 Sophie J. Gilbert1
Sophie J. Gilbert1 Lee B. Meakin2
Lee B. Meakin2 Cleo S. Bonnet1
Cleo S. Bonnet1 Mari A. Nowell3Warren C. Ladiges4John Morton4Victor C. Duance1
Mari A. Nowell3Warren C. Ladiges4John Morton4Victor C. Duance1 Deborah J. Mason1*
Deborah J. Mason1*- 1Pathophysiology and Repair Division, Arthritis Research UK Biomechanics and Bioengineering Centre, School of Biosciences, Cardiff University, Cardiff, UK
- 2School of Veterinary Sciences, University of Bristol, Bristol, UK
- 3Section of Inflammation, Skin and Joint Disease, Institute of Infection and Immunity, School of Medicine, Cardiff University, Cardiff, UK
- 4Department of Comparative Medicine, School of Medicine, University of Washington, Seattle, WA, USA
Objective: Protein kinase-like endoplasmic reticulum kinase (PERK) and protein kinase R (PKR) are implicated in endoplasmic reticulum stress-induced arthritis and pro-inflammatory cytokine-mediated cartilage degradation in vitro, respectively. We determined whether knockout of the cellular inhibitor of PERK and PKR, P58IPK causes joint degeneration in vivo and whether these molecules are activated in human osteoarthritis (OA).
Materials and Methods: Sections of knee joints from P58IPK-null and wild-type mice aged 12–13 and 23–25 months were stained with toluidine blue and scored for degeneration using the osteoarthritis research society international (OARSI) system. Bone changes were assessed by radiology and high-resolution micro-computed tomography of hind limbs. Sections from the medial tibial plateaus of two human knees, removed in total knee replacement surgery for OA, were immunolabelled for phosphorylated PERK and PKR and P58IPK.
Results: Knockout mice exhibited narrower tibiae (p = 0.0031) and smaller epiphyses in tibiae (p = 0.0004) and femora (p = 0.0214). Older knockout mice had reduced total volume inside the femoral periosteal envelope (p = 0.023), reduced tibial (p = 0.03), and femoral (p = 0.0012) bone volumes (BV) and reduced femoral BV fraction (p = 0.025). Compared with wild-types, younger P58IPK-null mice had increased OARSI scores in medial femoral condyles (p = 0.035). Thirty four percent of null mice displayed severe joint degeneration with complete articular cartilage loss from the medial compartment and heterotopic chondro-osseous tissue in the medial joint capsule. Phosphorylated PERK and PKR were localized throughout human osteoarthritic tibial plateaus but, in particular, in areas exhibiting the most degeneration. There was limited expression of P58IPK.
Conclusion: This study is the first to reveal a critical role for P58IPK in maintaining joint integrity in vivo, implicating the PKR and PERK stress signaling pathways in bony changes underlying the pathogenesis of joint degeneration.
Introduction
Osteoarthritis (OA) is a disease of the synovial joint associated with pathological changes in the bone and cartilage that cause pain and disability. Abnormal mechanical load and obesity represent primary risk factors for OA development and progression (1, 2). Several studies have implicated the stress-activated protein kinase R (PKR) and protein kinase-like endoplasmic reticulum kinase (PERK) signaling pathways in the pathogenesis of arthritic disease (3–9). PKR is constitutively expressed in all cells, with levels increasing upon cellular stress [reviewed in Ref. (10)]. In chondrocytes, constitutive PKR expression is known to be high (8), with further activation (phosphorylation) resulting from cellular stress leading to up-regulation of matrix degrading enzymes (3, 8), proteoglycan degradation (3, 4), phosphorylation of eukaryotic initiation factor 2 alpha (eIF2α) (11) and down-regulation of protein synthesis [reviewed in Ref. (10)]. Furthermore, the PKR pathway is mechano-responsive, gene expression being up-regulated (12) at the onset of load-induced OA (13) and mediating translational arrest induced by cyclic mechanical load in articular cartilage (14). The cellular level of PKR increases as tissues age, concomitant with a decrease in the rate of protein synthesis (15), implicating PKR in the pathogenesis of age-related diseases such as OA. PERK (16) and PKR (17, 18) are also phosphorylated during endoplasmic reticulum (ER) stress, leading to eIF2α phosphorylation and down-regulation of protein synthesis, thus, reducing the load of protein passing through the ER. Cells that have a high-secretory capacity, including pancreatic β cells, chondrocytes, and osteoblasts, are hyper-sensitive to ER stress induction (19–21).
P58IPK (58 kDa inhibitor protein kinase), is a member of the heat shock protein 40 family and has several known interacting partners allowing it to have diverse functions in the cell; it is localized to the cytosol where it acts as a cellular inhibitor of PKR and PERK activity and thus regulator of global protein synthesis and translocates to the ER during ER stress whereby it acts as a chaperone in the ER lumen. It is ubiquitously expressed, abundant in secretory cells of the pancreas and liver (15, 22), shows increased expression with age (15) and plays important roles in maintaining cellular homeostasis (23–27). In its protective role as a molecular chaperone, it interacts with unfolded proteins such as luciferase, rhodanese, and insulin to prevent their aggregation (28) and binds BiP to facilitate misfolded protein folding (29, 30). In addition, it has also been shown to bind to the ER protein translocation channel Sec61 to play a role in ER protein degradation (31). P58IPK mRNA and protein levels are increased in response to ER stress to inhibit PERK and PKR activity and restore homeostasis (23, 24, 32). Knockdown of P58IPK using siRNA in HEK-293 and HeLa cells increased levels of total PKR and phosphorylated PERK, with subsequent eIF2α phosphorylation, inhibition of protein synthesis and apoptosis (24). Knockdown of P58IPK increased release of tumor necrosis factor-alpha (TNF-α) in rat retinal cells (27), phosphorylation of eIF2α and expression of ER stress response proteins in embryonic stem cells (23). Furthermore, P58IPK-null cells have heightened basal levels of ER stress (26). P58IPK-null mice are smaller with lower body weights than heterozygous and wild-type mice, reduced body fat, hypoinsulinemia, and gradual onset diabetes (25, 26). However, up until now, skeletal phenotype has not been assessed in P58IPK-null mice.
Given the role of PERK and PKR in cell signaling events associated with arthritis, we tested the hypothesis that absence of P58IPK results in a degenerative joint phenotype in vivo. The aims of this observational study were to describe a phenotypic characterization of donated knee joints from a P58IPK knockout mouse colony.
Materials and Methods
Materials
All chemicals were obtained from Sigma (Poole, UK) unless otherwise stated and were of analytical grade.
Tissue
Human OA
The medial tibial plateau (MTP) was obtained from two randomly selected patients undergoing total knee replacement for OA (female, 83 and 59 years old). Informed consent was gained from each donor according to protocols approved by the Research Ethics Committee for Wales (Ref: 10/MRE0928; Cardiff & Vale Heath Board R&D approval Ref: 10/OAE/497).
P58IPK-null mice
Hind limbs were obtained from previously generated male P58IPK-null and age-matched wild-type mouse colonies as described (25) and approved by the University of Washington Institutional Animal Care and Use Committee. Two age groups were analyzed: 12–13 and 23–25 months. Mice were 100% C57Bl/6 and had been maintained on a C57Bl/6 background by alternate generation backcrossing. All comparisons are between null and wild-types of 12–13 months (four null, and three wild-type) and 23–25 months mice (five null, four wild-type). Where analyses deviate from this standard comparison, the numbers of animals used are clearly stated in the figure legends.
Specimen Preparation
Human material
Tissue was fixed (2 days, 10% neutral buffered formalin), decalcified (4°C, 10% EDTA, Fisher Scientific), paraffin embedded, and coronally sectioned (6 μm).
Mouse material
Hind limbs were immediately fixed post-mortem in formalin. For histology and immunohistochemistry, right knee joints were decalcified as described in Section “Human Material.” Joints were fixed at 90° and embedded frontally in paraffin blocks for coronal sectioning parallel to the tibia. Serial sections (5 μm) obtained at 100 μm intervals through the joint, were dewaxed and rehydrated prior to staining with toluidine blue or processing for immunohistochemistry. Left knee joints were stored in 70% ethanol prior to radiological and high-resolution micro-computed tomography (μCT) analysis.
Histological Scoring
Human MTP
The human MTPs had been previously stained with toluidine blue (33) and scored for degenerative changes. Sections were shown to have cartilage fibrillation, proteoglycan loss, and breach of the tidemark. Cartilage degradation severity (indicated by increasing Mankin score) increased toward the outer edge of the MTP (33).
Mouse knees
The medial femoral condyle (MFC), MTP, lateral femoral condyle (LFC), and lateral tibial plateau (LTP) from two sections (either side of the center of the joint, approximately 200 μm apart) were scored for degenerative changes by two independent observers blinded to genotype. For each mouse, a single score representing the mean value from both observers and sections was used for statistical comparisons. The osteoarthritis research society international (OARSI) scoring method (34) was used with toluidine blue staining utilized instead of safranin O. The OARSI system uses a 0–6 subjective score for osteoarthritic changes (Table S1 in Supplementary Material: parameter 1), 0–3 for subchondral bone changes (Table S1 in Supplementary Material: parameter 2), and 0–5 for proteoglycan depletion (Table S1 in Supplementary Material: parameter 3). This gives a total score of 14 for each quadrant (parameters 1–3).
Radiological Analysis
A KODAK In vivo Imaging System FX Pro (AMV, Lincoln, UK) was used to produce high-resolution digital radiographs of medial and frontal views of the left mouse hind legs. Capture settings were as follows: 2× binning, 0.4 mm aluminum filter, 30 s exposure, KVP 35, F-stop 3.99, FOV 22.2 mm. A region of interest (ROI), of the same size and shape, and located as near to the inferior tibio-fibular joint as possible, was defined (see Figure S1 in Supplementary Material). Placing the ROI as close to this joint as possible provided us with a landmark allowing us to make direct comparisons of the same region across animals. The exact location was defined from the Chi-square value, derived from the bone density analysis, which is a measure of the fitting quality in the non-linear least-squares fit of the cylinder to the bone segment. The ROI was analyzed for changes in long-bone density (g/cm3) using Carestream Molecular Imaging Software (version 5.3.3) with bone density software module (35). In addition, a ROI was drawn around any calcification within the Achilles to measure its area (pixels) and a linear ROI used to measure the height and width of the tibia at the tibio-fibular joint (millimeter) (Figure S1 in Supplementary Material).
High-Resolution μCT Analysis
Left mouse knees were imaged using the SkyScan 1172 (Bruker, Kontich, Belgium) with a voxel size of 4.8 μm (110 mm3) using previously reported scanning and reconstruction methods (36). The ROI incorporated 25 lines (0.25 mm) starting from the central line of the epiphyses and moving proximally for the femur and distally for the tibia. We evaluated the effect of genotype and age on epiphyseal total volume inside the periosteal envelope (TV), bone volume (BV), medullary volume (MV), and bone volume fraction (BV/TV) according to ASBMR guidelines (37). Femoral and tibial epiphyseal height, excluding the growth plate, is also reported (Figure S1 in Supplementary Material) and 3D reconstructions generated.
Immunohistochemistry
Active PERK and PKR and their endogenous inhibitor were immunolocalized in sequential sections from two human MTP using antibodies to phosphorylated PERK (Santa Cruz: sc-32577; 1:50), phosphorylated PKR (Santa Cruz: sc-101783; 1:50), and P58IPK (Abcam: ac70840; 1:20). Sections were deparaffinized and rehydrated prior to antigen retrieval (1 mg/mL trypsin for 1 h at 37°C). Each subsequent step was performed at room temperature unless stated otherwise and between each incubation step, sections were washed 3 min × 5 min in 0.01 M phosphate buffered saline (PBS, pH 7.4) containing 0.001% (v/v) Tween 20 (wash buffer). All antibodies were diluted in wash buffer. Endogenous peroxidase activity was blocked with 0.3% (v/v) hydrogen peroxide for 30 min. Sections were subsequently treated with 10% normal goat serum for 1 h prior to overnight incubation (4°C) with primary antibody. Biotinylated secondary antibody was applied and incubated for 30 min detection (Vectastain Elite ABC kit, nickel enhanced diaminobenzidine, Vector Laboratories). Sections were finally dehydrated, cleared in xylene, and mounted. Slides were viewed on a Leica DMRB microscope. IgG controls were negative (Figure S2 in Supplementary Material).
Data Analysis
For all statistics, unless stated otherwise, hindlimb characteristics were compared between null and wild-type mice of 12–13 months (four null, and three wild-type) and 23–25 months mice (five null, four wild-type). To assess and compare sample distributions, graphs show box and whisker plots of minimum and maximum values, 25th and 75th quartiles and median; outliers revealed by these plots were removed prior to further analysis. Data were tested for normality and equal variances prior to analysis (Minitab 16). Data were analyzed by general linear model analysis of variance (GLM ANOVA) with two factors (age and genotype) followed by Tukey’s post hoc test. Differences were considered significant at p ≤ 0.05.
Results
P58IPK-Null Mice have Narrower Tibias and Shorter Epiphyses
P58IPK-null mice exhibit a small body phenotype (Figure S3 in Supplementary Material) (25). Since no previous analysis of the long bones has been performed, we measured tibial length, width, and density, as well as height of the epiphyses. Loss of P58IPK appeared not to affect the overall length of the tibia (Figure 1A; p = 0.886). However, P58IPK-null mice had significantly shorter tibial (p = 0.0004) and femoral epiphyses (p = 0.0214) compared to wild-type mice (Figures 1B,C). This difference occurred in the tibial epiphyses of both 12–13-month-old (p = 0.032) and 23–25-month (p = 0.016) old null mice compared to the age-matched wild-types. In addition, null mice exhibited a significant reduction in the width of their tibiae at the tibia/fibula intersect when compared to wild-type mice (p = 0.0031; Figure 1D). There appeared to be no difference in the densities of the long-bone situated close to the inferior tibio-fibular joint of wild-type and null mice (Figure 1E).
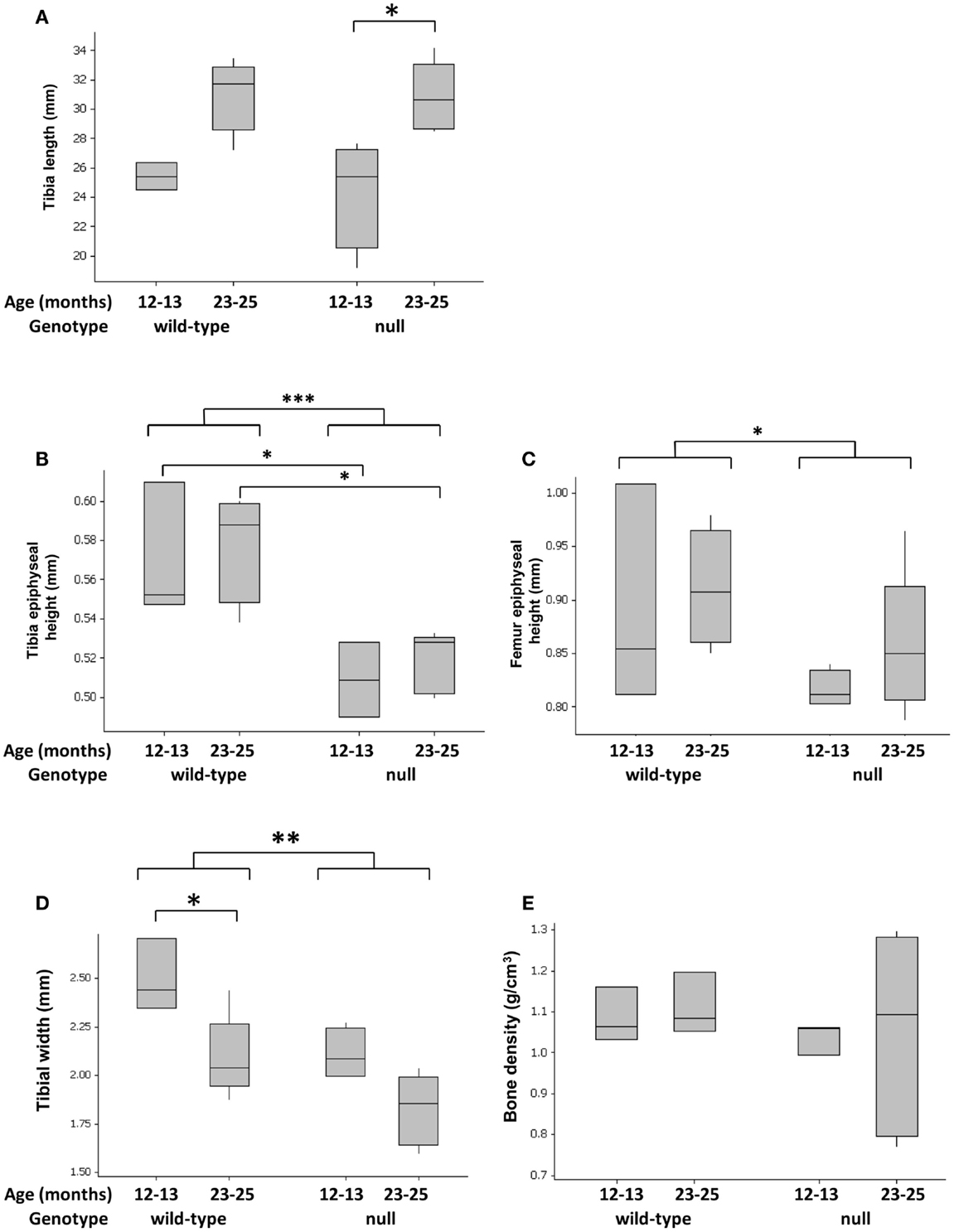
Figure 1. Tibial widths and epiphyseal heights are reduced in P58IPK-null mice. Radiographs and high-resolution μCT of the left limb from wild-type and null mice were used to measure tibial lengths (A), tibial epiphyseal heights (B), femoral lengths (C), and tibiae widths at the tibia/fibula intersect (D). Tibial bone density was measured close to the inferior tibio-fibular joint of wild-type and null mice (E), [Figure S1 in Supplementary Material]. Significant differences were detected by GLM ANOVA and Tukey’s post hoc test: *p ≤ 0.05; **p < 0.01; ***p < 0.001. For tibial length, width, and bone density measurements: 12–13-month-old mice n = 4 null, n = 3 wild-type and 23–25-month-old mice n = 5 null, n = 4 wild-type. For micro CT measurements: 12–13-month-old mice n = 3 null, n = 3 wild-type and 23–25-month-old mice n = 5 null, n = 4 wild-type.
Loss of P58IPK Causes Joint Degeneration
Joint degeneration was observed in both P58IPK-null (Figure 2A) and wild-type (Figure 2B) mice of both ages. These degenerative changes included cartilage tears (red arrows), subchondral bone changes (yellow arrows), and osteophyte formation (green arrows). To assess whether loss of P58IPK resulted in joint degeneration over and above that expected in aging C57Bl/6 mice (38–41), knees were scored using an adaptation of the OARSI system (34). Despite the presence of degeneration in both groups, GLM ANOVA of OARSI scores revealed that 12–13-month-old null mice had fourfold higher total scores (sum of all three parameters; Table S1 in Supplementary Material) in the MFC compartment than their wild-type, age-matched controls (Figure 2C; p = 0.035 following rank transformation of data). Analysis of individual parameters revealed that there was a weak, but inconclusive increase in the MFC bone score in null mice (parameter 2) (Figure 2D; GLM ANOVA genotype p = 0.051 following log transformation of data) and that 12–13-month-old null mice had 6.7-fold higher bone scores (parameter 2) than age-matched wild-types (Figure 2D; p = 0.024 following log transformation of data). P58IPK-null mice had 1.5-fold lower total scores (sum of all three parameters; Table S1 in Supplementary Material) in the LTP compared to wild-type mice, but this difference was not significant (Figure 2E; GLM ANOVA genotype p = 0.086). However, OA scores (parameter 1) were significantly reduced in the LTP (Figure 2F; GLM ANOVA genotype p = 0.033 following log transformation of data).
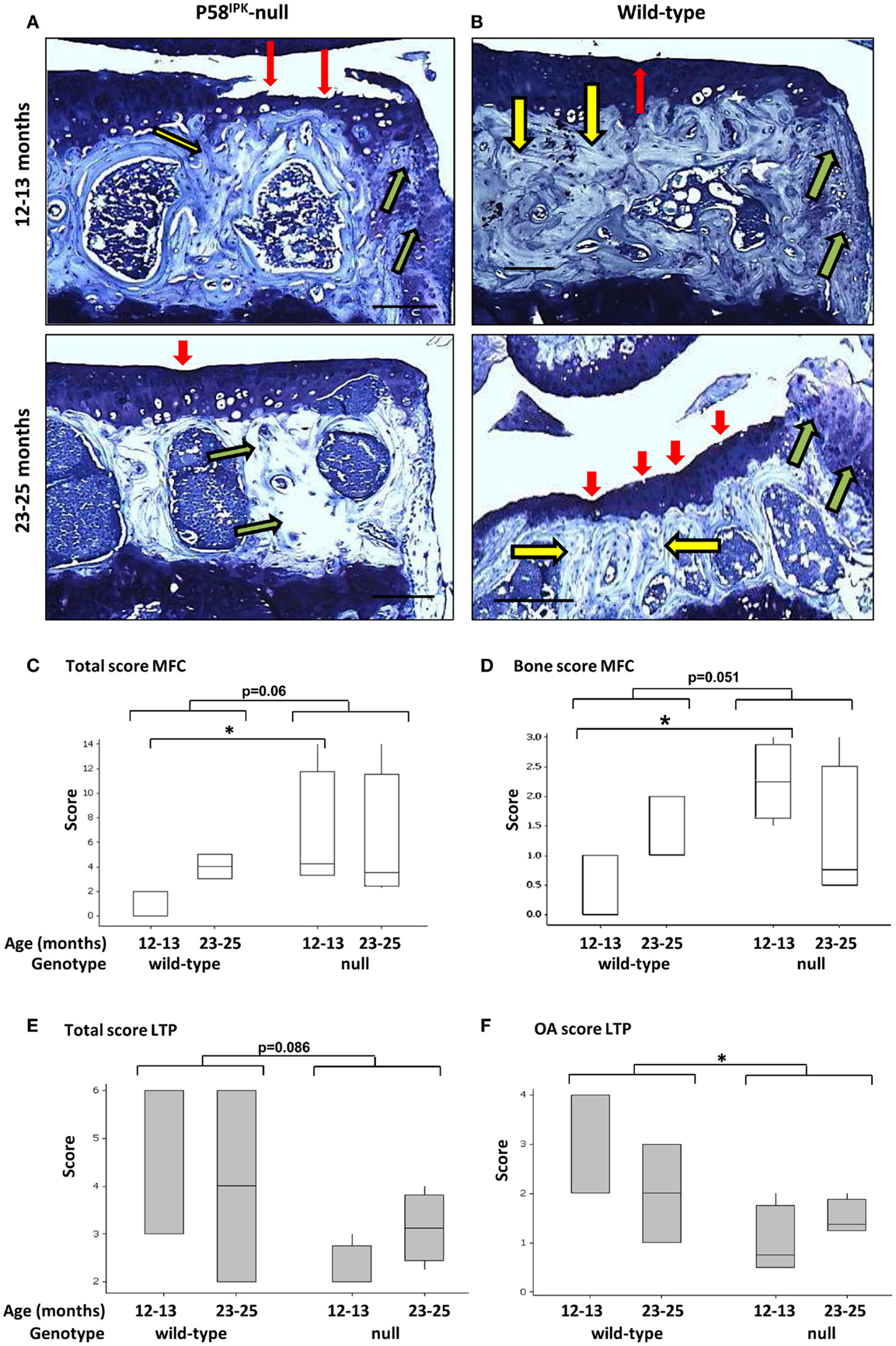
Figure 2. Knee joint degeneration is increased in P58IPK-null mice. Examples of toluidine blue stained coronal sections from the MTPs from P58IPK-null (A) and wild-type (B) mice from both age groups (12–13 months: top panels; 23–25 months: bottom panels) are shown representing typical signs of joint degeneration: cartilage tears and degeneration (red arrows); subchondral bone changes (yellow arrows); and osteophyte formation (green arrows). Scale bars = 500 μm. The OARSI scoring method (Table S1 in Supplementary Material) was used to determine whether null mice had degeneration over and above that commonly observed in C57Bl/6 wild-type mice. Total scores [parameters 1–3; (C)] and bone scores [parameter 2; (D)] from the MFC and total scores [parameters 1–3; (E)] and OA scores [parameter 1; (F)] from the LTP obtained using are shown. Significant differences were detected by GLM ANOVA and Tukey’s post hoc test: *p ≤ 0.05. LTP, lateral tibial plateau; MFC, medial femoral condyle. OARSI scores: 12–13-month-old mice n = 3 null, n = 3 wild-type and 23–25-month-old mice n = 4 null, n = 4 wild-type.
High-resolution μCT analysis revealed a significant reduction in bone mass in P58IPK-null mice (Table 1). GLM ANOVA revealed a significant effect of genotype on tibial bone with loss of P58IPK reducing TV (p = 0.002), BV (p < 0.0001), and BV/TV (p = 0.01). BV in 23–25-month-old null mice was significantly reduced compared to age-matched wild-types (p = 0.03). Individual μCT slices revealed that bone loss was largely confined to the medial plateau of the tibiae (Figure 3).
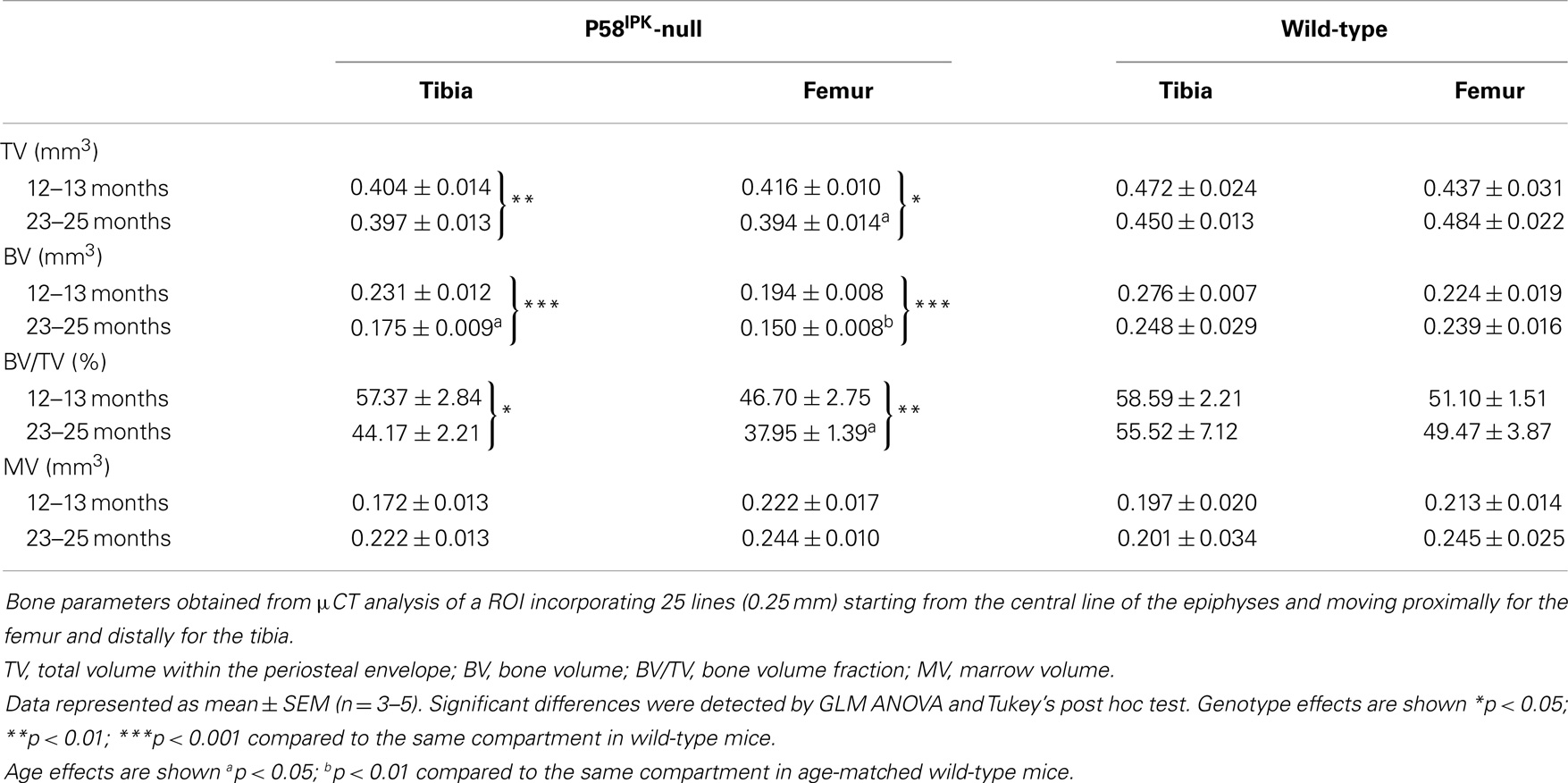
Table 1. Bone parameters measured using high-resolution μCT.
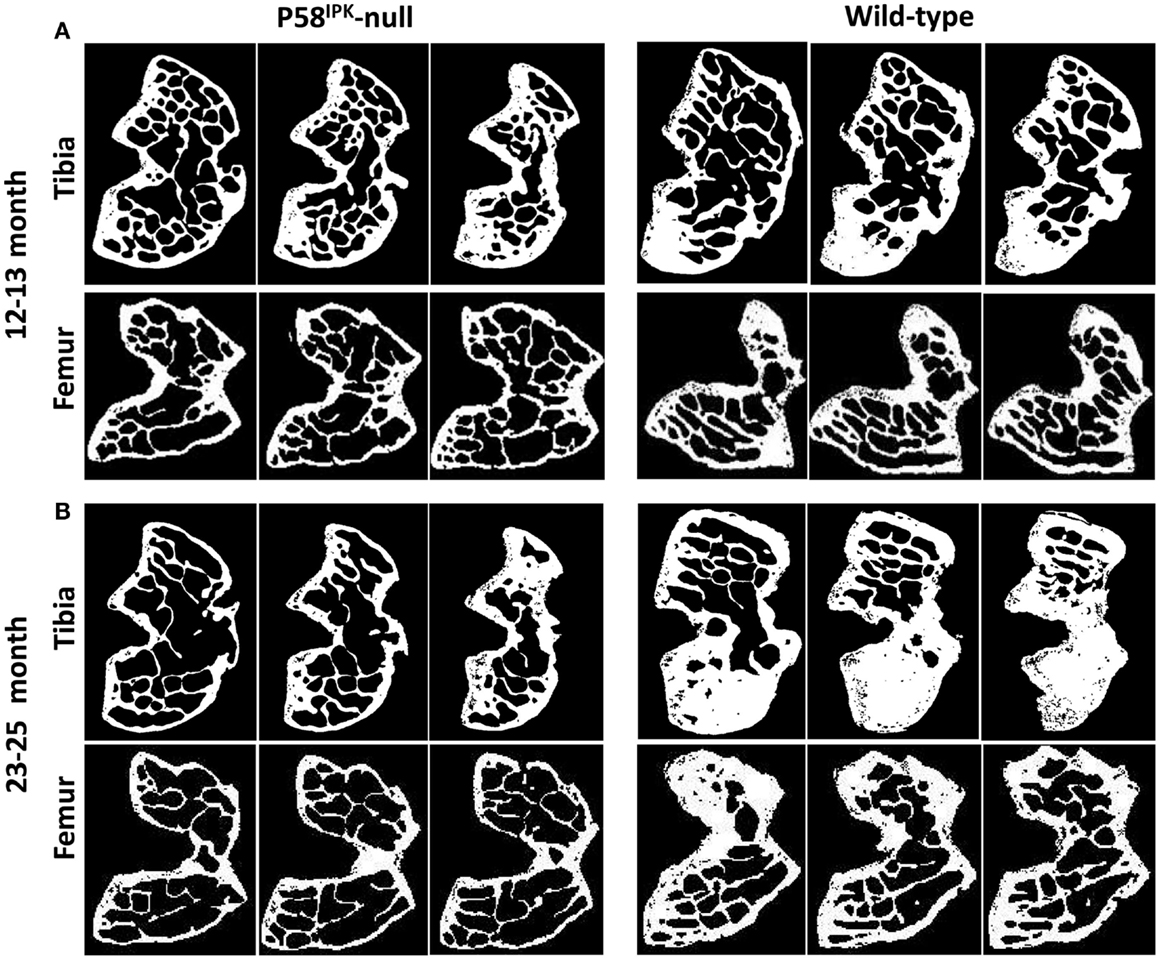
Figure 3. Significant bone loss occurs in the knees of P58IPK-null mice. MicroCT analysis of a ROI incorporating 25 lines (0.25 mm) starting from the central line of the epiphyses and moving proximally for the femur and distally for the tibia were obtained. Representative images are shown of slices taken from the top, middle and bottom of the 25 lines. (A) images from 12 to 13-month-old mice; (B) images from 23 to 25-month-old mice. In each slice, the lateral compartment is at the top and medial at the bottom.
General linear model analysis of variance also revealed a significant effect of genotype on the femoral bone with loss of P58IPK reducing TV (p = 0.013), BV (p = 0.0005), and BV/TV (p = 0.007). Femora from 23 to 25-month-old mice were smaller than those from age-matched wild-type mice with 18% reduction in TV (bone volume + marrow volume; p = 0.023), 37% reduction in BV (p = 0.0012), and a significant overall reduction of femoral BV fraction (BV/TV; p = 0.025). Individual slices reveal that bone loss occurred across the femoral condyle (Figure 3).
A Severe Degenerative Phenotype Occurs in a Subset of P58IPK-Null Mice
A very severe degenerative phenotype, not seen in any of the seven wild-type mice, was observed in a third of null mice; one from the 12–13 month group (Figures 4A–E) and two from the 23–25 month group (Figures 4F–O). The degeneration included loss of cartilage with erosion through to the subchondral bone, abnormal bone changes, and large osteophytes within the medial side of the joint. Moderate cartilage erosion was also observed in the lateral joint compartment of these mice. Heterotopic chondro-osseous tissue formed within the medial joint of each of the three mice with severe degeneration (Figures 4B,G,L), which was not observed in any other mice. Ectopic bone within the medial collateral ligaments and the joint capsule was observed within the contra-lateral leg of two of these mice (Figures 4C,M). High-resolution μCT images revealed changes in joint shape and osteophyte formation on the tibiae (Figures 4D,I,N) and femurs (Figures 4E,J,O) of these mice with severe degeneration. The extensive ectopic bone formation was further evident in frontal and side view 3D reconstructions from the μCT scans (Figure S4 in Supplementary Material). In addition to the two age groups included in this study, we also examined four 18-month-old mice (wild-type n = 2; null n = 2) and found that this severe phenotype was observed in one of the null mice (Figure S5 in Supplementary Material).
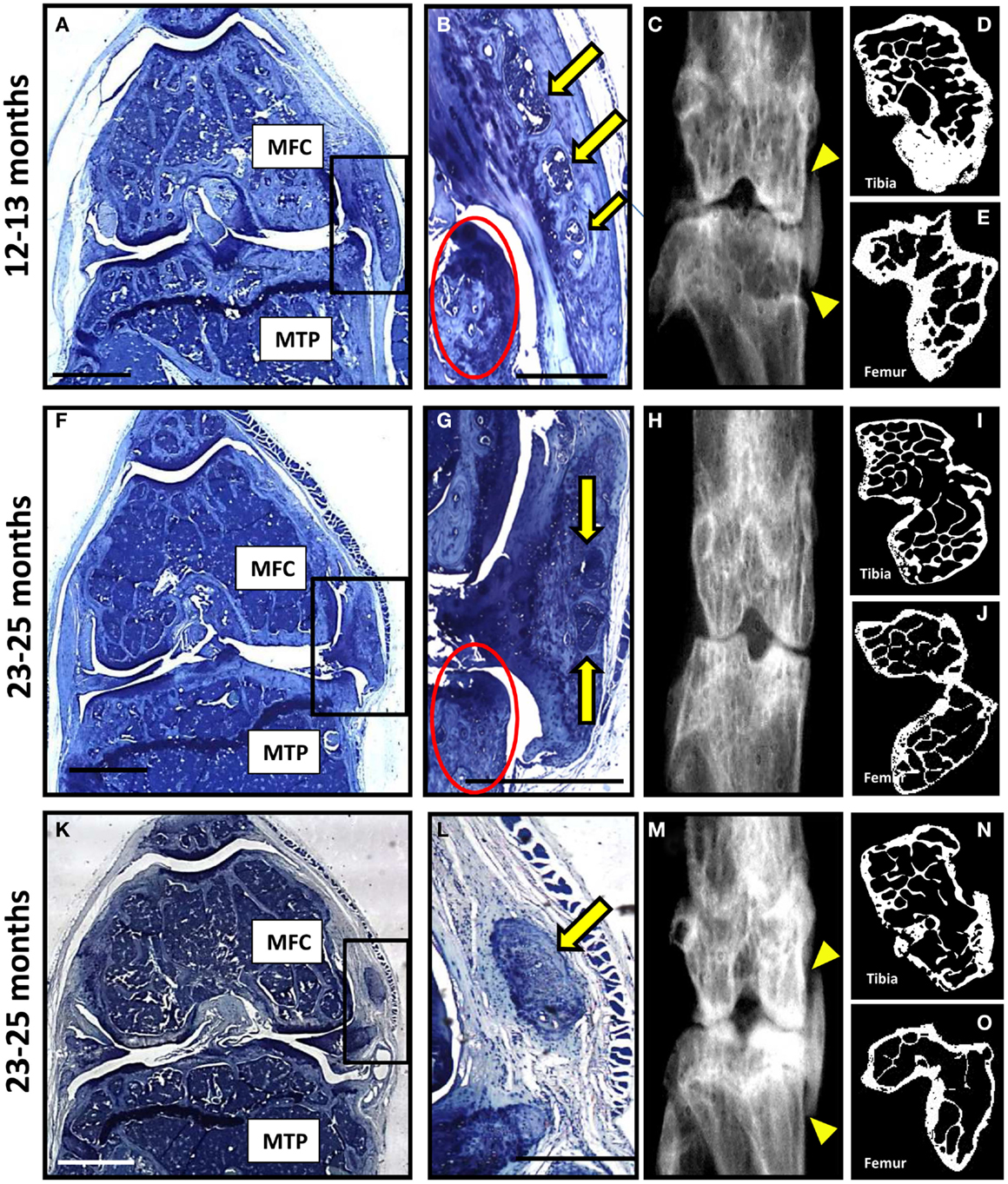
Figure 4. A subset of P58IPK-null mice reveal a severe degenerative joint phenotype. Coronal sections, stained with toluidine blue, are depicted from the right knee joints of three of the P58IPK-null mice aged 12–13 months (A) and 23–25 months (F,K) old with the severe phenotype. Significant bone remodeling and cartilage loss can be seen in the MTP and MFC. Magnified views of the heterotopic chondro-osseous tissue observed in the medial capsule of these sections (box) are depicted (B,G,L) showing areas of bone marrow formation (yellow arrows) along with osteophyte formation (red circles) in two of the mice. Coronal radiographs from the contra-lateral legs of each animal depicted in (A,F,K) are shown (C,H,M) highlighting the ectopic radio-dense areas in the medial collateral ligaments and the joint capsule of two of these mice [(C,M); yellow triangles]. High-resolution μCT images of the contra-lateral leg revealed changes in joint shape and osteophyte formation on the tibiae (D,I,N) and femurs (E,J,O). Scale bar = 500 μm except for (A,F,K) = 1 mm.
P58IPK-Null Mice have Less Calcification in Their Achilles Tendon
Achilles tendon calcification occurred in 95% of all mice (Figure 5A), with P58IPK-null mice exhibiting significantly less calcification than wild-types (p = 0.002, Figure 5B). There was an age-related increase in Achilles calcification in wild-type mice (3.3-fold increase in 23–25-month-old mice compared to 12–13-month-old mice, p = 0.0023). Null mice showed a threefold reduction in calcified tissue at 23–25 months compared to age-matched wild-types (p = 0.0009).
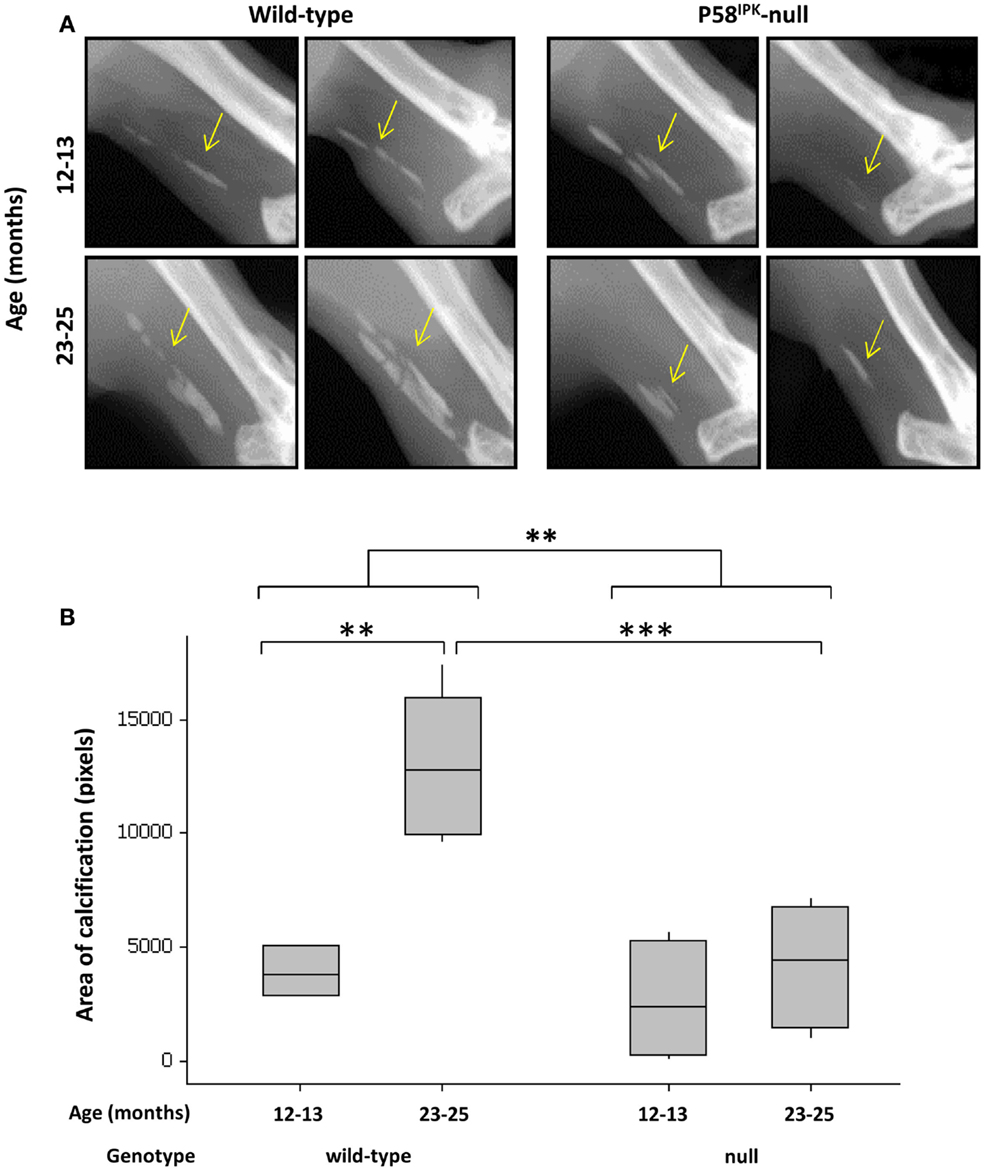
Figure 5. Achilles tendon calcification is reduced in P58IPK-null mice. (A) Radiographs from the left leg of wild-type and null mice show areas of calcification within the Achillles tendon (yellow arrow). A ROI was drawn around the calcified tissue and bone density software used to measure the area of calcification (B) with significant differences detected by GLM ANOVA and Tukey’s post hoc test: **p < 0.01; ***p < 0.001. Achilles calcification area: 12–13-month-old mice n = 5 null, n = 3 wild-type and 23–25-month-old mice n = 5 null, n = 4 wild-type.
Expression Levels of Phosphorylated PKR and PERK in Human OA Joint Tissues Correlate to the Severity of Degeneration
Sections from two human MTPs, with moderate to extensive OA degeneration (33), were examined for evidence of active stress signaling and representative images shown (Figure 6; Figure S6 in Supplementary Material). Toluidine blue stained sections show that both patients had more degeneration toward the outer edge of the plateau (Figures 6A,D). Patient 1 had extensive cartilage loss down to the subchondral bone and severe damage in what remained of the cartilage (Figure 6A). Patient 2 showed signs of proteoglycan loss, cartilage fibrillation, and a small region of significant cartilage fibrillation (Figure 6D). Immunohistochemistry revealed that active PERK and PKR were present in a range of joint tissues regardless of degenerative state (described in Figure S6 in Supplementary Material). However, in patient 1 (Figure 6A), more extensive staining was observed in the cartilage that was more severely damaged and, in particular, in the bone in regions underlying significant cartilage loss (Figures 6B,C). In patient 2 (Figure 6B), the staining for active PERK and PKR was more apparent in the cartilage as the damage increased in severity toward the outer edge but the extensive bone changes were not observed (Figures 6E,F). Staining for P58IPK was very limited throughout with very low or negative staining in osteocytes, bone lining cells, and chondrocytes (Figure S6 in Supplementary Material). However, positive staining was observed in the bone marrow (Figure S6O in Supplementary Material).
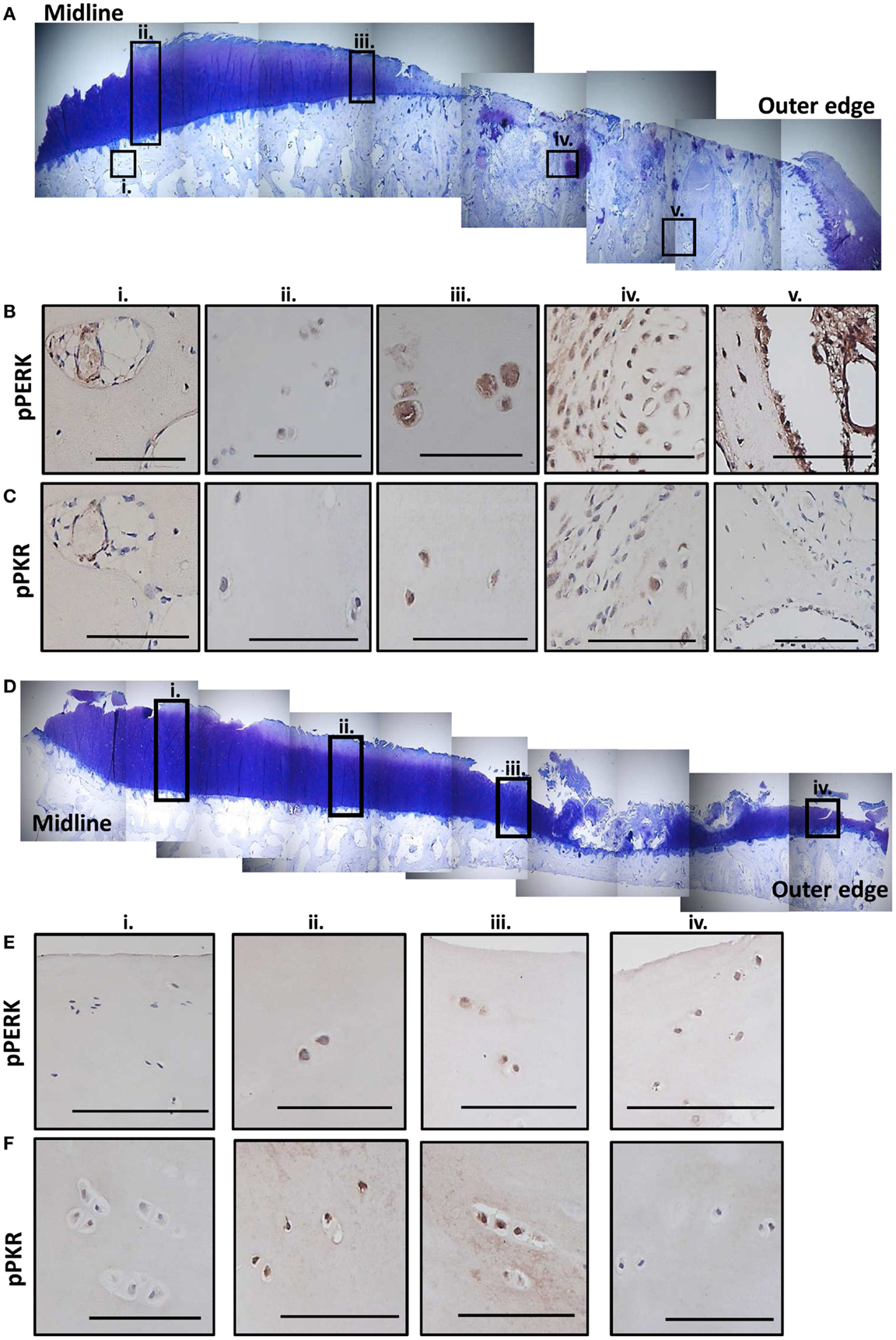
Figure 6. Expression of phosphorylated PKR and PERK correlate with the degree of degeneration in human OA tissue. Sections from the MTP of two patients undergoing total knee replacement for OA were stained with toluidine blue to establish the degree of tissue degeneration. The subcellular localization of phosphorylated PKR and PERK were determined by immunohistochemistry. Patient 1 had complete cartilage loss and severe bone changes toward the outer edge of the plateau with the remaining cartilage showing signs of significant fibrillation and proteoglycan loss (A). Five regions (black boxes; i–v) highlight areas of limited damage (joint middle) to severe damage (outer edge). Representative staining for phosphorylated PERK (B) and PKR (C) from these five regions are shown revealing more staining in the cartilage as the damage progresses outwards and significant activation in the remodeling bone in the areas underlying the complete loss of cartilage. Patient 2 had cartilage fibrillations and proteoglycan loss, which were more extensive toward the outer edge but the damage was less extensive that patient 1 (D). Four regions (black boxes; i–iv) highlight areas of limited damage (joint middle) to severe damage (outer edge). Representative staining for phosphorylated PERK (E) and PKR (F) from these four regions are shown revealing more staining in the cartilage as the damage progresses outwards but not the extensive activity in the bone as observed for patient 1. Scale bars = 100 μm.
Discussion
The aims of this observational study were to describe a phenotypic characterization of donated knee joints from a P58IPK knockout mouse colony. This study demonstrates for the first time that knockout of P58IPK, the cellular inhibitor of PKR and PERK, alters bone size and volume, and leads to a degenerative joint phenotype. Epiphyseal height was significantly reduced in the femora and tibiae of null mice, which also displayed significantly narrower tibiae than those of wild-type mice. These effects may be due to the reduced body fat and reduced body weight phenotype of P58IPK-null mice (25) (Figure S3 in Supplementary Material), either reducing load on the bones or reflecting direct effects of hypoinsulinemia and diabetes (25, 26) on bone growth.
These data also show that P58IPK plays a critical role in maintaining joint integrity. It has been well documented that C57Bl/6 mice develop spontaneous OA-like lesions as they age (38–41) and indeed this was observed in the wild-type mice used within this study. However, loss of P58IPK caused significantly increased joint degeneration scores that were greater than those which occur spontaneously in the aging wild-type C57Bl/6 mice. Furthermore, joint degeneration in P58IPK-null mice localized to the medial femoral compartment whereas C57Bl/6 mice develop spontaneous OA-like lesions predominantly in the lateral side of the tibia-femoral joint as they age (39, 40). The lateral compartment of P58IPK-null mice appeared relatively protected from age-related joint damage observed in wild-type mice, which may reflect a shift of loading from lateral to medial compartments in null mice. Such altered joint biomechanics is consistent with the increased subchondral bone OARSI scores in 12–13-month-old null mice, which were much greater in the medial and less in the lateral compartments, when compared to wild-types. A major risk factor for OA is abnormal loading and factors such as increased loading through the medial side of the joint (42) and altered contact surface areas on femoral condyles versus tibial plateau, mean that load-induced changes vary across different regions of the joint (43). There is some evidence that specific mechanical loading regimes can activate PKR in articular cartilage (14), although this has not been tested in bone.
High-resolution μCT analysis revealed significant bone loss from the region beneath the subchondral plate in the MTP and across the entire femoral condyle of mice lacking P58IPK. This may reflect osteoblast insufficiency, enhanced resorption (44) or a combination of these, associated with the diabetic phenotype of these null mice (45, 46). Both PERK and PKR are implicated in bone cell signaling. PERK plays a crucial role in bone development, osteoblast differentiation and maturation (47) whereas inactivation of PKR reduces osteoblast differentiation and calcification (48–50) and osteoclast differentiation (51). Thus, the changes in the subchondral bone and loss of bone below the subchondral plate in P58IPK-null mice may reflect site-specific effects of over-activated PERK and/or PKR on bone remodeling.
A severe phenotype, characterized by complete loss of the articular cartilage from the medial tibial and femoral compartments, subchondral bone remodeling, osteophyte formation and calcification of the collateral ligaments, was observed in a third of P58IPK-null mice. The reason for this variability is unknown but may reflect individual variations in immune status, ER stress, or joint loading. For example, virally infected P58IPK-null mice show enhanced inflammatory responses (26), and cells with a high-secretory burden from P58IPK-null mice have reduced capacity to cope with ER stress (31). In addition, ossification of collateral ligaments, as observed in all P58IPK-null mice with severely damaged joints, may cause, or be a consequence of altered joint loading. Such ossification occurs in the Dunkin–Hartley guinea pig (52) and STR/ort mouse models of spontaneous knee OA (53, 54), where ossification of the collateral and patellar ligaments and Achilles tendon is thought to alter the mechanical stability of the knee, contributing to cartilage breakdown and OA (55, 56). Transection of the anterior cruciate ligament (ACL) or non-invasive ACL rupture in mice also alters joint biomechanics and results in a remarkably similar severe phenotype to that observed in P58IPK-null mice with ectopic bone formation within the joint capsule, significant cartilage erosion and bone sclerosis (57–59). Of note, bone and proteoglycan deposition in the collateral ligaments, significant cartilage erosion on the femur and tibia and sclerosis in the underlying bone have also been observed in aging IL-6 knockout (60) and GM3 synthase knockout mice (61). Interestingly, absence of GM3 synathase prevents the conversion of GM3 to gangliosides, which are required to suppress MMP-13 and ADMATS-5 production. Whether PKR and/or PERK signaling pathways play a role in these models remains to be determined.
Although calcification of the periarticular structures only occurred in P58IPK-null mice with the severe phenotype, knee degeneration occurred in all P58IPK-null mice, suggesting that calcification is not the initiating factor in these mice. There is no doubt, however, that the calcified/ossified tissue will change the stiffness of the periarticular structures, alter joint biomechanics and accelerate degeneration. Calcification of the Achilles tendon increased with age in wild-type mice as expected (62), but interestingly, older P58IPK-null mice had significantly less calcification than age-matched wild-types. This may reflect the reduced body weight of these animals or abnormal biomechanics in the knee altering loading through the Achilles.
Loss of P58IPK increases levels of phosphorylated PERK and PKR (24). PERK and ER stress have been implicated in the pathogenesis of OA (5–7) and activation of PKR causes cartilage breakdown (3, 4, 63), is linked to load-induced OA (12) and is mechano-responsive (14). Consistent with this, and to our knowledge, for the first time, we demonstrate that active PKR and PERK are both expressed at the same time in a range of joint cells in human osteoarthritic tissue. Most notable is that the levels of these kinases appear to correlate with the degree of cartilage damage and that the highest expression is observed within the bone in areas underlying severe cartilage loss. In addition, there appears to be some evidence for region-specific activation of PKR and PERK in human OA, with PERK in surface zone cartilage cells and PKR in the mid zone. These observations are consistent with the notion that over-activation of the PKR pathway in P58IPK-null mice combined with increased loading of the medial compartment, may contribute to joint degeneration.
Given the functional cross-talk between PERK and PKR (64) and without the control of P58IPK, the extent of activation of these pathways may be exaggerated in these knockout mice. Stress, viral infection, and/or altered joint biomechanics in individual mice may increase inflammatory responses, heighten ER stress, and induce load related responses to cause the severe degenerative joint phenotype as seen in a cohort of these mice. In future studies, it would be advantageous to “challenge” younger P58IPK-null animals with an inflammatory or mechanical insult prior to the onset of overt degeneration to determine whether this initiates the severe degenerative joint phenotype. However, the only material currently available for skeletal analysis of P58IPK knockout mice is that from the hind limbs we describe in this paper. To our knowledge, there are no living colonies of this knockout animal and no frozen embryos available worldwide. Given the limited material available, our study does possess some limitations, namely, the lack of correlation of morphological changes directly with over-activation of the PKR/PERK pathway, the inability to determine the extent of ER stress, the effect of the loss of function of P58IPK on its role as a co-chaperone, and the relatively small sample size in each age group. That said this does not detract from the strong degenerative joint phenotype we have observed in these null animals providing the first in vivo evidence showing a critical role for P58IPK in OA-like joint degeneration. The major changes observed in P58IPK knockout bone is of particular interest in defining an important role for these pathways in degenerative joint disease.
Author Contributions
Conception and design: Sophie J. Gilbert, Victor C. Duance, and Deborah J. Mason. Collection and assembly of data: Sophie J. Gilbert, Lee B. Meakin, Mari A. Nowell, Cleo Selina Bonnet, Warren C. Ladiges, and John Morton. Analysis and interpretation of data: Sophie J. Gilbert, Lee B. Meakin, Mari A. Nowell, Cleo Selina Bonnet, and Deborah J. Mason. Drafting of the manuscript: Sophie J. Gilbert and Deborah J. Mason. Critical revision: Sophie J. Gilbert, Lee B. Meakin, Mari A. Nowell, Cleo Selina Bonnet, Victor C. Duance, and Deborah J. Mason. Final approval of the article: Sophie J. Gilbert, Lee B. Meakin, Mari A. Nowell, Cleo Selina Bonnet, Warren C. Ladiges, John Morton, Victor C. Duance, and Deborah J. Mason.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
This work was funded by Arthritis Research UK grants 16436 and 18461. Lee B. Meakin is funded by an Integrated Fellowship for Veterinarians from the Wellcome Trust. The authors thank Derek Scarborough for the processing and preparation of tissue for histology, Prof. Jo Price for guidance and expertise on CT and Rachel Hopkins, Hefina Williams, and Mitul Patel for technical assistance with the histology and x-ray analysis.
Supplementary Material
The Supplementary Material for this article can be found online at http://www.frontiersin.org/Journal/10.3389/fendo.2014.00174/abstract
Abbreviations
μCT, micro-computed tomography; BV, bone volume; BV/TV, bone volume fraction; eIF2α, eukaryotic initiation factor 2 alpha; ER, endoplasmic reticulum; GLM ANOVA, general linear model analysis of variance; IFN, interferon; LFC, lateral femoral condyle; LTP, lateral tibial plateau; MFC, medial femoral condyle; MTP, medial tibial plateau; MV, marrow volume; OA, osteoarthritis; OARSI, Osteoarthritis Research Society International; P58IPK, 58 kDa inhibitor protein kinase; PERK, protein kinase-like endoplasmic reticulum kinase; PKR, protein kinase R; RA, rheumatoid arthritis; TNF-α, tumor necrosis factor-alpha; TV, total tissue volume.
References
1. Griffin TM, Guilak F. The role of mechanical loading in the onset and progression of osteoarthritis. Exerc Sport Sci Rev (2005) 33:195–200. doi:10.1097/00003677-200510000-00008
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
2. Griffin TM, Guilak F. Why is obesity associated with osteoarthritis? Insights from mouse models of obesity. Biorheology (2008) 45:387–98. doi:10.3233/BIR-2008-0485
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
3. Gilbert SJ, Duance VC, Mason DJ. Does protein kinase R mediate TNF-alpha- and ceramide-induced increases in expression and activation of matrix metalloproteinases in articular cartilage by a novel mechanism? Arthritis Res Ther (2004) 6:R46–55. doi:10.1186/ar1381
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
4. Tam CL, Hofbauer M, Towle CA. Requirement for protein kinase R in interleukin-1alpha-stimulated effects in cartilage. Biochem Pharmacol (2007) 74:1636–41. doi:10.1016/j.bcp.2007.08.002
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
5. Ruiz-Romero C, Carreira V, Rego I, Remeseiro S, Lopez-Armada MJ, Blanco FJ. Proteomic analysis of human osteoarthritic chondrocytes reveals protein changes in stress and glycolysis. Proteomics (2008) 8:495–507. doi:10.1002/pmic.200700249
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
6. Nugent AE, Speicher DM, Gradisar I, McBurney DL, Baraga A, Doane KJ, et al. Advanced osteoarthritis in humans is associated with altered collagen VI expression and upregulation of ER-stress markers Grp78 and bag-1. J Histochem Cytochem (2009) 57:923–31. doi:10.1369/jhc.2009.953893
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
7. Takada K, Hirose J, Senba K, Yamabe S, Oike Y, Gotoh T, et al. Enhanced apoptotic and reduced protective response in chondrocytes following endoplasmic reticulum stress in osteoarthritic cartilage. Int J Exp Pathol (2011) 92:232–42. doi:10.1111/j.1365-2613.2010.00758.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
8. Gilbert SJ, Blain EJ, Al-Sabah A, Zhang Y, Duance VC, Mason DJ. Protein kinase R plays a pivotal role in oncostatin M and interleukin-1 signalling in bovine articular cartilage chondrocytes. Eur Cell Mater (2012) 23:41–57.
9. Yoo SA, You S, Yoon HJ, Kim DH, Kim HS, Lee K, et al. A novel pathogenic role of the ER chaperone GRP78/BiP in rheumatoid arthritis. J Exp Med (2012) 209:871–86. doi:10.1084/jem.20111783
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
10. Gilbert SJ, Duance VC, Mason D. Protein kinase R: a novel mediator of articular cartilage degradation in arthritis. Curr Rheumatol Rev (2006) 2:9–21. doi:10.2174/157339706775697026
11. Gilbert SJ, Duance VC, Mason DJ. Tumour necrosis factor alpha up-regulates protein kinase R (PKR)-activating protein (PACT) and increases phosphorylation of PKR and eukaryotic initiation factor 2-alpha in articular chondrocytes. Biochem Soc Trans (2002) 30:886–9. doi:10.1042/BST0300886
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
12. Gilbert SJ, Duance VC, Mason DJ. Differential expression in early stages of osteoarthritis in vivo. J Bone Miner Res (1999) 14:1044.
13. Bendele AM. Animal models of osteoarthritis in an era of molecular biology. J Musculoskelet Neuronal Interact (2002) 2:501–3.
14. Lomas C, Tang XD, Chanalaris A, Saklatvala J, Vincent TL. Cyclic mechanical load causes global translational arrest in articular chondrocytes: a process which is partially dependent upon PKR phosphorylation. Eur Cell Mater (2011) 22:178–89.
15. Ladiges W, Morton J, Blakely C, Gale M. Tissue specific expression of PKR protein kinase in aging B6D2F1 mice. Mech Ageing Dev (2000) 114:123–32. doi:10.1016/S0047-6374(00)00097-X
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
16. Harding HP, Zhang Y, Ron D. Protein translation and folding are coupled by an endoplasmic-reticulum-resident kinase. Nature (1999) 397:271–4. doi:10.1038/16729
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
17. Prostko CR, Dholakia JN, Brostrom MA, Brostrom CO. Activation of the double-stranded RNA-regulated protein kinase by depletion of endoplasmic reticular calcium stores. J Biol Chem (1995) 270:6211–5. doi:10.1074/jbc.270.11.6211
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
18. Lee ES, Yoon CH, Kim YS, Bae YS. The double-strand RNA-dependent protein kinase PKR plays a significant role in a sustained ER stress-induced apoptosis. FEBS Lett (2007) 581:4325–32. doi:10.1016/j.febslet.2007.08.001
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
19. Zhang P, McGrath B, Li S, Frank A, Zambito F, Reinert J, et al. The PERK eukaryotic initiation factor 2 alpha kinase is required for the development of the skeletal system, postnatal growth, and the function and viability of the pancreas. Mol Cell Biol (2002) 22:3864–74. doi:10.1128/MCB.22.11.3864-3874.2002
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
20. Yang L, Carlson SG, McBurney D, Horton WE Jr. Multiple signals induce endoplasmic reticulum stress in both primary and immortalized chondrocytes resulting in loss of differentiation, impaired cell growth, and apoptosis. J Biol Chem (2005) 280:31156–65. doi:10.1074/jbc.M501069200
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
21. Boot-Handford RP, Briggs MD. The unfolded protein response and its relevance to connective tissue diseases. Cell Tissue Res (2010) 339:197–211. doi:10.1007/s00441-009-0877-8
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
22. Korth MJ, Lyons CN, Wambach M, Katze MG. Cloning, expression, and cellular localization of the oncogenic 58-kDa inhibitor of the RNA-activated human and mouse protein kinase. Gene (1996) 170:181–8. doi:10.1016/0378-1119(95)00883-7
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
23. Yan W, Frank CL, Korth MJ, Sopher BL, Novoa I, Ron D, et al. Control of PERK eIF2alpha kinase activity by the endoplasmic reticulum stress-induced molecular chaperone P58IPK. Proc Natl Acad Sci U S A (2002) 99:15920–5. doi:10.1073/pnas.252341799
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
24. van Huizen R, Martindale JL, Gorospe M, Holbrook NJ. P58IPK, a novel endoplasmic reticulum stress-inducible protein and potential negative regulator of eIF2alpha signaling. J Biol Chem (2003) 278:15558–64. doi:10.1074/jbc.M212074200
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
25. Ladiges WC, Knoblaugh SE, Morton JF, Korth MJ, Sopher BL, Baskin CR, et al. Pancreatic beta-cell failure and diabetes in mice with a deletion mutation of the endoplasmic reticulum molecular chaperone gene P58IPK. Diabetes (2005) 54:1074–81. doi:10.2337/diabetes.54.4.1074
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
26. Goodman AG, Fornek JL, Medigeshi GR, Perrone LA, Peng X, Dyer MD, et al. P58(IPK): a novel “CIHD” member of the host innate defense response against pathogenic virus infection. PLoS Pathog (2009) 5:e1000438. doi:10.1371/journal.ppat.1000438
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
27. Yang H, Liu R, Cui Z, Chen ZQ, Yan S, Pei H, et al. Functional characterization of 58-kilodalton inhibitor of protein kinase in protecting against diabetic retinopathy via the endoplasmic reticulum stress pathway. Mol Vis (2011) 17:78–84.
28. Tao J, Sha B. Structural insight into the protective role of P58(IPK) during unfolded protein response. Methods Enzymol (2011) 490:259–70. doi:10.1016/B978-0-12-385114-7.00015-5
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
29. Rutkowski DT, Kang SW, Goodman AG, Garrison JL, Taunton J, Katze MG, et al. The role of p58IPK in protecting the stressed endoplasmic reticulum. Mol Biol Cell (2007) 18:3681–91. doi:10.1091/mbc.E07-03-0272
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
30. Petrova K, Oyadomari S, Hendershot LM, Ron D. Regulated association of misfolded endoplasmic reticulum lumenal proteins with P58/DNAJc3. EMBO J (2008) 27:2862–72. doi:10.1038/emboj.2008.199
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
31. Oyadomari S, Yun C, Fisher EA, Kreglinger N, Kreibich G, Oyadomari M, et al. Cotranslocational degradation protects the stressed endoplasmic reticulum from protein overload. Cell (2006) 126:727–39. doi:10.1016/j.cell.2006.06.051
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
32. Yan W, Gale MJ Jr, Tan SL, Katze MG. Inactivation of the PKR protein kinase and stimulation of mRNA translation by the cellular co-chaperone P58(IPK) does not require J domain function. Biochemistry (2002) 41:4938–45. doi:10.1021/bi0121499
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
33. Bonnet CS, Williams AS, Gilbert SJ, Harvey AK, Evans BA, Mason DJ. AMPA/kainate glutamate receptors contribute to inflammation, degeneration and pain related behaviour in inflammatory stages of arthritis. Ann Rheum Dis (2013). doi:10.1136/annrheumdis-2013-203670
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
34. Glasson SS, Chambers MG, van den Berg WB, Little CB. The OARSI histopathology initiative – recommendations for histological assessments of osteoarthritis in the mouse. Osteoarthritis Cartilage (2010) 18(Suppl 3):S17–23. doi:10.1016/j.joca.2010.05.025
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
35. Vizard DL, Wood DO, Papineni RV, Feke GD, Orton SP, McLaughlin WE. Analytical radiography for planar radiographic images implemented with a multi-modal system. Comput Methods Programs Biomed (2010) 99:88–97. doi:10.1016/j.cmpb.2010.01.003
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
36. Meakin LB, Sugiyama T, Galea GL, Browne WJ, Lanyon LE, Price JS. Male mice housed in groups engage in frequent fighting and show a lower response to additional bone loading than females or individually housed males that do not fight. Bone (2013) 54:113–7. doi:10.1016/j.bone.2013.01.029
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
37. Bouxsein ML, Boyd SK, Christiansen BA, Guldberg RE, Jepsen KJ, Muller R. Guidelines for assessment of bone microstructure in rodents using micro-computed tomography. J Bone Miner Res (2010) 25:1468–86. doi:10.1002/jbmr.141
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
38. Wilhelmi G, Faust R. Suitability of C57 black mouse as an experimental animal for study of skeletal changes due to aging, with special reference to osteo-arthrosis and its response to tribenoside. Pharmacology (1976) 14:289–96. doi:10.1159/000136607
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
39. Stoop R, van der Kraan PM, Buma P, Hollander AP, Billinghurst RC, Poole AR, et al. Type II collagen degradation in spontaneous osteoarthritis in C57Bl/6 and BALB/c mice. Arthritis Rheum (1999) 42:2381–9. doi:10.1002/1529-0131(199911)42:11<2381::AID-ANR17>3.0.CO;2-E
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
40. van der Kraan PM, Stoop R, Meijers TH, Poole AR, van den Berg WB. Expression of type X collagen in young and old C57Bl/6 and Balb/c mice. Relation with articular cartilage degeneration. Osteoarthritis Cartilage (2001) 9:92–100. doi:10.1053/joca.2000.0364
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
41. Helminen HJ, Saamanen AM, Salminen H, Hyttinen MM. Transgenic mouse models for studying the role of cartilage macromolecules in osteoarthritis. Rheumatology (2002) 41:848–56. doi:10.1093/rheumatology/41.8.848
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
42. Chang A, Hurwitz D, Dunlop D, Song J, Cahue S, Hayes K, et al. The relationship between toe-out angle during gait and progression of medial tibiofemoral osteoarthritis. Ann Rheum Dis (2007) 66:1271–5. doi:10.1136/ard.2006.062927
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
43. Poulet B, Hamilton RW, Shefelbine S, Pitsillides AA. Characterizing a novel and adjustable noninvasive murine joint loading model. Arthritis Rheum (2011) 63:137–47. doi:10.1002/art.27765
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
44. Bar-Shira-Maymon B, Coleman R, Steinhagen-Thiessen E, Silbermann M. Correlation between alkaline and acid phosphatase activities and age-related osteopenia in murine vertebrae. Calcif Tissue Int (1989) 44:99–107. doi:10.1007/BF02556468
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
45. Jehle PM, Jehle DR, Mohan S, Bohm BO. Serum levels of insulin-like growth factor system components and relationship to bone metabolism in Type 1 and Type 2 diabetes mellitus patients. J Endocrinol (1998) 159:297–306. doi:10.1677/joe.0.1590297
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
46. Tuominen JT, Impivaara O, Puukka P, Ronnemaa T. Bone mineral density in patients with type 1 and type 2 diabetes. Diabetes Care (1999) 22:1196–200. doi:10.2337/diacare.22.7.1196
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
47. Wei J, Sheng X, Feng D, McGrath B, Cavener DR. PERK is essential for neonatal skeletal development to regulate osteoblast proliferation and differentiation. J Cell Physiol (2008) 217:693–707. doi:10.1002/jcp.21543
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
48. Yoshida K, Okamura H, Amorim BR, Ozaki A, Tanaka H, Morimoto H, et al. Double-stranded RNA-dependent protein kinase is required for bone calcification in MC3T3-E1 cells in vitro. Exp Cell Res (2005) 311:117–25. doi:10.1016/j.yexcr.2005.09.006
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
49. Yoshida K, Okamura H, Amorim BR, Hinode D, Yoshida H, Haneji T. PKR-mediated degradation of STAT1 regulates osteoblast differentiation. Exp Cell Res (2009) 315:2105–14. doi:10.1016/j.yexcr.2009.02.003
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
50. Yoshida K, Okamura H, Ochiai K, Hoshino Y, Haneji T, Yoshioka M, et al. PKR plays a positive role in osteoblast differentiation by regulating GSK-3beta activity through a beta-catenin-independent pathway. Mol Cell Endocrinol (2012) 361(1–2):99–105. doi:10.1016/j.mce.2012.03.019
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
51. Teramachi J, Morimoto H, Baba R, Doi Y, Hirashima K, Haneji T. Double stranded RNA-dependent protein kinase is involved in osteoclast differentiation of RAW264.7 cells in vitro. Exp Cell Res (2010) 316:3254–62. doi:10.1016/j.yexcr.2010.08.006
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
52. Jimenez PA, Glasson SS, Trubetskoy OV, Haimes HB. Spontaneous osteoarthritis in Dunkin Hartley guinea pigs: histologic, radiologic, and biochemical changes. Lab Anim Sci (1997) 47:598–601.
53. Walton M. Degenerative joint disease in the mouse knee; histological observations. J Pathol (1977) 123:109–22. doi:10.1002/path.1711230403
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
54. Walton M. Degenerative joint disease in the mouse knee; radiological and morphological observations. J Pathol (1977) 123:97–107. doi:10.1002/path.1711230207
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
55. Collins C, Evans RG, Ponsford F, Miller P, Elson CJ. Chondro-osseous metaplasia, bone density and patellar cartilage proteoglycan content in the osteoarthritis of STR/ORT mice. Osteoarthritis Cartilage (1994) 2:111–8. doi:10.1016/S1063-4584(05)80061-5
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
56. Evans RG, Collins C, Miller P, Ponsford FM, Elson CJ. Radiological scoring of osteoarthritis progression in STR/ORT mice. Osteoarthritis Cartilage (1994) 2:103–9. doi:10.1016/S1063-4584(05)80060-3
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
57. Glasson SS, Blanchet TJ, Morris EA. The surgical destabilization of the medial meniscus (DMM) model of osteoarthritis in the 129/SvEv mouse. Osteoarthritis Cartilage (2007) 15:1061–9. doi:10.1016/j.joca.2007.03.006
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
58. Christiansen BA, Anderson MJ, Lee CA, Williams JC, Yik JH, Haudenschild DR. Musculoskeletal changes following non-invasive knee injury using a novel mouse model of post-traumatic osteoarthritis. Osteoarthritis Cartilage (2012) 20(7):773–82. doi:10.1016/j.joca.2012.04.014
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
59. Onur TS, Wu R, Chu S, Chang W, Kim HT, Dang AB. Joint instability and cartilage compression in a mouse model of posttraumatic osteoarthritis. J Orthop Res (2014) 32:318–23. doi:10.1002/jor.22509
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
60. de Hooge AS, van de Loo FA, Bennink MB, Arntz OJ, De Hooge P, van den Berg WB. Male IL-6 gene knock out mice developed more advanced osteoarthritis upon aging. Osteoarthritis Cartilage (2005) 13:66–73. doi:10.1016/j.joca.2004.09.011
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
61. Sasazawa F, Onodera T, Yamashita T, Seito N, Tsukuda Y, Fujitani N, et al. Depletion of gangliosides enhances cartilage degradation in mice. Osteoarthritis Cartilage (2014) 22:313–22. doi:10.1016/j.joca.2013.11.015
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
62. Smith RK, Birch HL, Goodman S, Heinegard D, Goodship AE. The influence of ageing and exercise on tendon growth and degeneration – hypotheses for the initiation and prevention of strain-induced tendinopathies. Comp Biochem Physiol A Mol Integr Physiol (2002) 133:1039–50. doi:10.1016/S1095-6433(02)00148-4
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
63. Gilbert SJ, Duance VC, Mason DJ. Tumour necrosis factor alpha up-regulates protein kinase R (PKR)-activating protein (PACT) and increases phosphorylation of PKR and eukaryotic initiation factor 2-alpha in articular chondrocytes. Biochem Soc Trans (2001) 30:886–9. doi:10.1042/BST0300886
64. Baltzis D, Qu LK, Papadopoulou S, Blais JD, Bell JC, Sonenberg N, et al. Resistance to vesicular stomatitis virus infection requires a functional cross talk between the eukaryotic translation initiation factor 2alpha kinases PERK and PKR. J Virol (2004) 78:12747–61. doi:10.1128/JVI.78.23.12747-12761.2004
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Keywords: P58IPK, bone, articular cartilage, osteoarthritis, PERK, PKR
Citation: Gilbert SJ, Meakin LB, Bonnet CS, Nowell MA, Ladiges WC, Morton J, Duance VC and Mason DJ (2014) Deletion of P58IPK, the cellular inhibitor of the protein kinases PKR and PERK, causes bone changes and joint degeneration in mice. Front. Endocrinol. 5:174. doi: 10.3389/fendo.2014.00174
Received: 06 August 2014; Accepted: 01 October 2014;
Published online: 17 October 2014.
Edited by:
Alison Gartland, The University of Sheffield, UKReviewed by:
Paula H. Stern, Northwestern University Feinberg School of Medicine, USAAntonia Sophocleous, The University of Edinburgh, UK
Adam M. Taylor, Lancaster University, UK
Copyright: © 2014 Gilbert, Meakin, Bonnet, Nowell, Ladiges, Morton, Duance and Mason. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Deborah J. Mason, Arthritis Research UK Biomechanics and Bioengineering Centre, School of Biosciences, Cardiff University, Museum Avenue, Cardiff CF10 3AX, UK e-mail:bWFzb25kakBjYXJkaWZmLmFjLnVr