 Felicitas Mungenast
Felicitas Mungenast Theresia Thalhammer
Theresia Thalhammer- Department of Pathophysiology and Allergy Research, Center for Pathophysiology, Infectiology and Immunology, Medical University of Vienna, Vienna, Austria
Ovarian cancer is still the deadliest of all gynecologic malignancies in women worldwide. This is attributed to two main features of these tumors, namely, (i) a diagnosis at an advanced tumor stage, and, (ii) the rapid onset of resistance to standard chemotherapy after an initial successful therapy with platin- and taxol-derivatives. Therefore, novel targets for an early diagnosis and better treatment options for these tumors are urgently needed. Epidemiological data show that induction and biology of ovarian cancer is related to life-time estrogen exposure. Also experimental data reveal that ovarian cancer cells share a number of estrogen regulated pathways with other hormone-dependent cancers, e.g., breast and endometrial cancer. However, ovarian cancer is a heterogeneous disease and the subtypes are quite different with respect to mutations, origins, behaviors, markers, and prognosis and respond differently to standard chemotherapy. Therefore, a characterization of ovarian cancer subtypes may lead to better treatment options for the various subtypes and in particular for the most frequently observed high-grade serous ovarian carcinoma. For this intention, further studies on estrogen-related pathways and estrogen formation in ovarian cancer cells are warranted. The review gives an overview on ovarian cancer subtypes and explains the role of estrogen in ovarian cancer. Furthermore, enzymes active to synthesize and metabolize estrogens are described and strategies to target these pathways are discussed.
General Introduction
Currently, ovarian cancer is the fifth most common cancer in women in industrialized countries. It is mainly a disease of postmenopausal women, because more than 80% of all cases are being diagnosed in women older than 50 years. Although a falling rate of new ovarian cancer cases of 1.1% each year and an increase in the relative 5-year survival time from 33.6% in 1975 to 45.2% in 2010 was observed in industrialized countries, ovarian cancer is still the deadliest of all gynecologic malignancies worldwide. The alarming data are greatly attributed to the generally late diagnosis of the disease. More than 80% of all newly detected cases are advanced epithelial ovarian cancers (EOC) with peritoneal metastases and/or metastases in distant organs [Féderation Internationale de Gynécologie et d’Obstétrique (FIGO) stage III–IV]. The FIGO staging system was established by the FIGO and is most commonly used in EOC diagnosis (1). Effective preventative measures and reliable screening tools for an early detection are not yet available. Although the majority of women experience a variety of non-specific symptoms in the year before diagnosis, the disease is not commonly recognized until the tumor reaches an advanced stage. Another problem is the early development of resistance to the standard chemotherapy regimens with cisplatin/oxaliplatin in combination with paclitaxel. This leads to an early relapse and tumor progression. Considering these problems, it is clear that reliable diagnostic tools for an early detection of these cancers and more treatment options are urgently needed (2–4).
Epidemiological data show that induction and biology of ovarian cancer is related to estrogen exposure and metabolism. Experimental data demonstrated that ovarian cancer cells share a number of estrogen regulated pathways with other hormone-dependent cancers such as breast and endometrial cancer. Such pathways were studied in more details already in these tumors (5–7).
This review gives an overview on ovarian cancer heterogeneity and estrogen-related mechanisms in ovarian cancer biology. Thereby, data on enzymes active to synthesize and inactivate estrogens as well as on estrogen receptors (ER) are shown and strategies to target these pathways are discussed.
Ovarian Cancer as a Heterogeneous Disease
Classification
More and more, the classification of the heterogeneous group of ovarian carcinomas is coming into focus of research and ovarian cancer subtypes with different behaviors, mutations, origins, markers, and prognosis are characterized at a molecular level for their molecular signature. Since subtypes of ovarian cancer respond differently to the common therapies, a characterization of the tumor type is very important for a successful treatment (8).
Epithelial ovarian cancer is the most frequent tumor in the ovary as up to 98% of all cases are classified as EOCs. To this group belongs the most common and also the most deadly ovarian cancer, the high-grade serous ovarian cancer (HGSC), showing a frequency of 70%. It is followed by endometrial ovarian carcinomas (EC) and clear cell carcinomas (CCC) with frequencies of 10% for each entity, low-grade serous ovarian carcinomas (LGSC) with 5%, and mucinous carcinomas (MC) with 3% frequency (9). Other tumors, e.g., stromal cord tumors, teratomas, etc., are only rarely observed (10).
A summary of the predicted origins of different ovarian cancer subtypes and the mutations frequently found in these tumor types is given in Table 1.

Table 1. Origins and significant mutations of the EOC subtypes.
Several immunohistochemical and genetic analyses have been done to detect differences and identify features of ovarian cancer subtypes. Köbel et al. (13) did biomarker analyses and came to the conclusion that some biomarkers, e.g., Ki-67 as a cell proliferation marker, Williams tumor protein 1 and also CA125 show significant differences in expression rates between the subtypes. Therefore, they can be used as subtype specific biomarkers.
The serous subtypes are classified as low-grade (LGSC) and high-grade (HGSC). LGSC and HGSC represent two distinct tumor types with a different underlying pathogenesis rather than low-grade and high-grade variants of the same neoplasm. Both are usually at advanced stage at the time of diagnosis (FIGO stage III or IV). BRAF (v-raf murine sarcoma viral oncogene homolog B) and KRAS (Kirsten rat sarcoma viral oncogene homolog) mutations are important molecular events in LGSC, while HGSC as the most common subtype of ovarian cancer is almost always associated with p53 mutations. HGSC arises from the epithelium of the distal fallopian tube and not from the ovarian epithelial cells (9). In contrast to the serous tumors, EC and CCC are typically present as low-stage neoplasms and usually arise from endometriosis. Primary ovarian MCs are almost always unilateral and FIGO stage I tumors. This group consists mainly of so-called intestinal or enteric type MCs. Generally, MCs arise in a step-wise manner from a pre-existing mucinous cystadenoma or mucinous borderline tumor.
Another system classifies different ovarian cancer subtypes (mentioned above) into type I and type II tumors. Type I tumors (LGSC, low-grade EC, CCC, and MC) are generally slow growing and genetically more stable than type II tumors. A step-wise progression from a benign precursor lesion (endometriosis in the case of endometrioid tumors) to borderline tumors and next to the invasive tumors is characterized by genetic aberrations targeting specific cell signaling pathways, e.g., KRAS or BRAF mutations. Type II tumors (HGSC, high-grade EC, and undifferentiated carcinomas) are clinically aggressive and exhibit high genetic instability with frequent p53 mutations (14).
Serous Ovarian Carcinoma Subtypes
High-grade serous carcinoma
High-grade serous ovarian carcinoma is a highly aggressive tumor, which is usually detected in an advanced stage. After initially responding to standard platin- and taxane-based chemotherapy, the majority of patients will experience recurrence and develop resistance to therapeutic drugs within 24 months (8, 15). Early peritoneal metastasis is also common (16). The pathological morphology of HGSC is heterogeneous showing a papillary, glandular, or solid architecture. The tumor cells form large multi-layered epithelial areas, which are surrounded by tumor stroma. Mononuclear giant cells with large nuclei are commonly found in these tumors (13).
In contrast to LGSCs, HGSCs have a very high mitotic rate and usually carry mutations in the p53 gene. HGSCs are characterized by a high chromosomal instability. Loss of function mutations in the breast cancer type susceptibility genes 1 and 2 (BRCA1/2) are also associated with HGSC. Somatic mutations in, e.g., neurofibromin (NF1) and cyclin-dependent kinase (CDK)12 genes are common (9, 12, 16).
While previous models predict that HGSC develops from inclusion cysts of the ovarian epithelium, it is now agreed that HGSC arises from the serous tubal intraepithelial carcinomas (STICs). The latter develop from cells on the junction of the fallopian tube epithelium with the mesothelium of the tubal serosa. Cells there undergo malignant transformation and metastasize to the nearby ovary and later into the surrounding pelvic peritoneum.
Serous tubal intraepithelial carcinomas have mostly the same p53 mutations, express the same oncogenes and also have similar phenotypic characteristics as HGSCs (15). STICs are found in 67% of all HGSC cases (17). They are also associated with BRCA1/2 mutations (18).
High-grade serous ovarian cancer tumors in patients with mutated BRCA1/2 have a more aggressive behavior and high-grade histology, but they are frequently responsive to chemotherapy. In many cases, their high sensitivity to the platinum-based regimens, may lead to a slightly improved 5-year survival (19).
Low-grade serous ovarian carcinoma
Low-grade serous ovarian cancer is rather rare with <5% of all EOCs. LGSCs are thought to develop stepwise from benign serous cystadenomas via the formation of serous borderline tumors to the final carcinoma. However, LGSCs rarely transform to HGSC tumors (9, 20). If LGSCs are detected at an earlier stage, the prognosis after treatment is favorable. Even for patients with advanced stage tumors, the 5-year survival is longer than that for HGSC patients, although LGSCs are quite resistant to conventional chemotherapy. Similar to HGSC, this subtype often spreads intraperitoneally (21). In the histological picture, micropapillary structures and psammoma bodies (which are calcium incorporations that are formed from necrotic tumor cells) are frequently seen. LGSC cells have rather uniform nuclei and a much lower mitotic rate than HGSC tumor cells. Genetically, there is less chromosomal instability in LGSC than in HGSC. However, the presence of BRAF and KRAS mutations, as well as mutations in other genes (Table 1) is common (11, 16).
Ovarian Cancer as a Hormone-Dependent Cancer
Epidemiological Data
There is strong epidemiological evidence that etiology, pathogenesis, and progression of ovarian cancers are greatly dependent on the activity of estrogens. Furthermore, the balance between estrogen and progesterone is critical for the formation of ovarian cancers (22).
Statistical analyses show that the incidence of ovarian cancer is much higher in industrial countries than in developing countries. The birth rates in industrial countries are low compared to developing countries (23). There is strong evidence that reproductive factors including multiple pregnancies, breastfeeding, and use of oral conceptive pill (OCP) protect against ovarian cancer. With each pregnancy, the risk of developing ovarian cancer decreases by 10–16% and a pregnancy at the age of 35 years is twice as protective as at the age of 25 years (24, 25). Also, a significant protective effect is seen in women that do breastfeeding for more than 18 months (26, 27). Similarly, application of OCP for more than 3 years causes a 30–50% reduced risk of developing ovarian cancer (28).
In contrast to these protective factors, women with an early first period and a late menopause as well as women that receive drugs for the treatment of infertility (gonadotropin releasing-hormone antagonists or clomiphene) have an increased risk of developing ovarian cancers. The latter is thought to be caused by high concentrations of estrogen after stimulation of the sex-steroid hormone synthesis in the ovary (29). Also, application of hormone replacement therapy (HRT) was found to be a risk factor for ovarian cancer. An approximately 22% increased risk of ovarian cancer over 5 years was seen in postmenopausal women using unopposed estrogen as HRT. The risk was still significantly increased (by approximately 10%) by the application of a combination of estrogen and progestin (30, 31). Data from a study in a large patient cohort in England revealed that the incidence of ovarian cancer increased with longer duration of HRT therapy, especially if HRT was taken for 10 or more years. There was a higher relative risk for developing EOCs rather than MCs, ECs, or CCCs. But the composition of the HRT did not influence the risk (32). A more recent study revealed that women taking unopposed oral estrogen therapy have an increased risk of HGSCs, LGSCs, and ECs. Only the risk for MCs was decreased. Similar, an increased risk for serous carcinomas and ECs was found in women receiving an estrogen/progestin combination as HRT (33). On the other hand, women with previous HRT have a better prognosis when diagnosed with ovarian cancer. They are more likely to be diagnosed at younger age and lower tumor stage allowing a complete surgical removal of the tumor mass (complete debulking without any signs of a residual tumor mass). An increased overall survival, specifically in the subgroup of patients, which had a complete debulking, was found in these studies. No correlation was seen with the progression free survival (34). Data from another study, however, showed that if HRT was administered following tumor debulking, the prognosis remained unchanged. Furthermore, the survival time was independent on the expression of estrogen and progesterone receptors (PGR) in the cancer tissues. This study was done with a small number of patients only, and therefore, data may not be representative for a larger cohort of ovarian cancer patients (35). After tumor operation, especially younger ovarian cancer patients may suffer from estrogen withdrawal symptoms, and therefore, they will consider HRT treatment. Since data are not clear yet, studies with more patients suffering from different types of ovarian cancer are urgently needed.
Ovarian Cancer Hypothesis
To explain the effects of estrogens in the etiology of ovarian cancer, different hypothesis are available. The “incessant ovulation hypothesis” was developed already in 1971, but the more recent “incessant menstruation hypothesis” is now favored (36–38).
The incessant ovulation hypothesis
This hypothesis attributes ovarian cancer formation to repetitive wounding during ovulation and the subsequent activation of repair mechanisms. These processes are associated with an increased number of mutations accumulating in epithelial cells. This finally drives tumor formation and progression (39).
The association between sex steroids and cancer can be explained by processes that take place during the menstrual cycles, in which the ovarian surface epithelium (OSE) plays pivotal roles during ovulation and postovulatory wound repair. Indeed, most of the total proliferative activity of the OSE is related to ovulation repair and formation of the corpus luteum. In the menstrual cycle, the OSE covering growing follicles enters into the proliferative phase during pro-estrus/estrus transition. After the ovulation, the proliferation rate of OSE cells covering the newly formed corpus luteum decreases. Also, the exposure of the OSE to high doses of the gonadotropins luteinizing hormone (LH) and follicle stimulating hormone (FSH) during the menstrual cycle promotes cell proliferation and tumor growth (40). As a positive effect, progesterone, which is increased during pregnancy and during OCP application, promotes clearing of transformed cells from the ovarian surface epithelial layers (37, 41).
The incessant menstruation hypothesis
High-grade serous ovarian cancer are sought to derive from cells in the fimbriae of the fallopian tubes, which are floating in bloody peritoneal fluid. Thereby, they are exposed to iron-induced oxidative stress derived from retrograde menstruation. The genotoxic effect of reactive oxygen species, generated from hemolysis of erythrocytes by pelvic macrophages would explain the distal site of tubal intraepithelial neoplasia (37, 38, 41).
Estrogens and Ovarian Cancer
At the cellular level, tumor promoting effects of estrogen are conferred in a (i) receptor-dependent and (ii) -independent way.
i) Receptor-dependent ways: binding of estrogen to the nuclear estrogen receptor-α (ER-α) leads to the transcriptional activation of estrogen-responsive genes, which provide signaling systems for cell division and differentiation. Among these genes are proto-oncogenes, such as c-fos, c-myc, and HER2/neu; cell cycle regulating cyclins, growth factors, and others (42). Binding to membrane-bound G-protein-coupled estrogen receptor (GPER, formerly known as GPR30) activates second messenger systems. Thereby, GPER confers rapid non-genomic effects of estrogens (43).
ii) In a receptor independent way, formation of reactive metabolites via cytochrome P450 enzymes (CYPs) may lead to the generation of mutagenic DNA adducts. Free radicals generated by the metabolic activation of estrogens cause mutations. Accumulation of mutations in various genes in cells in the fallopian tubes and in the ovary will lead to the neoplastic transformation of cells (Figure 1) (10, 44, 45).
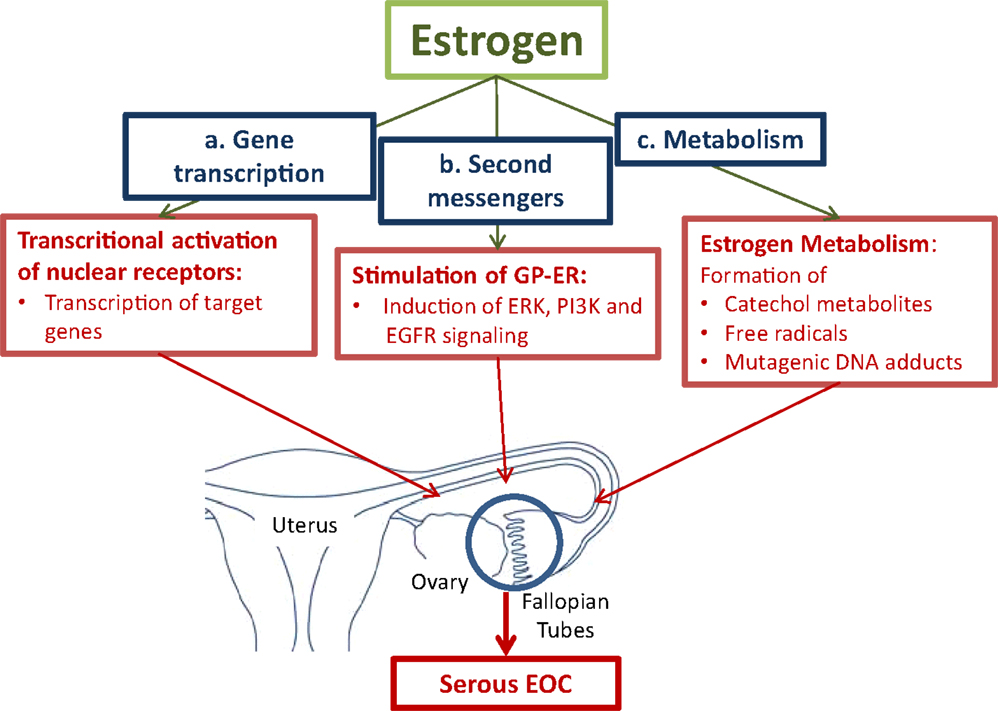
Figure 1. Pathways for estrogen to convert tumor promoting effects in cells in the fallopian tubes and the ovaries. (a.) Activation of the nuclear estrogen receptor-α (ER-α) leads to the transcriptional activation of estrogen-responsive genes, which stimulate cell proliferation. (b.) Binding to membrane-bound G-protein-coupled estrogen receptor (GPER) activates second messenger systems. In cancer cells, estrogen induces extracellular-signal regulated kinase (ERK), phosphoinositide 3-kinase (PI3K), and epidermal growth factor receptor (EGFR) leading to enhanced cell proliferation. (c.) The formation of reactive metabolites leads to the generation of mutagenic DNA adducts. Free radicals from the metabolic activation of estrogens will cause mutations. Accumulation of mutations will lead to neoplastic transformation of proliferating cells (10, 42, 44–46)
Description of pathways
i) Transcriptional effects of estrogens on target genes are mediated by activation of nuclear receptors, the estrogen receptor-α (ER-α), and estrogen receptor-β (ER-β). Upon binding of estrogens to ER-α, transcription of a battery of genes, which stimulate cell proliferation, is induced. Enhanced proliferation is associated with an increased risk of mutations that accumulate in cells finally leading to malignant transformation. ER-β was found to counteract the growth stimulating effects of ER-α in ovarian cancer cells (47).
Another member of the nuclear receptor superfamily, which confers estrogenic effects in ovarian cancer is the estrogen-related receptor-α (ERR-α). It is known to regulate metabolic homeostasis under conditions of high energy demand, e.g., in brown adipocytes. The increased expression and activity of ERR-α was associated with a less favorable clinical outcome of ovarian cancer (42).
Estrogens also promote tumor progression by influencing signaling pathways. Via the seven-transmembrane spanning G-protein-coupled receptor, named GPER, estrogen rapidly activates the extracellular signal-regulated kinases (ERK)-1 and ERK-2, and confers estrogenic effects to cells devote of the classical nuclear ER (48). A recent study in endometrial cancer cells showed that GPER mediates the estrogen stimulated induction of the kinases ERK-1 and -2 and the phosphatidylinositol-4, 5-bisphosphate 3-kinase (PI3K) via activation of matrix metalloproteinase. This is followed by the subsequent transactivation of the epidermal growth factor receptor (EGFR) (46).
ii) Metabolism of estrogen may cause DNA damage by the formation of mutagenic purinergic DNA adducts and by generation of free radicals from the metabolic activation to reactive catechol estrogens. Catechol estrogens are formed by aromatic hydroxylation of primary estrogens at either the C-2 or C-4 position. The catechol metabolites are inactivated by their conjugation with active sulfate or uridine-diphosphate (UDP)-glucuronic acid by steroid sulfotransferases (SULTs) and UDP-glucuronosyltransferases, respectively. Before their conjugation to more water-soluble metabolites, hydroxylation of the steroid moiety via specific CYP isoenzymes, namely CYP1A1 and CYP1A2, which catalyze hydroxylation in position 2, and CYP1B1, which is an estrogen 4-hydroxylase, occurs (44). Especially, 4-hydroxyestrogens can be oxidized to quinone intermediates, which react with purine bases of the DNA. This results in depurinating adducts, which generate highly mutagenic, apurinic sites. 2-hydroxyestrogens produce less genotoxic DNA adducts. Studies in rodents demonstrated that E2 and E1, as well as their catechol metabolites, in particular 4-hydroxy E2/E1 (4-OH E2/E1), have carcinogenic effects. In a redox cycle, 4-OH E2/E1 is converted to the quinone derivatives. The conversion back to 4-OH E2/E1 is associated with the formation of oxygen radicals. DNA mutations caused by free radicals will lead to the neoplastic transformation of cells (45).
Estrogen and BRCA1/2 Mutations
Breast and ovarian cancer mostly arise sporadically, but a small number of cases (approximately 10%) of these cancers are associated with mutations in BRCA1/2 genes. Defects in the DNA damage response or in the DNA repair pathways in patients with the BRCA1/2 mutation are responsible for the high penetrance of these cancers in the breast and/or ovary. It was shown that carriers of BRCA1/2 mutations have also increased levels of estrogen, which may trigger breast and ovarian cancers (49). Indeed, in premenopausal patients with BRCA1/2 mutations, removal of both ovaries and of the fallopian tubes reduces the risk of these cancers (50).
Estrogen Synthesis in Ovarian Cancer Cells
During the reproductive years, ovaries produce and release progesterone and the estrogens estrone (E1), 17β-estradiol (E2), and estriol (E3).
E2, the most active form of natural estrogens, together with progesterone is critical for normal uterine function, establishment and maintenance of pregnancy, and mammary gland development. Furthermore, it is responsible for endocrine, paracrine, and autocrine actions in various tissues and organs. Ovarian steroid hormone formation takes place in the ovarian granulosa and theca cells, which work in a collaborative way for the synthesis after stimulation by the gonadotropins LH and FSH. Theca cells respond to LH signaling by increasing the expression of steroid synthesizing enzymes for the transformation of cholesterol to the androgens (5-androstenedione and testosterone). Granulosa cells respond to FSH signaling by stimulating the expression of enzymes for the synthesis of estrogens (E2 and E1) from androgen precursors (51).
While in premenopausal women, the main part of active estrogens derives from the synthesis in the ovary, after the menopause, estrogens are formed locally in various tissues such as in liver, brain, and adipose tissue. There, E2 is produced from circulating androgen and estrogen precursors. These precursors are bound to sex-steroid binding globulins in the blood. They are transported to the ovary, where they are taken up into ovarian epithelial cells by transporters, e.g., from the family of organic anion transporting peptides (OATPs) (52). The importance of the visceral adipose tissue of postmenopausal women for E2 production is reflected by the high concentrations of estrone-sulfate (E1-S), 5-androstendione, and dehydroepiandrosterone-sulfate (DHEA-S) in these cells. The local concentrations are up to 60× higher than in serum, while E2 and testosterone levels are increased by sevenfold only (53, 54).
The biological activity of estrogen is regulated independent on the expression and activity of receptors by the expression and function of steroid (estrogen)-metabolizing enzymes expressed locally at the target tissues. Among these enzymes, aromatase (CYP19A1), steroid sulfatase (STS), and 17β-hydroxysteroid dehydrogenases (17β-HSD) are highly important.
In hormone-dependent cancer, formation of the biological most active estrogen E2 from steroid precursors is mediated via the aromatase and the sulfatase pathway (Figure 2).
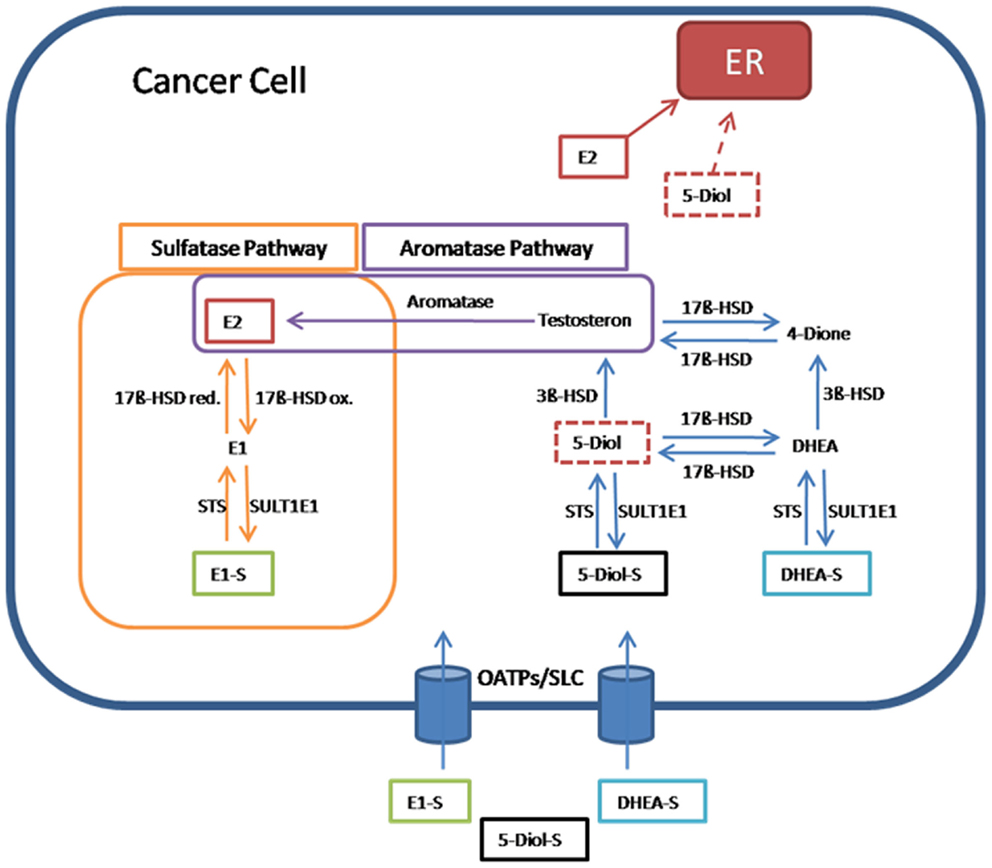
Figure 2. Pathway for 17β-estradiol formation in cancer cells. Estrone-sulfate (E1-S), a precursor for the most active estrogen 17β-estradiol (E2), androstenediol (5-Diol-S), and dehydroepiandrosterone sulfate (DHEA-S) are taken up from the blood into cancer cells by transporters from the organic anion transporting polypeptide family (OATPs) and other members of the solute carriers (SLCs). In the sulfatase pathway, E1-S is converted through steroid sulfatase (STS) to estrone (E1), which is transformed via the reductive 17β-hydroxysteroid dehydrogenases (17β-HSD red) to E2. E2 as the most active estrogen binds and activates estrogen receptors (ERs). In the reverse pathway from E2 to E1-S, the oxidative 17β-hydroxysteroid dehydrogenases (17β-HSDox) convert E2 to E1. The estrogen sulfotransferase SULT1E1 inactivates estrogens by adding sulfate to hydroxyl-groups on the steroid ring. In the aromatase pathway, E2 is produced from testosterone by the aromatase. Testosterone itself is formed from 5-androstenediol (5-Diol) via 3β-HSD. 5-Diol is generated by the removal of the sulfate from 5-Diol-S via STS. Also 5-Diol can activate ER, but with lower affinity than E2. In an alternative way, testosterone is derived from the conversion of DHEA-S to androstenedione (4-Dione) via DHEA. Finally, 17β-HSD transforms 4-Dione to testosterone (55).
In the aromatase pathway, steroid precursors derived from the circulation, like dehydroepiandrosterone (DHEA), are first converted to testosterone and, subsequently, by the aromatase, E2 is generated.
In the sulfatase pathway, the sulfate moiety is removed by STS from the inactive E1-S to form the active estrogen E1. E1 is converted to E2 by the reducing 17β-HSD isoenzymes. In the inactivating pathway, E2 is oxidized to E1 by 17β-HSD enzymes. E1, which exerts also estrogenic effects via binding to ERs, can be inactivated by estrogen sulfotransferase (SULT1E1). The resulting inactive E1-S can again become activated by the removal of sulfate (7, 55).
The precursor of all steroid hormones is cholesterol, which is mainly derived from the synthesis, particularly in the liver, or from the nutrition. Cholesterol is converted over a few steps to pregnenolone, and next to progesterone. Progesterone acts as the starting point for all steroid hormones in the adrenal cortex and in various other peripheral organs, e.g., the liver. The steroid hormone precursors DHEA, its sulfate metabolite DHEA-S, 5α-androstenediol sulfate (5-Diol-S), and E1-S are also synthesized in large quantity there. They are released into the circulation.
In the group of postmenopausal women, the levels of E2 and progesterone are up to 90% lower than those from premenopausal women. However, the concentration of other steroid hormones (DHEA, 5-Diol-S) is similar in pre- and postmenopausal women. After the menopause, E1-S is the most abundant estrogen in the peripheral blood of women. Ten to thirty percent of estrogens in serum are bound to sex-steroid binding globulins in order to provide a reservoir for peripheral formation of E2. These proteins are synthesized in the liver after the stimulation with estrogen and thyroid hormones. Progesterone inhibits their production (56, 57).
Estrogen Synthesis in Hormone-Dependent Cancers
Epidemiological and experimental studies showed that higher endogenous estrogen exposure through early menarche (<12 years), late menopause (>55 years), nulliparity, obesity (postmenopausal), use of postmenopausal HRT, and increased plasma E2 levels lead to an increased risk of breast cancer. Of particular importance is the circulating inactive plasma estrogen precursor E1-S, which has been reported to serve as the predominant source for tumor E2 in postmenopausal patients with breast cancer. E1-S is derived from peripheral tissues, including the adrenal gland, adipocytes, liver, muscle, skin, and bone (54). Following its cellular uptake by transporters of the OATPs family (52), E1-S is desulfonated to E1 by STS and E1 is further converted to E2 by 17β-HSDs. This intracellular production of E2 stimulates the proliferation of estrogen-dependent tumor cells (58). As compared to the other sources of unconjugated estrogens (which act as precursors for the aromatase pathway), E1-S (precursor for the sulfatase pathway) has about 5–10 times higher plasma circulating levels than other estrogens (59). Moreover, sulfatase activity is 130–200 times higher than aromatase activity (60) and the concentration of sulfatase is three times higher in breast cancer tissues than normal tissues (61). Two-thirds of all breast cancers have a positive ER status and as consequence, they are sensitive to estrogens. These tumors respond well to hormone therapy (62). Also in endometrial cancer, biosynthesis of active estrogen is achieved from E1-S, which is transported into endometrial cells, where it is converted to E2 by STS and 17β-HSDs (63).
Enzymes in the Estrogen Metabolism Pathway
Steroid Sulfatase
Steroid sulfatase is a member of the arylsulfatase family, which is known for the hydrolysis of sulfate ester bonds in a wide range of substrates. The corresponding gene is located on the X-chromosome and contains 10 exons. The group of Hernandez-Guzman et al. (64) was the first to isolate STS from the human placenta, to purify and crystallize it. This enzyme consists of two membrane-spanning hydrophobic alpha helices that are arranged anti-parallel. The amino acid proline on position 212 acts on the cytosolic side of the endoplasmatic reticulum membrane as turning point. STS is present in the ER of many tissues and is especially high in ovarian granulose cells (65, 66). It plays an essential role in the synthesis of E2, by converting E1-S into active E1 (Figure 2).
Estrogen Sulfotransferase
Estrogen sulfotransferase belongs to the family of SULTs, phase II detoxification enzymes. Ten isoforms of SULT are known to be expressed in humans. These enzymes have a wide range of substrates including hydroxysteroids, thyroid hormones, phenols, arylamines, and primary alcohols. Because SULT1E1 has the highest affinity of all SULTs for E1 and E2, it is also called estrogen specific sulfotransferase. It catalyzes the transfer of a sulfuryl group from 3′-phosphoadenosine 5′-phosphosulfate (PAPS) to nucleophilic groups of SULT1E1 substrates. PAPS are synthesized by PAPS synthesizing enzymes (PAPS synthetases).
Estrogen sulfotransferase acts in an antagonistic way to STS by converting E1 to E1-S. Therefore, it is inactivating the estrogen. The corresponding gene is located on chromosome 4q3.12, has eight exons and a length of 20 kB. SULT1E1 is present in various tissues, including liver, testis, mammary epithelium cells, and endometrium (67–69).
17β-Hydroxysteroid Dehydrogenases
17β-HSDs form a family of enzymes (14 isoforms in vertebrates) that catalyze the conversion between highly active 17β-hydroxy steroid hormones and 17-keto steroid hormones with lower or even missing activity. 17β-HSD isoenzymes have different enzymatic properties and a characteristic cell-specific expression pattern according to their different physiological functions. They are multifunctional and modulate other steroid structures as well. Among their substrates are bile acids, retinoids, fatty acids, and others.
The reductive 17β2-HSD isoenzyme activates E1 to E2 and 4-Dione to testosterone. Also the isoforms 7 and 12 are reductive estrogenic enzymes. In contrast, isoforms 4, 8, 10, and 14 are oxidative enzymes responsible for E2 inactivation (63).
In the normal ovary, 17β-HSDs are detected in granulosa cells of developing follicles, but not in the normal OSE. However, a variety of EOC have been reported to be positive for different 17β-HSD isoenzymes.
For example, a recent study showed that reducing 17β12-HSD is overexpressed in many human carcinomas including ovarian carcinoma, while it is not present in the normal OSE. Importantly, patients with EOC tumors with a weak or moderate expression of 17β12-HSD had a better overall survival than those with strongly 17β12-HSD positive tumors (55).
Estrogen Receptors
Estrogen Receptors-α/-β
Currently, two isoforms of the nuclear ER, namely ER-α and ER-β are known. In 1996, Kuiper et al. (70) cloned the second ER, named ER-β from rat testis. As a consequence, ER was renamed to ER-α. The two ERs are encoded by the ESR1 and ESR2 genes. Alternative promoter usage and splicing produces various transcript variants. However, expression pattern and function of many of these variants has not been determined so far.
The ERs are members of the nuclear receptor superfamily of steroid receptors and function as ligand inducible transcription factors (71). The nuclear receptor superfamily structure is defined through five different domains, namely A/B, C, D, E, and F. Each domain fulfills another obligation, which is essential for the correct function of the receptor. The A/B domain, also named N-terminal domain includes a ligand independent activation function (AF-1). This domain also has a rather poor (20%) homology with the other ER isoform (72). This gives a hint that this region takes mainly part to the ER subtype specific actions on target genes. Moreover ER-α seems to have a stronger corresponding AF-1 function than ER-β (73). The DNA-binding domain C is responsible for specific DNA-binding and receptor dimerization. This highly conserved domain shows a high homology between ER-α and ER-β. The D-domain is a flexible hinge with the ligand-binding domain and is called “Hinge domain.” ER-α shares only 30% homology in this domain with ER-β. It contains a nuclear localization signal, which is essential for the transport of ERs to the nucleus. The ligand-binding E-domain is responsible for hormone-dependent activation (AF-2), ligand binding, and together with the DNA-binding domain for receptor dimerization. ER-α and ER-β show a homology of 55% in the ligand-binding domain (74, 75). The amino acid sequence of the ligand-binding cavity varies only in two positions, but this leads to a significant smaller cavity in ER-β. This may be an advantage for receptor subtype specific drugs (76, 77). The function of the F domain is still not clear. A dimerization of ER is necessary for transcriptional activation. Either a homodimer of one ER isoform or a heterodimer of ER-α and ER-β is active (73). The regulation of the transcription of target genes is achieved by binding of the receptors to estrogen response elements in target genes. This is followed by recruitment of a variety of coregulators to alter chromatin structure and facilitate the recruitment of the RNA polymerase II transcriptional machinery (78).
Estrogen receptors mediate many estrogenic effects on gene regulation and, therefore, ERs are essential for various developmental and functional processes in several tissues/cells (79). ERs are important for sexual development and reproductive function, but also play a role in other tissues such as bone.
ER-α is the predominant ER in the uterus, mammary gland, testis, pituitary, liver, kidney, heart, and skeletal muscle (80–82). Some ER-α target genes mediate proliferation and cell survival (83). Importantly, sustained estrogenic exposure and activation of ER-α may increase the risk and/or the progression of various cancers, including cancers in the breast and endometrium.
The ER-β gene consists of eight exons and through alternative splicing five isoform, namely ER-β1-5 are generated by deletion of one or more exons. Human ER-β1 protein has a length of 530 amino acids (84, 85). ER-β is predominantly expressed in ovary and prostate (80–82). In contrast to ER-α, ER-β activates antiproliferative and pro-apoptotic pathways in many cancer cells (83).
The Membrane-Bound G-Protein-Coupled Estrogen Receptor
G-protein-coupled estrogen receptor functions as a G coupled plasma membrane-associated receptor and works independent of nuclear ERs. It binds various estrogens including E2, E1 and E3, ER antagonists, phytoestrogens, and xenoestrogens (86). GPER, which mediates rapid estrogen signaling via stimulation of adenylyl cyclase, is expressed in normal ovary, where it regulates physiological processes such as follicle maturation. The receptor is expressed in many EOC samples. However, the prognostic impact of GPER expression in EOC is controversial. In an earlier study, GPER was seen to be preferentially expressed in “high risk” tumors with a worse prognosis (87). Later, no relation between GPER expression and survival rates of EOC patients was found (88). Recently, GPER expression was shown to correlate with gonadotropin (LH and FSH) receptors. Only in EOCs, which are negative for gonadotropin receptors, a higher GPER expression was associated with a more favorable outcome for the patients. These findings suggest that GPER may reduce ovarian cancer cell proliferation in the absence of gonadotropin signaling only (89). Therefore, synthetic agonists and antagonists for GPER, which are now available, might be tested in gonadotropin receptor-negative tumors of EOC patients (90).
Progesterone and Progesterone Receptors
Progesterone is synthesized from cholesterol in the corpus luteum, follicles, placenta, and in other organs, e.g., the adrenal gland. During transport through the blood plasma, it is bound to cortisol binding globulin, because of its lipophilic nature and also to avoid degradation.
In the ovary, progesterone plays an important role in the follicle maturation and moreover, it is responsible for preparing the female genital tract for pregnancy. It also maintains the pregnancy after fertilization. Progesterone promotes the growth of the uterus musculature and changes the endometrium from a proliferative to a secretory tissue. It also decreases the myometrium activity during pregnancy and changes the quality and quantity of the cervix mucus, thus preventing the entrance of sperms into the uteri and the tubes. In the ovary, it works in concert with estrogen to promote follicle maturation, ovulation, and formation of the corpus luteum.
The effects of progesterone are mediated by the two members of nuclear PGR isoforms. Like other receptors of this family, PGR consists of four domains, namely the ligand-binding domain, the Hinge-Region as the flexible link between the ligand-binding domain and the DNA-binding domain, and the N-terminal domain (91). The two isoforms PR-A and PR-B are encoded by one gene but the transcription is carried out by two different promoters. Apart from the lack of a 164 amino-sequence at the N-terminal end in PR-A, the two PGRs are identical. PR-B is responsible for the transcriptional activation of progesterone responsive genes, but it can be inhibited by PR-A (92–95). PR-A and PR-B act either as homo- or heterodimers and both are synthesized in equal amounts in normal epithelial cells (96).
Progesterone as an antagonist of estrogen has an antiproliferative effect on specific cells. It acts in part by decreasing the production of ERs, and through activation of 17β-HSD and SULT1E1. Indeed, elevated PGR levels were associated with a significantly better survival rate in EOC patients as data from a recent meta-analysis showed (97).
Estrogen Synthesizing Enzymes and Receptors in Ovarian Cancer
There is evidence that estrogens play a role in the progression of ovarian cancer. An overexpression of STS will lead to an increased level of E2 and this may contribute to cancer progression. In postmenopausal women, the local conversion from circulating steroid hormone precursors, e.g., E1-S and DHEA-S to active E2 could promote ovarian cancer progression.
Indeed, the expression levels of key enzymes vary between normal tissues and different subtypes of EOC. For example, STS was detected in 30% of serous carcinomas, in 70% of CCCs, and in 8% of MCs (98). Ovarian cancer studies further showed that a longer progression free survival is significantly associated with lower STS levels. That can be explained by the fact that through a high expression of STS, more E2 is synthesized (99). Similar effects of STS were also reported from breast cancer studies, where STS activity correlated with the E2 serum levels. On the opposite, high estrogen inactivating SULT1E1 levels were associated with smaller tumors, a better prognosis, and a negative lymph node status in ovarian cancer patients (100–103). In the same way, in breast cancer a high expression of SULT1E1 together with a decreased level of STS correlates with a better prognosis, smaller tumor size, and a negative lymph status at the time of diagnosis. Another evidence that SULT1E1 acts as a tumor suppressor is based on a study with xenograft models carried out by Xu et al. (69) showing that an overexpression of SULT1E1 inhibited estrogen-dependent cell growth and induced tumor cell apoptosis.
High E2 levels were often observed in ovarian cancer patients (102). E2 was shown to increase the mobility of ovarian cancer cells via the inhibition of cell–cell adhesion. This promotes metastasis (104), and a similar effect of E2 was also observed in breast cancer (105).
Mostly earlier studies in ovarian cancer patients reported that high levels of ER-α and low levels ER-β are associated with a worse prognosis (106–110). However, in other studies, high expression of ER-α was found to be associated with a better prognosis (111, 112). Also higher ER-β levels were significantly associated with longer disease-free survival and a longer overall survival in one study. The reduction of ER-β significantly correlated with the hyper-methylation of the ER-β promoter, causing an inhibition of gene translation (113). A proof for the protective effect of ER-β is that this isoform is the dominant ER isoform in healthy ovaries. But in all serous tumors and also in metastasis, ER-α is usually dominant, and ER-β expression is rather weak. This leads to the conclusion that the gradual reduction of ER-β during tumor progression (from normal to borderline to malign tissue to metastasis) is a continuous process and important for malignant transformation and cancer cell proliferation (114, 115).
Progesterone has been proved to decrease the proliferative effect of estrogens and inhibit inflammation and cancer penetration by suppressing ovulation. It also initiates apoptosis in tumor cells. PGR is a biomarker for a better prognosis and longer overall survival in ovarian cancer (116). This is in line with findings that PGR levels are significantly lower in benign, borderline, and malignant ovarian tumors than in healthy tissue (108, 117). Recently, a multi-center investigation in 2933 women with invasive EOCs showed that PGR and ER are positive prognostic biomarkers for ECs and HGSCs (118).
Ovarian Cancer and Hormonal Treatment
There is evidence that estrogen has an influence on the progression of the EOCs at least in some subgroups of patients (119). To combat estrogen stimulated tumor cell growth, selective estrogen receptor modulators (SERMs) that function as agonists or antagonists for ERs, have been developed. However, modulation of the tissue-specific expression of ER subtypes, expression of co-regulatory proteins, and varying ER conformational changes induced by ligand binding may change the activity of the hormonal therapy. The best studied SERM is tamoxifen that is highly effective to block the ER-signaling pathways. It prevents breast cancer recurrence in many patients with positive ER status. In postmenopausal patients failing tamoxifen therapy, the synthesis of estrogen can be blocked by aromatase inhibitors anastrozole, letrozole, and exemestane (120).
In ovarian cancer, the therapeutic value for SERMs and aromatase inhibitors to block tumor progression and recurrence is not thoroughly established, yet. Only small-scale studies were done so far. Additionally, in the few studies, the patients were not selected based on their hormone-receptor status (ER positive or negative) or age (pre- vs. postmenopausal). Moreover, in some studies, patients were only selected after resistance to standard chemotherapy. Later, the group of Tropé et al. (121) compared data from different studies with tamoxifen (in total a collective of 647 patients) and found a response rate ranging from 0 to 56%, with a mean response rate of 11%. The treatment with aromatase inhibitor letrozole showed a response rate ranging from 0 to 35.7%, with stable disease rates ranging from 20 to 42% (122–124). For anastrozole, response rates of 1.9–4.3% and stable disease rates of 42–61% were reported (125, 126).
Whether ovarian cancer patients, who express ERs and estrogen synthesizing enzymes such as STS and 17β-HSDs in their tumors, may have a better response rate to hormonal therapy, should be investigated. Nevertheless, inhibition of estrogen activating STS would offer a novel approach to combat ovarian cancer. Among already available STS inhibitors, the cyclopentane carboxylate derivate STX64 (irosustat) is currently undergoing clinical trials for therapy of prostate, endometrial and breast cancer. With this drug, serum levels of E1, E2, 4-Dione and DHEA were decreased and stable disease for up to 7 months was even seen in breast cancer patients with advanced disease. However, for a more efficient depletion of tumor estrogen, application of STS inhibitors together with an aromatase inhibitor could also be of benefit in postmenopausal women in order to block both, E2 formation via the aromatase and sulfatase pathway (57). Also, subgroups of ovarian cancers should be studied in different therapeutic settings. This would help to identify patients, for which hormonal therapy might offer an additional possibility to prevent recurrence of ovarian cancer.
Summary
Ovarian cancer is the deadliest of all gynecological malignancies in women and these tumors are usually seen in women after the age of 50 years. The still poor prognosis for ovarian cancer is partly attributed to the fact that the diagnosis is usually made at a late stage, when the cancer has already spread to other organs. There are only limited options for a successful chemotherapeutic treatment so far and novel strategies are needed.
Several epidemiological and experimental data revealed that ovarian cancer shares many estrogen regulated pathways with other hormone-dependent cancers, e.g., breast cancer. Therefore, local estrogen synthesis from circulating steroid hormone precursors by steroid-forming and steroid-inactivating enzymes may be important to drive ovarian cancer progression in women after the menopause. Indeed, these enzymes and receptors were identified in ovarian cancer cells and their expression was shown to be related to clinical parameters. So far, such studies were mostly done in a small group of patients, which were not selected based on their age, ovarian cancer subtype, hormone-receptor status, and resistance to standard chemotherapy. Because ovarian cancer is a heterogeneous disease and tumors vary with respect to their origin, behavior, and prognosis, they may also differ in their sensitivity to hormonal therapy. At least in subgroups of patients, who express enzymes for estrogen biosynthesis and receptors for estrogen signaling in their tumors, hormonal therapy might offer an additional possibility to prevent recurrence of ovarian cancer.
The review explains the role of estrogen in ovarian cancer and it gives an overview on ovarian cancer subtypes. Furthermore, enzymes active to synthesize and metabolize estrogens as well as estrogen signaling pathways are described. Strategies to target these pathways are discussed.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
The work was in part supported by the FP-6 STREP project LSHC-CT-2005-018698.
Abbreviations
CCC, clear cell carcinoma; CYP, cytochrome P450 enzyme; DHEA(-S), dehydroepiandrosterone(-sulfate); EC, endometrial ovarian carcinoma; EOC, epithelial ovarian cancer; ER-α, estrogen receptor-α; ER-β, estrogen receptor-β; ERR-α, estrogen-related receptor-α; E1, estrone; E1-S, estrone-sulfate; E2, 17ß-estradiol; E3, estriol; FIGO, Féderation Internationale de Gynécologie et d’Obstétrique; FSH, follicle stimulating hormone; GPER, G-protein-coupled estrogen receptor; HGSC, high-grade serous ovarian cancer; HRT, hormone replacement therapy; LGSC, low-grade serous ovarian cancer; LH, luteinizing hormone; MC, mucinous carcinomas; OATP, organic anion transporting polypeptide; OCP, oral conceptive pill; OSE, ovarian surface epithelium; PGR, progesterone receptor; SERM, selective estrogen receptor modulators; STIC, serous tubal intraepithelial carcinoma; STS, steroid sulfatase; SULT, sulfotransferase; SULT1E1, estrogen sulfotransferase; 4-OH-E1/E2, 4-hydroxy E2/E1; 5-Diol-S, 5α-androstenediol sulfate; 17β-HSD, 17β-hydroxysteroid dehydrogenases.
References
1. FIGO Committee on Gynecologic Oncology. Current FIGO staging for cancer of the vagina, fallopian tube, ovary, and gestational trophoblastic neoplasia. Int J Gynaecol Obstet (2009) 105:3–4. doi: 10.1016/j.ijgo.2008.12.015
2. Cancer Research UK. (2014). Available from: www.cancerresearchuk.org/cancer-help/type/ovariancancer.
3. American Cancer Society. (2014). Available from: www.cancer.org/cancer/ovariancancer/detailedguide/ovarian-cancer-key-statistics
4. National Cancer Institute. (2014). Available from: http://seer.cancer.gov/statfacts/html/ovary.html
5. Modugno F, Laskey R, Smith AL, Andersen CL, Haluska P, Oesterreich S. Hormone response in ovarian cancer: time to reconsider as a clinical target? Endocr Relat Cancer (2012) 19:R255–79. doi:10.1530/ERC-12-0175
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
6. Schuler S, Ponnath M, Engel J, Ortmann O. Ovarian epithelial tumors and reproductive factors: a systematic review. Arch Gynecol Obstet (2013) 287:1187–204. doi:10.1007/s00404-013-2784-1
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
7. Labrie F. All sex steroids are made intracellularly in peripheral tissues by the mechanisms of intracrinology after menopause. J Steroid Biochem Mol Biol (2014). doi:10.1016/j.jsbmb.2014.06.001
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
8. Erickson BK, Conner MG, Landen CN Jr. The role of the fallopian tube in the origin of ovarian cancer. Am J Obstet Gynecol (2013) 209:409–14. doi:10.1016/j.ajog.2013.04.019
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
9. Gilks CB, Prat J. Ovarian carcinoma pathology and genetics: recent advances. Hum Pathol (2009) 40:1213–23. doi:10.1016/j.humpath.2009.04.017
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
10. Gajjar K, Martin-Hirsch PL, Martin FL. CYP1B1 and hormone-induced cancer. Cancer Lett (2012) 324:13–30. doi:10.1016/j.canlet.2012.04.021
11. Singer G, Kurman RJ, Chang HW, Cho SK, Ie M. Diverse tumorigenic pathways in ovarian serous carcinoma. Am J Pathol (2002) 160:1223–8. doi:10.1016/S0002-9440(10)62549-7
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
12. Shih Ie M, Kurman RJ. Ovarian tumorigenesis: a proposed model based on morphological and molecular genetic analysis. Am J Pathol (2004) 164:1511–8. doi:10.1016/S0002-9440(10)63708-X
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
13. Köbel M, Kalloger SE, Boyd N, Mckinney S, Mehl E, Palmer C, et al. Ovarian carcinoma subtypes are different diseases: implications for biomarker studies. PLoS Med (2008) 5:e232. doi:10.1371/journal.pmed.0050232
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
14. McCluggage WG. Morphological subtypes of ovarian carcinoma: a review with emphasis on new developments and pathogenesis. Pathology (2011) 43:420–32. doi:10.1097/PAT.0b013e328348a6e7
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
15. Kessler M, Fotopoulou C, Meyer T. The molecular fingerprint of high grade serous ovarian cancer reflects its fallopian tube origin. Int J Mol Sci (2013) 14:6571–96. doi:10.3390/ijms14046571
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
16. Prat J. Ovarian carcinomas: five distinct diseases with different origins, genetic alterations, and clinicopathological features. Virchows Arch (2012) 460:237–49. doi:10.1007/s00428-012-1203-5
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
17. Carlson JW, Miron A, Jarboe EA, Parast MM, Hirsch MS, Lee Y, et al. Serous tubal intraepithelial carcinoma: its potential role in primary peritoneal serous carcinoma and serous cancer prevention. J Clin Oncol (2008) 26:4160–5. doi:10.1200/JCO.2008.16.4814
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
18. Cass I, Holschneider C, Datta N, Barbuto D, Walts AE, Karlan BY. BRCA-mutation-associated fallopian tube carcinoma: a distinct clinical phenotype? Obstet Gynecol (2005) 106:1327–34. doi:10.1097/01.AOG.0000187892.78392.3f
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
19. Alsop K, Fereday S, Meldrum C, Defazio A, Emmanuel C, George J, et al. BRCA mutation frequency and patterns of treatment response in BRCA mutation-positive women with ovarian cancer: a report from the Australian ovarian cancer study group. J Clin Oncol (2012) 30:2654–63. doi:10.1200/JCO.2011.39.8545
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
20. Koshiyama M, Matsumura N, Konishi I. Recent concepts of ovarian carcinogenesis: type I and type II. Biomed Res Int (2014) 2014:934261. doi:10.1155/2014/934261
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
21. Crispens MA, Bodurka D, Deavers M, Lu K, Silva EG, Gershenson DM. Response and survival in patients with progressive or recurrent serous ovarian tumors of low malignant potential. Obstet Gynecol (2002) 99:3–10. doi:10.1016/S0029-7844(01)01649-0
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
22. Folkerd EJ, Dowsett M. Influence of sex hormones on cancer progression. J Clin Oncol (2010) 28:4038–44. doi:10.1200/JCO.2009.27.4290
23. International Agency for Research on Cancer. (2013). Available from: http://globocan.iarc.fr/ia/World/atlas.html
24. Purdie DM, Bain CJ, Siskind V, Webb PM, Green AC. Ovulation and risk of epithelial ovarian cancer. Int J Cancer (2003) 104:228–32. doi:10.1002/ijc.10927
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
25. Whiteman DC, Siskind V, Purdie DM, Green AC. Timing of pregnancy and the risk of epithelial ovarian cancer. Cancer Epidemiol Biomarkers Prev (2003) 12:42–6.
26. Titus-Ernstoff L, Rees JR, Terry KL, Cramer DW. Breast-feeding the last born child and risk of ovarian cancer. Cancer Causes Control (2010) 21:201–7. doi:10.1007/s10552-009-9450-8
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
27. Jordan SJ, Cushing-Haugen KL, Wicklund KG, Doherty JA, Rossing MA. Breast-feeding and risk of epithelial ovarian cancer. Cancer Causes Control (2012) 23:919–27. doi:10.1007/s10552-012-9963-4
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
28. Narod SA, Risch H, Moslehi R, Dorum A, Neuhausen S, Olsson H, et al. Oral contraceptives and the risk of hereditary ovarian cancer. Hereditary ovarian cancer clinical study group. N Engl J Med (1998) 339:424–8. doi:10.1056/NEJM199808133390702
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
29. Choi JH, Wong AS, Huang HF, Leung PC. Gonadotropins and ovarian cancer. Endocr Rev (2007) 28:440–61. doi:10.1210/er.2006-0036
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
30. Zhou B, Sun Q, Cong R, Gu H, Tang N, Yang L, et al. Hormone replacement therapy and ovarian cancer risk: a meta-analysis. Gynecol Oncol (2008) 108:641–51. doi:10.1016/j.ygyno.2007.12.003
31. Pearce CL, Near AM, Van Den Berg DJ, Ramus SJ, Gentry-Maharaj A, Menon U, et al. Validating genetic risk associations for ovarian cancer through the international ovarian cancer association consortium. Br J Cancer (2009) 100:412–20. doi:10.1038/sj.bjc.6604820
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
32. Beral V, Bull D, Green J, Reeves G. Ovarian cancer and hormone replacement therapy in the million women study. Lancet (2007) 369:1703–10. doi:10.1016/S0140-6736(07)60534-0
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
33. Morch LS, Lokkegaard E, Andreasen AH, Kjaer SK, Lidegaard O. Hormone therapy and different ovarian cancers: a national cohort study. Am J Epidemiol (2012) 175:1234–42. doi:10.1093/aje/kwr446
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
34. Hein A, Thiel FC, Bayer CM, Fasching PA, Haberle L, Lux MP, et al. Hormone replacement therapy and prognosis in ovarian cancer patients. Eur J Cancer Prev (2013) 22:52–8. doi:10.1097/CEJ.0b013e328355ec22
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
35. Li L, Pan Z, Gao K, Zhang W, Luo Y, Yao Z, et al. Impact of post-operative hormone replacement therapy on life quality and prognosis in patients with ovarian malignancy. Oncol Lett (2012) 3:244–9.
36. Fathalla MF. Incessant ovulation – a factor in ovarian neoplasia? Lancet (1971) 2:163. doi:10.1016/S0140-6736(71)92335-X
37. Vercellini P, Crosignani P, Somigliana E, Vigano P, Buggio L, Bolis G, et al. The ‘incessant menstruation’ hypothesis: a mechanistic ovarian cancer model with implications for prevention. Hum Reprod (2011) 26:2262–73. doi:10.1093/humrep/der211
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
38. Seidman JD. The presence of mucosal iron in the fallopian tube supports the “incessant menstruation hypothesis” for ovarian carcinoma. Int J Gynecol Pathol (2013) 32:454–8. doi:10.1097/PGP.0b013e31826f5ce2
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
39. Wright JW, Jurevic L, Stouffer RL. Dynamics of the primate ovarian surface epithelium during the ovulatory menstrual cycle. Hum Reprod (2011) 26:1408–21. doi:10.1093/humrep/der057
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
40. Levanon K, Crum C, Drapkin R. New insights into the pathogenesis of serous ovarian cancer and its clinical impact. J Clin Oncol (2008) 26:5284–93. doi:10.1200/JCO.2008.18.1107
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
41. Smith ER, Xu XX. Ovarian ageing, follicle depletion, and cancer: a hypothesis for the aetiology of epithelial ovarian cancer involving follicle depletion. Lancet Oncol (2008) 9:1108–11. doi:10.1016/S1470-2045(08)70281-X
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
42. Chang CY, Mcdonnell DP. Molecular pathways: the metabolic regulator estrogen-related receptor alpha as a therapeutic target in cancer. Clin Cancer Res (2012) 18:6089–95. doi:10.1158/1078-0432.CCR-11-3221
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
43. Filardo EJ, Quinn JA, Bland KI, Frackelton AR Jr. Estrogen-induced activation of Erk-1 and Erk-2 requires the G protein-coupled receptor homolog, GPR30, and occurs via trans-activation of the epidermal growth factor receptor through release of HB-EGF. Mol Endocrinol (2000) 14:1649–60. doi:10.1210/mend.14.10.0532
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
44. Cavalieri EL, Rogan EG. Unbalanced metabolism of endogenous estrogens in the etiology and prevention of human cancer. J Steroid Biochem Mol Biol (2011) 125:169–80. doi:10.1016/j.jsbmb.2011.03.008
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
45. Yager JD. Mechanisms of estrogen carcinogenesis: the role of E2/E1-quinone metabolites suggests new approaches to preventive intervention – a review. Steroids (2014). doi:10.1016/j.steroids.2014.08.006
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
46. Petrie WK, Dennis MK, Hu C, Dai D, Arterburn JB, Smith HO, et al. G protein-coupled estrogen receptor-selective ligands modulate endometrial tumor growth. Obstet Gynecol Int (2013) 2013:472720. doi:10.1155/2013/472720
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
47. Bossard C, Busson M, Vindrieux D, Gaudin F, Machelon V, Brigitte M, et al. Potential role of estrogen receptor beta as a tumor suppressor of epithelial ovarian cancer. PLoS One (2012) 7:e44787. doi:10.1371/journal.pone.0044787
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
48. Filardo EJ, Quinn JA, Sabo E. Association of the membrane estrogen receptor, GPR30, with breast tumor metastasis and transactivation of the epidermal growth factor receptor. Steroids (2008) 73:870–3. doi:10.1016/j.steroids.2007.12.025
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
49. Widschwendter M, Rosenthal AN, Philpott S, Rizzuto I, Fraser L, Hayward J, et al. The sex hormone system in carriers of BRCA1/2 mutations: a case-control study. Lancet Oncol (2013) 14:1226–32. doi:10.1016/S1470-2045(13)70448-0
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
50. Kim J, Oktay K. Baseline E(2) levels are higher in BRCA2 mutation carriers: a potential target for prevention? Cancer Causes Control (2013) 24:421–6. doi:10.1007/s10552-012-0127-3
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
51. Kyoto Encyclopedia of Genes and Genomes (KEGG) (2014). Available from: www.kegg.jp/dbget-bin/www_bget?hsa04913
52. Secky L, Svoboda M, Klameth L, Bajna E, Hamilton G, Zeillinger R, et al. The sulfatase pathway for estrogen formation: targets for the treatment and diagnosis of hormone-associated tumors. J Drug Deliv (2013) 2013:957605. doi:10.1155/2013/957605
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
53. Labrie F. Extragonadal synthesis of sex steroids: intracrinology. Ann Endocrinol (Paris) (2003) 64:95–107.
54. Suzuki T, Miki Y, Fukuda T, Nakata T, Moriya T, Sasano H. Analysis for localization of steroid sulfatase in human tissues. Methods Enzymol (2005) 400:303–16. doi:10.1016/S0076-6879(05)00018-2
55. Labrie F. Intracrinology. Mol Cell Endocrinol (1991) 78:C113–8. doi:10.1016/0303-7207(91)90116-A
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
56. Audet-Walsh E, Lepine J, Gregoire J, Plante M, Caron P, Tetu B, et al. Profiling of endogenous estrogens, their precursors, and metabolites in endometrial cancer patients: association with risk and relationship to clinical characteristics. J Clin Endocrinol Metab (2011) 96:E330–9. doi:10.1210/jc.2010-2050
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
57. Purohit A, Foster PA. Steroid sulfatase inhibitors for estrogen- and androgen-dependent cancers. J Endocrinol (2012) 212:99–110. doi:10.1530/JOE-11-0266
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
58. Samavat H, Kurzer MS. Estrogen metabolism and breast cancer. Cancer Lett (2014). doi:10.1016/j.canlet.2014.04.018
59. Santner SJ, Leszczynski D, Wright C, Manni A, Feil PD, Santen RJ. Estrone sulfate: a potential source of estradiol in human breast cancer tissues. Breast Cancer Res Treat (1986) 7:35–44. doi:10.1007/BF01886734
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
60. Purohit A, Woo LW, Potter BV. Steroid sulfatase: a pivotal player in estrogen synthesis and metabolism. Mol Cell Endocrinol (2011) 340:154–60. doi:10.1016/j.mce.2011.06.012
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
61. Utsumi T, Yoshimura N, Takeuchi S, Maruta M, Maeda K, Harada N. Elevated steroid sulfatase expression in breast cancers. J Steroid Biochem Mol Biol (2000) 73:141–5. doi:10.1016/S0960-0760(00)00060-1
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
62. Roodi N, Bailey LR, Kao WY, Verrier CS, Yee CJ, Dupont WD, et al. Estrogen receptor gene analysis in estrogen receptor-positive and receptor-negative primary breast cancer. J Natl Cancer Inst (1995) 87:446–51. doi:10.1093/jnci/87.6.446
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
63. Rizner TL. Estrogen biosynthesis, phase I and phase II metabolism, and action in endometrial cancer. Mol Cell Endocrinol (2013) 381:124–39. doi:10.1016/j.mce.2013.07.026
64. Hernandez-Guzman FG, Higashiyama T, Osawa Y, Ghosh D. Purification, characterization and crystallization of human placental estrone/dehydro- epiandrosterone sulfatase, a membrane-bound enzyme of the endoplasmic reticulum. J Steroid Biochem Mol Biol (2001) 78:441–50. doi:10.1016/S0960-0760(01)00119-4
65. Clemens JW, Kabler HL, Sarap JL, Beyer AR, Li PK, Selcer KW. Steroid sulfatase activity in the rat ovary, cultured granulosa cells, and a granulosa cell line. J Steroid Biochem Mol Biol (2000) 75:245–52. doi:10.1016/S0960-0760(00)00171-0
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
66. Ghosh D. Human sulfatases: a structural perspective to catalysis. Cell Mol Life Sci (2007) 64:2013–22. doi:10.1007/s00018-007-7175-y
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
67. Honma S, Shimodaira K, Shimizu Y, Tsuchiya N, Saito H, Yanaihara T, et al. The influence of inflammatory cytokines on estrogen production and cell proliferation in human breast cancer cells. Endocr J (2002) 49:371–7. doi:10.1507/endocrj.49.371
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
68. Adjei AA, Thomae BA, Prondzinski JL, Eckloff BW, Wieben ED, Weinshilboum RM. Human estrogen sulfotransferase (SULT1E1) pharmacogenomics: gene resequencing and functional genomics. Br J Pharmacol (2003) 139:1373–82. doi:10.1038/sj.bjp.0705369
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
69. Xu Y, Liu X, Guo F, Ning Y, Zhi X, Wang X, et al. Effect of estrogen sulfation by SULT1E1 and PAPSS on the development of estrogen-dependent cancers. Cancer Sci (2012) 103:1000–9. doi:10.1111/j.1349-7006.2012.02258.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
70. Kuiper GG, Enmark E, Pelto-Huikko M, Nilsson S, Gustafsson JA. Cloning of a novel receptor expressed in rat prostate and ovary. Proc Natl Acad Sci U S A (1996) 93:5925–30. doi:10.1073/pnas.93.12.5925
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
71. Beato M. Gene regulation by steroid hormones. Cell (1989) 56:335–44. doi:10.1016/0092-8674(89)90237-7
72. Zhao C, Dahlman-Wright K, Gustafsson JA. Estrogen receptor beta: an overview and update. Nucl Recept Signal (2008) 6:e003. doi:10.1621/nrs.06003
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
73. Delaunay F, Pettersson K, Tujague M, Gustafsson JA. Functional differences between the amino-terminal domains of estrogen receptors alpha and beta. Mol Pharmacol (2000) 58:584–90.
74. Tora L, White J, Brou C, Tasset D, Webster N, Scheer E, et al. The human estrogen receptor has two independent nonacidic transcriptional activation functions. Cell (1989) 59:477–87. doi:10.1016/0092-8674(89)90031-7
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
75. Picard D, Kumar V, Chambon P, Yamamoto KR. Signal transduction by steroid hormones: nuclear localization is differentially regulated in estrogen and glucocorticoid receptors. Cell Regul (1990) 1:291–9.
76. Brzozowski AM, Pike AC, Dauter Z, Hubbard RE, Bonn T, Engstrom O, et al. Molecular basis of agonism and antagonism in the oestrogen receptor. Nature (1997) 389:753–8. doi:10.1038/39645
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
77. Pike AC, Brzozowski AM, Hubbard RE, Bonn T, Thorsell AG, Engstrom O, et al. Structure of the ligand-binding domain of oestrogen receptor beta in the presence of a partial agonist and a full antagonist. EMBO J (1999) 18:4608–18. doi:10.1093/emboj/18.17.4608
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
78. Kumar R, Thompson EB. The structure of the nuclear hormone receptors. Steroids (1999) 64:310–9. doi:10.1016/S0039-128X(99)00014-8
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
79. Kumar R, Zakharov MN, Khan SH, Miki R, Jang H, Toraldo G, et al. The dynamic structure of the estrogen receptor. J Amino Acids (2011) 2011:812540. doi:10.4061/2011/812540
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
80. Byers M, Kuiper GG, Gustafsson JA, Park-Sarge OK. Estrogen receptor-beta mRNA expression in rat ovary: down-regulation by gonadotropins. Mol Endocrinol (1997) 11:172–82. doi:10.1210/me.11.2.172
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
81. Couse JF, Lindzey J, Grandien K, Gustafsson JA, Korach KS. Tissue distribution and quantitative analysis of estrogen receptor-alpha (ERalpha) and estrogen receptor-beta (ERbeta) messenger ribonucleic acid in the wild-type and ERalpha-knockout mouse. Endocrinology (1997) 138:4613–21. doi:10.1210/en.138.11.4613
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
82. Brandenberger AW, Lebovic DI, Tee MK, Ryan IP, Tseng JF, Jaffe RB, et al. Oestrogen receptor (ER)-alpha and ER-beta isoforms in normal endometrial and endometriosis-derived stromal cells. Mol Hum Reprod (1999) 5:651–5. doi:10.1093/molehr/5.7.651
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
83. Bardin A, Hoffmann P, Boulle N, Katsaros D, Vignon F, Pujol P, et al. Involvement of estrogen receptor beta in ovarian carcinogenesis. Cancer Res (2004) 64:5861–9. doi:10.1158/0008-5472.CAN-04-0552
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
84. Kuiper GG, Gustafsson JA. The novel estrogen receptor-beta subtype: potential role in the cell- and promoter-specific actions of estrogens and anti-estrogens. FEBS Lett (1997) 410:87–90. doi:10.1016/S0014-5793(97)00413-4
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
85. Ogawa S, Inoue S, Watanabe T, Hiroi H, Orimo A, Hosoi T, et al. The complete primary structure of human estrogen receptor beta (hER beta) and its heterodimerization with ER alpha in vivo and in vitro. Biochem Biophys Res Commun (1998) 243:122–6. doi:10.1006/bbrc.1997.7893
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
86. Filardo EJ, Thomas P. Minireview: G protein-coupled estrogen receptor-1, GPER-1: its mechanism of action and role in female reproductive cancer, renal and vascular physiology. Endocrinology (2012) 153:2953–62. doi:10.1210/en.2012-1061
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
87. Smith HO, Arias-Pulido H, Kuo DY, Howard T, Qualls CR, Lee SJ, et al. GPR30 predicts poor survival for ovarian cancer. Gynecol Oncol (2009) 114:465–71. doi:10.1016/j.ygyno.2009.05.015
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
88. Kolkova Z, Casslen V, Henic E, Ahmadi S, Ehinger A, Jirstrom K, et al. The G protein-coupled estrogen receptor 1 (GPER/GPR30) does not predict survival in patients with ovarian cancer. J Ovarian Res (2012) 5:9. doi:10.1186/1757-2215-5-9
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
89. Heublein S, Mayr D, Vrekoussis T, Friese K, Hofmann SS, Jeschke U, et al. The G-protein coupled estrogen receptor (GPER/GPR30) is a gonadotropin receptor dependent positive prognosticator in ovarian carcinoma patients. PLoS One (2013) 8:e71791. doi:10.1371/journal.pone.0071791
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
90. Prossnitz ER, Barton M. Estrogen biology: new insights into GPER function and clinical opportunities. Mol Cell Endocrinol (2014) 389:71–83. doi:10.1016/j.mce.2014.02.002
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
91. Hill KK, Roemer SC, Churchill ME, Edwards DP. Structural and functional analysis of domains of the progesterone receptor. Mol Cell Endocrinol (2012) 348:418–29. doi:10.1016/j.mce.2011.07.017
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
92. Kastner P, Bocquel MT, Turcotte B, Garnier JM, Horwitz KB, Chambon P, et al. Transient expression of human and chicken progesterone receptors does not support alternative translational initiation from a single mRNA as the mechanism generating two receptor isoforms. J Biol Chem (1990) 265:12163–7.
93. Vegeto E, Shahbaz MM, Wen DX, Goldman ME, O’Malley BW, Mcdonnell DP. Human progesterone receptor A form is a cell- and promoter-specific repressor of human progesterone receptor B function. Mol Endocrinol (1993) 7:1244–55. doi:10.1210/me.7.10.1244
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
94. Li X, O’Malley BW. Unfolding the action of progesterone receptors. J Biol Chem (2003) 278:39261–4. doi:10.1074/jbc.R300024200
95. Lonard DM, O’Malley BW. Nuclear receptor coregulators: modulators of pathology and therapeutic targets. Nat Rev Endocrinol (2012) 8:598–604. doi:10.1038/nrendo.2012.100
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
96. Mote PA, Bartow S, Tran N, Clarke CL. Loss of co-ordinate expression of progesterone receptors A and B is an early event in breast carcinogenesis. Breast Cancer Res Treat (2002) 72:163–72. doi:10.1023/A:1014820500738
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
97. Zhao D, Zhang F, Zhang W, He J, Zhao Y, Sun J. Prognostic role of hormone receptors in ovarian cancer: a systematic review and meta-analysis. Int J Gynecol Cancer (2013) 23:25–33. doi:10.1097/IGC.0b013e3182788466
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
98. Okuda T, Saito H, Sekizawa A, Shimizu Y, Akamatsu T, Kushima M, et al. Steroid sulfatase expression in ovarian clear cell adenocarcinoma: immunohistochemical study. Gynecol Oncol (2001) 82:427–34. doi:10.1006/gyno.2001.6322
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
99. Chura JC, Blomquist CH, Ryu HS, Argenta PA. Estrone sulfatase activity in patients with advanced ovarian cancer. Gynecol Oncol (2009) 112:205–9. doi:10.1016/j.ygyno.2008.08.037
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
100. Utsumi T, Yoshimura N, Takeuchi S, Ando J, Maruta M, Maeda K, et al. Steroid sulfatase expression is an independent predictor of recurrence in human breast cancer. Cancer Res (1999) 59:377–81.
101. Miyoshi Y, Ando A, Hasegawa S, Ishitobi M, Taguchi T, Tamaki Y, et al. High expression of steroid sulfatase mRNA predicts poor prognosis in patients with estrogen receptor-positive breast cancer. Clin Cancer Res (2003) 9:2288–93.
102. Kirilovas D, Schedvins K, Naessen T, Von Schoultz B, Carlstrom K. Conversion of circulating estrone sulfate to 17beta-estradiol by ovarian tumor tissue: a possible mechanism behind elevated circulating concentrations of 17beta-estradiol in postmenopausal women with ovarian tumors. Gynecol Endocrinol (2007) 23:25–8. doi:10.1080/09513590601058333
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
103. Poisson Pare D, Song D, Luu-The V, Han B, Li S, Liu G, et al. Expression of estrogen sulfotransferase 1E1 and steroid sulfatase in breast cancer: a immunohistochemical study. Breast Cancer (Auckl) (2009) 3:9–21.
104. Park SH, Cheung LW, Wong AS, Leung PC. Estrogen regulates snail and slug in the down-regulation of E-cadherin and induces metastatic potential of ovarian cancer cells through estrogen receptor alpha. Mol Endocrinol (2008) 22:2085–98. doi:10.1210/me.2007-0512
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
105. Al Sarakbi W, Mokbel R, Salhab M, Jiang WG, Reed MJ, Mokbel K. The role of STS and OATP-B mRNA expression in predicting the clinical outcome in human breast cancer. Anticancer Res (2006) 26:4985–90.
106. Fujimoto J, Hirose R, Sakaguchi H, Tamaya T. Clinical significance of expression of estrogen receptor alpha and beta mRNAs in ovarian cancers. Oncology (2000) 58:334–41. doi:10.1159/000012121
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
107. Li AJ, Baldwin RL, Karlan BY. Estrogen and progesterone receptor subtype expression in normal and malignant ovarian epithelial cell cultures. Am J Obstet Gynecol (2003) 189:22–7. doi:10.1067/mob.2003.328
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
108. Lindgren PR, Cajander S, Backstrom T, Gustafsson JA, Makela S, Olofsson JI. Estrogen and progesterone receptors in ovarian epithelial tumors. Mol Cell Endocrinol (2004) 221:97–104. doi:10.1016/j.mce.2004.02.020
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
109. Burges A, Bruning A, Dannenmann C, Blankenstein T, Jeschke U, Shabani N, et al. Prognostic significance of estrogen receptor alpha and beta expression in human serous carcinomas of the ovary. Arch Gynecol Obstet (2010) 281:511–7. doi:10.1007/s00404-009-1185-y
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
110. Halon A, Materna V, Drag-Zalesinska M, Nowak-Markwitz E, Gansukh T, Donizy P, et al. Estrogen receptor alpha expression in ovarian cancer predicts longer overall survival. Pathol Oncol Res (2011) 17:511–8. doi:10.1007/s12253-010-9340-0
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
111. Darb-Esfahani S, Wirtz RM, Sinn BV, Budczies J, Noske A, Weichert W, et al. Estrogen receptor 1 mRNA is a prognostic factor in ovarian carcinoma: determination by kinetic PCR in formalin-fixed paraffin-embedded tissue. Endocr Relat Cancer (2009) 16:1229–39. doi:10.1677/ERC-08-0338
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
112. Aust S, Bachmayr-Heyda A, Pateisky P, Tong D, Darb-Esfahani S, Denkert C, et al. Role of TRAP1 and estrogen receptor alpha in patients with ovarian cancer – a study of the OVCAD consortium. Mol Cancer (2012) 11:69. doi:10.1186/1476-4598-11-69
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
113. Suzuki F, Akahira J, Miura I, Suzuki T, Ito K, Hayashi S, et al. Loss of estrogen receptor beta isoform expression and its correlation with aberrant DNA methylation of the 5′-untranslated region in human epithelial ovarian carcinoma. Cancer Sci (2008) 99:2365–72. doi:10.1111/j.1349-7006.2008.00988.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
114. Chan KK, Wei N, Liu SS, Xiao-Yun L, Cheung AN, Ngan HY. Estrogen receptor subtypes in ovarian cancer: a clinical correlation. Obstet Gynecol (2008) 111:144–51. doi:10.1097/01.AOG.0000296715.07705.e9
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
115. Rutherford T, Brown WD, Sapi E, Aschkenazi S, Munoz A, Mor G. Absence of estrogen receptor-beta expression in metastatic ovarian cancer. Obstet Gynecol (2000) 96:417–21. doi:10.1016/S0029-7844(00)00917-0
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
116. Lee P, Rosen DG, Zhu C, Silva EG, Liu J. Expression of progesterone receptor is a favorable prognostic marker in ovarian cancer. Gynecol Oncol (2005) 96:671–7. doi:10.1016/j.ygyno.2005.01.043
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
117. De Stefano I, Zannoni GF, Prisco MG, Fagotti A, Tortorella L, Vizzielli G, et al. Cytoplasmic expression of estrogen receptor beta (ERbeta) predicts poor clinical outcome in advanced serous ovarian cancer. Gynecol Oncol (2011) 122:573–9. doi:10.1016/j.ygyno.2011.05.025
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
118. Sieh W, Kobel M, Longacre TA, Bowtell DD, Defazio A, Goodman MT, et al. Hormone-receptor expression and ovarian cancer survival: an ovarian tumor tissue analysis consortium study. Lancet Oncol (2013) 14:853–62. doi:10.1016/S1470-2045(13)70253-5
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
119. Yokoyama Y, Mizunuma H. Recurrent epithelial ovarian cancer and hormone therapy. World J Clin Cases (2013) 1:187–90. doi:10.12998/wjcc.v1.i6.187
120. Brueggemeier RW. Aromatase inhibitors: new endocrine treatment of breast cancer. Semin Reprod Med (2004) 22:31–43. doi:10.1055/s-2004-823025
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
121. Tropé C, Marth C, Kaern J. Tamoxifen in the treatment of recurrent ovarian carcinoma. Eur J Cancer (2000) 36(Suppl 4):S59–61. doi:10.1016/S0959-8049(00)00228-8
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
122. Miller WR. Aromatase inhibitors – where are we now? Br J Cancer (1996) 73:415–7. doi:10.1038/bjc.1996.73
123. Bowman A, Gabra H, Langdon SP, Lessells A, Stewart M, Young A, et al. CA125 response is associated with estrogen receptor expression in a phase II trial of letrozole in ovarian cancer: identification of an endocrine-sensitive subgroup. Clin Cancer Res (2002) 8:2233–9.
124. Kavanagh JJ. Intraperitoneal chemotherapy in the treatment of ovarian cancer. J Surg Oncol (2007) 96:541–2.
125. Del Carmen MG, Fuller AF, Matulonis U, Horick NK, Goodman A, Duska LR, et al. Phase II trial of anastrozole in women with asymptomatic mullerian cancer. Gynecol Oncol (2003) 91:596–602. doi:10.1016/j.ygyno.2003.08.021
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
126. Krasner CN, Mcmeekin DS, Chan S, Braly PS, Renshaw FG, Kaye S, et al. A Phase II study of trabectedin single agent in patients with recurrent ovarian cancer previously treated with platinum-based regimens. Br J Cancer (2007) 97:1618–24. doi:10.1038/sj.bjc.6604088
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Keywords: ovarian cancer, estrogen synthesis, estrogen sulfotransferase, estrogen sulfatase, estrogen receptor alpha/beta, G-protein-coupled estrogen receptor, progesterone
Citation: Mungenast F and Thalhammer T (2014) Estrogen biosynthesis and action in ovarian cancer. Front. Endocrinol. 5:192. doi: 10.3389/fendo.2014.00192
Received: 20 September 2014; Paper pending published: 08 October 2014;
Accepted: 25 October 2014; Published online: 12 November 2014.
Edited by:
Lena Ekström, Karolinska Institutet, SwedenReviewed by:
Toshinobu Tokumoto, Shizuoka University, JapanTakayoshi Ubuka, Waseda University, Japan
Copyright: © 2014 Mungenast and Thalhammer. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Theresia Thalhammer, Department of Pathophysiology and Allergy Research, AKH-Waehringer Guertel 18-20/3Q, Vienna A-1090, Austria e-mail:dGhlcmVzaWEudGhhbGhhbW1lckBtZWR1bml3aWVuLmFjLmF0