouse model of ASD exhibits social impairment and stereotypic behavior suggestive of mTOR overactivation (363). The BTBR model shows extensive anatomical abnormalities in the white matter of the corpus callosum and the hippocampal commissure (364). Uchino and Waga (365) identified novel SHANK3 transcripts whose transcription started at the vicinity of the CpG-island 2 in the mouse brain and developed the Shank3 mutant mice that exhibit autistic-like behaviors. Waga et al. (366) identified two different amino-terminus truncated Shank3 transcripts, Shank3c-3 and Shank3c-4, expressed from the intron 10 of the Shank3 gene, and suggested the epigenetic regulation of the expression of these transcripts via methyl CpG-binding protein 2 (MeCP2). Interestingly, MeCP2 mediates activity-dependent regulation of synaptic strength during the process of circuit formation and prevents uncontrolled recurrent excitation that may result in a pathophysiological increase of neuronal excitability, aberrant network activity, and seizures, which are common Rett patients (182).
The valproic acid model of ASD has become widely used (367–371). However, it is not widely known that valproic acid at the usual therapeutic doses used for the treatment of epilepsy has anti-thyroid effects (372) and induces hearing loss in patients (373).
Conclusion
Thyroid hormones exert both genomic and non-genomic actions in many tissues, organs, and systems over the course of a lifetime. In particular, they are crucial during early neurodevelopment, since key phases of the CNS development depend of the expression of thyroid hormones regulated genes. These genes affect, among other things, proliferation, migration, and maturation of neurons and glial cells, which under certain circumstances can result in abnormal connectivity, and consequently in behavioral dysfunction. Morphofunctional alterations caused during pregnancy and early postnatal are permanent, and thus they are a risk factor for the development of behavioral and mental disorders later in life. The knowledge of how thyroid hormones regulate these phases of development may help to understand altered regulatory mechanisms in neurodevelopmental diseases such as ASD, ADHD, schizophrenia, and epilepsy with cytoarchitectonic alterations similar to those found in hypothyroidism and hypothyroxinemia and vice versa. By combining basic and clinical investigation, new data will be obtained to better understand the basic phases of brain development and the genetic and physiological events underlying some of the human diseases mentioned above. Despite of obvious differences between humans and other mammals in cortical organization and function, animal models might be a useful tool to approach the understanding of common etiological factors in hypothyroidism and ASD since, as the evo-devo tell us, both rodents and humans share homologous gene pathways involved in these diseases.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
Supported by grants from the Spanish Ministerio de Ciencia e Innovación SAF2009-10689 and the Universidad Miguel Hernández Institutional Funding for Research to Pere Berbel, and from the Nancy Lurie Marks Family Foundation, Wellesley, MA, USA, to Gustavo C. Román.
References
1. De Robertis EM. Evo-devo: variations on ancestral themes. Cell (2008) 132:185–95. doi: 10.1016/j.cell.2008.01.003
2. Rakic P. Evolution of the neocortex: a perspective from developmental biology. Nat Rev Neurosci (2009) 10:724–35. doi:10.1038/nrn2719
3. Román GC, Ghassabian A, Bongers-Shokking JJ, Jaddoe VW, Hofman A, de Rijke YB, et al. Association of gestational maternal hypothyroxinemia and increased autism risk. Ann Neurol (2013) 74:733–42. doi:10.1002/ana.23976
4. Stiles J. Brain development and the nature versus nurture debate. Prog Brain Res (2011) 189:3–22. doi:10.1016/B978-0-444-53884-0.00015-4
5. Román GC. Nutritional disorders in tropical neurology. Handb Clin Neurol (2013) 114:381–404. doi:10.1016/B978-0-444-53490-3.00030-3
6. Legrand J. Hormones thyroïdiennes et maturation du système nerveux. J Physiol (Paris) (1983) 78:603–52.
7. Morreale de Escobar G, Escobar del Rey F. Maternal thyroid deficiency during pregnancy and subsequent neuropsychological development of the child. N Engl J Med (1999) 341:2015–6. doi:10.1056/NEJM199912233412613
8. Haddow JE, Palomaki GE, Allan WC, Williams JR, Knight GJ, Gagnon J, et al. Maternal thyroid deficiency during pregnancy and subsequent neuropsychological development of the child. N Eng Med J (1999) 341:549–95. doi:10.1056/NEJM199908193410801
9. Zoeller RT, Rovet J. Timing of thyroid hormone action in the developing brain: clinical observations and experimental findings. J Neuroendocrinol (2004) 16:809–18. doi:10.1111/j.1365-2826.2004.01243.x
10. Berbel P, Obregón MJ, Bernal J, Escobar del Rey F, Morreale de Escobar G. Iodine supplementation during pregnancy: a public health challenge. Trends Endocrinol Metab (2007) 18:338–43. doi:10.1016/j.tem.2007.08.009
11. Berbel P, Bernal J. Hypothyroxinemia: a subclinical condition affecting neurodevelopment. Expert Rev Endocrinol Metab (2010) 5:563–75. doi:10.1586/eem.10.37
12. Ahmed OM, El-Gareib AW, El-Bakry AM, Abd El-Tawab SM, Ahmed RG. Thyroid hormones states and brain development interactions. Int J Dev Neurosci (2008) 26:147–209. doi:10.1016/j.ijdevneu.2007.09.011
13. Morreale de Escobar G, Ares S, Berbel P, Obregón MJ, Escobar del Rey F. The changing role of maternal thyroid hormone in fetal brain development. Semin Perinatol (2008) 32:380–6. doi:10.1053/j.semperi.2008.09.002
14. Williams GR. Neurodevelopmental and neurophysiological actions of thyroid hormone. J Neuroendocrinol (2008) 20:784–94. doi:10.1111/j.1365-2826.2008.01733.x
15. Stagnaro-Green A, Pearce E. Thyroid disorders in pregnancy. Nat Rev Endocrinol (2012) 8:650–8. doi:10.1038/nrendo.2012.171
16. Cheng SY, Leonard JL, Davis PJ. Molecular aspects of thyroid hormone actions. Endocr Rev (2010) 31:139–70. doi:10.1210/er.2009-0007
17. Aranda A, Pascual A. Nuclear hormone receptors and gene expression. Physiol Rev (2001) 81:1269–304.
18. Forrest D, Reh TA, Rüsch A. Neurodevelopmental control by thyroid hormone receptors. Curr Opin Neurobiol (2002) 12:49–56. doi:10.1016/S0959-4388(02)00289-1
19. Anderson GW, Schoonover CM, Jones SA. Control of thyroid hormone action in the developing rat brain. Thyroid (2003) 13:1039–56. doi:10.1089/105072503770867219
20. Bernal J. Thyroid hormones and brain development. Vitam Horm (2005) 71:95–122. doi:10.1016/S0083-6729(05)71004-9
21. Flamant F, Gauthier K, Samarut J. Thyroid hormones signaling is getting more complex: STORMs are coming. Mol Endocrinol (2007) 21:321–33. doi:10.1210/me.2006-0035
22. Bernal J, Morte B. Thyroid hormone receptor activity in the absence of ligand: physiological and developmental implications. Biochem Biophys Acta (2013) 1830:3893–9. doi:10.1016/j.bbagen.2012.04.014
23. Chatonnet F, Guyot R, Benoît G, Flamant F. Genome-wide analysis of thyroid hormone receptors shared and specific functions in neural cells. Proc Natl Acad Sci U S A (2013) 110:E766–75. doi:10.1073/pnas.1210626110
24. Dumitrescu AM, Refetoff S. The syndromes of reduced sensitivity to thyroid hormone. Biochim Biophys Acta (2013) 1830:3987–4003. doi:10.1016/j.bbagen.2012.08.005
25. Refetoff S, Bassett JH, Beck-Peccoz P, Bernal J, Brent G, Chatterjee K, et al. Classification and proposed nomenclature for inherited defects of thyroid hormone action, cell transport, and metabolism. Thyroid (2014) 24:407–9. doi:10.1089/thy.2013.3393.nomen
26. Friesema EC, Jansen J, Milici C, Visser TJ. Thyroid hormone transporters. Vitam Horm (2005) 70:137–67. doi:10.1016/S0083-6729(05)70005-4
27. Abe T, Suzuki T, Unno M, Tokui T, Ito S. Thyroid hormone transporters: recent advances. Trends Endocrinol Metab (2002) 13:215–20. doi:10.1016/S1043-2760(02)00599-4
28. Sugiyama D, Kusuhara H, Taniguchi H, Ishikawa S, Nozaki Y, Aburatani H, et al. Functional characterization of rat brain-specific organic anion transporter (Oatp14) at the blood-brain barrier: high affinity transporter for thyroxine. J Biol Chem (2003) 278:43489–95. doi:10.1074/jbc.M306933200
29. Bernal J. Role of monocarboxylate anion transporter 8 (MCT8) in thyroid hormone transport: answers from mice. Endocrinology (2006) 147:4034–5. doi:10.1210/en.2006-0695
30. Braun D, Kinne A, Bräuer AU, Sapin R, Klein MO, Köhrle J, et al. Developmental and cell type-specific expression of thyroid hormone transporters in the mouse brain and in primary brain cells. Glia (2011) 59:463–71. doi:10.1002/glia.21116
31. Rodrigues TB, Ceballos A, Grijota-Martínez C, Nuñez B, Refetoff S, Cerdán S, et al. Increased oxidative metabolism and neurotransmitter cycling in the brain of mice lacking the thyroid hormone transporter SLC16A2 (MCT8). PLoS One (2013) 8(10):e74621. doi:10.1371/journal.pone.0074621
32. Müller J, Heuer H. Expression pattern of thyroid hormone transporters in the postnatal mouse brain. Front Endocrinol (2014) 5:92. doi:10.3389/fendo.2014.00092
33. Wirth EK, Schweizer U, Köhrle J. Transport of thyroid hormone in brain. Front Endocrinol (2014) 5:98. doi:10.3389/fendo.2014.00098
34. Guadaño-Ferraz A, Escámez MJ, Morte B, Vargiu P, Bernal J. Transcriptional induction of RC3/neurogranin by thyroid hormone: differential neuronal sensitivity is not correlated with thyroid hormone receptor distribution in the brain. Brain Res Mol Brain Res (1997) 49:37–44. doi:10.1016/S0169-328X(97)00119-8
35. Bianco AC, Salvatore D, Gereben B, Berry MJ, Larsen PR. Biochemistry, cellular and molecular biology, and physiological roles of the iodothyronine-selenodeiodinases. Endocr Rev (2002) 23:38–89. doi:10.1210/edrv.23.1.0455
36. Bianco AC, Kim BW. Deiodinases: implications of the local control of thyroid hormone action. J Clin Invest (2006) 116:2571–9. doi:10.1172/JCI29812
37. Morte B, Bernal J. Thyroid hormone action: astrocyte-neuron communication. Front Endocrinol (2014) 5:82. doi:10.3389/fendo.2014.00082
38. Visser TJ, van Haasteren GA, Linkels E, Kaptein E, van Toor H, de Greef WJ. Gender-specific changes in thyroid hormone-glucuronidating enzymes in rat liver during short-term fasting and long-term food restriction. Eur J Endocrinol (1996) 135:489–97. doi:10.1530/eje.0.1350489
39. Flamant F, Baxter JD, Forrest D, Refetoff S, Samuels H, Scanlan TS, et al. International union of pharmacology. LIX. The pharmacology and classification of the nuclear receptor superfamily: thyroid hormone receptors. Pharmacol Rev (2006) 58:705–11. doi:10.1124/pr.58.4.3
40. Oetting A, Yen PM. New insights into thyroid hormone action. Best Pract Res Clin Endocrinol Metab (2007) 21:193–208. doi:10.1016/j.beem.2007.04.004
41. Bernal J, Pekonen F. Ontogenesis of the nuclear 3,5,3’-triiodothyronine receptor in the human fetal brain. Endocrinology (1984) 114:677–9. doi:10.1210/endo-114-2-677
42. Perez-Castillo A, Bernal J, Ferreiro B, Pans T. The early ontogenesis of thyroid hormone receptor in the rat fetus. Endocrinology (1985) 117:2457–61. doi:10.1210/endo-117-6-2457
43. Bradley DJ, Towle HC, Young WS III. Spatial and temporal expression of alpha- and beta-thyroid hormone receptor mRNAs, including the beta 2-subtype, in the developing mammalian nervous system. J Neurosci (1992) 12:2288–302.
44. Morte B, Díez D, Ausó E, Belinchón MM, Gil-Ibáñez P, Grijota-Martínez C, et al. Thyroid hormone regulation of gene expression in the developing rat fetal cerebral cortex: prominent role of the Ca2+/calmodulin-dependent protein kinase pathway. Endocrinology (2010) 151:810–20. doi:10.1210/en.2009-0958
45. Casas F, Rochard P, Rodier A, Cassar-Malek I, Marchal-Victorion S, Wiesner RJ, et al. A variant form of the nuclear triiodothyronine receptor c-ErbAalpha1 plays a direct role in regulation of mitochondrial RNA synthesis. Mol Cell Biol (1999) 19:7913–24.
46. Casas F, Pessemesse L, Grandemange S, Seyer P, Baris O, Gueguen N, et al. Overexpression of the mitochondrial T3 receptor induces skeletal muscle atrophy during aging. PLoS One (2009) 4(5):e5631. doi:10.1371/journal.pone.0005631
47. Wang D, Xia X, Liu Y, Oetting A, Walker RL, Zhu Y, et al. Negative regulation of TSHalpha target gene by thyroid hormone involves histone acetylation and corepressor complex dissociation. Mol Endocrinol (2009) 23:600–9. doi:10.1210/me.2008-0389
48. Xu M, Iwasaki T, Shimokawa N, Sajdel-Sulkowska EM, Koibuchi N. The effect of low dose lipopolysaccharide on thyroid hormone-regulated actin cytoskeleton modulation and type 2 iodothyronine deiodinase activity in astrocytes. Endocr J (2013) 60:1221–30. doi:10.1507/endocrj.EJ13-0294
49. Calvo R, Obregón MJ, Ruiz de Oña C, Escobar del Rey F, Morreale de Escobar G. Congenital hypothyroidism, as studied in rats. Crucial role of maternal thyroxine but not of 3,5,3’-triiodothyronine in the protection of the fetal brain. J Clin Invest (1990) 86:889–99. doi:10.1172/JCI114790
50. Kester MH, Martínez de Mena R, Obregón MJ, Marinkovic D, Howatson A, Visser TJ, et al. Iodothyronine levels in the human developing brain: major regulatory roles of iodothyronine deiodinases in different areas. J Clin Endocrinol Metab (2004) 89:3117–28. doi:10.1210/jc.2003-031832
51. Huang CB, Chen FS, Chung MY. Transient hypothyroxinemia of prematurity is associated with abnormal cranial ultrasound and illness severity. Am J Perinatol (2002) 19:139–47. doi:10.1055/s-2002-25308
52. Chan SY, Vasilopoulou E, Kilby MD. The role of the placenta in thyroid hormone delivery to the fetus. Nat Clin Pract Endocrinol Metab (2009) 5:45–54. doi:10.1038/ncpendmet1026
53. Glinoer D. The importance of iodine nutrition during pregnancy. Public Health Nutr (2007) 10:1542–6. doi:10.1017/S1368980007360886
54. Gaitan JE, Mayoral LG, Gaitan E. Defective thyroidal iodine concentration in protein-calorie malnutrition. J Clin Endocrinol Metab (1983) 57:327–33. doi:10.1210/jcem-57-2-327
55. Vanderpas J. Nutritional epidemiology and thyroid hormone metabolism. Annu Rev Nutr (2006) 26:293–322. doi:10.1146/annurev.nutr.26.010506.103810
56. Leung AM, Pearce EN, Braverman LE. Iodine nutrition in pregnancy and lactation. Endocrinol Metab Clin North Am (2011) 40:765–77. doi:10.1016/j.ecl.2011.08.001
57. Berbel P, Morreale de Escobar G. Iodine and brain development. In: Preedy VR, Watson RR, Martin CR, editors. International Handbook of Behavior, Food and Nutrition. New York, NY: Springer Press (2011). p. 2105–34.
58. Köhrle J. Selenium and the thyroid. Curr Opin Endocrinol Diabetes Obes (2013) 20:441–8. doi:10.1097/01.med.0000433066.24541.88
59. Howdeshell KL. A model of the development of the brain as a construct of the thyroid system. Environ Health Perspect (2002) 110:337–48. doi:10.1289/ehp.02110s3337
60. Zoeller TR, Dowling AL, Herzig CT, Iannacone EA, Gauger KJ, Bansal R. Thyroid hormone, brain development, and the environment. Environ Health Perspect (2002) 110:355–61. doi:10.1289/ehp.02110s3355
61. Koibuchi N, Iwasaki T. Regulation of brain development by thyroid hormone and its modulation by environmental chemicals. Endocr J (2006) 53:295–303. doi:10.1507/endocrj.KR-69
62. Kimura-Kuroda J, Nagata I, Kuroda Y. Disrupting effects of hydroxy-polychlorinated biphenyl (PCB) congeners on neuronal development of cerebellar Purkinje cells: a possible causal factor for developmental brain disorders? Chemosphere (2007) 67:S412–20. doi:10.1016/j.chemosphere.2006.05.137
63. Román GC. Autism: transient in utero hypothyroxinemia related to maternal flavonoid ingestion during pregnancy and to other environmental antithyroid agents. J Neurol Sci (2007) 262:15–26. doi:10.1016/j.jns.2007.06.023
64. Parent AS, Naveau E, Gerard A, Bourguignon JP, Westbrook GL. Early developmental actions of endocrine disruptors on the hypothalamus, hippocampus, and cerebral cortex. J Toxicol Environ Health B Crit Rev (2011) 14:328–45. doi:10.1080/10937404.2011.578556
65. Gilbert ME, Rovet J, Chen Z, Koibuchi N. Developmental thyroid hormone disruption: prevalence, environmental contaminants and neurodevelopmental consequences. Neurotoxicology (2012) 33:842–52. doi:10.1016/j.neuro.2011.11.005
66. de Cock M, Maas YG, van de Bor M. Does perinatal exposure to endocrine disruptors induce autism spectrum and attention deficit hyperactivity disorders? Acta Paediatr (2012) 101:811–8. doi:10.1111/j.1651-2227.2012.02693.x
67. Zoeller RT, Brown TR, Doan LL, Gore AC, Skakkebaek NE, Soto AM, et al. Endocrine-disrupting chemicals and public health protection: a statement of principles from the endocrine society. Endocrinology (2012) 153:4097–110. doi:10.1210/en.2012-1422
68. Bizhanova A, Kopp P. Controversies concerning the role of pendrin as an apical iodide transporter in thyroid follicular cells. Cell Physiol Biochem (2009) 28:485–90. doi:10.1159/000335103
69. Langer P. The impacts of organochlorines and other persistent pollutants on thyroid and metabolic health. Front Neuroendocrinol (2010) 31:497–518. doi:10.1016/j.yfrne.2010.08.001
70. Iavicoli I, Fontana L, Leso V, Bergamaschi A. The effects of nanomaterials as endocrine disruptors. Int J Mol Sci (2013) 14:16732–801. doi:10.3390/ijms140816732
71. Hinther A, Vawda S, Skirrow RC, Veldhoen N, Collins P, Cullen JT, et al. Nanometals induce stress and alter thyroid hormone action in Amphibia at or below North American water quality guidelines. Environ Sci Technol (2010) 44:8314–21. doi:10.1021/es101902n
72. Hetzel BS. Iodine deficiency disorders (IDD) and their eradication. Lancet (1983) 2:1126–9. doi:10.1016/S0140-6736(83)90636-0
73. Delange F. The disorders induced by iodine deficiency. Thyroid (1994) 4:107–28. doi:10.1089/thy.1994.4.107
74. Morreale de Escobar G, Obregón MJ, Escobar del Rey F. Maternal thyroid hormones early in pregnancy and fetal brain development. Best Pract Res Clin Endocrinol Metab (2004) 18:225–48. doi:10.1016/j.beem.2004.03.012
75. Glinoer D. The regulation of thyroid function during normal pregnancy: importance of the iodine nutrition status. Best Pract Res Clin Endocrinol Metab (2004) 18:133–52. doi:10.1016/j.beem.2004.03.001
76. Dumont JE, Optiz R, Christophe D, Vassart G, Roger PP, Maenhaut C. The phylogeny, ontogeny, anatomy and regulation of the iodine metabolizing thyroid. In: DeGroot LJ, editor. Thyroid Disease Manager. South Dartmouth, MA: Endocrine Education, Inc (2008). p. 1–109.
77. Leung AM, Pearce EN, Braverman LE. Perchlorate, iodine and the thyroid. Best Pract Res Clin Endocrinol Metab (2010) 24:133–41. doi:10.1016/j.beem.2009.08.009
78. Azizi F, Smyth P. Breast feeding and maternal and infant iodine nutrition. Clin Endocrinol (Oxf) (2009) 70:803–9. doi:10.1111/j.1365-2265.2008.03442.x
79. Stagnaro-Green A, Abalovich M, Alexander E, Azizi F, Mestman J, Negro R, et al. Guidelines of the American thyroid association for the diagnosis and management of thyroid disease during pregnancy and postpartum. Thyroid (2011) 21:1081–125. doi:10.1089/thy.2011.0087
80. Azizi F, Aminorroya A, Hedayati M, Rezvanian H, Amini M, Mirmiran P. Urinary iodine excretion in pregnant women residing in areas with adequate iodine intake. Public Health Nutr (2003) 6:95–8. doi:10.1079/PHN2002366
81. Travers CA, Guttikonda K, Norton CA, Lewis PR, Mollart LJ, Wiley V, et al. Iodine status in pregnant women and their newborns: are our babies at risk of iodine deficiency? Med J Aust (2006) 184:617–20.
82. Lazarus JH, Smyth PPA. Iodine deficiency in the UK and Ireland. Lancet (2008) 372:888. doi:10.1016/S0140-6736(08)61390-2
83. Andersson M, Karumbunathan V, Zimmermann MB. Global iodine status in 2011 and trends over the past decade. J Nutr (2012) 142:744–50. doi:10.3945/jn.111.149393
84. Leung AM, Pearce EN, Braverman LE. Sufficient iodine intake during pregnancy: just do it. Thyroid (2013) 23:7–8. doi:10.1089/thy.2012.0491
85. World Health Organization. Trace Elements in Human Nutrition and Health. Geneva: World Health Organization (1996).
87. Pop VJ, Brouwers EP, Vader HL, Vulsma T, van Baar AL, de Vijlder JJ. Maternal hypothyroxinaemia during early pregnancy and subsequent child development: a 3-year follow-up study. Clin Endocrinol (Oxf) (2003) 59:282–8. doi:10.1046/j.1365-2265.2003.01822.x
88. Vermiglio F, Lo Presti VP, Moleti M, Sidoti M, Tortorella G, Scaffidi G, et al. Attention deficit and hyperactivity disorders in the offspring of mothers exposed to mild-moderate iodine deficiency: a possible novel iodine deficiency disorder in developed countries. J Clin Endocrinol Metab (2004) 89:6054–60. doi:10.1210/jc.2004-0571
89. Morreale de Escobar G, Obregón MJ, Escobar del Rey F. Iodine deficiency and brain development in the first half of pregnancy. Public Health Nutr (2007) 10:1554–70. doi:10.1017/S1368980007360928
90. Zimmermann MB, Jooste PL, Pandav CS. Iodine-deficiency disorders. Lancet (2008) 372:1251–62. doi:10.1016/S0140-6736(08)61005-3
91. Andersson M, Aeberli I, Wüst N, Piacenza AM, Bucher T, Henschen I, et al. The Swiss iodized salt program provides adequate iodine for school children and pregnant women, but weaning infants not receiving iodine-containing complementary foods as well as their mothers are iodine deficient. J Clin Endocrinol Metab (2010) 95:5217–24. doi:10.1210/jc.2010-0975
92. Santos NC, Costa P, Ruano D, Macedo A, Soares MJ, Valente J, et al. Revisiting thyroid hormones in schizophrenia. J Thyroid Res (2012) 2012:569147. doi:10.1155/2012/569147
93. Bath SC, Steer CD, Golding J, Emmett P, Rayman MP. Effect of inadequate iodine status in UK pregnant women on cognitive outcomes in their children: results from the Avon longitudinal study of parents and children (ALSPAC). Lancet (2013) 382:331–7. doi:10.1016/S0140-6736(13)60436-5
94. Hamza RT, Hewedi DH, Sallam MT. Iodine deficiency in Egyptian autistic children and their mothers: relation to disease severity. Arch Med Res (2013) 44:555–61. doi:10.1016/j.arcmed.2013.09.012
95. Kooistra L, Crawford S, van Baar AL, Brouwers EP, Pop VJ. Neonatal effects of maternal hypothyroxinemia during early pregnancy. Pediatrics (2006) 117:161–7. doi:10.1542/peds.2005-0227
96. Kasatkina EP, Samsonova LN, Ivakhnenko VN, Ibragimova GV, Ryabykh AV, Naumenko LL, et al. Gestational hypothyroxinemia and cognitive function in offspring. Neurosci Behav Physiol (2006) 36:619–24. doi:10.1007/s11055-006-0066-0
97. Li Y, Shan Z, Tengm W, Yu X, Li Y, Fan C, et al. Abnormalities of maternal thyroid function during pregnancy affect neuropsychological development of their children at 25-30 months. Clin Endocrinol (Oxf) (2010) 72:825–9. doi:10.1111/j.1365-2265.2009.03743.x
98. Berbel P, Mestre JL, Santamaría A, Palazón I, Franco A, Graells M, et al. Delayed neurobehavioral development in children born to pregnant women with mild hypothyroxinemia during the first month of gestation: the importance of early iodine supplementation. Thyroid (2009) 19:511–9. doi:10.1089/thy.2008.0341
99. Suárez-Rodríguez M, Azcona-San Julián C, Alzina de Aguilar V. Hypothyroxinemia during pregnancy: the effect on neurodevelopment in the child. Int J Dev Neurosci (2012) 30:435–8. doi:10.1016/j.ijdevneu.2012.07.004
100. Moreno-Reyes R, Suetens C, Mathieu F, Begaux F, Zhu D, Rivera MT, et al. Kashin-Beck osteoarthropathy in rural Tibet in relation to selenium and iodine status. N Engl J Med (1998) 339:1112–20. doi:10.1056/NEJM199810153391604
101. Thilly CH, Swennen B, Bourdoux P, Ntambue K, Moreno-Reyes R, Gillies J, et al. The epidemiology of iodine-deficiency disorders in relation to goitrogenic factors and thyroid-stimulating-hormone regulation. Am J Clin Nutr (1993) 57:267S–70S.
102. Kaas JH. Evolution of columns, modules, and domains in the neocortex of primates. Proc Natl Acad Sci U S A (2012) 109:10655–60. doi:10.1073/pnas.1201892109
103. Luders E, Narr KL, Thompson PM, Rex DE, Jancke L, Steinmetz H, et al. Gender differences in cortical complexity. Nat Neurosci (2004) 7:799–800. doi:10.1038/nn1277
104. Kaas JH. The evolution of brains from early mammals to humans. Wiley Interdiscip Rev Cogn Sci (2013) 4:33–45. doi:10.1002/wcs.1206
105. Stenzel D, Huttner WB. Role of maternal thyroid hormones in the developing neocortex and during human evolution. Front Neuroanat (2013) 7:19. doi:10.3389/fnana.2013.00019
106. Innocenti GM. Development and evolution: two determinants of cortical connectivity. Prog Brain Res (2011) 189:65–75. doi:10.1016/B978-0-444-53884-0.00018-X
107. Amaral DG, Schumann CM, Nordahl CW. Neuroanatomy of autism. Trends Neurosci (2008) 31:137–45. doi:10.1016/j.tins.2007.12.005
108. Coghlan S, Horder J, Inkster B, Mendez MA, Murphy DG, Nutt DJ. GABA system dysfunction in autism and related disorders: from synapse to symptoms. Neurosci Biobehav Rev (2012) 36:2044–55. doi:10.1016/j.neubiorev.2012.07.005
109. Mountcastle V. The evolution of ideas concerning the function of the neocortex. Cereb Cortex (1995) 5:289–95. doi:10.1093/cercor/5.4.289
110. Marín-Padilla M. Dual origin of the mammalian neocortex and evolution of the cortical plate. Anat Embryol (1978) 152:109–26. doi:10.1007/BF00315920
111. Angevine JB Jr, Sidman RL. Autoradiographic study of cell migration during histogenesis of cerebral cortex in the mouse. Nature (1961) 192:766–8. doi:10.1038/192766b0
112. Zecevic N, Rakic P. Development of layer I neurons in the primate cerebral cortex. J Neurosci (2001) 21:5607–19.
113. Rakic P. Mode of cell migration to the superficial layers of fetal monkey neocortex. J Comp Neurol (1972) 145:61–83. doi:10.1002/cne.901450105
114. Rakic P, Cameron RS, Komuro H. Recognition, adhesión, transmembrane signaling and cell motility in guided neuronal migration. Curr Opin Neurobiol (1994) 4:63–9. doi:10.1016/0959-4388(94)90033-7
115. Rakic P. Specification of cerebral cortical areas. Science (1988) 241:170–6. doi:10.1126/science.3291116
116. O’Leary DD, Sahara S. Genetic regulation of arealization of the neocortex. Curr Opin Neurobiol (2008) 18:90–100. doi:10.1016/j.conb.2008.05.011
117. Krubitzer LA, Seelke AM. Cortical evolution in mammals: the bane and beauty of phenotypic variability. Proc Natl Acad Sci U S A (2012) 109:10647–54. doi:10.1073/pnas.1201891109
118. Cholfin JA, Rubenstein JL. Frontal cortex subdivision patterning is coordinately regulated by Fgf8, Fgf17, and Emx2. J Comp Neurol (2008) 509:144–55. doi:10.1002/cne.21709
119. Mallamaci A. Molecular bases of cortico-cerebral regionalization. Prog Brain Res (2011) 189:37–64. doi:10.1016/B978-0-444-53884-0.00017-8
120. Caviness VS Jr, Rakic P. Mechanisms of cortical development: a view from mutations in mice. Annu Rev Neurosci (1978) 1:297–326. doi:10.1146/annurev.ne.01.030178.001501
121. McConnell SK. Fates of visual cortical neurons in the ferret after isochronic and heterochronic transplantation. J Neurosci (1988) 8:945–74.
122. Krubitzer L, Campi KL, Cooke DF. All rodents are not the same: a modern synthesis of cortical organization. Brain Behav Evol (2011) 78:51–93. doi:10.1159/000327320
123. Kaas JH. The evolution of neocortex in primates. Prog Brain Res (2012) 195:91–102. doi:10.1016/B978-0-444-53860-4.00005-2
124. O’Rourke NA, Dailey ME, Smith SJ, McConnell SK. Diverse migratory pathways in the developing cerebral cortex. Science (1992) 258:299–302. doi:10.1126/science.1411527
125. Anderson SA, Eisenstat DD, Shi L, Rubenstein JL. Interneuron migration from basal forebrain to neocortex: dependence on Dlx genes. Science (1997) 278:474–6. doi:10.1126/science.278.5337.474
126. Marin O. Cellular and molecular mechanisms controlling the migration of neocortical interneurons. Eur J Neurosci (2013) 38:2019–29. doi:10.1111/ejn.12225
127. Letinic K, Rakic P. Telencephalic origin of human thalamic GABAergic neurons. Nat Neurosci (2001) 4:931–6. doi:10.1038/nn0901-931
128. Letinic K, Zoncu R, Rakic P. Origin of GABAergic neurons in the human neocortex. Nature (2002) 417:645–9. doi:10.1038/nature00779
129. Jones EG. The origins of cortical interneurons: mouse versus monkey and human. Cereb Cortex (2009) 19:1953–6. doi:10.1093/cercor/bhp088
130. Le Magueresse C, Monyer H. GABAergic interneurons shape the functional maturation of the cortex. Neuron (2013) 77:388–405. doi:10.1016/j.neuron.2013.01.011
131. Rubenstein JL. Annual research review: development of the cerebral cortex: implications for neurodevelopmental disorders. J Child Psychol Psychiatry (2011) 52:339–55. doi:10.1111/j.1469-7610.2010.02307.x
132. Westerink RH, Beekwilder JP, Wadman WJ. Differential alterations of synaptic plasticity in dentate gyrus and CA1 hippocampal area of calbindin-D28K knockout mice. Brain Res (2012) 1450:1–10. doi:10.1016/j.brainres.2012.02.036
134. Innocenti GM. Exuberant development of connections, and its possible permissive role in cortical evolution. Trends Neurosci (1995) 18:397–402. doi:10.1016/0166-2236(95)93936-R
135. Rakic P. A small step for the cell, a giant leap for mankind: a hypothesis of neocortical expansion during evolution. Trends Neurosci (1995) 18:383–8. doi:10.1016/0166-2236(95)93934-P
136. Obregon MJ, Mallol J, Pastor R, Morreale de Escobar G, Escobar del Rey F. L-thyroxine and 3,5,3’-triiodo-L-thyronine in rat embryos before onset of fetal thyroid function. Endocrinology (1984) 114:305–7. doi:10.1210/endo-114-1-305
137. Kratzsch J, Pulzer F. Thyroid gland development and defects. Best Pract Res Clin Endocrinol Metab (2008) 22:57–75. doi:10.1016/j.beem.2007.08.006
138. Biebermann H, Grüters A, Schöneberg T, Gudermann T. Congenital hypothyroidism caused by mutations in the thyrotropin-receptor gene. N Engl J Med (1997) 336:1390–1. doi:10.1056/NEJM199705083361914
139. Pasca di Magliano M, Di Lauro R, Zannini M. Pax8 has a key role in thyroid cell differentiation. Proc Natl Acad Sci U S A (2000) 97:13144–9. doi:10.1073/pnas.240336397
140. Dumitrescu AM, Liao XH, Weiss RE, Millen K, Refetoff S. Tissue-specific thyroid hormone deprivation and excess in monocarboxylate transporter (mct) 8-deficient mice. Endocrinology (2006) 147:4036–43. doi:10.1210/en.2006-0390
141. Friesema EC, Grueters A, Biebermann H, Krude H, von Moers A, Reeser M, et al. Association between mutations in a thyroid hormone transporter and severe X-linked psychomotor retardation. Lancet (2004) 364:1435–7. doi:10.1016/S0140-6736(04)17226-7
142. Trajkovic M, Visser TJ, Mittag J, Horn S, Lukas J, Darras VM, et al. Abnormal thyroid hormone metabolism in mice lacking the monocarboxylate transporter 8. J Clin Invest (2007) 117:627–35. doi:10.1172/JCI28253
143. Braun D, Wirth EK, Wohlgemuth F, Reix N, Klein MO, Gruters A, et al. Aminoaciduria, but normal thyroid hormone levels and signalling, in mice lacking the amino acid and thyroid hormone transporter Slc7a8. Biochem J (2011) 439:249–55. doi:10.1042/BJ20110759
144. Allan W, Herndon CN, Dudley FC. Some examples of the inheritance of mental deficiency: apparently sex-linked idiocy and microcephaly. Am J Ment Defic (1944) 48:325–34.
145. Dumitrescu AM, Liao XH, Best TB, Brockmann K, Refetoff S. A novel syndrome combining thyroid and neurological abnormalities is associated with mutations in a monocarboxylate transporter gene. Am J Hum Genet (2004) 74:168–75. doi:10.1086/380999
146. Wirth EK, Roth S, Blechschmidt C, Hölter SM, Becker L, Racz I, et al. Neuronal 3’,3,5-triiodothyronine (T3) uptake and behavioral phenotype of mice deficient in Mct8, the neuronal T3 transporter mutated in Allan-Herndon-Dudley syndrome. J Neurosci (2009) 29:9439–49. doi:10.1523/JNEUROSCI.6055-08.2009
147. Hernandez A, Martinez ME, Fiering S, Galton VA, St Germain D. Type 3 deiodinase is critical for the maturation and function of the thyroid axis. J Clin Invest (2006) 116:476–84. doi:10.1172/JCI26240
148. Hernandez A, Morte B, Belinchón MM, Ceballos A, Bernal J. Critical role of types 2 and 3 deiodinases in the negative regulation of gene expression by T3 in the mouse cerebral cortex. Endocrinology (2012) 153:2919–28. doi:10.1210/en.2011-1905
149. Morte B, Ceballos A, Diez D, Grijota-Martínez C, Dumitrescu AM, Di Cosmo C, et al. Thyroid hormone-regulated mouse cerebral cortex genes are differentially dependent on the source of the hormone: a study in monocarboxylate transporter-8- and deiodinase-2-deficient mice. Endocrinology (2010) 151:2381–7. doi:10.1210/en.2009-0944
150. Flamant F, Samarut J. Thyroid hormone receptors: lessons from knockout and knock-in mutant mice. Trends Endocrinol Metab (2003) 14:85–90. doi:10.1016/S1043-2760(02)00043-7
151. Refetoff S, DeWind LT, DeGroot LJ. Familial syndrome combining deaf-mutism, stuppled epiphyses, goiter and abnormally high PBI: possible target organ refractoriness to thyroid hormone. J Clin Endocrinol Metab (1967) 27:279–94. doi:10.1210/jcem-27-2-279
152. Beck-Peccoz P, Chatterjee VK. The variable clinical phenotype in thyroid hormone resistance syndrome. Thyroid (1994) 4:225–32. doi:10.1089/thy.1994.4.225
153. Rosenberg IN. The antithyroid activity of some compounds that inhibit peroxidase. Science (1952) 116:503–5. doi:10.1126/science.116.3019.503
154. Escobar del Rey F, Morreale de Escobar G. The effect of propylthiouracil, methylthiouracil and thiouracil on the peripheral metabolism of 1-thyroxine in thyroidectomized, 1-thyroxine maintained rats. Endocrinology (1961) 69:456–65. doi:10.1210/endo-69-3-456
155. De Groot LJ, Davis AM. Studies on the biosynthesis of iodotyrosines: a soluble thyroidal iodide-peroxidase tyrosine-iodinase system. Endocrinology (1962) 70:492–504. doi:10.1210/endo-70-4-492
156. Oppenheimer JH, Schwartz H, Surks L. MIPropylthiouracil inhibits the conversion of L-thyroxine to L-triiodothyronine. An explanation of the antithyroxine effect of propylthiouracil and evidence supporting the concept that triiodothyronine is the active thyroid hormone. J Clin Invest (1972) 51:2493–7. doi:10.1172/JCI107063
157. Azizi F, Amouzegar A. Management of hyperthyroidism during pregnancy and lactation. Eur J Endocrinol (2011) 164:871–6. doi:10.1530/EJE-10-1030
158. Ausó E, Lavado-Autric R, Cuevas E, Escobar del Rey F, Morreale de Escobar G, Berbel P. A moderate and transient deficiency of maternal thyroid function at the beginning of fetal neocorticogenesis alters neuronal migration. Endocrinology (2004) 145:4037–47. doi:10.1210/en.2004-0274
159. Cuevas E, Ausó E, Telefont M, Morreale de Escobar G, Sotelo C, Berbel P. Transient maternal hypothyroxinemia at onset of corticogenesis alters tangential migration of medial ganglionic eminence-derived neurons. Eur J Neurosci (2005) 22:541–51. doi:10.1111/j.1460-9568.2005.04243.x
160. Mano MT, Potter BJ, Belling GB, Chavadej J, Hetzel BS. Fetal brain in response to iodine deficiency in a primate model (Callithrix jacchus). J Neurol Sci (1987) 9:287–300. doi:10.1016/0022-510X(87)90236-X
161. Potter BJ, Mano MT, Belling GB, McIntosh GH, Hua C, Cragg BG, et al. Retarded fetal brain development resulting from severe dietary iodine deficiency in sheep. Neuropathol Appl Neurobiol (1982) 8:303–13. doi:10.1111/j.1365-2990.1982.tb00299.x
162. Li JQ, Wang X, Yan YQ, Wang KW, Qin DK, Xin ZF, et al. The effects on fetal brain development in the rat of a severely iodine deficient diet derived from an endemic area: observations on the first generation. Neuropathol Appl Neurobiol (1986) 12:261–76.
163. Martínez-Galán JR, Pedraza P, Santacana M, Escobar del Rey F, Morreale de Escobar G, Ruiz-Marcos A. Early effects of iodine deficiency on radial glial cells of the hippocampus of the rat fetus. A model of neurological cretinism. J Clin Invest (1997) 99:2701–9. doi:10.1172/JCI119459
164. Lavado-Autric R, Ausó E, García-Velasco JV, Arufe M, Escobar del Rey F, Berbel P, et al. Early maternal hypothyroxinemia alters histogenesis and cerebral cortex cytoarchitecture of the progeny. J Clin Invest (2003) 111:1073–82. doi:10.1172/JCI200316262
165. Yu F, Wang Y, Xu H, Dong J, Wei W, Wang Y, et al. Developmental iodine deficiency delays the maturation of newborn granule neurons associated with downregulation of p35 in postnatal rat hippocampus. Environ Toxicol (2012) 29:847–55. doi:10.1002/tox.21811
166. Wang Y, Wei W, Wang Y, Dong J, Song B, Min H, et al. Neurotoxicity of developmental hypothyroxinemia and hypothyroidism in rats: impairments of long-term potentiation are mediated by phosphatidylinositol 3-kinase signaling pathway. Toxicol Appl Pharmacol (2013) 271:257–65. doi:10.1016/j.taap.2013.04.034
167. Scow RW, Simpson ME. Thyroidectomy in the newborn rat. Anat Rec (1945) 91:209–26. doi:10.1002/ar.1090910305
168. Morreale de Escobar G, Obregon MJ, Escobar del Rey F. Fetal and maternal thyroid hormones. Horm Res (1987) 26:12–27. doi:10.1159/000180681
169. Berbel P, Navarro D, Ausó E, Varea E, Rodríguez AE, Ballesta JJ, et al. Role of late maternal thyroid hormones in cerebral cortex development: an experimental model for human prematurity. Cereb Cortex (2010) 20:1462–75. doi:10.1093/cercor/bhp212
170. Dowling AL, Martz GU, Leonard JL, Zoeller RT. Acute changes in maternal thyroid hormone induce rapid and transient changes in gene expression in fetal rat brain. J Neurosci (2000) 20:2255–65.
171. Dowling AL, Zoeller RT. Thyroid hormone of maternal origin regulates the expression of RC3/neurogranin mRNA in the fetal rat brain. Brain Res Mol Brain Res (2000) 82:126–32. doi:10.1016/S0169-328X(00)00190-X
172. Alvarez-Dolado M, Ruiz M, Del Rio JA, Alcantara S, Burgaya F, Sheldon M. Thyroid hormone regulates reelin and dab-1 expression during brain development. J Neurosci (1999) 19:6979–93.
173. Pathak A, Sinha RA, Mohan V, Mitra K, Godbole MM. Maternal thyroid hormone before the onset of fetal thyroid function regulates reelin and downstream signaling cascade affecting neocortical neuronal migration. Cereb Cortex (2011) 21:11–21. doi:10.1093/cercor/bhq052
174. Miller LD, Park KS, Guo QM, Alkharouf NW, Malek RL, Lee NH, et al. Silencing of Wnt signaling and activation of multiple metabolic pathways in response to thyroid hormone-stimulated cell proliferation. Mol Cell Biol (2001) 21:6626–39. doi:10.1128/MCB.21.19.6626-6639.2001
175. Matthews RP, Guthrie CR, Wailes LM, Zhao X, Means AR, McKnight GS. Calcium/calmodulin-dependent protein kinase types II and IV differentially regulate CREB-dependent gene expression. Mol Cell Biol (1994) 14:6107–16. doi:10.1128/MCB.14.9.6107
176. Watterson DM, Mirzoeva S, Guo L, Whyte A, Bourguignon JJ, Hibert M, et al. Ligand modulation of glial activation: cell permeable, small molecule inhibitors of serine-threonine protein kinases can block induction of interleukin 1 beta and nitric oxide synthase II. Neurochem Int (2001) 39:459–68. doi:10.1016/S0197-0186(01)00053-5
177. Murray PD, Kingsbury TJ, Krueger BK. Failure of Ca2+-activated, CREB-dependent transcription in astrocytes. Glia (2009) 57:828–34. doi:10.1002/glia.20809
178. Kuno-Murata M, Koibuchi N, Fukuda H, Murata M, Chin WW. Augmentation of thyroid hormone receptor-mediated transcription by Ca2+/calmodulin-dependent protein kinase type IV. Endocrinology (2000) 141:2275–8. doi:10.1210/endo.141.6.7612
179. McKenzie GJ, Stevenson P, Ward G, Papadia S, Bading H, Chawla SJ, et al. Nuclear Ca2+ and CaM kinase IV specify hormonal- and Notch-responsiveness. J Neurochem (2005) 93:171–85. doi:10.1111/j.1471-4159.2005.03010.x
180. Navarro D, Alvarado M, Navarrete F, Giner M, Pacheco P, Morreale de Escobar G, et al. Maternal and fetal rat’s hypothyroidism during gestation and lactation unbalances cortical VGluT1-VGAT immunoreactivity and alters attention déficit and anxiety. Eur Thyroid J (2012) 1(Suppl 1):208. doi:10.1159/000339890
181. Carlezon WA Jr, Duman RS, Nestler EJ. The many faces of CREB. Trends Neurosci (2005) 28:436–45. doi:10.1016/j.tins.2005.06.005
182. Qiu Z, Sylwestrak EL, Lieberman DN, Zhang Y, Liu XY, Ghosh A. The Rett syndrome protein MeCP2 regulates synaptic scaling. J Neurosci (2012) 32:989–94. doi:10.1523/JNEUROSCI.0175-11.2012
183. Lv J, Xin Y, Zhou W, Qiu Z. The epigenetic switches for neural development and psychiatric disorders. J Genet Genomics (2013) 40:339–46. doi:10.1016/j.jgg.2013.04.007
184. Wang H, Fukushima H, Kida S, Zhuo M. Ca2+/calmodulin-dependent protein kinase IV links group I metabotropic glutamate receptors to fragile X mental retardation protein in cingulate cortex. J Biol Chem (2009) 284:18953–62. doi:10.1074/jbc.M109.019141
185. Wang H, Morishita Y, Miura D, Naranjo JR, Kida S, Zhuo M. Roles of CREB in the regulation of FMRP by group I metabotropic glutamate receptors in cingulate cortex. Mol Brain (2012) 5:27. doi:10.1186/1756-6606-5-27
186. Krueger DD, Bear MF. Toward fulfilling the promise of molecular medicine in fragile X syndrome. Annu Rev Med (2011) 62:411–29. doi:10.1146/annurev-med-061109-134644
187. Tuchman R, Hirtz D, Mamounas LA. NINDS epilepsy and autism spectrum disorders workshop report. Neurology (2013) 29:1630–6. doi:10.1212/WNL.0b013e3182a9f482
188. Lu J, Wu Y, Sousa N, Almeida OF. SMAD pathway mediation of BDNF and TGF beta 2 regulation of proliferation and differentiation of hippocampal granule neurons. Development (2005) 132:3231–42. doi:10.1242/dev.01893
189. Castrén ML, Castrén E. BDNF in fragile X syndrome. Neuropharmacology (2014) 76(Pt C):729–36. doi:10.1016/j.neuropharm.2013.05.018
190. Eayrs JT, Taylor SH. The effect of thyroid deficiency induced by methyl thiouracil on the maturation of the central nervous system. J Anat (1951) 85:350–8.
191. Mohan V, Sinha RA, Pathak A, Rastogi L, Kumar P, Pal A, et al. Maternal thyroid hormone deficiency affects the fetal neocorticogenesis by reducing the proliferating pool, rate of neurogenesis and indirect neurogenesis. Exp Neurol (2012) 237:477–88. doi:10.1016/j.expneurol.2012.07.019
192. Berbel P, Ausó E, Garcia-Velasco JV, Molina ML, Camacho M. Role of thyroid hormones in the maturation and organisation of rat barrel cortex. Neuroscience (2001) 107:383–94. doi:10.1016/S0306-4522(01)00368-2
193. Rami A, Rabié A, Patel AJ. Thyroid hormone and development of the rat hippocampus: cell acquisition in the dentate gyrus. Neuroscience (1986) 19:1207–16. doi:10.1016/0306-4522(86)90134-X
194. Perez-Juste G, Aranda A. The cyclin-dependent kinase inhibitor p27(Kip1) is involved in thyroid hormone-mediated neuronal differentiation. J Biol Chem (1999) 274:5026–31. doi:10.1074/jbc.274.8.5026
195. Qiu R, Wang J, Tsark W, Lu Q. Essential role of PDZ-RGS3 in the maintenance of neural progenitor cells. Stem Cells (2010) 28:1602–10. doi:10.1002/stem.478
196. O’Roak BJ, Vives L, Fu W, Egertson JD, Stanaway IB, Phelps IG, et al. Multiplex targeted sequencing identifies recurrently mutated genes in autism spectrum disorders. Science (2012) 338:1619–22. doi:10.1126/science.1227764
197. Barr AR, Kilmartin JV, Gergely F. CDK5RAP2 functions in centrosome to spindle pole attachment and DNA damage response. J Cell Biol (2010) 189:23–39. doi:10.1083/jcb.200912163
198. Wang Y, Wang Y, Dong J, Wei W, Song B, Min H, et al. Developmental hypothyroxinemia and hypothyroidism reduce proliferation of cerebellar granule neuron precursors in rat offspring by downregulation of the sonic hedgehog signaling pathway. Mol Neurobiol (2014) 49:1143–52. doi:10.1007/s12035-013-8587-3
199. Kuida K, Haydar TF, Kuan CY, Gu Y, Taya C, Karasuyama H, et al. Reduced apoptosis and cytochrome c-mediated caspase activation in mice lacking caspase 9. Cell (1998) 94:325–37. doi:10.1016/S0092-8674(00)81476-2
200. Chenn A, Walsh CA. Regulation of cerebral cortical size by control of cell cycle exit in neural precursors. Science (2002) 297:365–9. doi:10.1126/science.1074192
201. Singh R, Upadhyay G, Godbole MM. Hypothyroidism alters mitochondrial morphology and induces release of apoptogenic proteins during rat cerebellar development. J Endocrinol (2003) 176:321–9. doi:10.1677/joe.0.1760321
202. Babu S, Sinha RA, Mohan V, Rao G, Pal A, Pathak A, et al. Effect of hypothyroxinemia on thyroid hormone responsiveness and action during rat postnatal neocortical development. Exp Neurol (2011) 228:91–8. doi:10.1016/j.expneurol.2010.12.012
203. Sugimoto C, Maekawa S, Miyata S. OBCAM, an immunoglobulin superfamily cell adhesion molecule, regulates morphology and proliferation of cerebral astrocytes. J Neurochem (2010) 112:818–28. doi:10.1111/j.1471-4159.2009.06513.x
204. Martínez-Galán JR, Escobar del Rey F, Morreale de Escobar G, Santacana M, Ruíz-Marcos A. Hypothyroidism alters the development of radial glial cells in the term fetal and postnatal neocortex of the rat. Brain Res Dev Brain Res (2004) 153:109–14. doi:10.1016/j.devbrainres.2004.08.002
205. Lucio RA, García JV, Ramón Cerezo J, Pacheco P, Innocenti GM, Berbel P. The development of auditory callosal connections in normal and hypothyroid rats. Cereb Cortex (1997) 7:303–16. doi:10.1093/cercor/7.4.303
206. Goodman JH, Gilbert ME. Modest thyroid hormone insufficiency during development induces a cellular malformation in the corpus callosum: a model of cortical dysplasia. Endocrinology (2007) 148:2593–7. doi:10.1210/en.2006-1276
207. Tan XJ, Fan XT, Kim HJ, Butler R, Webb P, Warner M, et al. Liver X receptor beta and thyroid hormone receptor alpha in brain cortical layering. Proc Natl Acad Sci U S A (2010) 107:12305–10. doi:10.1073/pnas.1006162107
208. Fan X, Kim HJ, Bouton D, Warner M, Gustafsson JA. Expression of liver X receptor beta is essential for formation of superficial cortical layers and migration of later-born neurons. Proc Natl Acad Sci U S A (2008) 105:13445–50. doi:10.1073/pnas.0806974105
209. Guo L, Xu P, Tang X, Wu Q, Xing Y, Gustafsson JA, et al. Liver X receptor β delays transformation of radial glial cells into astrocytes during mouse cerebral cortical development. Neurochem Int (2014) 71:8–16. doi:10.1016/j.neuint.2014.03.009
210. Berkenstam A, Färnegårdh M, Gustafsson JA. Convergence of lipid homeostasis through liver X and thyroid hormone receptors. Mech Ageing Dev (2004) 125:707–17. doi:10.1016/j.mad.2004.05.005
211. Berbel P, Marco P, Cerezo JR, DeFelipe J. Distribution of parvalbumin immunoreactivity in the neocortex of hypothyroid adult rats. Neurosci Lett (1996) 204:65–8. doi:10.1016/0304-3940(96)12318-1
212. Morte B, Manzano J, Scanlan T, Vennström B, Bernal J. Deletion of the thyroid hormone receptor alpha 1 prevents the structural alterations of the cerebellum induced by hypothyroidism. Proc Natl Acad Sci U S A (2002) 99:3985–9. doi:10.1073/pnas.062413299
213. Ceballos A, Belinchon MM, Sanchez-Mendoza E, Grijota-Martinez C, Dumitrescu AM, Refetoff S, et al. Importance of monocarboxylate transporter 8 for the blood-brain barrier-dependent availability of 3,5,3’-triiodo-L-thyronine. Endocrinology (2009) 150:2491–6. doi:10.1210/en.2008-1616
214. Nicholson JL, Altman J. The effects of early hypo- and hyperthyroidism on the development of the rat cerebellar cortex. Synaptogenesis in the molecular layer. Brain Res (1972) 44:25–36. doi:10.1016/0006-8993(72)90362-9
215. Lauder JM. Thyroid influences on the developing cerebellum and hippocampus of the rat. In: DeLong GR, Robbins J, Condliffe PG, editors. Iodine and the Brain. New York, NY: Plenum Press (1989). p. 79–90.
216. Berbel P, Guadaño-Ferraz A, Martínez M, Quiles JA, Balboa R, Innocenti GM. Organization of auditory callosal connections in hypothyroid rats. Eur J Neurosci (1993) 5:1465–78. doi:10.1111/j.1460-9568.1993.tb00214.x
217. Gilbert ME, Sui L, Walker MJ, Anderson W, Thomas S, Smoller SN, et al. Thyroid hormone insufficiency during brain development reduces parvalbumin immunoreactivity and inhibitory function in the hippocampus. Endocrinology (2007) 148:92–102. doi:10.1210/en.2006-0164
218. Wallis K, Sjögren M, van Hogerlinden M, Silberberg G, Fisahn A, Nordström K, et al. Locomotor deficiencies and aberrant development of subtype- specific GABAergic interneurons caused by an unliganded thyroid hormone receptor alpha1. J Neurosci (2008) 28:1904–15. doi:10.1523/JNEUROSCI.5163-07.2008
219. Venero C, Guadaño-Ferraz A, Herrero AI, Nordström K, Manzano J, de Escobar GM, et al. Anxiety, memory impairment, and locomotor dysfunction caused by a mutant thyroid hormone receptor alpha1 can be ameliorated by T3 treatment. Genes Dev (2005) 19:2152–63. doi:10.1101/gad.346105
220. Patel LS, Wenzel HJ, Schwartzkroin PA. Physiological and morphological characterization of dentate granule cells in the p35 knock-out mouse hippocampus: evidence for an epileptic circuit. J Neurosci (2004) 24:9005–14. doi:10.1523/JNEUROSCI.2943-04.2004
221. Van Middlesworth L, Norris CH. Audiogenic seizures and cochlear damage in rats after perinatal antithyroid treatment. Endocrinology (1980) 106:1686–90. doi:10.1210/endo-106-6-1686
222. Eayrs JT, Goohead B. Postnatal development of the cerebral cortex in the rat. J Anat (1959) 93:385–402.
223. Vincent J, Legrand C, Rabié A, Legrand J. Effects of thyroid hormone on synaptogenesis in the molecular layer of the developing rat cerebellum. J Physiol (1982/1983) 78:729–38.
224. Yu X, Malenka RC. Beta-catenin is critical for dendritic morphogenesis. Nat Neurosci (2003) 6:1169–77. doi:10.1038/nn1132
225. Gravel C, Sasseville R, Hawkes R. Maturation of the corpus callosum of the rat: II. Influence of thyroid hormones on the number and maturation of axons. J Comp Neurol (1990) 291:147–61. doi:10.1002/cne.902910109
226. Gravel C, Hawkes R. Maturation of the corpus callosum of the rat: I. Influence of thyroid hormones on the topography of callosal projections. J Comp Neurol (1990) 291:128–46. doi:10.1002/cne.902910109
227. Guadaño-Ferraz A, Escobar del Rey F, Morreale de Escobar G, Innocenti GM, Berbel P. The development of the anterior commissure in normal and hypothyroid rats. Brain Res Dev Brain Res (1994) 81:293–308. doi:10.1016/0165-3806(94)90315-8
228. Berbel P, Guadaño-Ferraz A, Angulo A, Ramón Cerezo J. Role of thyroid hormones in the maturation of interhemispheric connections in rats. Behav Brain Res (1994) 64:9–14. doi:10.1016/0166-4328(94)90114-7
229. Nunez J, Couchie D, Aniello F, Bridoux AM. Regulation by thyroid hormone of microtubule assembly and neuronal differentiation. Neurochem Res (1991) 16:975–82. doi:10.1007/BF00965840
230. Muñoz A, Rodriguez-Peña A, Perez-Castillo A, Ferreiro B, Sutcliffe JG, Bernal J. Effects of neonatal hypothyroidism on rat brain gene expression. Mol Endocrinol (1991) 5:273–80. doi:10.1210/mend-5-2-273
231. Li CP, Olavarria JF, Greger BE. Occipital cortico-pyramidal projection in hypothyroid rats. Brain Res Dev Brain Res (1995) 89:227–34. doi:10.1016/0165-3806(95)00119-X
232. Innocenti GM, Price DJ. Exuberance in the development of cortical networks. Nat Rev Neurosci (2005) 6:955–65. doi:10.1038/nrn1790
233. Friauf E, McConnell SK, Shatz CJ. Functional synaptic circuits in the subplate during fetal and early postnatal development of cat visual cortex. J Neurosci (1990) 10:2601–13.
234. Oeschger FM, Wang WZ, Lee S, García-Moreno F, Goffinet AM, Arbonés ML, et al. Gene expression analysis of the embryonic subplate. Cereb Cortex (2012) 22:1343–59. doi:10.1093/cercor/bhr197
235. Torres-Reveron J, Friedlander MJ. Properties of persistent postnatal cortical subplate neurons. J Neurosci (2007) 27:9962–74. doi:10.1523/JNEUROSCI.1536-07.2007
236. Kanold PO, Kara P, Reid RC, Shatz CJ. Role of subplate neurons in functional maturation of visual cortical columns. Science (2003) 301:521–5. doi:10.1126/science.1084152
237. Kanold PO, Shatz CJ. Subplate neurons regulate maturation of cortical inhibition and outcome of ocular dominance plasticity. Neuron (2006) 51:627–38. doi:10.1016/j.neuron.2006.07.008
238. Piñon MC, Jethwa A, Jacobs E, Campagnoni A, Molnár Z. Dynamic integration of subplate neurons into the cortical barrel field circuitry during postnatal development in the Golli-tau-eGFP (GTE) mouse. J Physiol (2009) 587:1903–15. doi:10.1113/jphysiol.2008.167767
239. Navarro D, Alvarado M, Morte B, Berbel D, Sesma J, Pacheco P, et al. Late maternal hypothyroidism alters the expression of Camk4 in neocortical subplate neurons. A comparison with Nurr1 labeling. Cereb Cortex (2013). doi:10.1093/cercor/bht129
240. Volpe JJ. Subplate neurons – missing link in brain injury of the premature infant? Pediatrics (1996) 97:112–3.
241. Volpe JJ. Electroencephalography may provide insight into timing of premature brain injury. Pediatrics (2009) 124:e542–4. doi:10.1542/peds.2009-1244
242. Eastwood SL, Harrison PJ. Cellular basis of reduced cortical reelin expression in schizophrenia. Am J Psychiatry (2006) 163:540–2. doi:10.1176/appi.ajp.163.3.540
243. McQuillen PS, Ferriero DM. Perinatal subplate neuron injury: implications for cortical development and plasticity. Brain Pathol (2005) 15:250–60. doi:10.1111/j.1750-3639.2005.tb00528.x
244. Stolp H, Neuhaus A, Sundramoorthi R, Molnár Z. The long and the short of it: gene and environment interactions during early cortical development and consequences for long-term neurological disease. Front Psychiatry (2012) 3:50. doi:10.3389/fpsyt.2012.00050
245. Hoerder-Suabedissen A, Oeschger FM, Krishnan ML, Belgard TG, Wang WZ, Lee S, et al. Expression profiling of mouse subplate reveals a dynamic gene network and disease association with autism and schizophrenia. Proc Natl Acad Sci U S A (2013) 110:3555–60. doi:10.1073/pnas.1218510110
246. Ausó E, Cases O, Fouquet C, Camacho M, García-Velasco JV, Gaspar P, et al. Protracted expression of serotonin transporter and altered thalamocortical projections in the barrelfield of hypothyroid rats. Eur J Neurosci (2001) 14:1968–80. doi:10.1046/j.0953-816x.2001.01815.x
247. Blue ME, Erzurumlu RS, Jhaveri S. A comparison of pattern formation by thalamocortical and serotonergic afferents in the rat barrel field cortex. Cereb Cortex (1991) 1:380–9. doi:10.1093/cercor/1.5.380
248. Osterheld-Haas MC, Hornung JP. Laminar development of the mouse barrel cortex: effects of neurotoxins against monoamines. Exp Brain Res (1996) 110:183–95. doi:10.1007/BF00228550
249. Bennett-Clarke CA, Leslie MJ, Lane RD, Rhoades RW. Effect of serotonin depletion on vibrissa-related patterns of thalamic afferents in the rat’s somatosensory cortex. J Neurosci (1994) 14:7594–607.
250. Sanchez-Toscano F, Escobar del Rey F, Morreale de Escobar G, Ruiz-Marcos A. Measurement of the effects of hypothyroidism on the number and distribution of spines along the apical shaft of pyramidal neurons of the rat cerebral cortex. Brain Res (1977) 126:547–50. doi:10.1016/0006-8993(77)90606-0
251. Wei W, Wang Y, Wang Y, Dong J, Min H, Song B, et al. Developmental hypothyroxinaemia induced by maternal mild iodine deficiency delays hippocampal axonal growth in the rat offspring. J Neuroendocrinol (2013) 25:852–62. doi:10.1111/jne.12058
252. Berbel P, Innocenti GM. The development of the corpus callosum in cats: a light- and electron-microscopic study. J Comp Neurol (1988) 276:132–56. doi:10.1002/cne.902760109
253. Savage DD, Otero MA, Montano CY, Razani-Boroujerdi S, Paxton LL, Kasarskis EJ. Perinatal hypothyroidism decreases hippocampal mossy fiber zinc density in rats. Neuroendocrinology (1992) 55:20–7. doi:10.1159/000126092
254. Madeira MD, Paula-Barbosa MM. Reorganization of mossy fiber synapses in male and female hypothyroid rats: a stereological study. J Comp Neurol (1993) 337:334–52. doi:10.1002/cne.903370213
255. Melo CV, Mele M, Curcio M, Comprido D, Silva CG, Duarte CB. BDNF regulates the expression and distribution of vesicular glutamate transporters in cultured hippocampal neurons. PLoS One (2013) 8(1):e53793. doi:10.1371/journal.pone.0053793
256. Chakraborty G, Magagna-Poveda A, Parratt C, Umans JG, MacLusky NJ. Reduced hippocampal brain-derived neurotrophic factor (BDNF) in neonatal rats after prenatal exposure to propylthiouracil (PTU). Endocrinology (2012) 153:1311–6. doi:10.1210/en.2011-1437
257. Wong CC, Leung MS. Effects of neonatal hypothyroidism on the expressions of growth cone proteins and axon guidance molecules related genes in the hippocampus. Mol Cell Endocrinol (2001) 184:143–50. doi:10.1016/S0303-7207(01)00592-5
258. Koibuchi N, Chin WW. Thyroid hormone action and brain development. Trends Endocrinol Metab (2000) 11:123–8. doi:10.1016/S1043-2760(00)00238-1
259. Cortés C, Eugenin E, Aliaga E, Carreño LJ, Bueno SB, Gonzalez P, et al. Hypothyroidism in the adult rat causes an incremento of Bdnf in the brain, neuronal and astrocytes apoptosis, gliosis and deterioration of the postsunaptic density. Thyroid (2012) 22:951–63. doi:10.1089/thy.2010.0400
260. Sajdel-Sulkowska EM, Xu M, Koibuchi N. Increase in cerebellar neurotrophin-3 and oxidative stress markers in autism. Cerebellum (2009) 8:366–72. doi:10.1007/s12311-009-0105-9
261. Sánchez-Ponce D, DeFelipe J, Garrido JJ, Muñoz A. In vitro maturation of the cisternal organelle in the hippocampal neuron’s axon initial segment. Mol Cell Neurosci (2011) 48:104–16. doi:10.1016/j.mcn.2011.06.010
262. Bridi MS, Abel T. The NR4A orphan nuclear receptors mediate transcription-dependent hippocampal synaptic plasticity. Neurobiol Learn Mem (2013) 105:151–8. doi:10.1016/j.nlm.2013.06.020
263. Modregger J, Ritter B, Witter B, Paulsson M, Plomann M. All three PACSIN isoforms bind to endocytic proteins and inhibit endocytosis. J Cell Sci (2000) 113:4511–21.
264. Gilbert ME, Sui L. Dose-dependent reductions in spatial learning and synaptic function in the dentate gyrus of adult rats following developmental thyroid hormone insufficiency. Brain Res (2006) 1069:10–22. doi:10.1016/j.brainres.2005.10.049
265. Alzoubi KH, Gerges NZ, Aleisa AM, Alkadhi KA. Levothyroxin restores hypothyroidism-induced impairment of hippocampus-dependent learning and memory: behavioral, electrophysiological, and molecular studies. Hippocampus (2009) 19:66–78. doi:10.1002/hipo.20476
266. Opazo MC, Gianini A, Pancetti F, Azkcona G, Alarcón L, Lizana R, et al. Maternal hypothyroxinemia impairs spatial learning and synaptic nature and function in the offspring. Endocrinology (2008) 149:5097–106. doi:10.1210/en.2008-0560
267. Sánchez-Alonso JL, Muñoz-Cuevas J, Vicente-Torres MA, Colino A. Role of low-voltage-activated calcium current on the firing pattern alterations induced by hypothyroidism in the rat hippocampus. Neuroscience (2010) 171:993–1005. doi:10.1016/j.neuroscience.2010.10.003
268. Caminiti R, Ghaziri H, Galuske R, Hof PR, Innocenti GM. Evolution amplified processing with temporally dispersed slow neuronal connectivity in primates. Proc Natl Acad Sci U S A (2009) 106:19551–6. doi:10.1073/pnas.0907655106
269. Ng L, Pedraza PE, Faris JS, Vennström B, Curran T, Morreale de Escobar G, et al. Audiogenic seizure susceptibility in thyroid hormone receptor beta-deficient mice. Neuroreport (2001) 12:2359–62. doi:10.1097/00001756-200108080-00015
270. Negishi T, Kawasaki K, Sekiguchi S, Ishii Y, Kyuwa S, Kuroda Y, et al. Attention-deficit and hyperactive neurobehavioural characteristics induced by perinatal hypothyroidism in rats. Behav Brain Res (2005) 159:323–31. doi:10.1016/j.bbr.2004.11.012
271. Gilbert ME, Lasley SM. Developmental thyroid hormone insufficiency and brain development: a role for brain-derived neurotrophic factor (BDNF)? Neuroscience (2013) 239:253–70. doi:10.1016/j.neuroscience.2012.11.022
272. Suo Z, Cox AA, Bartelli N, Rasul I, Festoff BW, Premont RT, et al. GRK5 deficiency leads to early Alzheimer-like pathology and working memory impairment. Neurobiol Aging (2007) 28:1873–88. doi:10.1016/j.neurobiolaging.2006.08.013
273. Rao JS, Rapoport SI, Kim HW. Decreased GRK3 but not GRK2 expression in frontal cortex from bipolar disorder patients. Int J Neuropsychopharmacol (2009) 12:851–60. doi:10.1017/S146114570900025X
274. Wang L, Zhou C, Wang Z, Liu J, Jing Z, Zhang Z, et al. Dynamic variation of genes profiles and pathways in the hippocampus of ischemic mice: a genomic study. Brain Res (2011) 1372:13–21. doi:10.1016/j.brainres.2010.11.099
275. Klugmann M, Leichtlein CB, Symes CW, Klaussner BC, Brooks AI, Young D, et al. A novel role of circadian transcription factor DBP in hippocampal plasticity. Mol Cell Neurosci (2006) 31:303–14. doi:10.1016/j.mcn.2005.09.019
276. Bailey A, Luthert P, Dean A, Harding B, Janota I, Montgomery M, et al. A clinicopathological study of autism. Brain (1998) 121:889–905. doi:10.1093/brain/121.5.889
277. Palmen SJ, van Engeland H, Hof PR, Schmitz C. Neuropathological findings in autism. Brain (2004) 127:2572–83. doi:10.1093/brain/awh287
278. Wegiel J, Kuchna I, Nowicki K, Imaki H, Wegiel J, Marchi E, et al. The neuropathology of autism: defects of neurogenesis and neuronal migration, and dysplastic changes. Acta Neuropathol (2010) 119:755–70. doi:10.1007/s00401-010-0655-4
279. Castermans D, Vermeesch JR, Fryns JP, Steyaert JG, Van de Ven WJ, Creemers JW, et al. Identification and characterization of the TRIP8 and REEP3 genes on chromosome 10q21.3 as novel candidate genes for autism. Eur J Hum Genet (2007) 15:422–31. doi:10.1038/sj.ejhg.5201785
280. Courchesne E, Pierce K, Schumann CM, Redcay E, Buckwalter JA, Kennedy DP, et al. Mapping early brain development in autism. Neuron (2007) 56:399–413. doi:10.1016/j.neuron.2007.10.016
281. Kern JK, Geier DA, Sykes LK, Geier MR. Evidence of neurodegeneration in autism spectrum disorder. Transl Neurodegener (2013) 2:17. doi:10.1186/2047-9158-2-17
282. Petropoulos H, Friedman SD, Shaw DWW, Artru AA, Dawson G, Dager SR. Gray matter abnormalities in autism spectrum disorder revealed by T2 relaxation. Neurology (2006) 67:632–6. doi:10.1212/01.wnl.0000229923.08213.1e
283. Hardan AY, Girgis RR, Lacerda ALT, Yorbik O, Kilpatrick M, Keshavan MS. Magnetic resonance imaging study of the orbitofrontal cortex in autism. J Child Neurol (2006) 21:866–71. doi:10.1177/08830738060210100701
284. Courchesne E, Mouton PR, Calhoun ME, Semendeferi K, Ahrens-Barbeau C, Hallet MJ, et al. Neuron number and size in prefrontal cortex of children with autism. JAMA (2011) 306:2001–10. doi:10.1001/jama.2011.1638
285. McAlonan GM, Cheung V, Cheung C, Suckling J, Lam GY, Tai KS, et al. Mapping the brain in autism. A voxel-based MRI study of volumetric differences and intercorrelations in autism. Brain (2005) 128:268–76. doi:10.1093/brain/awh332
286. Theoharides TC, Asadi S, Patel AB. Focal brain inflammation and autism. J Neuroinflammation (2013) 10:46. doi:10.1186/1742-2094-10-46
287. Vargas DL, Nascimbene C, Krishnan C, Zimmerman AW, Pardo CA. Neuroglial activation and neuroinflammation in the brain of patients with autism. Ann Neurol (2005) 57:67–81. doi:10.1002/ana.20315
288. Uppal N, Gianatiempo I, Wicinski B, Schmeidler J, Heinsen H, Schmitz C, et al. Neuropathology of the posteroinferior occipitotemporal gyrus in children with autism. Mol Autism (2014) 5(1):17. doi:10.1186/2040-2392-5-17
289. Jacot-Descombes S, Uppal N, Wicinski B, Santos M, Schmeidler J, Giannakopoulos P, et al. Decreased pyramidal neuron size in Brodmann areas 44 and 45 in patients with autism. Acta Neuropathol (2012) 124:67–79. doi:10.1007/s00401-012-0976-6
290. Bauman ML, Kemper TL. Neuroanatomic observations of the brain in autism: a review and future directions. Int J Dev Neurosci (2005) 23:183–7. doi:10.1016/j.ijdevneu.2004.09.006
291. Bauman ML, Kemper TL. Histoanatomic observations of the brain in early infantile autism. Neurology (1985) 35:866–7. doi:10.1212/WNL.35.6.866
292. Casanova MF, El-Baz A, Vanbogaert E, Narahari P, Switala A. A topographic study of minicolumnar core width by lamina comparison between autistic subjects and controls: possible minicolumnar disruption due to an anatomical element in-common to multiple laminae. Brain Pathol (2010) 20:451–8. doi:10.1111/j.1750-3639.2009.00319.x
293. Opris I, Casanova MF. Prefrontal cortical minicolumn: from executive control to disrupted cognitive processing. Brain (2014) 137:1863–75. doi:10.1093/brain/awt359
294. Casanova MF, El-Baz AS, Kamat SS, Dombroski BA, Khalifa F, Elnakib A, et al. Focal cortical dysplasias in autism spectrum disorders. Acta Neuropathol Commun (2013) 1:67. doi:10.1186/2051-5960-1-67
295. Alarcón M, Abraham BS, Stone JL, Duvall JA, Perederiy JV, Bomar JM, et al. Linkage, association, and gene expression analyses identify CNTNAP2 as an autism-susceptibility gene. Am J Hum Genet (2008) 82:150–9. doi:10.1016/j.ajhg.2007.09.005
296. Piggot J, Shirinyan D, Shemmassian S, Vazirian S, Alarcón M. Neural systems approaches to the neurogenetics of autism spectrum disorders. Neuroscience (2009) 164:247–56. doi:10.1016/j.neuroscience.2009.05.054
297. Kotagiri P, Chance SA, Szele FG, Esiri MM. Subventricular zone cytoarchitecture changes in autism. Dev Neurobiol (2014) 74:25–41. doi:10.1002/dneu.22127
298. Gozzi M, Nielson DM, Lenroot RK, Ostuni JL, Luckenbaugh DA, Thurm AE, et al. A magnetization transfer imaging study of corpus callosum myelination in young children with autism. Biol Psychiatry (2012) 72:215–20. doi:10.1016/j.biopsych.2012.01.026
299. Fatemi SH, Aldinger KA, Ashwood P, Bauman ML, Blaha CD, Blatt GJ, et al. Consensus paper: pathological role of the cerebellum in autism. Cerebellum (2012) 11:777–807. doi:10.1007/s12311-012-0355-9
300. Ritvo ER, Freeman BJ, Scheibel AB, Duong T, Robinson H, Guthrie D, et al. Lower Purkinje cell counts in the cerebella of four autistic subjects: initial findings of the UCLA-NSAC autopsy research report. Am J Psychiatry (1986) 143:862–6.
301. Gaffney GR, Tsai LY, Kuperman S, Minchin S. Cerebellar structure in autism. Am J Dis Child (1987) 141:1330–2.
302. Whitney ER, Kemper TL, Bauman ML, Rosene DL, Blatt GJ. Cerebellar Purkinje cells are reduced in a subpopulation of autistic brains: a stereological experiment using calbindin-D28k. Cerebellum (2008) 7:406–16. doi:10.1007/s12311-008-0043-y
303. Scott JA, Schumann CM, Goodlin-Jones BL, Amaral DG. A comprehensive volumetric analysis of the cerebellum in children and adolescents with autism spectrum disorder. Autism Res (2009) 2:246–57. doi:10.1002/aur.97
304. Courchesne E, Saitoh O, Yeung-Courchesne R, Press GA, Lincoln AJ, Haas RH, et al. Abnormality of cerebellar vermian lobules VI and VII in patients with infantile autism: identification of hypoplastic and hyperplastic subgroups by MR imaging. AJR Am J Roentgenol (1994) 162:123–30. doi:10.2214/ajr.162.1.8273650
305. Jeong JW, Tiwari VN, Behen ME, Chugani HT, Chugani DC. In vivo detection of reduced Purkinje cell fibers with diffusion MRI tractography in children with autistic spectrum disorders. Front Hum Neurosci (2014) 8:110. doi:10.3389/fnhum.2014.00110
306. Perry EK, Lee ML, Martin-Ruiz CM, Court JA, Volsen SG, Merrit J. Cholinergic activity in autism: abnormalities in the cerebral cortex and basal forebrain. Am J Psychiatry (2001) 158:1058–66. doi:10.1176/appi.ajp.158.7.1058
307. Lee M, Martin-Ruiz C, Graham A, Court J, Jaros E, Perry R. Nicotinic receptor abnormalities in the cerebellar cortex in autism. Brain (2002) 125:1483–95. doi:10.1093/brain/awf160
308. Blatt GJ, Fitzgerald CM, Guptill JT, Booker AB, Kemper TL, Bauman ML. Density and distribution of hippocampal neurotransmitter receptors in autism: an autoradiographic study. J Autism Dev Disord (2001) 31:537–43. doi:10.1023/A:1013238809666
309. Oblak AL, Gibbs TT, Blatt GJ. Decreased GABA(B) receptors in the cingulate cortex and fusiform gyrus in autism. J Neurochem (2010) 114:1414–23. doi:10.1111/j.1471-4159.2010.06858.x
310. Oblak AL, Gibbs TT, Blatt GJ. Reduced GABA(A) receptors and benzodiazepine binding sites in the posterior cingulate cortex and fusiform gyrus in autism. Brain Res (2011) 1380:218–28. doi:10.1016/j.brainres.2010.09.021
311. Fatemi SH, Reutiman TJ, Folsom TD, Thuras PD. GABAA receptor down-regulation in brains of subjects with autism. J Autism Dev Disord (2009) 39:223–30. doi:10.1007/s10803-008-0646-7
312. Fatemi SH, Halt AR, Stary JM, Kanodia R, Schulz SC, Realmuto GR. Glutamic acid decarboxylase 65 and 67 kDa proteins are reduced in autistic parietal and cerebellar cortices. Biol Psychiatry (2002) 52:805–10. doi:10.1016/S0006-3223(02)01430-0
313. Fatemi SH, Folsom TD. Dysregulation of fragile x mental retardation protein and metabotropic glutamate receptor 5 in superior frontal cortex of individuals with autism: a postmortem brain study. Mol Autism (2011) 2:6. doi:10.1186/2040-2392-2-6
314. Fatemi SH, Folsom TD, Kneeland RE, Liesch SB. Metabotropic glutamate receptor 5 up-regulation in children with autism is associated with underexpression of both fragile X mental retardation protein and GABAA receptor beta 3 in adults with autism. Anat Rec (2011) 294:1635–45. doi:10.1002/ar.21299
315. Rustan OG, Folsom TD, Yousefi MK, Fatemi SH. Phosphorylated fragile X mental retardation protein at serine 499, is reduced in cerebellar vermis and superior frontal cortex of subjects with autism: implications for fragile X mental retardation protein-metabotropic glutamate receptor 5 signaling. Mol Autism (2013) 14(1):41. doi:10.1186/2040-2392-4-41
316. Oblak A, Gibbs TT, Blatt GJ. Reduced serotonin receptor subtypes in a limbic and a neocortical region in autism. Autism Res (2013) 6:571–83. doi:10.1002/aur.1317
317. Kang JQ, Barnes G. A common susceptibility factor of both autism and epilepsy: functional deficiency of GABA A receptors. J Autism Dev Disord (2013) 43:68–79. doi:10.1007/s10803-012-1543-7
318. Betancur C, Buxbaum JD. SHANK3 haploinsufficiency: a “common” but underdiagnosed highly penetrant monogenic cause of autism spectrum disorders. Mol Autism (2013) 4:17. doi:10.1186/2040-2392-4-17
319. Abdallah MW, Mortensen EL, Greaves-Lord K, Larsen N. Neonatal levels of neurotrophic factors and risk of autism spectrum disorders. Acta Psychiatr Scand (2013) 128:61–9. doi:10.1111/acps.12020
320. Moessner R, Marshall CR, Sutcliffe JS, Skaug J, Pinto D, Vincent J, et al. Contribution of SHANK3 mutations to autism spectrum disorder. Am J Hum Genet (2007) 81:1289–97. doi:10.1086/522590
321. Betancur C. Etiological heterogeneity in autism spectrum disorders: more than 100 genetic and genomic disorders and still counting. Brain Res (2011) 1380:42–77. doi:10.1016/j.brainres.2010.11.078
322. Chatonnet F, Flamant F, Morte B. A temporary compendium of thyroid hormone target genes in brain. Biochim Biophys Acta (2014). doi:10.1016/j.bbagrm.2014.05.023
323. Pescucci C, Meloni I, Bruttini M, Ariani F, Longo I, Mari F, et al. Chromosome 2 deletion encompassing the MAP2 gene in a patient with autism and Rett-like features. Clin Genet (2003) 64:497–501. doi:10.1046/j.1399-0004.2003.00176.x
324. Kalkman HO. Potential opposite roles of the extracellular signal-regulated kinase (ERK) pathway in autism spectrum and bipolar disorders. Neurosci Biobehav Rev (2012) 36:2206–13. doi:10.1016/j.neubiorev.2012.07.008
325. Waltes R, Duketis E, Knapp M, Anney RJ, Huguet G, Schlitt S, et al. Common variants in genes of the postsynaptic FMRP signalling pathway are risk factors for autism spectrum disorders. Hum Genet (2014) 133:781–92. doi:10.1007/s00439-013-1416-y
326. Cao F, Yin A, Wen G, Sheikh AM, Tauqeer Z, Malik M, et al. Alteration of astrocytes and Wnt/β-catenin signaling in the frontal cortex of autistic subjects. J Neuroinflammation (2012) 9(1):223. doi:10.1186/1742-2094-9-223
327. Chen YZ, Matsushita M, Girirajan S, Lisowski M, Sun E, Sul Y, et al. Evidence for involvement of GNB1L in autism. Am J Med Genet B Neuropsychiatr Genet (2012) 159B:61–71. doi:10.1002/ajmg.b.32002
328. Emanuele E, Orsi P, Barale F, di Nemi SU, Bertona M, Politi P. Serum levels of vascular endothelial growth factor and its receptors in patients with severe autism. Clin Biochem (2010) 43:317–9. doi:10.1016/j.clinbiochem.2009.10.005
329. Kim SJ, Gonen D, Hanna GL, Leventhal BL, Cook EH Jr. Deletion polymorphism in the coding region of the human NESP55 alternative transcript of GNAS1. Mol Cell Probes (2000) 14:191–4. doi:10.1006/mcpr.2000.0300
330. Corbett BA, Kantor AB, Schulman H, Walker WL, Lit L, Ashwood P, et al. A proteomic study of serum from children with autism showing differential expression of apolipoproteins and complement proteins. Mol Psychiatry (2007) 12:292–306. doi:10.1038/sj.mp.4001943
331. Garbett K, Ebert PJ, Mitchell A, Lintas C, Manzi B, Mirnics K, et al. Immune transcriptome alterations in the temporal cortex of subjects with autism. Neurobiol Dis (2008) 30:303–11. doi:10.1016/j.nbd.2008.01.012
332. Stoner R, Chow ML, Boyle MP, Sunkin SM, Mouton PR, Roy S, et al. Patches of disorganization in the neocortex of children with autism. N Engl J Med (2014) 370:1209–19. doi:10.1056/NEJMoa1307491
333. Pickett J, London E. The neuropathology of autism: a review. J Neuropathol Exp Neurol (2005) 64:925–35. doi:10.1097/01.jnen.0000186921.42592.6c
334. Li J, Liu J, Zhao L, Ma Y, Jia M, Lu T, et al. Association study between genes in reelin signaling pathway and autism identifies DAB1 as a susceptibility gene in a Chinese Han population. Prog Neuropsychopharmacol Biol Psychiatry (2013) 44:226–32. doi:10.1016/j.pnpbp.2013.01.004
335. Du X, An Y, Yu L, Liu R, Qin Y, Guo X, et al. A genomic copy number variant analysis implicates the MBD5 and HNRNPU genes in Chinese children with infantile spasms and expands the clinical spectrum of 2q23.1 deletion. BMC Med Genet (2014) 15:62. doi:10.1186/1471-2350-15-62
336. Fatemi SH. Reelin glycoprotein: structure, biology and roles in health and disease. Mol Psychiatry (2005) 10:251–7. doi:10.1038/sj.mp.4001613
337. Persico AM, D’Agruma L, Maiorano N, Totaro A, Militerni R, Bravaccio C, et al. Collaborative linkage study of autism. Reelin gene alleles and haplotypes as a factor predisposing to autistic disorder. Mol Psychiatry (2001) 6:150–9. doi:10.1038/sj.mp.4000850
338. Zhang H, Liu X, Zhang C, Mundo E, Macciardi F, Grayson DR. Reelin gene alleles and susceptibility for autism spectrum disorders. Mol Psychiatry (2002) 7:1012–7. doi:10.1038/sj.mp.4001124
339. Skaar DA, Shao Y, Haines JL, Stenger JE, Jaworski J, Martin ER, et al. Analysis of the RELN gene as a genetic risk factor for autism. Mol Psychiatry (2005) 10:563–71. doi:10.1038/sj.mp.4001614
340. Serajee FJ, Zhong H, Mahbubul Huq AH. Association of reelin gene polymorphisms with autism. Genomics (2006) 87:75–83. doi:10.1016/j.ygeno.2005.09.008
341. Krebs MO, Betancur C, Leroy S, Bourdel MC, Gillberg C, Leboyer M, et al. Absence of association between a polymorphic GGC repeat in the 50 untranslated region of the reelin gene and autism. Mol Psychiatry (2002) 7:801–4. doi:10.1038/sj.mp.4001071
342. Bonora E, Beyer KS, Lamb JA, Parr JR, Klauck SM, Benner A, et al. International molecular genetic study of autism (IMGSAC). Analysis of reelin as a candidate gene for autism. Mol Psychiatry (2003) 10:885–92. doi:10.1038/sj.mp.4001310
343. Devlin B, Bennett P, Dawson G, Figlewicz DA, Grigorenko EL, McMahon W, et al. Alleles of a reelin CGG repeat do not convey liability to autism in a sample from the CPEA network. Am J Med Genet (2004) 126B:46–50. doi:10.1002/ajmg.b.20125
344. Li J, Nguyen L, Gleason C, Lotspeich L, Spiker D, Risch N. Lack of evidence for an association between WNT2 and RELN polymorphisms and autism. Am J Med Genet (2004) 126B:51–7. doi:10.1002/ajmg.b.20122
345. Wang Z, Hong Y, Zou L, Zhong R, Zhu B, Shen N, et al. Reelin gene variants and risk of autism spectrum disorders: an integrated meta-analysis. Am J Med Genet B Neuropsychiatr Genet (2014) 165:192–200. doi:10.1002/ajmg.b.32222
346. Bi C, Wu J, Jiang T, Liu Q, Cai W, Yu P, et al. Mutations of ANK3 identified by exome sequencing are associated with autism susceptibility. Hum Mutat (2012) 33:1635–8. doi:10.1002/humu.22174
347. Turner G, Partington M, Kerr B, Mangelsdorf M, Gecz J. Variable expression of mental retardation, autism, seizures, and dystonic hand movements in two families with an identical ARX gene mutation. Am J Med Genet (2002) 112:405–11. doi:10.1002/ajmg.10714
348. Egger G, Roetzer KM, Noor A, Lionel AC, Mahmood H, Schwarzbraun T, et al. Identification of risk genes for autism spectrum disorder through copy number variation analysis in Austrian families. Neurogenetics (2014) 15:117–27. doi:10.1007/s10048-014-0394-0
349. Nishimura K, Nakamura K, Anitha A, Yamada K, Tsujii M, Iwayama Y, et al. Genetic analyses of the brain-derived neurotrophic factor (BDNF) gene in autism. Biochem Biophys Res Commun (2007) 356:200–6. doi:10.1016/j.bbrc.2007.02.135
350. Zuko A, Kleijer KT, Oguro-Ando A, Kas MJ, van Daalen E, van der Zwaag B, et al. Contactins in the neurobiology of autism. Eur J Pharmacol (2013) 719:63–74. doi:10.1016/j.ejphar.2013.07.016
351. Kim HW, Cho SC, Kim JW, Cho IH, Kim SA, Park M, et al. Family-based association study between NOS-I and -IIA polymorphisms and autism spectrum disorders in Korean trios. Am J Med Genet B Neuropsychiatr Genet (2009) 150B:300–6. doi:10.1002/ajmg.b.30798
352. Yang W, Liu J, Zheng F, Jia M, Zhao L, Lu T, et al. The evidence for association of ATP2B2 polymorphisms with autism in Chinese Han population. PLoS One (2013) 8(4):e61021. doi:10.1371/journal.pone.0061021
353. Morte B, Martínez de Arrieta C, Manzano J, Coloma A, Bernal J. Identification of a cis-acting element that interferes with thyroid hormone induction of the neurogranin (NRGN) gene. FEBS Lett (1999) 464:179–83. doi:10.1016/S0014-5793(99)01706-8
354. Kelleher RJ III, Geigenmüller U, Hovhannisyan H, Trautman E, Pinard R, Rathmell B, et al. High-throughput sequencing of mGluR signaling pathway genes reveals enrichment of rare variants in autism. PLoS One (2012) 7(4):e35003. doi:10.1371/journal.pone.0035003
355. Sicca F, Imbrici P, D’Adamo MC, Moro F, Bonatti F, Brovedani P, et al. Autism with seizures and intellectual disability: possible causative role of gain-of-function of the inwardly-rectifying K+ channel Kir4.1. Neurobiol Dis (2011) 43:239–47. doi:10.1016/j.nbd.2011.03.016
356. Lassig JP, Vachirasomtoon K, Hartzell K, Leventhal M, Courchesne E, Courchesne R, et al. Physical mapping of the serotonin 5-HT(7) receptor gene (HTR7) to chromosome 10 and pseudogene (HTR7P) to chromosome 12, and testing of linkage disequilibrium between HTR7 and autistic disorder. Am J Med Genet (1999) 88:472–5. doi:10.1002/(SICI)1096-8628(19991015)88:5<472::AID-AJMG7>3.0.CO;2-G
357. Gadad BS, Hewitson L, Young KA, German DC. Neuropathology and animal models of autism: genetic and environmental factors. Autism Res Treat (2013) 2013:731935. doi:10.1155/2013/731935
358. Provenzano G, Zunino G, Genovesi S, Sgadó P, Bozzi Y. Mutant mouse models of autism spectrum disorders. Dis Markers (2012) 33:225–39. doi:10.3233/DMA-2012-0917
359. Sadamatsu M, Kanai H, Xu X, Liu Y, Kato N. Review of animal models for autism: implication of thyroid hormone. Congenit Anom (Kyoto) (2006) 46:1–9. doi:10.1111/j.1741-4520.2006.00094.x
360. Moy SS, Riddick NV, Nikolova VD, Teng BL, Agster KL, Nonneman RJ, et al. Repetitive behavior profile and supersensitivity to amphetamine in the C58/J mouse model of autism. Behav Brain Res (2014) 259:200–14. doi:10.1016/j.bbr.2013.10.052
361. Sgadò P, Genovesi S, Kalinovsky A, Zunino G, Macchi F, Allegra M, et al. Loss of GABAergic neurons in the hippocampus and cerebral cortex of engrailed-2 null mutant mice: implications for autism spectrum disorders. Exp Neurol (2013) 247:496–505. doi:10.1016/j.expneurol.2013.01.021
362. Sgadò P, Provenzano G, Dassi E, Adami V, Zunino G, Genovesi S, et al. Transcriptome profiling in engrailed-2 mutant mice reveals common molecular pathways associated with autism spectrum disorders. Mol Autism (2013) 4(1):5. doi:10.1186/2040-2392-4-51
363. Burket JA, Benson AD, Tang AH, Deutsch SI. Rapamycin improves sociability in the BTBR T(+)Itpr3(tf)/J mouse model of autism spectrum disorders. Brain Res Bull (2014) 100:70–5. doi:10.1016/j.brainresbull.2013.11.005
364. Ellegood J, Babineau BA, Henkelman RM, Lerch JP, Crawley JN. Neuroanatomical analysis of the BTBR mouse model of autism using magnetic resonance imaging and diffusion tensor imaging. Neuroimage (2012) 70:288–300. doi:10.1016/j.neuroimage.2012.12.029
365. Uchino S, Waga C. SHANK3 as an autism spectrum disorder-associated gene. Brain Dev (2013) 35:106–10. doi:10.1016/j.braindev.2012.05.013
366. Waga C, Asano H, Sanagi T, Suzuki E, Nakamura Y, Tsuchiya A, et al. Identification of two novel Shank3 transcripts in the developing mouse neocortex. J Neurochem (2014) 128:280–93. doi:10.1111/jnc.12505
367. Almeida LE, Roby CD, Krueger BK. Increased BDNF expression in fetal brain in the valproic acid model of autism. Mol Cell Neurosci (2014) 59C:57–62. doi:10.1016/j.mcn.2014.01.007
368. Martin HG, Manzoni OJ. Late onset deficits in synaptic plasticity in the valproic acid rat model of autism. Front Cell Neurosci (2014) 8:23. doi:10.3389/fncel.2014.00023
369. Chomiak T, Hung J, Cihal A, Dhaliwal J, Baghdadwala M, Dzwonek A, et al. Auditory-cued sensorimotor task reveals disengagement deficits in rats exposed to the autism-associated teratogen valproic acid. Neuroscience (2014) 268:212–20. doi:10.1016/j.neuroscience.2014.02.049
370. Chomiak T, Hu B. Alterations of neocortical development and maturation in autism: insight from valproic acid exposure and animal models of autism. Neurotoxicol Teratol (2013) 36:57–66. doi:10.1016/j.ntt.2012.08.005
371. Engineer CT, Centanni TM, Im KW, Borland MS, Moreno NA, Carraway RS, et al. Degraded auditory processing in a rat model of autism limits the speech representation in non-primary auditory cortex. Dev Neurobiol (2014). doi:10.1002/dneu.22175
372. Sahu JK, Gulati S, Kabra M, Arya R, Sharma R, Gupta N, et al. Evaluation of subclinical hypothyroidism in ambulatory children with controlled epilepsy on valproate monotherapy. J Child Neurol (2012) 27:594–7. doi:10.1177/0883073811421985
Keywords: evo-devo, cortical development, autism, thyroid hormones, hypothyroidism
Citation: Berbel P, Navarro D and Román GC (2014) An evo-devo approach to thyroid hormones in cerebral and cerebellar cortical development: etiological implications for autism. Front. Endocrinol. 5:146. doi: 10.3389/fendo.2014.00146
Received: 12 April 2014; Accepted: 25 August 2014;
Published online: 09 September 2014.
Edited by:
Frédéric Flamant, Ecole Normale Supérieure de Lyon, FranceReviewed by:
Giorgio Innocenti, Karolinska Institutet, SwedenConstantino Sotelo, University Pierre and Marie Curie, France
Joseph V. Martin, Rutgers University, USA
Robert Thomas Zoeller, University of Massachusetts Amherst, USA
Copyright: © 2014 Berbel, Navarro and Román. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Pere Berbel, Instituto de Neurociencias, Universidad Miguel Hernández, Av. Ramón y Cajal s/n, Sant Joan d’Alacant, Alicante 03550, Spain e-mail:cGVyZS5iZXJiZWxAdW1oLmVz