 Rabi Yacoub
Rabi Yacoub Kyung Lee
Kyung Lee John Cijiang He
John Cijiang He- Division of Nephrology, Department of Medicine, Icahn School of Medicine at Mount Sinai, New York, NY, USA
Sirtuins (SIRTs) are members of the silent information regulator 2 family. In mammals, of the seven known SIRTs, SIRT1 function is most studied and has been shown to regulate wide range of cellular functions that affect metabolic homeostasis and aging. SIRT1 exerts anti-apoptotic, anti-oxidative, and anti-inflammatory effects against cellular injury, and protects the cells through the regulation of mitochondrial biogenesis, autophagy, and metabolism in response to the cellular energy and redox status. SIRT1 also promotes vasodilation and protects vascular tissues. In humans and animal models with diabetic kidney disease (DKD), its expression tends to be decreased in renal cells, and increased expression of SIRT1 was found to play a renal protective role in animal models with DKD. In this review, we discuss the role and potential mechanisms by which SIRT1 protects against DKD.
Introduction
Diabetes mellitus (DM) is a major medical problem worldwide. It is the underlying cause of microvascular disorders such as diabetic nephropathy and retinopathy and macrovascular diseases such as coronary artery and peripheral vascular diseases. Currently, more than 347 million people worldwide are suffering from DM (1), and the World Health Organization projects that it will be the seventh leading cause of death by 2030. The increased prevalence of DM has led to a significant increase in the prevalence of diabetic kidney disease (DKD) with estimates that 44% of all new end stage renal disease (ESRD) cases in US are due to DKD (2, 3). Several factors including hyperglycemia, insulin resistance, renal lipid accumulation, inflammation, and activation of the renin–angiotensin system (RAS) are involved in the pathogenesis of DKD (4) and they activate multiple signaling pathways resulting in kidney cell injury and the development and progression of the disease (5, 6).
Since the discovery of the silent information regulator 2 (Sir2) family and its beneficial effects on aging (7, 8), scientists have shown that the homologs of Sir2 in higher eukaryotic organisms, known as Sirtuins (SIRTs), are a conserved family of a nicotinamide adenine dinucleotide (NAD+)-dependent deacetylases/mono-ADP ribosyltransferases that are associated with numerous cellular signaling pathways that include senescence (9–12), apoptosis (13), DNA damage repair (14), and autophagy (12, 15). By far, SIRT1 is the most studied member of this family and its protective roles against kidney injury are well established, making it a promising candidate for targeted therapies to halt disease progression.
General Cellular Functions of SIRT1
SIRT1 exerts its cytoprotective effects through various mechanisms. It has anti-apoptotic, anti-oxidative, and anti-inflammatory effects, along with its regulation of mitochondrial biogenesis and autophagy (Figure 1).
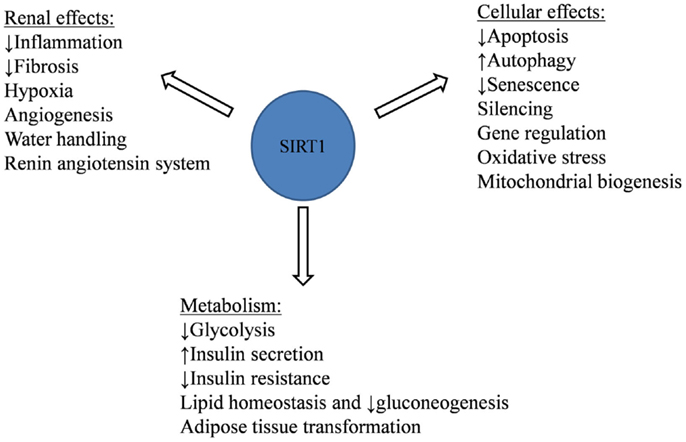
Figure 1. SIRT1 cellular, renal, and metabolic protective effects against aging-related and metabolic diseases.
Cellular Senescence, Oxidative Stress, and Mitochondrial Dysfunction
Aging is considered the most important contributor to the etiologies of metabolic decline and related diseases (16). This process is thought to be due mainly to the accumulation of oxidative stress related mitochondrial DNA (mtDNA) damages, leading to mitochondrial dysfunction (17). Increasing Sirt1 expression by calorie restriction (CR) in mice abrogated the dysmorphic mitochondrial appearances associated with aging (e.g., mitochondrial swelling and disintegration of cristae), whereas decreasing Sirt1 expression resulted in an early aging phenotype in mice, revealing the important role of SIRT1 on cellular senescence and other aging-related diseases (9–12).
Increased oxidative stress has been associated with aging, and SIRT1 has been shown to combat oxidative stress by modulating transcriptional activities of several key proteins involved in oxidative stress response and mitochondrial biogenesis. Peroxisome proliferator-activated receptor-gamma coactivator-1α (PGC-1α) is a transcriptional factor involved in lipid homeostasis and in mitochondrial biogenesis, which prevents and protects against oxidative stress (18). PGC-1α activity appears to be regulated by SIRT1 deacetylation in a tissue specific manner (19). Deacetylation of PGC-1α by SIRT1 has been observed in skeletal muscles, where PGC-1α deacetylation increases mitochondrial biogenesis, mass, and improves exercise endurance (20), and in brown adipose tissue (BAT), and endothelial cells (21).
Recent evidence suggests that SIRT1 can regulate the expression of a longevity gene p66Shc. p66Shc is one of three isoforms encoded by the proto-oncogene SHC (Src homologous and collagen) and has been shown to promote oxidative stress, leading to mitochondrial dysfunction, senescence, and apoptosis (22, 23). Deletion of p66Shc in Akita diabetic mice showed a renoprotective phenotype that included the attenuation of oxidative stress and glomerular/tubular injury and reduction in albuminuria (24, 25).
Several studies have shown that SIRT1 negatively regulates expression of p66Shc through deacetylation of histone H3 on its promoter (26). Treating different cell lines that do not usually express p66Shc with histone deacetylase inhibitors induced p66Shc expression (23, 26), and deletion of p66Shc in Akita diabetic mice resulted in upregulation in SIRT1 expression in mice kidneys and ameliorated kidney fibrosis and preserved podocytes mass and function (24).
Autophagy and Apoptosis
Autophagy and apoptosis are two closely related processes that are triggered by common upstream signaling pathways to constitute a stress adaptation, where in general autophagy inhibits apoptosis to maintain survival (27, 28). SIRT1 exerts an anti-apoptotic and pro-autophagic responses in cells under stress conditions by directly deacetylating essential autophagy proteins (Atg), such as Atg5, Atg7, and Atg8 (29), and by deacetylation of transcription factors, such as FOXO3a, to increase the expression of autophagy proteins (12, 15). Deacetylation of FOXO3a by SIRT1 also prevents apoptosis by enhancing the expression of p27Kip1, a cyclin-dependent kinase inhibitor that causes G1 arrest to maintain cell viability (12, 30).
In addition, SIRT1 has been shown to deacetylate and inactivate the transcriptional activity of p53, a tumor suppressor responsible of maintaining cellular integrity by inducing cell-cycle arrest, and if necessary evoking apoptotic cell death (31). With aging, the depletion of NAD+ storage attenuates SIRT1 activity, leading to hyperacetylation of p53. p53 has been shown to stimulate or repress autophagy depending on its subcellular localization, where cytoplasmic p53 promotes apoptosis and inhibits autophagy (32). While it is known that SIRT1 regulates p53 function by deacetylation, whether it affects its cytoplasmic localization is not known (33).
Adipose Tissue Transformation
One of the mechanism by which CR is thought to extend life span is through fat mobilization. Upon CR, SIRT1 binds and represses the fat regulator peroxisome proliferator-activated receptor-gamma (PPARγ), attenuating adipogenesis, and triggering lipolysis (34). It also selectively decreases white adipose genes Angiotensinogen (Agt), Resistin, Wdnm1L, Chemerin, and Pank3 (35). PPARγ deacetylation by SIRT1 causes also a lipid transformation from white adipose tissue (WAT) to BAT through regulating ligand-dependent coactivator/corepressor exchange at the PPARγ transcriptional complex (35). WAT distribution affects metabolic risk and is linked to metabolic diseases as obesity, diabetes, and dyslipidemia (36). The metabolic benefits of this conversion include prevention of diet-induced obesity and increased insulin sensitivity (37).
Role of SIRT1 in DM
It is well established that the risk of micro and macrovascular complications in patients with DM is closely related to the glycemic control. In the fasting state, hyperglycemia is directly related to hepatic glucose production, which in turn, along with the decreased insulin production or increased insulin resistance, is responsible for the hyperglycemia in the postprandial state (38). SIRT1 participates in regulating glucose homeostasis through regulating hepatic glucose production, lipid metabolism and insulin production, and sensitivity (39–42).
Hyperglycemia
SIRT1 decreases hepatic glucose production via deacetylation and activation of the AMPK kinase LKB1 (39). When activated, AMPK switches off hepatic glucose, cholesterol, and triglyceride productions and promotes fatty acid oxidation. AMPK in turn also activates SIRT1 via increasing its substrate NAD+ (43). This reciprocal activation/dynamic interaction between AMPK and SIRT1 is disrupted by hyperglycemia, which decreases AMPK expression, leading to reduced SIRT1 expression (44).
Lipid Metabolism and Insulin Production and Sensitivity
Under the fasting state, hepatic SIRT1 regulates lipid homeostasis and gluconeogenesis by positively regulating PPARα and its coactivator PGC-1α (10). In addition, SIRT1 also suppresses glycolysis via deacetylation of phosphoglycerate mutase-1 (PGAM1) and decreasing the expression of glycolysis genes glucokinase (GK) and liver pyruvate kinase (LPK), while PGC-1α increases the expressions of GK and LPK (10, 40). In contrast, under the feeding state hepatic SIRT1 negatively regulates gluconeogenesis via mTorc2/Akt signaling pathway, resulting in decreased transcription of gluconeogenic genes glucose-6-phosphatase (G6pase) and phosphoenolpyruvate carboxykinase (Pepck) (45). Experimental mice model of hepatic Sirt1 deficiency displayed hyperglycemia, glucose intolerance, hepatic insulin resistance, and oxidative stress in insulin-sensitive organs through disrupted mTorc2/Akt signaling (45). This bimodal regulation of gluconeogenesis under feeding/fasting states by SIRT1 promotes adaptation to nutrient deprivation (10, 41).
SIRT1 also enhances insulin secretion from the pancreatic beta cells by regulating the expression of uncoupling protein 2 (Ucp2) (42), and decreases insulin resistance via reducing the expression of the proteins in the insulin receptor signaling pathway, such as protein tyrosine phosphatase 1B (PTP1B), and by inhibiting insulin-induced IRS-2 (insulin receptor substrate-2) tyrosine phosphorylation by deacetylation (46, 47).
Expression of SIRT1 in Diabetic Kidney
SIRT1 expression changes under different physiological and morbid conditions. It is decreased in conditions of chronic metabolic stress, oxidative stress, or hypoxia that drives the pathophysiologies of age-related diseases including diabetes, cardiovascular, and renal diseases. In aging kidneys both the expression and activity of SIRT1 is decreased due to age-associated reduction in systemic NAD+ biosynthesis (12). Similarly, reduction in SIRT1 expression was observed in kidney glomeruli and tubulointerstitial compartments of patients with mild to severe DKD, which was inversely correlated with the histopathological severity of the renal disease and with the amount of proteinuria (48, 49). Experimental mouse models of DM similarly showed a loss of Sirt1 in renal proximal tubules (PT) and podocytes (48, 49), where the loss of PT Sirt1 preceded the loss of podocyte Sirt1 and the concomitant albuminuria. Interestingly, restoration of PT Sirt1 was sufficient to increase pore densities in podocytes and to mitigate albuminuria and worsening of DKD (49), suggesting that PT Sirt1 confers protection in maintaining the glomerular structure and function during the early stages of DKD and that therapeutic agents that increase the renal SIRT1 expression and activity may have a favorable impact in slowing the disease progression of DKD.
Role and Cellular Mechanisms of SIRT1 in DKD
Among many studies indicating SIRT1’s protective role in numerous different cell types, several studies have described its protective role in different kidney cells. Some of these effects are in the general context of SIRT1’s cytoprotective actions, while others seem to have a unique specificity to specialized renal cells. In the following section, we will discuss the deleterious effects of reduced SIRT1 expression on kidney cells, and the mechanisms by which SIRT1 is found to exert its benefits on renal cells and on the concomitant inflammation, angiogenesis, and fibrosis, all of which contributes to the progression of DKD (Table 1).
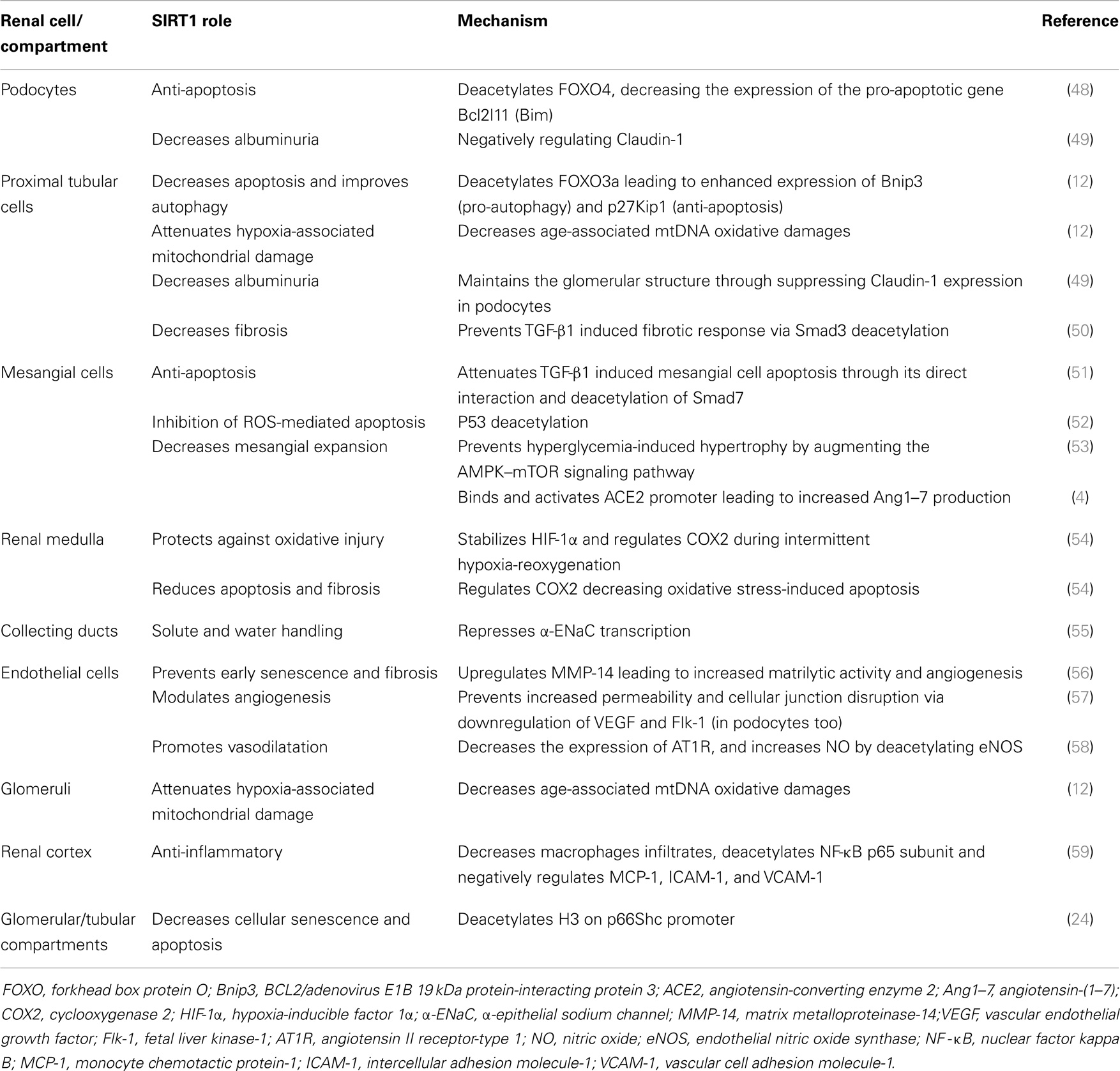
Table 1. Role and cellular mechanisms of SIRT1 in DKD.
Podocytes
One of the earliest changes in DKD is the loss of podocytes, leading to proteinuria and further kidney damage (60–62). In DKD, podocyte apoptosis is aggravated by hyperglycemia via increasing the production of advanced glycation end products (AGEs), which in turn increases FOXO4 acetylation and suppresses SIRT1 expression (48). This decreased SIRT1 expression leads to the accumulation of acetylated FOXO4 and to the expression of the pro-apoptotic gene Bcl2l11 (also known as Bim), resulting in apoptosis (48). Hyperglycemia also stimulates the generation of intracellular reactive oxygen species (ROS) through NADPH oxidase and mitochondrial pathways, leading to activation of the pro-apoptotic p38 mitogen-activated protein kinase (p38 MAPK) and Caspase 3 in podocytes (5).
Proximal Tubular Cells
Tubular SIRT1 has been shown to affect podocyte function via maintaining a high glomerular nicotinamide mononucleotide (NMN) concentration via diffusion of the NMN produced in the PTs (49, 63). Decreased expression of NMN has been observed in DKD (64), which is likely to be the first pathological changes preceding proteinuria (65). PT SIRT1 communicates with podocytes through the release of NMN. PT SIRT1 appears to negatively modulate the expression of the tight junction protein Claudin-1 (49). In healthy conditions, Claudin-1 is not expressed in podocytes but primarily expressed on glomerular parietal cells. However, in diabetic mice models podocytes express Claudin-1, possibly as a part of a podocyte dedifferentiation mechanism that occurs in DKD or by reorganization of the podocyte actin cytoskeleton (63). SIRT1 also protects PTs under hypoxic conditions by inducing autophagy and inhibiting apoptosis (12, 24).
Renal Medulla
In a healthy individual, a quarter of the cardiac output is directed to the kidneys, with most of this flow going to the cortex to optimize glomerular filtration. However, the renal medulla’s blood flow is low to preserve osmotic gradient and enhance concentration ability. The renal medulla is also under chronic and constant oxidative stress due to the rapid change in interstitial tonicity and the low oxygen tension (66, 67). A small percentage of oxygen consumed by the mitochondria is incompletely reduced to ROS, which then targets the other mitochondrial components and augments the generation of increased ROS by the injured mitochondria. Under these conditions, renal mitochondria undergo a constant autophagy process. To cope with hypoxia, higher organisms’ adaptive mechanisms includes switching energy metabolism from oxygen phosphorylation to HIF-1 mediated anaerobic glycolysis (68), which concurrently blocks mitochondrial energy metabolism and biogenesis (69). SIRT1 is normally expressed in the inner medulla and is upregulated during intermittent hypoxia-reoxygenation and protects against oxidative stress via stabilizing HIF-1α and regulating of cyclooxygenase 2 (COX2) (54, 70). However in chronic hypoxic state, the renal medulla endures in DKD, SIRT1 activity is inhibited due to decreased NAD+ (58).
Mesangial Cells
Mesangial injury and expansion mark early histological changes in DKD, and they are correlated closely with the degree of albuminuria. SIRT1 attenuates TGF-β1 induced mesangial cell apoptosis through its direct interaction and deacetylation of Smad7, enhancing its ubiquitin-mediated proteasome degradation (51). SIRT1 also prevents high glucose-induced mesangial cells hypertrophy by augmenting the AMPK–mTOR signaling pathway (53) and subsequently blocking the activation of Akt signaling (71). NAD+ treatment mitigates the high glucose-induced Akt and mTOR phosphorylation in cultured mesangial cells (53).
Endothelial Cells and Angiogenesis
As in diabetic retinopathy, new vessel formation is observed in DKD patients (72) and in animal models (73), contributing to its pathogenesis. Early on in DKD, there is an increase in endothelial cells number, caused by the imbalance between proliferation and apoptosis, where VEGF-A appears to be the major driver of this imbalance. Other factors affecting angiogenesis include nitric oxide deficiency, glomerular hypertension (74), altered expressions of VEGF receptors 1 and 2, Angiopoietin 2, and Tie-2 (57, 75, 76). In vivo and in vitro studies have shown that resveratrol (RSV), a SIRT1 activator, downregulates high glucose-induced VEGF-A and Flk-1 (VEGFR-2) expressions in both glomerular podocytes and endothelial cells. RSV also inhibits VEGF-A induced increased permeability and cellular junction disruption of cultured endothelial cells (57). SIRT1 also maintains endothelial cells function and prevents early senescence via upregulating matrix metalloproteinase-14 (MMP-14), an important factor for endothelial cells regeneration after injury. MMP-14 cleavage products serve as a ligand for epidermal growth factor (EGF) receptors (56). Endothelial cells have also been shown to express early senescence features in presence of high glucose through the down regulation of SIRT1 expression, leading to an increased acetylation of FOXO1 by p300 (77).
Inflammation
Microinflammation of the glomeruli and tubulointerstitial regions and subsequent extracellular matrix expansion are common pathways for the progression of DKD, which occurs in response to renal damage as a defense mechanism. This dynamic process of pro-inflammatory macrophages M1 and anti-inflammatory macrophages M2 recruitment to the kidneys eventually leads to kidney fibrosis, highlighting the importance of inflammation as a therapy target to slow the DKD progression (78). SIRT1 can deacetylate NF-κB p65 subunit and negatively regulate the NF-κB signaling mediated expression of the inflammation-related proteins monocyte chemotactic protein-1 (MCP-1), intercellular adhesion molecule-1 (ICAM-1), and vascular cell adhesion protein-1 (VCAM-1) (59). Dietary restriction in Wistar fatty rates restores Sirt1 expression and ameliorates diabetic nephropathy abnormalities (i.e., albuminuria, mesangial matrix expansion, and renal fibrosis), which is in part mediated by Sirt1’s anti-inflammatory effects as evidenced by decreased macrophages infiltrates and changes in expression of NF-κB p65, MCP-1, ICAM-1, and VCAM-1 (59).
Fibrosis
Kidney fibrosis is the final outcome of progressive DKD, and it results in a significant destruction of normal kidney structure accompanied by functional deterioration. TGF-β1 is upregulated in response to various kidney injury stimuli, causing renal fibrosis and epithelial–mesenchymal transformation of the renal tubules (79). Recent studies have identified Smad2 and Smad3 acetylation in response to TGF-β1 stimulation (50, 80, 81) and shown that the RSV treatment of cultured PTs leads to deacetylation of Smad3 (50). In addition, RSV administration abolished TGF-β1/Smad3 induced up-regulation of α-SMA, collagen IV, and fibronectin in UUO mouse model of kidney fibrosis, suggesting that SIRT1 activity may be essential in preventing TGF-β1 induced fibrotic response via Smad3 deacetylation (50).
Renin-Angiotensin System and SIRT1
Renin–angiotensin system, especially Angiotensin II (AngII) is closely associated with the development and progression of DKD. RAS inhibition by angiotensin-converting enzyme inhibitors (ACEi), which inhibits the conversion of AngI to AngII, or angiotensin receptors blockers (ARBs) decreases proteinuria in patients with diabetic nephropathy and halts the disease progression to ESRD, thus improving patient survival (82). Angiotensin-converting enzyme 2 (ACE2), a homolog of ACE not inhibited by ACEi, counteracts AngII effects by hydrolyzing AngII into Angiotensin 1–7 (Ang 1–7), which in turn protects against DKD by increasing tissue triglyceride degradation and decreasing kidney weight, mesangial expansion, proteinuria, and renal fibrosis (4). SIRT1 regulates RAS by binding and activating ACE2 promoter, leading to increased Ang 1–7 concentrations (4, 44). It also promotes vasodilatation by decreasing the expression of the potent vasoconstrictor angiotensin II receptor-type I (AT1R), and protects vascular tissues through increased nitric oxide production by deacetylating eNOS in endothelial cells (58).
Water Handling
Diabetes mellitus is associated with a significant polyuria and natriuresis, as well as increased plasma aldosterone and anti-diuretic hormone arginine vasopressin (AVP) levels. Studies have identified serum and glucocorticoid induced kinase-1 (SGK-1) as a key signaling element in diabetic nephropathy (83, 84). Under the hyperglycemic state the increase in Ca2+ and TGF-β1 leads to upregulation of SGK-1 in kidney, which regulates epithelial Na+ channel (ENaC) activation, leading to increased sodium absorption (85). A physiological role for SIRT1 in regulating the α-ENaC expression has been reported in cultured renal inner medullary collecting duct cells (mIMCD3) (55). SIRT1 interacts with Dot (disruptor of telomeric silencing)-1, a histone H3K79 methyltransferase, and enhances its activities on histone H3K79 methylation in chromatin along the α-ENaC promoter, and thereby repressing α-ENaC transcription in mIMCD3 cells (55).
Conclusion and Prospects
Since the first description of the Sir2 family and its effects on longevity in yeast, our understanding of the specific actions and role of SIRT1 on different kidney diseases have increased tremendously. Administration of SIRT1 activators showed a restoration of SIRT1 levels, decreased albuminuria, glomerular hypertrophy, and kidney fibrosis in DKD models (86–90). The mechanisms by which these activators exert their beneficiary effects are currently under extensive research, highlighting the importance of developing therapies to increase either SIRT1 expression or activity in patients with DKD to prevent disease progression. In addition, single nucleotide polymorphisms (SNPs) within the gene encoding SIRT1 have shown to have a directionally consistent association with diabetic nephropathy leading to the assumption that SIRT1 not only play a protective role, but certain SNPs variations of SIRT1 might predispose an individual to DKD (91). These findings warrant further investigations into the functions of these SNPs variations, and to develop new strategies for protection against renal diseases.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1. Danaei G, Finucane MM, Lu Y, Singh GM, Cowan MJ, Paciorek CJ, et al. National, regional, and global trends in fasting plasma glucose and diabetes prevalence since 1980: systematic analysis of health examination surveys and epidemiological studies with 370 country-years and 2.7 million participants. Lancet (2011) 378(9785):31–40. doi: 10.1016/S0140-6736(11)60679-X
2. Mauer M, Zinman B, Gardiner R, Suissa S, Sinaiko A, Strand T, et al. Renal and retinal effects of enalapril and losartan in type 1 diabetes. N Engl J Med (2009) 361(1):40–51. doi:10.1056/NEJMoa0808400
3. Foley RN, Collins AJ. End-stage renal disease in the United States: an update from the United States Renal Data System. J Am Soc Nephrol (2007) 18(10):2644–8. doi:10.1681/ASN.2007020220
4. Mori J, Patel VB, Ramprasath T, Alrob OA, DesAulniers J, Scholey JW, et al. Angiotensin 1-7 mediates renoprotection against diabetic nephropathy by reducing oxidative stress, inflammation, and lipotoxicity. Am J Physiol Renal Physiol (2014) 306(8):F812–21. doi:10.1152/ajprenal.00655.2013
5. Susztak K, Raff AC, Schiffer M, Bottinger EP. Glucose-induced reactive oxygen species cause apoptosis of podocytes and podocyte depletion at the onset of diabetic nephropathy. Diabetes (2006) 55(1):225–33. doi:10.2337/diabetes.55.01.06.db05-0894
6. Chuang PY, Yu Q, Fang W, Uribarri J, He JC. Advanced glycation endproducts induce podocyte apoptosis by activation of the FOXO4 transcription factor. Kidney Int (2007) 72(8):965–76. doi:10.1038/sj.ki.5002456
7. Sinclair DA, Guarente L. Extrachromosomal rDNA circles – a cause of aging in yeast. Cell (1997) 91(7):1033–42. doi:10.1016/S0092-8674(00)80493-6
8. Kaeberlein M, McVey M, Guarente L. The SIR2/3/4 complex and SIR2 alone promote longevity in Saccharomyces cerevisiae by two different mechanisms. Genes Dev (1999) 13(19):2570–80. doi:10.1101/gad.13.19.2570
9. Cohen HY, Miller C, Bitterman KJ, Wall NR, Hekking B, Kessler B, et al. Calorie restriction promotes mammalian cell survival by inducing the SIRT1 deacetylase. Science (2004) 305(5682):390–2. doi:10.1126/science.1099196
10. Rodgers JT, Lerin C, Haas W, Gygi SP, Spiegelman BM, Puigserver P. Nutrient control of glucose homeostasis through a complex of PGC-1alpha and SIRT1. Nature (2005) 434(7029):113–8. doi:10.1038/nature03354
11. Bordone L, Cohen D, Robinson A, Motta MC, van Veen E, Czopik A, et al. SIRT1 transgenic mice show phenotypes resembling calorie restriction. Aging Cell (2007) 6(6):759–67. doi:10.1111/j.1474-9726.2007.00335.x
12. Kume S, Uzu T, Horiike K, Chin-Kanasaki M, Isshiki K, Araki S, et al. Calorie restriction enhances cell adaptation to hypoxia through Sirt1-dependent mitochondrial autophagy in mouse aged kidney. J Clin Invest (2010) 120(4):1043–55. doi:10.1172/JCI41376
13. Vaziri H, Dessain SK, Ng Eaton E, Imai SI, Frye RA, Pandita TK, et al. hSIR2(SIRT1) functions as an NAD-dependent p53 deacetylase. Cell (2001) 107(2):149–59. doi:10.1016/S0092-8674(01)00527-X
14. Guarente L. Sir2 links chromatin silencing, metabolism, and aging. Genes Dev (2000) 14(9):1021–6. doi:10.1101/gad.14.9.1021
15. Nath KA. The role of Sirt1 in renal rejuvenation and resistance to stress. J Clin Invest (2010) 120(4):1026–8. doi:10.1172/JCI42184
16. Barzilai N, Huffman DM, Muzumdar RH, Bartke A. The critical role of metabolic pathways in aging. Diabetes (2012) 61(6):1315–22. doi:10.2337/db11-1300
17. Hiona A, Sanz A, Kujoth GC, Pamplona R, Seo AY, Hofer T, et al. Mitochondrial DNA mutations induce mitochondrial dysfunction, apoptosis and sarcopenia in skeletal muscle of mitochondrial DNA mutator mice. PLoS One (2010) 5(7):e11468. doi:10.1371/journal.pone.0011468
18. Valle I, Alvarez-Barrientos A, Arza E, Lamas S, Monsalve M. PGC-1alpha regulates the mitochondrial antioxidant defense system in vascular endothelial cells. Cardiovasc Res (2005) 66(3):562–73. doi:10.1016/j.cardiores.2005.01.026
19. Xu Q, Xia P, Li X, Wang W, Liu Z, Gao X. Tetramethylpyrazine ameliorates high glucose-induced endothelial dysfunction by increasing mitochondrial biogenesis. PLoS One (2014) 9(2):e88243. doi:10.1371/journal.pone.0088243
20. Higashida K, Kim SH, Jung SR, Asaka M, Holloszy JO, Han DH. Effects of resveratrol and SIRT1 on PGC-1alpha activity and mitochondrial biogenesis: a reevaluation. PLoS Biol (2013) 11(7):e1001603. doi:10.1371/journal.pbio.1001603
21. Lagouge M, Argmann C, Gerhart-Hines Z, Meziane H, Lerin C, Daussin F, et al. Resveratrol improves mitochondrial function and protects against metabolic disease by activating SIRT1 and PGC-1alpha. Cell (2006) 127(6):1109–22. doi:10.1016/j.cell.2006.11.013
22. Menini S, Amadio L, Oddi G, Ricci C, Pesce C, Pugliese F, et al. Deletion of p66Shc longevity gene protects against experimental diabetic glomerulopathy by preventing diabetes-induced oxidative stress. Diabetes (2006) 55(6):1642–50. doi:10.2337/db05-1477
23. Ventura A, Luzi L, Pacini S, Baldari CT, Pelicci PG. The p66Shc longevity gene is silenced through epigenetic modifications of an alternative promoter. J Biol Chem (2002) 277(25):22370–6. doi:10.1074/jbc.M200280200
24. Vashistha H, Meggs L. Diabetic nephropathy: lessons from the mouse. Ochsner J (2013) 13(1):140–6. doi:10.1043/1524-5012-13.1.140
25. Vashistha H, Singhal PC, Malhotra A, Husain M, Mathieson P, Saleem MA, et al. Null mutations at the p66 and bradykinin 2 receptor loci induce divergent phenotypes in the diabetic kidney. Am J Physiol Renal Physiol (2012) 303(12):F1629–40. doi:10.1152/ajprenal.00246.2012
26. Zhou S, Chen HZ, Wan YZ, Zhang QJ, Wei YS, Huang S, et al. Repression of P66Shc expression by SIRT1 contributes to the prevention of hyperglycemia-induced endothelial dysfunction. Circ Res (2011) 109(6):639–48. doi:10.1161/CIRCRESAHA.111.243592
27. Marino G, Niso-Santano M, Baehrecke EH, Kroemer G. Self-consumption: the interplay of autophagy and apoptosis. Nat Rev Mol Cell Biol (2014) 15(2):81–94. doi:10.1038/nrm3735
28. Maiuri MC, Zalckvar E, Kimchi A, Kroemer G. Self-eating and self-killing: crosstalk between autophagy and apoptosis. Nat Rev Mol Cell Biol (2007) 8(9):741–52. doi:10.1038/nrm2239
29. Lee IH, Cao L, Mostoslavsky R, Lombard DB, Liu J, Bruns NE, et al. A role for the NAD-dependent deacetylase Sirt1 in the regulation of autophagy. Proc Natl Acad Sci USA (2008) 105(9):3374–9. doi:10.1073/pnas.0712145105
30. Hiromura K, Pippin JW, Fero ML, Roberts JM, Shankland SJ. Modulation of apoptosis by the cyclin-dependent kinase inhibitor p27(Kip1). J Clin Invest (1999) 103(5):597–604. doi:10.1172/JCI5461
31. Kitada M, Kume S, Takeda-Watanabe A, Kanasaki K, Koya D. Sirtuins and renal diseases: relationship with aging and diabetic nephropathy. Clin Sci (2013) 124(3):153–64. doi:10.1042/CS20120190
32. Tasdemir E, Maiuri MC, Galluzzi L, Vitale I, Djavaheri-Mergny M, D’Amelio M, et al. Regulation of autophagy by cytoplasmic p53. Nat Cell Biol (2008) 10(6):676–87. doi:10.1038/ncb1730
33. Salminen A, Kaarniranta K. SIRT1: regulation of longevity via autophagy. Cell Signal (2009) 21(9):1356–60. doi:10.1016/j.cellsig.2009.02.014
34. Picard F, Kurtev M, Chung N, Topark-Ngarm A, Senawong T, Machado De Oliveira R, et al. Sirt1 promotes fat mobilization in white adipocytes by repressing PPAR-gamma. Nature (2004) 429(6993):771–6. doi:10.1038/nature02583
35. Qiang L, Wang L, Kon N, Zhao W, Lee S, Zhang Y, et al. Brown remodeling of white adipose tissue by SirT1-dependent deacetylation of Ppargamma. Cell (2012) 150(3):620–32. doi:10.1016/j.cell.2012.06.027
36. Gesta S, Tseng YH, Kahn CR. Developmental origin of fat: tracking obesity to its source. Cell (2007) 131(2):242–56. doi:10.1016/j.cell.2007.10.004
37. Seale P, Conroe HM, Estall J, Kajimura S, Frontini A, Ishibashi J, et al. Prdm16 determines the thermogenic program of subcutaneous white adipose tissue in mice. J Clin Invest (2011) 121(1):96–105. doi:10.1172/JCI44271
38. Inzucchi SE, Bergenstal RM, Buse JB, Diamant M, Ferrannini E, Nauck M, et al. Management of hyperglycemia in type 2 diabetes: a patient-centered approach: position statement of the American Diabetes Association (ADA) and the European Association for the Study of Diabetes (EASD). Diabetes Care (2012) 35(6):1364–79. doi:10.2337/dc12-0413
39. Price NL, Gomes AP, Ling AJ, Duarte FV, Martin-Montalvo A, North BJ, et al. SIRT1 is required for AMPK activation and the beneficial effects of resveratrol on mitochondrial function. Cell Metab (2012) 15(5):675–90. doi:10.1016/j.cmet.2012.04.003
40. Hallows WC, Yu W, Denu JM. Regulation of glycolytic enzyme phosphoglycerate mutase-1 by Sirt1 protein-mediated deacetylation. J Biol Chem (2012) 287(6):3850–8. doi:10.1074/jbc.M111.317404
41. Purushotham A, Schug TT, Xu Q, Surapureddi S, Guo X, Li X. Hepatocyte-specific deletion of SIRT1 alters fatty acid metabolism and results in hepatic steatosis and inflammation. Cell Metab (2009) 9(4):327–38. doi:10.1016/j.cmet.2009.02.006
42. Moynihan KA, Grimm AA, Plueger MM, Bernal-Mizrachi E, Ford E, Cras-Meneur C, et al. Increased dosage of mammalian Sir2 in pancreatic beta cells enhances glucose-stimulated insulin secretion in mice. Cell Metab (2005) 2(2):105–17. doi:10.1016/j.cmet.2005.07.001
43. Canto C, Gerhart-Hines Z, Feige JN, Lagouge M, Noriega L, Milne JC, et al. AMPK regulates energy expenditure by modulating NAD+ metabolism and SIRT1 activity. Nature (2009) 458(7241):1056–60. doi:10.1038/nature07813
44. Clarke NE, Belyaev ND, Lambert DW, Turner AJ. Epigenetic regulation of angiotensin-converting enzyme 2 (ACE2) by SIRT1 under conditions of cell energy stress. Clin Sci (2014) 126(7):507–16. doi:10.1042/CS20130291
45. Wang RH, Kim HS, Xiao C, Xu X, Gavrilova O, Deng CX. Hepatic Sirt1 deficiency in mice impairs mTorc2/Akt signaling and results in hyperglycemia, oxidative damage, and insulin resistance. J Clin Invest (2011) 121(11):4477–90. doi:10.1172/JCI46243
46. Sun C, Zhang F, Ge X, Yan T, Chen X, Shi X, et al. SIRT1 improves insulin sensitivity under insulin-resistant conditions by repressing PTP1B. Cell Metab (2007) 6(4):307–19. doi:10.1016/j.cmet.2007.08.014
47. Zhang J. The direct involvement of SirT1 in insulin-induced insulin receptor substrate-2 tyrosine phosphorylation. J Biol Chem (2007) 282(47):34356–64. doi:10.1074/jbc.M706644200
48. Chuang PY, Dai Y, Liu R, He H, Kretzler M, Jim B, et al. Alteration of forkhead box O (foxo4) acetylation mediates apoptosis of podocytes in diabetes mellitus. PLoS One (2011) 6(8):e23566. doi:10.1371/journal.pone.0023566
49. Hasegawa K, Wakino S, Simic P, Sakamaki Y, Minakuchi H, Fujimura K, et al. Renal tubular Sirt1 attenuates diabetic albuminuria by epigenetically suppressing Claudin-1 overexpression in podocytes. Nat Med (2013) 19(11):1496–504. doi:10.1038/nm.3363
50. Li J, Qu X, Ricardo SD, Bertram JF, Nikolic-Paterson DJ. Resveratrol inhibits renal fibrosis in the obstructed kidney: potential role in deacetylation of Smad3. Am J Pathol (2010) 177(3):1065–71. doi:10.2353/ajpath.2010.090923
51. Kume S, Haneda M, Kanasaki K, Sugimoto T, Araki S, Isshiki K, et al. SIRT1 inhibits transforming growth factor beta-induced apoptosis in glomerular mesangial cells via Smad7 deacetylation. J Biol Chem (2007) 282(1):151–8. doi:10.1074/jbc.M605904200
52. Kume S, Haneda M, Kanasaki K, Sugimoto T, Araki S, Isono M, et al. Silent information regulator 2 (SIRT1) attenuates oxidative stress-induced mesangial cell apoptosis via p53 deacetylation. Free Radic Biol Med (2006) 40(12):2175–82. doi:10.1016/j.freeradbiomed.2006.02.014
53. Zhuo L, Fu B, Bai X, Zhang B, Wu L, Cui J, et al. NAD blocks high glucose induced mesangial hypertrophy via activation of the sirtuins-AMPK-mTOR pathway. Cell Physiol Biochem (2011) 27(6):681–90. doi:10.1159/000330077
54. He W, Wang Y, Zhang MZ, You L, Davis LS, Fan H, et al. Sirt1 activation protects the mouse renal medulla from oxidative injury. J Clin Invest (2010) 120(4):1056–68. doi:10.1172/JCI41563
55. Zhang D, Li S, Cruz P, Kone BC. Sirtuin 1 functionally and physically interacts with disruptor of telomeric silencing-1 to regulate alpha-ENaC transcription in collecting duct. J Biol Chem (2009) 284(31):20917–26. doi:10.1074/jbc.M109.020073
56. Vasko R, Xavier S, Chen J, Lin CH, Ratliff B, Rabadi M, et al. Endothelial sirtuin 1 deficiency perpetrates nephrosclerosis through downregulation of matrix metalloproteinase-14: relevance to fibrosis of vascular senescence. J Am Soc Nephrol (2014) 25(2):276–91. doi:10.1681/ASN.2013010069
57. Wen D, Huang X, Zhang M, Zhang L, Chen J, Gu Y, et al. Resveratrol attenuates diabetic nephropathy via modulating angiogenesis. PLoS One (2013) 8(12):e82336. doi:10.1371/journal.pone.0082336
58. Miyazaki R, Ichiki T, Hashimoto T, Inanaga K, Imayama I, Sadoshima J, et al. SIRT1, a longevity gene, downregulates angiotensin II type 1 receptor expression in vascular smooth muscle cells. Arterioscler Thromb Vasc Biol (2008) 28(7):1263–9. doi:10.1161/ATVBAHA.108.166991
59. Kitada M, Takeda A, Nagai T, Ito H, Kanasaki K, Koya D. Dietary restriction ameliorates diabetic nephropathy through anti-inflammatory effects and regulation of the autophagy via restoration of Sirt1 in diabetic Wistar fatty (fa/fa) rats: a model of type 2 diabetes. Exp Diabetes Res (2011) 2011:908185. doi:10.1155/2011/908185
60. Matsusaka T, Sandgren E, Shintani A, Kon V, Pastan I, Fogo AB, et al. Podocyte injury damages other podocytes. J Am Soc Nephrol (2011) 22(7):1275–85. doi:10.1681/ASN.2010090963
61. Mundel P, Shankland SJ. Podocyte biology and response to injury. J Am Soc Nephrol (2002) 13(12):3005–15. doi:10.1097/01.ASN.0000039661.06947.FD
62. Eid AA, Gorin Y, Fagg BM, Maalouf R, Barnes JL, Block K, et al. Mechanisms of podocyte injury in diabetes: role of cytochrome P450 and NADPH oxidases. Diabetes (2009) 58(5):1201–11. doi:10.2337/db08-1536
63. Nihalani D, Susztak K. Sirt1-Claudin-1 crosstalk regulates renal function. Nat Med (2013) 19(11):1371–2. doi:10.1038/nm.3386
64. Yoshino J, Mills KF, Yoon MJ, Imai S. Nicotinamide mononucleotide, a key NAD(+) intermediate, treats the pathophysiology of diet- and age-induced diabetes in mice. Cell Metab (2011) 14(4):528–36. doi:10.1016/j.cmet.2011.08.014
65. Thomas MC, Tikellis C, Kantharidis P, Burns WC, Cooper ME, Forbes JM. The role of advanced glycation in reduced organic cation transport associated with experimental diabetes. J Pharmacol Exp Ther (2004) 311(2):456–66. doi:10.1124/jpet.104.070672
66. Chou SY, Porush JG, Faubert PF. Renal medullary circulation: hormonal control. Kidney Int (1990) 37(1):1–13. doi:10.1038/ki.1990.1
67. Brezis M, Rosen S. Hypoxia of the renal medulla – its implications for disease. N Engl J Med (1995) 332(10):647–55. doi:10.1056/NEJM199503093321006
68. Lim JH, Lee YM, Chun YS, Chen J, Kim JE, Park JW. Sirtuin 1 modulates cellular responses to hypoxia by deacetylating hypoxia-inducible factor 1alpha. Mol Cell (2010) 38(6):864–78. doi:10.1016/j.molcel.2010.05.023
69. Kim JW, Tchernyshyov I, Semenza GL, Dang CV. HIF-1-mediated expression of pyruvate dehydrogenase kinase: a metabolic switch required for cellular adaptation to hypoxia. Cell Metab (2006) 3(3):177–85. doi:10.1016/j.cmet.2006.02.002
70. Rane S, He M, Sayed D, Vashistha H, Malhotra A, Sadoshima J, et al. Downregulation of miR-199a derepresses hypoxia-inducible factor-1alpha and Sirtuin 1 and recapitulates hypoxia preconditioning in cardiac myocytes. Circ Res (2009) 104(7):879–86. doi:10.1161/CIRCRESAHA.108.193102
71. Chan AY, Dolinsky VW, Soltys CL, Viollet B, Baksh S, Light PE, et al. Resveratrol inhibits cardiac hypertrophy via AMP-activated protein kinase and Akt. J Biol Chem (2008) 283(35):24194–201. doi:10.1074/jbc.M802869200
72. Nakagawa T, Kosugi T, Haneda M, Rivard CJ, Long DA. Abnormal angiogenesis in diabetic nephropathy. Diabetes (2009) 58(7):1471–8. doi:10.2337/db09-0119
73. Cooper ME, Vranes D, Youssef S, Stacker SA, Cox AJ, Rizkalla B, et al. Increased renal expression of vascular endothelial growth factor (VEGF) and its receptor VEGFR-2 in experimental diabetes. Diabetes (1999) 48(11):2229–39. doi:10.2337/diabetes.48.11.2229
74. Hohenstein B, Hausknecht B, Boehmer K, Riess R, Brekken RA, Hugo CP. Local VEGF activity but not VEGF expression is tightly regulated during diabetic nephropathy in man. Kidney Int (2006) 69(9):1654–61. doi:10.1038/sj.ki.5000294
75. Suri C, Jones PF, Patan S, Bartunkova S, Maisonpierre PC, Davis S, et al. Requisite role of angiopoietin-1, a ligand for the TIE2 receptor, during embryonic angiogenesis. Cell (1996) 87(7):1171–80. doi:10.1016/S0092-8674(00)81813-9
76. Maisonpierre PC, Suri C, Jones PF, Bartunkova S, Wiegand SJ, Radziejewski C, et al. Angiopoietin-2, a natural antagonist for Tie2 that disrupts in vivo angiogenesis. Science (1997) 277(5322):55–60. doi:10.1126/science.277.5322.55
77. Mortuza R, Chen S, Feng B, Sen S, Chakrabarti S. High glucose induced alteration of SIRTs in endothelial cells causes rapid aging in a p300 and FOXO regulated pathway. PLoS One (2013) 8(1):e54514. doi:10.1371/journal.pone.0054514
78. Wada J, Makino H. Inflammation and the pathogenesis of diabetic nephropathy. Clin Sci (2013) 124(3):139–52. doi:10.1042/CS20120198
79. Sato M, Muragaki Y, Saika S, Roberts AB, Ooshima A. Targeted disruption of TGF-beta1/Smad3 signaling protects against renal tubulointerstitial fibrosis induced by unilateral ureteral obstruction. J Clin Invest (2003) 112(10):1486–94. doi:10.1172/JCI19270
80. Simonsson M, Kanduri M, Gronroos E, Heldin CH, Ericsson J. The DNA binding activities of Smad2 and Smad3 are regulated by coactivator-mediated acetylation. J Biol Chem (2006) 281(52):39870–80. doi:10.1074/jbc.M607868200
81. Inoue Y, Itoh Y, Abe K, Okamoto T, Daitoku H, Fukamizu A, et al. Smad3 is acetylated by p300/CBP to regulate its transactivation activity. Oncogene (2007) 26(4):500–8. doi:10.1038/sj.onc.1209826
82. Brenner BM, Cooper ME, de Zeeuw D, Keane WF, Mitch WE, Parving HH, et al. Effects of losartan on renal and cardiovascular outcomes in patients with type 2 diabetes and nephropathy. N Engl J Med (2001) 345(12):861–9. doi:10.1056/NEJMoa011161
83. Terada Y, Kuwana H, Kobayashi T, Okado T, Suzuki N, Yoshimoto T, et al. Aldosterone-stimulated SGK1 activity mediates profibrotic signaling in the mesangium. J Am Soc Nephrol (2008) 19(2):298–309. doi:10.1681/ASN.2007050531
84. Panchapakesan U, Pollock C, Saad S. Renal epidermal growth factor receptor: its role in sodium and water homeostasis in diabetic nephropathy. Clin Exp Pharmacol Physiol (2011) 38(2):84–8. doi:10.1111/j.1440-1681.2010.05472.x
85. Hills CE, Bland R, Bennett J, Ronco PM, Squires PE. High glucose up-regulates ENaC and SGK1 expression in HCD-cells. Cell Physiol Biochem (2006) 18(6):337–46. doi:10.1159/000097611
86. Chen KH, Hung CC, Hsu HH, Jing YH, Yang CW, Chen JK. Resveratrol ameliorates early diabetic nephropathy associated with suppression of augmented TGF-beta/smad and ERK1/2 signaling in streptozotocin-induced diabetic rats. Chem Biol Interact (2011) 190(1):45–53. doi:10.1016/j.cbi.2011.01.033
87. Lee MJ, Feliers D, Sataranatarajan K, Mariappan MM, Li M, Barnes JL, et al. Resveratrol ameliorates high glucose-induced protein synthesis in glomerular epithelial cells. Cell Signal (2010) 22(1):65–70. doi:10.1016/j.cellsig.2009.09.011
88. Tikoo K, Singh K, Kabra D, Sharma V, Gaikwad A. Change in histone H3 phosphorylation, MAP kinase p38, SIR 2 and p53 expression by resveratrol in preventing streptozotocin induced type I diabetic nephropathy. Free Radic Res (2008) 42(4):397–404. doi:10.1080/10715760801998646
89. Wu L, Zhang Y, Ma X, Zhang N, Qin G. The effect of resveratrol on FoxO1 expression in kidneys of diabetic nephropathy rats. Mol Biol Rep (2012) 39(9):9085–93. doi:10.1007/s11033-012-1780-z
90. Xu Y, Nie L, Yin YG, Tang JL, Zhou JY, Li DD, et al. Resveratrol protects against hyperglycemia-induced oxidative damage to mitochondria by activating SIRT1 in rat mesangial cells. Toxicol Appl Pharmacol (2012) 259(3):395–401. doi:10.1016/j.taap.2011.09.028
Keywords: SIRT1, diabetic kidney disease, senescence, deacetylation, apoptosis, autophagy, diabetes mellitus, sirtuin
Citation: Yacoub R, Lee K and He JC (2014) The role of SIRT1 in diabetic kidney disease. Front. Endocrinol. 5:166. doi: 10.3389/fendo.2014.00166
Received: 29 August 2014; Accepted: 25 September 2014;
Published online: 09 October 2014.
Edited by:
Sonia Q. Doi, Uniformed Services University, USAReviewed by:
Subrata Chakrabarti, The University of Western Ontario, CanadaThierry Durroux, Centre National de la Recherche Scientifique, France
Copyright: © 2014 Yacoub, Lee and He. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: John Cijiang He, Division of Nephrology, Department of Medicine, Icahn School of Medicine at Mount Sinai, One Gustave L Levy Place, Box 1243, New York, NY 10029, USA e-mail:Y2lqaWFuZy5oZUBtc3NtLmVkdQ==