 John M. Hamlyn
John M. Hamlyn- Department of Physiology, University of Maryland School of Medicine, Baltimore, MD, USA
The work of deWardener and colleagues stimulated longstanding interest in natriuretic hormones (NHs). In addition to the atrial peptides (APs), the circulation contains unidentified physiologically relevant NHs. One NH is controlled by the central nervous system (CNS) and likely secreted by the pituitary. Its circulating activity is modulated by salt intake and the prevailing sodium concentration of the blood and intracerebroventricular fluid, and contributes to postprandial and dehydration natriuresis. The other NH, mobilized by atrial stretch, promotes natriuresis by increasing the production of intrarenal dopamine and/or nitric oxide (NO). Both NHs have short (<35 min) circulating half lives, depress renotubular sodium transport, and neither requires the renal nerves. The search for NHs led to endogenous cardiotonic steroids (CTS) including ouabain-, digoxin-, and bufadienolide-like materials. These CTS, given acutely in high nanomole to micromole amounts into the general or renal circulations, inhibit sodium pumps and are natriuretic. Among these CTS, only bufalin is cleared sufficiently rapidly to qualify for an NH-like role. Ouabain-like CTS are cleared slowly, and when given chronically in low daily nanomole amounts, promote sodium retention, augment arterial myogenic tone, reduce renal blood flow and glomerular filtration, suppress NO in the renal vasa recta, and increase sympathetic nerve activity and blood pressure. Moreover, lowering total body sodium raises circulating endogenous ouabain. Thus, ouabain-like CTS have physiological actions that, like aldosterone, support renal sodium retention and blood pressure. In conclusion, the mammalian circulation contains two non-AP NHs. Identification of the CNS NH should be a priority.
Introduction
Natriuretic hormones (NHs) can be defined as substances whose circulating levels and effects fluctuate in a parallel manner with dietary sodium intake (1). NHs have long been implicated in sodium balance and are likely to be of the most significance in western acculturated societies where sodium intake typically is >100 meq/day (2). Indeed, ingestion of high salt meals raises the osmolarity of the circulation, stimulates secretion of antidiuretic hormone (ADH), and raises the natriuretic activity of the blood. In principle, the mode of action of NHs includes suppression of primary active sodium transport in the kidney and/or damping of secondary active transport systems involving sodium (1) or even potassium (3), effects on renal vascular tone and glomerular filtration rate (GFR), and activation of intrarenal natriuretic factors, such as prostaglandins, nitric oxide (NO), or dopamine. This article presents a personal and condensed overview of known and unknown non-atrial NHs and addresses the role of endogenous sodium pump inhibitors as NHs.
Searching for Natriuretic Hormones
It is well accepted that sodium balance is not fully explained by the up and downregulation of glomerular filtration and mineralocorticoid-stimulated reabsorption (4, 5). The first clear evidence for a “third factor” arose from the pioneering experiments of deWardener in which dogs that received excess mineralocorticoid and vasopressin increased their urinary sodium excretion in response to blood volume expansion with saline at a time when glomerular filtration was being lowered experimentally (6). Thus, the increase in sodium excretion was mediated by diminished tubular reabsorption of sodium and water. Cross-circulation studies, as well as work using isolated kidney studies in dogs and rats (6–10) excluded significant alterations in the composition of the blood, changes in renal nerve activity, glomerular filtration, renal blood flow, or renal perfusion pressure as mediators. A humoral “NH” was required.
The discovery of the atrial peptides (APs) and their natriuretic activity initially promised to explain some of the outstanding functions of an NH (11–13). APs augment sodium excretion (14–16) and saline infusions raise plasma AP (17, 18). However, in dogs, the effects of physiological changes in plasma APs and low dose infusions on sodium excretion were less obvious and, under certain experimental conditions, circulating APs and sodium excretion changed diametrically or, were temporally unconnected (19–21). Thus, some other NH was required.
The search for humoral agents that trigger salt excretion has relied on a variety of assays that range from isolated enzymes all the way to whole kidneys and animals (22). Table 1 lists some tissues and fluids from which a variety of natriuretic factors were obtained. It is a significant accomplishment that numerous factors with natriuretic activity including guanylin, uroguanylin, urodilatin, LLu-α, xanthurenic acid, and a number of steroidal sodium pump inhibitors have been isolated and identified (14, 22–27). These materials likely account for some of the bioactivity in some, but not all, studies where natriuretic activity has been demonstrated. It is less clear that any of these materials fits the physiological profile expected for a NH as will be apparent from the discussion that follows.
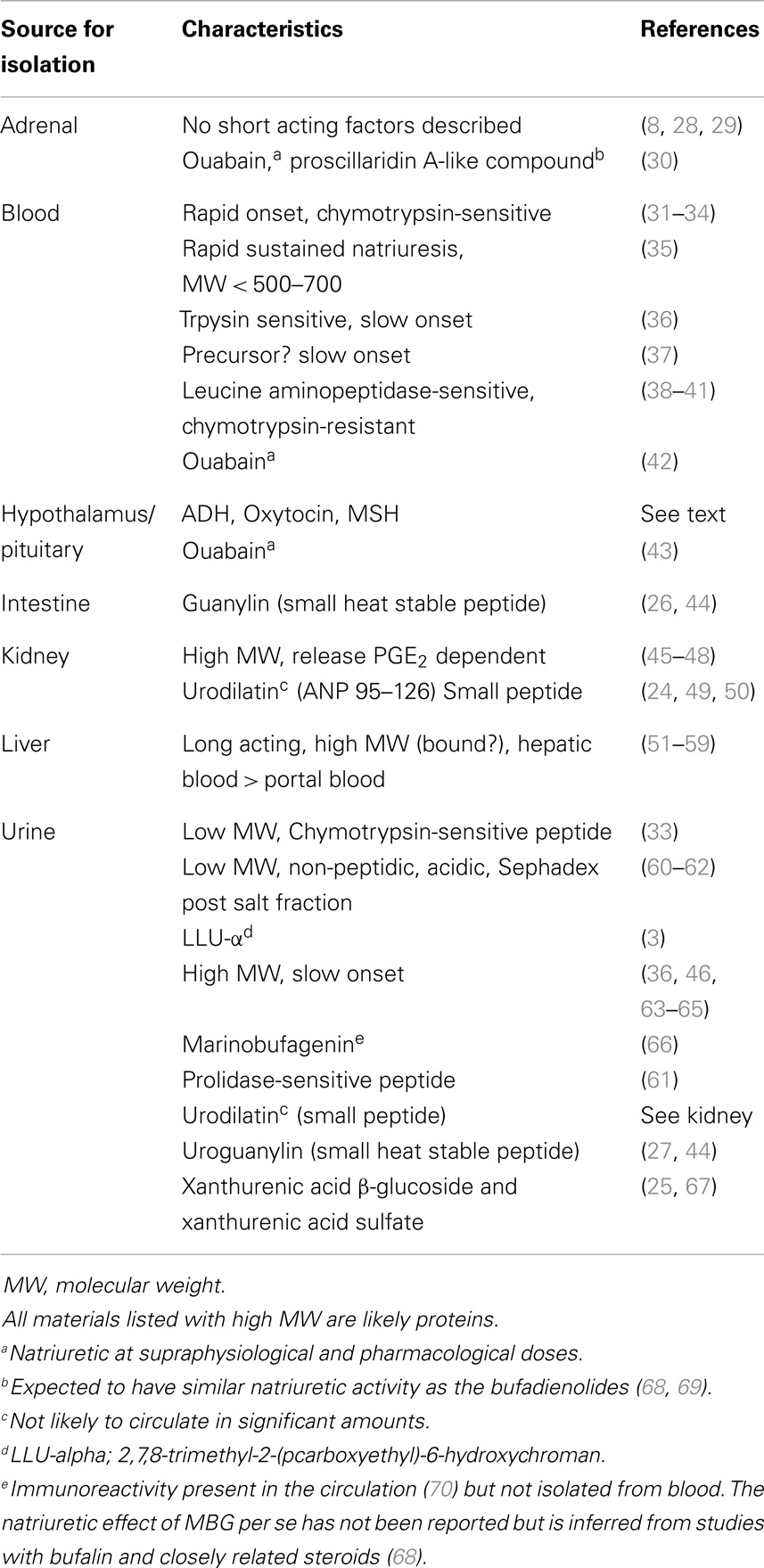
Table 1. An overview of sources and characteristics of natriuretic factors.
Natriuretic Hormones: How Many?
Other than the APs, there are numerous hormones and endogenous materials that are known natriuretic agents. These include melanocyte stimulating hormone, dopamine, certain phospholipids, prostaglandins, kinins, and parathyroid hormone (71). These are not discussed here.
Evidence based upon pharmacological interventions, as well as an analysis of the kinetics of salt excretion mentioned below, suggests there are at least two major NH mechanisms unrelated to the APs. One mechanism is activated by the central nervous system (CNS) and the other involves maneuvers that increase atrial stretch. Pharmacological inhibition of renal NO blunts the magnitude of saline natriuresis (72) and both specific and non-selective dopamine antagonists attenuate volume expansion and water immersion (i.e., atrial stretch mediated) natriuresis but not that activated by CNS sodium (73–77). Yet another key factor that distinguishes these two NH systems is their kinetics; the rates of the decline in sodium excretion when the natriuretic stimuli are abruptly removed differ markedly for CNS- and atrial distention natriuresis. The kinetic features are potentially diagnostic; they can be used to evaluate candidate NHs.
The atrial distention arising from balloon inflation requires intact cardiac but not renal nerves, the stretch can be reversed in seconds, and the evoked natriuresis declines rapidly (21). Critically, the kinetics of the decline in natriuresis are uncontaminated by residual volume that typically would remain following a saline load (78). The second experimental paradigm is the natriuresis evoked by infusion of hypertonic saline into the brain. As the flow rates in the cerebral ventricles are much higher than the rates at which hypertonic stimuli are typically infused, simply stopping the infusion exposes the kinetics of the decline in salt excretion. Accordingly, Figure 1 compares the decline in renal sodium excretion evoked by either atrial distension or CNS sodium. Three points are apparent: (1) the decay kinetics in both instances are first order; for CNS natriuresis, they remain linear for well over 1 h. The kinetics demonstrate that a single reaction likely is the dominant rate limiting step for the natriuresis evoked by each stimulus. (2) The CNS natriuresis, when activated by hypertonic saline (79–83), dehydration (84), or norepinephrine (85), produces similar rate constants with no major species differences. (3) The rate constants for the decline in CNS natriuresis are ~2–3-fold less (slower) than that evoked by atrial distension. Thus, the combined evidence derived from the sensitivity to pharmacological agents and the kinetic observations indicate that CNS- and atrial distension natriuresis must be mediated by different mechanisms.
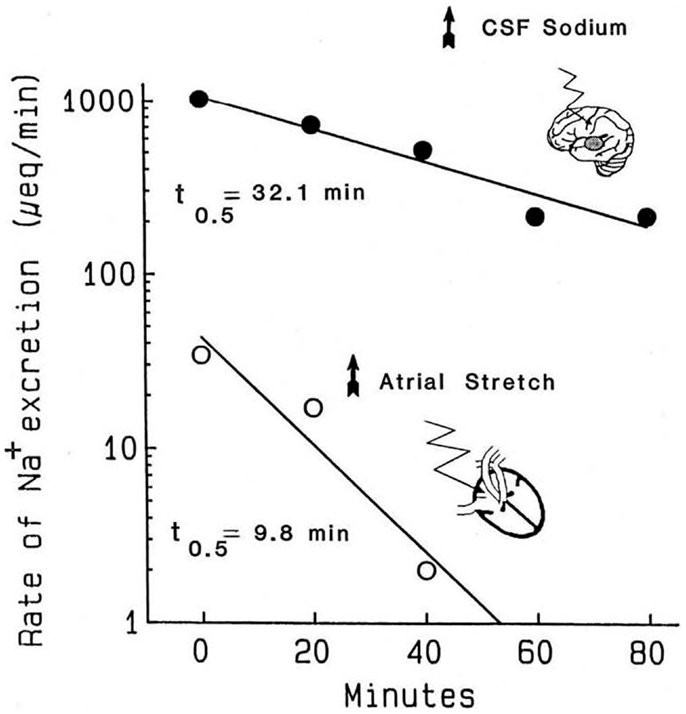
Figure 1. Kinetics of the decline in natriuresis following abrupt removal of the natriuretic stimulus. Reproduced from Ref. (71) with permission. Data are adapted from Ref. (21) in conscious dogs and Ref. (86) in conscious goats. Reported values for the t1/2 of the decline in CNS natriuresis range from 24 to 32 min in the goat, 32 min in the sheep, and 15 min in the rat (71). These half times are likely to represent the clearance of their respective humoral mediators from the circulation.
Compensatory mechanisms might conceivably alter the kinetics in Figure 1, especially if significant salt and water loss were to occur along with declining blood pressures. During the 40 min atrial distention in Figure 1, blood pressure increased modestly. Plasma renin was suppressed in one set of experiments but not another. Following the distension, in one set of experiments, blood pressure remained elevated even though the natriuresis declined rapidly and aldosterone was unchanged or increased. Nevertheless, changes in aldosterone would have been too slow to have had impact. Under the conditions used, and among the measured hormonal and hemodynamic variables, the only changes convincingly associated with the decline in natriuresis following atrial distension were the return of left, right, and pulmonary pressures (i.e., cardiac nerve activity) to normal. With regard to CNS natriuresis, the decline in natriuresis is an extended first-order process; the absence of curvature over the time course implies no major influence by a compensatory process.
Among the candidate NHs in Table 1, there is, unfortunately, no readily interpretable information regarding the halftimes for the decline in their natriuretic effects. Most of the unidentified materials were impure, with variable onset times, and, reminiscent of urodilatin (49), some produced a natriuresis that lasted many hours following infusion. The absence of kinetic information is understandable; the primary experimental emphasis was the demonstration of natriuresis per se. And for decay kinetics to be informative, a near steady-state natriuresis would ideally be desirable prior to stimulus removal. This is not always an easy condition to meet. Regarding the recently identified materials in Table 1, no kinetic information is available. However, among all the materials, urodilatin shows a most interesting physiological correlate; in human beings, urinary urodilatin excretion closely paralleled the circadian rhythm for sodium excretion over many days (49). As urodilatin itself is not found in the circulation, it is not, by definition, an NH; although the unknown substance (?) that presumably links sodium intake with urinary urodilatin and sodium excretion could be. Thus, for all listed materials in Table 1, there is currently no compelling evidence that their behaviors fit the definition of a physiologically relevant NH given in the introduction.
Hereafter, I focus primarily on CNS natriuresis and consider the potential role of sodium pump inhibitors as NHs.
CNS Natriuresis
The brain, via an unknown humoral NH, mediates the natriuresis evoked by increased plasma sodium concentration, intracerebroventricular (icv) sodium, and dehydration (79, 81, 87). The natriuresis may be damped but is not eliminated by renal denervation (88), is activated by small increases of plasma sodium (1–2 mM). The CNS NH may have a dominant influence in postprandial natriuresis (89) and is a blood-bourne factor distinct from APs (90, 91), ADH (92), or dopamine (74).
Central nervous system natriuresis can be activated by the elevation of either blood-bourne or cerebrospinal fluid sodium; both dehydration and postprandial natriuresis are blocked or reversed by hyponatremic CSF (93–95) or rehydration (96). Push–pull perfusion techniques suggest a discrete area of the third ventricle is near the sodium sensing apparatus (97). Further, the ablation of central structures, including the anteroventral and posterior hypothalamus in a variety of species, or decapitation, profoundly influence the ability to regulate osmotic balance, tolerate hyperosmotic challenge, and excrete sodium (80, 83, 98–106). The lesioned areas have included the median eminence, medial preoptic nucleus, organum vasculosum of the lamina terminalis, and the periventricular preoptic area. The consequences of these lesions are impaired thirst and ADH secretion, reduced renal natriuretic response, and hypernatremia. In contrast, this same system, when overactivated, can lead to profound hyponatremia. This phenomenon, sometimes termed “cerebral salt wasting,” and resembling some of the features of the syndrome of inappropriate ADH secretion, has been noted in some CNS disorders (107–111).
The observation that dehydration results in hypernatremia and provokes a compensatory natriuresis in the face of reduced extracellular fluid volumes, and that the natriuresis subsides with rehydration, suggests that the tendency to hypernatremia during dehydration and following a high salt meal is actively opposed by an unknown osmotically sensitive mechanism [see in Ref. (84)]. In each instance, the natriuretic response to these stimuli is present in animals with denervated kidneys but absent in animals with hypothalamic lesions (84, 112). Further, CNS natriuresis is not explained by blood pressure changes and persists when renal artery pressures are servo controlled (92, 113).
In each of the aforementioned situations, changes in circulating ADH have been implicated as the efferent mediator of CNS natriuresis (114, 115). Indeed, CNS natriuresis is either absent or slowed in rats congenitally deficient in AVP (116, 117), is absent in hypophysectomized rats but reappears in rats pretreated with large amounts of ADH and in rats given a dD-AVP analog (88). ADH certainly contributes to the control of sodium excretion in rats, dogs, and man (84, 89, 118–123). ADH infusions are natriuretic, and specifically implicated in CNS (114, 115) but not saline natriuresis. However, ADH is not sufficient to account for CNS natriuresis (92, 117), although it may be permissive (124, 125). For example, AV3V-lesioned sheep and dehydrated normal sheep both lost similar amounts of body water, although the hypernatremia was much worse in the lesioned animals (112). Thus, something other than ADH was lacking in the lesioned animals to explain the greater hypernatremia with the same overall water loss.
Little is known about the chemical nature of the CNS NH other than it appears to be heat stable (126). Its actions have an interesting temporal association with ADH and/or oxytocin (92, 127). For example, plasma ADH rises during the prehypertensive period associated with mineralocorticoid escape; a period when increased CNS NH would be expected (128). Consistent with the latter supposition, urinary sodium excretion in sheep given 3–4 day infusions of aldosterone was almost entirely blocked by the acute CNS administration of a low sodium cerebrospinal fluid during mineralocorticoid escape (129). Further, mineralocorticoids also augment the osmotic sensitivity of ADH secretion (130).
Oxytocin also has a role in renal sodium excretion (131–134) and restores the ability of hypophysectomized dogs and rats to excrete sodium at a brisk rate during saline expansion (20, 135). Yet other humoral factors implicated during CNS and ADH natriuresis include an inhibitor of prostacyclin synthesis (136) and a humoral substance that inhibits active sodium transport in toad bladder (137). Hemorrhage, paradoxically, also evokes a natriuresis that depends on the CNS (118). The natriuresis is blocked when intrarenal prostaglandin synthesis is inhibited (119). The simplest interpretation is that activation of intrarenal V1 receptors stimulates prostaglandin synthesis and the resultant products influence sodium reabsorption at distal tubular sites (138). Overall, the phenomenon ascribed to CNS natriuresis has complex interdependencies and is associated with the diminution of renal tubular sodium transport.
Of significant relevance, CNS-mediated natriuresis depends upon the prevailing level of dietary sodium intake. In sodium depleted dogs, infusion of hypertonic saline into the carotid artery is not natriuretic (139). Moreover, the phenomenon of postprandial natriuresis in the sheep is activated only when dietary sodium intakes reach a threshold of 50–75 mmol of sodium/24 h (140), i.e., when plasma renin and aldosterone are largely suppressed. Thus, the CNS NH system is likely of great physiological relevance; it is appropriately integrated with other key factors that govern long-term sodium balance.
The CNS also has a permissive role in the response to saline expansion of blood volume (79, 102, 103). Hypophysectomy reduces saline natriuresis; the deficit is reversed partially by administration of oxytocin and ADH (141). Furthermore, the application of a constricting vice to the neck of anesthetized dogs so as to exclude the brain and pituitary factors from the circulation impairs saline natriuresis (102). In view of the abovementioned role of the CNS, it is surprising that remarkably little attention has been focused on the natriuretic activity associated with extracts from brain and pituitary (Table 1). The little that is known is that the bioactivity of natriuretic extracts from hypothalamus persists following treatment with thioglycollate (to exclude oxytocin or ADH), and that an unidentified tridecapeptide was found in bioactive fractions from the posterior pituitary (142, 143).
Are Sodium Pump Inhibitors Natriuretic Hormones?
There is much evidence linking sodium pump inhibitors with salt balance and cardiovascular and renal disease (144, 145). The Na,K-ATPase inhibitory activity of plasma from normal individuals on a high sodium diet was 25 times greater than that when the individuals were on a low sodium intake (146). Further, the plasma from individuals on high sodium diets, purified natriuretic material from urine, and ouabain, all stimulated glucose-6-phosphate dehydrogenase (G6PD) activity. G6PD activity is claimed to be inversely related to Na,K-ATPase activity (147) and related to inhibition of proximal tubular Na,K-ATPase (148), although the G6PD assay is not considered a surrogate method for the Na,K-ATPase.
Nevertheless, increased blood levels of sodium pump inhibitors, as measured by traditional well-accepted means, have been repeatedly associated with acute volume expansion, high dietary salt, mineralocorticoid excess, chronic renal failure, and CNS natriuresis (31, 32, 35, 38, 149–155). Haddy and coworkers using animal models of low renin hypertension observed that sodium pump inhibition could be reproduced in normal animals given a rapid volume expansion and that this effect could be transferred to the arteries of another animal via the plasma (156). Further, in acutely saline-expanded dogs, the plasma levels of a polar Na,K-ATPase inhibitor and a digoxin immunoreactive material were elevated at a time when endogenous ouabain (EO) was unchanged (37, 157). Moreover, the plasma of dogs undergoing atrial distension strongly inhibited the ouabain-sensitive 86Rb uptake into human red cells. Notably, the bioactivity of the plasma declined substantially when retested a few days later, and was undetectable after 10 days (Hamlyn and Goetz, unpublished observations). This indicates that the inhibitor is unstable in plasma and is reminiscent of the labile digoxin-like material described by Graves et al. (158). Other work implicated the CNS in the control of humoral sodium pump inhibitors; Buckalew et al. (159) found that the jugular effluent inhibited active sodium transport to a greater extent than the blood from the femoral vein. Further, increased levels of circulating sodium pump inhibitors depend upon the integrity of hypothalamic structures within the AV3V area (103, 160). Moreover, the lesion sites overlap those whose integrity is required for CNS natriuresis. Thus, the interrelationship between increased circulating sodium pump inhibitors and natriuresis continues to be of interest. When taken together, there is no doubt that the circulation contains inhibitors of sodium transport, but what are these materials, do their levels change appropriately with salt, and are they natriuretic? Below we focus on sodium pump inhibitors that have been isolated and that have been previously linked with the aforementioned criteria.
Identification of Sodium Pump Inhibitors
Starting from either human plasma or urine (brain, adrenal, and the eye are not discussed here), four groups isolated sodium pump inhibitors and identified them as ouabain- (42, 161, 162), digoxin- (163), marinobufagenin- [MBG, (66)], and telocinobufagin-like steroids (162, 164), respectively. There are altered levels of these materials in numerous experimental and clinical studies (70, 164–169). All these steroids inhibit the sodium pump and, when bound, at least one evokes biased signaling in a manner strikingly reminiscent of the β-adrenergic receptor (168, 170–172). These cardiotonic steroids (CTS) typically are natriuretic and variably kaliuretic when infused acutely at pharmacological (micromolar) doses into anesthetized animals or the renal artery and, in the case of ouabain, selectively inhibit sodium transport in the distal tubules (68, 69, 173, 174). The natriuretic response is linearly related to the inhibition of Na pumps in the dog (175). But are they physiologically relevant NHs?
What Do the Kinetics of the Decline in Natriuresis Tell Us about the Role of Known Sodium Pump Inhibitors?
By comparing the circulating half lives of any putative NH with the half times in Figure 1, it is possible to determine whether it is a plausible mediator of natriuresis. Here, I examine the circulatory half lives of a number of well-known sodium pump inhibitors and compare them with the information in Figure 1. For example, in the dog, the plasma half lives for intravenous ouabain, digoxin, resibufagenin, and bufalin were ~18 h, ~30 h, 21 min, and 25 min, respectively [Ref. (176–178)]. In the rat, the circulating half lives for intravenous cinobufagin, resibufagenin, and bufalin were 44, 42, and 25 min, respectively (179). Therefore, it is apparent that, among these known steroidal sodium pump inhibitors all, with the exception of bufalin, are simply cleared too slowly from the circulation to be kinetically plausible humoral mediators of CNS natriuresis. In the case of atrial distention natriuresis, the kinetic analysis reveals that none of the abovementioned sodium pump inhibitors are likely primary humoral mediators. With regard to CNS natriuresis, only the clearance of bufalin is sufficiently fast in both dogs and rats to warrant further investigation. The kinetic analysis does not prove bufalin as the humoral mediator in CNS natriuresis, but simply suggests that this steroid (or those that are closely related but for which no clearance data are available, e.g., MBG) cannot, as yet, be excluded. A lingering concern with bufalin, or any CTS sodium pump inhibitor, as a NH is the potentially serious conceptual problem that their acute vasoconstrictive action within the renal vasculature will oppose their tubular effects (180).
Renal Sodium Pump Isoforms: Is Their Ouabain Sensitivity Important?
Nearly all mammalian tissues express the α-1 catalytic subunit of the sodium pump; muscle and muscle and nerve also express sodium pump isoforms with α-2 and α-3 subunits (181). In the rat kidney, sodium pumps with the α-1 catalytic subunit are insensitive to micromolar ouabain but are somewhat sensitive to bufalin and marinobufagenin; the acute natriuretic effect of bufalin is greater than that of ouabain (69). For many years, it was believed that the kidney expressed only the α1 isoform even though the ouabain sensitivity of the renal Na pump increases progressively along the nephron (182); the distal tubules are believed to be ~50–100-fold more sensitive than their proximal tubule counterparts. More recently, small numbers of highly ouabain-sensitive α-2 sodium pumps have been detected in rat kidney and they are functionally significant. For example, in response to acute low doses of ouabain, the α2 sodium pumps trigger enhanced Ca2+ signaling and NO generation in the descending vasa recta (183). It is not known if these signaling effects extend to the renal epithelia, but if they do then the acute natriuretic effects of ouabain could involve short-term NO-mediated events. In contrast, the acute natriuretic effects of bufalin and other bufadienolides are thought to be mediated by inhibition of α1 sodium pumps (184).
In the kidney, the renal ouabain-insensitive α-1 sodium pumps far outnumber their ouabain-sensitive α-2 cousins. Interestingly, saline natriuresis was augmented when rodent α-1 sodium pumps were made highly ouabain-sensitive (185). Further, the augmented component of the natriuresis was blocked by digoxin antibody fragments (Fab). However, the kinetic analysis in Figure 1 makes it clear that neither ouabain nor digoxin are viable mediators of atrial distention (saline) natriuresis; the digoxin Fab fragments must, therefore, have interacted with an unknown material that preferred ouabain-sensitive sodium pumps. Thus, occupation of the ouabain binding site by this material can contribute to, but does not fully account for, the phenomenon of saline natriuresis.
Ouabain as a Salt Retaining Steroid
In contrast to the well-accepted acute natriuretic effects of high doses of sodium pump inhibitors, the chronic effects of low concentrations can be diametrically opposite. In the case of ouabain, the prolonged daily administration of low nanomole amounts in the rat suppresses Ca2+ signaling and NO generation in the endothelium of the descending vasa recta, reduces renal blood flow and glomerular filtration, raises sympathetic nerve activity, directly augments vascular myogenic tone and contractility, and raises blood pressure (186–193). Further, chronically reduced total body sodium in human beings is associated with elevated circulating levels of EO (194, 195), i.e., the chronic relationship between plasma EO and salt intake is, like aldosterone and renin, roughly “L”-shaped (196). In addition, and as might be anticipated from the above noted chronic observations, clinical studies have shown that among salt-loaded EH patients, renal tubular sodium reabsorption was highest in the group with elevated circulating EO (197). Thus, the behavior of circulating EO under physiological circumstances, as well as its long-term vascular and renal tubular actions, all appear to favor sodium retention.
Dramatic increases in circulating EO have been reported during exercise, a state associated with increased sympathetic activity and a decline in renal blood flow (198). The circulating levels of EO rise acutely in response to the stress of cardiac surgery (199) and the preoperative plasma levels of EO enhance the identification of those patients who will develop acute kidney injury postsurgery (200). Once again, the behavior and actions of EO in these stressful situations is associated directly or indirectly with salt and water retention, rather than salt excretion. When taken together, the current evidence strongly favors the view that EO is a physiologically relevant hormone with a variety of interesting actions that augment vascular tone and promote renal sodium retention.
In summary, the hunt for NHs has led recently to the complete identification of numerous natriuretic materials. In spite of these notable successes, none of the materials seems to fit the anticipated physiological profile for a mammalian NH. Much evidence indicates there are two major non-AP NHs that remain to be isolated and identified. It may be argued that identification of the CNS NH should be a priority in view of its broad physiological relevance, relationship to dietary sodium intake, and the implication of a profound role in salt balance in a number of pathological disorders.
Conflict of Interest Statement
The Review Editor Frans H. H. Leenen declares that, despite having collaborated with the author John Hamlyn, the review process was handled objectively and no conflict of interest exists. The author declares that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
I thank Mordecai P. Blaustein for helpful comments. Supported in part by the US National Institutes of Health HL107555.
References
2. Brown IJ, Tzoulaki I, Candeias V, Elliott P. Salt intakes around the world: implications for public health. Int J Epidemiol (2009) 38(3):791–813. doi: 10.1093/ije/dyp139
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
3. Wechter WJ, Kantoci D, Murray ED Jr, D’Amico DC, Jung ME, Wang WH. A new endogenous natriuretic factor: LLU-alpha. Proc Natl Acad Sci USA (1996) 93(12):6002–7. doi:10.1073/pnas.93.12.6002
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
5. Slatopolsky E, Elkan IO, Weerts C, Bricker NS. Studies on the characteristics of the control system governing sodium excretion in uremic man. J Clin Invest (1968) 47:521–30. doi:10.1172/JCI105877
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
6. de Wardener HE, Mills IH, Clapham WF, Hayter CJ. Studies on the efferent mechanism of the sodium diuresis which follows the administration of intravenous saline in the dog. Clin Sci (1961) 21:249–58.
7. Bahlman JS, McDonald J, Ventom MG, de Wardener HE. The effect on urinary sodium excretion of blood volume expansion without changing the composition of the blood. Clin Sci (1967) 32:403–13.
8. Tobian L, Coffee K, McCrea P. Evidence for a humoral factor of non-renal and non-adrenal origin which influences sodium excretion. Trans Assoc Am Physicians (1967) 80:200–6.
9. Kaloyanides GJ, Azer M. Evidence of a humoral mechanism in volume expansion natriuresis. J Clin Invest (1971) 50:1603–12. doi:10.1172/JCI106648
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
10. Knock CA. Further evidence in vivo for a circulating natriuretic substance after expanding the blood volume in rats. Clin Sci (1980) 59:423–33.
12. Goetz KL. Evidence that atriopeptin is not a physiological regular of sodium excretion. Hypertension (1990) 15:9–19. doi:10.1161/01.HYP.15.1.9
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
13. Rubattu S, Sciarretta S, Valenti V, Stanzione R, Volpe M. Natriuretic peptides: an update on bioactivity, potential therapeutic use, and implication in cardiovascular diseases. Am J Hypertens (2008) 21(7):733–41. doi:10.1038/ajh.2008.174
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
14. De Bold AJ, Borenstein HB, Veress AT, Sonnenberg H. A rapid and potent natriuretic response to intravenous injection of atrial myocardial extract in rats. Life Sci (1981) 28:89–94. doi:10.1016/0024-3205(81)90370-2
15. Richards AM, McDonald D, Fitzpatrick MA, Nicholls MG, Espiner EA, Ikram H, et al. Atrial natriuretic hormone has biological effects in man at physiological plasma concentrations. J Clin Endocrinol Metab (1988) 67(6):1134–9. doi:10.1210/jcem-67-6-1134
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
16. Anderson JV, Struthers AD, Christofides ND, Bloom SR. Atrial natriuretic peptides: an endogenous factor enhancing sodium excretion in man. Clin Sci (1986) 70:327–31.
17. Lang RE, Tholken H, Ganten D, Luft FC, Ruskoaho H, Unger T. Atrial natriuretic factor – a circulating hormone stimulated by volume loading. Nature (1985) 314:264–6. doi:10.1038/314264a0
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
18. Richards AM, Tonolo G, Polonia J, Montorsi P. Contrasting plasma atrial natriuretic factor concentrations during comparable natriuresis with infusions of atrial natriuretic factor and saline in normal man. Clin Sci (Lond) (1988) 75(5):455–62.
19. Goetz KL, Wang BC, Geer PG, Leadley RJ Jr, Reinhardt HW. Atrial stretch increases sodium excretion independently of release of atrial peptides. Am J Physiol (1986) 250:R946–50.
20. Goetz KL, Wang BC, Geer PG, Sundet WD, Needleman P. Effects of atriopeptin infusion versus effects of left atrial stretch in awake dogs. Am J Physiol (1986) 250:R221–6.
21. Goetz KL, Wang BC, Bie P, Leadley RJ, Geer PG. Natriuresis during atrial distension and a concurrent decline in plasma atriopeptin. Am J Physiol (1988) 255:R259–67.
23. De Bold AJ. On the shoulders of giants: the discovery of atrial natriuretic factor. Can J Physiol Pharmacol (1987) 65(10):2007–12. doi:10.1139/y87-314
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
24. Schulz-Knappe P, Forssmann K, Herbst F, Hock D, Pipkorn R, Forssmann WG. Isolation and structural analysis of “urodilatin”, a new peptide of the cardiodilatin-(ANP)-family, extracted from human urine. Klin Wochenschr (1988) 66(17):752–9. doi:10.1007/BF01726570
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
25. Cain CD, Schroeder FC, Shankel SW, Mitchnick M, Schmertzler M, Bricker NS. Identification of xanthurenic acid 8-O-beta-d-glucoside and xanthurenic acid 8-O-sulfate as human natriuretic hormones. Proc Natl Acad Sci U S A (2007) 104(45):17873–8. doi:10.1073/pnas.0705553104
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
26. Currie MG, Fok KF, Kato J, Moore RJ, Hamra FK, Duffin KL, et al. Guanylin: an endogenous activator of intestinal guanylate cyclase. Proc Natl Acad Sci U S A (1992) 89:947–51. doi:10.1073/pnas.89.3.947
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
27. Hamra FK, Forte LR, Eber SL, Pidhorodeckyj NV, Krause WJ, Freeman RH, et al. Uroguanylin: structure and activity of a second endogenous peptide that stimulates intestinal guanylate cyclase. Proc Natl Acad Sci USA (1993) 90:10464–8. doi:10.1073/pnas.90.22.10464
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
28. Veress AT, Pearce JW, Solomon S. The effect of acute adrenalectomy on volume natriuresis in the rat. Proc Soc Exp Biol Med (1974) 145(2):533–6. doi:10.3181/00379727-145-37846
29. Kaloyanides GJ, Cohen L, DiBona GF. Failure of selected end organ ablation to modify the natriuresis of blood volume expansion in the dog. Clin Sci Mol Med (1977) 52:351–6.
30. Schneider R, Wray V, Nimtz M, Lehmann WD, Kirch U, Antolovic R, et al. Bovine adrenals contain, in addition to ouabain, a second inhibitor of the sodium pump. J Biol Chem (1998) 273(2):784–92. doi:10.1074/jbc.273.2.784
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
31. Kramer HJ, Gonick HC. Effect of extracellular volume expansion on renal Na,K-ATPase and cell metabolism. Nephron (1974) 12:281–96. doi:10.1159/000180341
32. Kramer HJ. Natriuretic activity in plasma following extracellular volume expansion. In: Kauffman W, Krause DK, editors. Central Nervous Control of Na+ Balance-Relations to the Renin-Angiotensin System. Stuttgart: Georg Thieme Verlag (1976). p. 126–35.
33. Sedlakova E, Lichardus B, Cort JH. Plasma saluretic activity: its nature and relation to oxytocin analogues. Science (1969) 164:580–92. doi:10.1126/science.164.3879.580
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
34. Kramer HJ, Rietzel C, Klingmuller D, Dusing R. Further studies on the isolation and purification of a small molecular weight natriuretic hormone. In: Lichardus B, Schrier RW, Ponec J, editors. Hormonal Regulation of Sodium Excretion. North Holland: Elsevier (1980). p. 313–22.
35. Buckalew VM Jr, Nelson DB. Natriuretic and sodium transport inhibitory activity in plasma of volume-expanded dogs. Kidney Int (1974) 5:12–22. doi:10.1038/ki.1974.2
36. Sealey JE, Kirshman JD, Laragh JH. Natriuretic activity in plasma and urine of salt-loaded man and sheep. J Clin Invest (1969) 48:2210–24. doi:10.1172/JCI106187
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
37. Gruber KA, Whitaker JM, Buckalew VM Jr. Endogenous digitalis-like substance in plasma of volume expanded dogs. Nature (1980) 287:743–5. doi:10.1038/287743a0
38. Bourgoignie JJ, Hwang KH, Espinel C, Klahr S, Bricker NS. A natriuretic factor in the serum of patients with chronic uremia. J Clin Invest (1972) 51:1514–27. doi:10.1172/JCI106948
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
39. Bourgoignie JJ, Weisser F, Rolf D, Klahr S, Bricker NS. Demonstration of a low molecular weight natriuretic factor in uremic serum. Trans Assoc Am Physicians (1970) 83:277–87.
40. Bourgoignie JJ, Klahr S, Bricker NS. Inhibition of transepithelial sodium transport in the frog skin by a low molecular weight fraction of uremic serum. J Clin Invest (1971) 50:303–11. doi:10.1172/JCI106495
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
41. Bourgoignie JJ, Hwang KH, Ipakchi EJ, Bricker NS. The presence of a natriuretic factor in the urine of patients with chronic uremia. J Clin Invest (1974) 53:1559–67. doi:10.1172/JCI107706
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
42. Hamlyn JM, Blaustein MP, Bova S, DuCharme DW, Harris DW, Mandel F, et al. Identification and characterization of a ouabain-like compound from human plasma. Proc Natl Acad Sci USA (1991) 88(14):6259–63. doi:10.1073/pnas.88.21.9907-d
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
43. Kawamura A, Guo J, Itagaki Y, Bell C, Wang Y, Haupert GT Jr, et al. On the structure of endogenous ouabain. Proc Natl Acad Sci U S A (1999) 96(12):6654–9. doi:10.1073/pnas.96.12.6654
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
44. Forte LR, Fan X, Hamra FK. Salt and water homeostasis: uroguanylin is a circulating peptide hormone with natriuretic activity. Am J Kidney Dis (1996) 28(2):296–304. doi:10.1016/S0272-6386(96)90318-2
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
45. Cambier P, Godon JP. Role of prostaglandins in the production of natriuretic factor by the isolated rat kidney. Ren Physiol (1984) 7(3):163–75.
46. Godon JP, Nizet A. Release by isolated dog kidney of a natriuretic material following saline loading. Arch Int Physiol Biochim (1974) 82:309–11. doi:10.3109/13813457409070478
47. Gonick HJ, Saldanha LF. A natriuretic principle derived from kidney tissue of volume expanded rats. J Clin Invest (1975) 56:247–55. doi:10.1172/JCI108087
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
48. Louis F, Favre H. Basal activity of the natriuretic factor extracted from the rat kidney as a function of the diet and its role in the regulation of the acute sodium balance. Clin Sci (1980) 58:385–91.
49. Drummer C, Fiedler F, König A, Gerzer R. Urodilatin, a kidney-derived natriuretic factor, is excreted with a circadian rhythm and is stimulated by saline infusion in man. J Am Soc Nephrol (1991) 1(9):1109–13.
50. Feller SM, Gagelmann M, Forssmann WG. Urodilatin: a newly described member of the ANP family. Trends Pharmacol Sci (1989) 10(3):93–4. doi:10.1016/0165-6147(89)90199-5
51. Ivanov YI. The evidence of production or activation of a natriuretic factor in the liver. Endocrinol Exp (1979) 13:195–200.
53. Zubiaur M, Fernandez Munoz MD, Hernando L, Lopez-Novoa JM. Role of the natriuretic hormone in the specific natriuresis induced by intraportal infusion of hypertonic saline in dogs. Miner Electrolyte Metab (1987) 13:13–8.
54. Sealey JE, Laragh JH. Further studies of a natriuretic substance occurring in human urine and plasma. Circ Res (1971) 28(Suppl II):II32–43. doi:10.1161/01.RES.28.5.II-32
55. Passo SS, Thornborough JR, Rothballer AB. Hepatic receptors in the control of sodium excretion in anesthetized cats. Am J Physiol (1973) 224:373–5.
56. Perlmutt JH, Aziz O, Haberich FJ. A comparison of sodium excretion in response to infusion of isotonic saline into the vena porta and vena cava of conscious dogs. Pflugers Arch (1975) 357:1–14. doi:10.1007/BF00584540
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
57. Daly JJ, Roe JW, Horrocks P. A comparison of sodium excretion following the infusion of saline into systemic and portal veins in the dog: evidence for a hepatic role in the control of sodium excretion. Clin Sci (1967) 33:481–7.
58. Potkay S, Gilmore JP. Renal response to vena cava and portal venous infusions of sodium chloride in unanesthetized dogs. Clin Sci (1970) 39:13–20.
59. Strandhoy JW, Williamson HE. Evidence for an hepatic role in the control of sodium excretion. Proc Soc Exp Biol Med (1970) 133:419–22. doi:10.3181/00379727-133-34487
60. Clarkson EM, Raw SM, de Wardener HE. Further observations on a low-molecular weight natriuretic substance in the urine of normal man. Kidney Int (1979) 16:710–21. doi:10.1038/ki.1979.187
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
61. Clarkson EM, Young DR, Raw SM, de Wardener HE. Chemical properties, physiological action and further separation of a low molecular weight natriuretic substance in the urine of normal man. In: Lichardus B, Schrier RW, Ponec J, editors. Hormonal Regulation of Sodium Excretion. North Holland: Elsevier (1980). p. 333–40.
62. Fine LG, Bourgoignie JJ, Hwang KH, Bricker NS. On the influence of the natriuretic factor from patients with chronic uremia on the bioelectric properties and sodium transport of the isolated mammalian collecting tubule. J Clin Invest (1976) 58(3):590–7. doi:10.1172/JCI108505
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
63. Clarkson EM, Raw SM, de Wardener HE. Two natriuretic substances in extracts of urine from normal man when salt-depleted and salt-loaded. Kidney Int (1976) 10:381–94. doi:10.1038/ki.1976.124
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
64. Kruck F. Influence of humoral factors on renal tubular sodium handling. Nephron (1969) 6:205–16. doi:10.1159/000179729
65. Viskoper JR, Czaczkeo JW, Schwartz N, Ullmann TD. Natriuretic activity of a substance isolated from human urine during the excretion of a salt load. Nephron (1971) 8:540–8. doi:10.1159/000179959
66. Bagrov AY, Fedorova OV, Dmitrieva RI, Howald WN, Hunter AP, Kuznetsova EA, et al. Characterization of a urinary bufodienolide Na+, K+-ATPase inhibitor in patients after acute myocardial infarction. Hypertension (1998) 31:1097–103. doi:10.1161/01.HYP.31.5.1097
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
67. Hoffman A, Okun-Gurevich M, Ovcharenko E, Goltsman I, Karram T, Cain C, et al. Renal effects of a novel endogenous natriuretic agent xanthurenic acid 8-o-β-d-glucoside in rats. Physiol Rep (2013) 1(6):e00155. doi:10.1002/phy2.155
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
68. Eliades D, Pamnani MB, Swindall BT, Haddy FJ. Effects of bufalin on renal venous outflow, urine flow and natriuresis in the anesthetized dog. Adv Exp Med Biol (1991) 308:205–10. doi:10.1007/978-1-4684-6015-5_17
69. Pamnani MB, Chen S, Bryant HJ, Schooley JF Jr, Eliades DC, Yuan CM, et al. Effects of three sodium-potassium adenosine triphosphatase inhibitors. Hypertension (1991) 18(3):316–24. doi:10.1161/01.HYP.18.3.316
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
70. Bagrov AY, Shapiro JI, Fedorova OV. Endogenous cardiotonic steroids: physiology, pharmacology, and novel therapeutic targets. Pharmacol Rev (2009) 61(1):9–38. doi:10.1124/pr.108.000711
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
71. Hamlyn JM, Ludens JH. Nonatrial natriuretic hormones. In: Seldin DW, Giebisch G, editors. The Kidney, Physiology and Pathophysiology. New York, NY: Raven Press (1992). p. 1885–924.
72. Alberola A, Pinilla JM, Quesada T, Romero JC, Salom MG, Salazar FJ. Role of nitric oxide in mediating renal response to volume expansion. Hypertension (1992) 19(6 Pt 2):780–4. doi:10.1161/01.HYP.19.6.780
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
73. Coruzzi P, Novarini A, Musiari L, Ravanetti C, Ghielmi S, Rodella A, et al. The antinatriuretic effect of dopaminergic blockade during volume expansion is independent of circulating atrial natriuretic factor. Clin Sci (Lond) (1989) 77(5):479–84.
74. Hansell P, Sjoquist M, Fasching A, Isaksson B, Karlsson M, Ulfendahl HR. CNS-induced natriuresis during dopamine receptor blockade. Further support for the existence of, at least, two separate natriuretic hormonal systems. Acta Physiol Scand (1988) 133:373–80. doi:10.1111/j.1748-1716.1988.tb08419.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
75. Jose PA, Holloway RR, Campbell TW, Eisner GM. Dopamine blockade attenuates the natriuresis of saline loading in the adrenalectomized rat. Nephron (1988) 48:54–7. doi:10.1159/000184869
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
76. McClanahan M, Sowers JR, Beck FWJ, Mohanty PK, McKenzie T. Dopaminergic regulation of natriuretic response to acute volume expansion in dogs. Clin Sci (1985) 68:263–9.
77. Hegde S, Jadhav AL, Lokhandwala MF. Role of kidney dopamine in the natriuretic response to volume expansion in rats. Hypertension (1989) 13:828–34. doi:10.1161/01.HYP.13.6.828
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
78. Buckalew VM Jr, Adkins TG. Effects of interrupting intravenous saline infusion on sodium excretion in the dog. Ren Physiol (1978) 1:11–8.
79. Andersson B, Jobin M, Olsson K. Stimulation of urinary salt excretion following injections of hypertonic NaCl solution into the 3rd brain ventricle. Acta Physiol Scand (1966) 67:127–8. doi:10.1111/j.1748-1716.1966.tb03293.x
80. Andersson B, Dallman MF, Olsson K. Evidence for a hypothalamic control of renal sodium excretion. Acta Physiol Scand (1969) 75:496–510. doi:10.1111/j.1748-1716.1969.tb04403.x
81. Andersson B, Westbye O. Synergistic action of sodium and angiotension on brain mechanisms controlling fluid balance. Life Sci (1970) 9:601–8. doi:10.1016/0024-3205(70)90090-1
82. Blaine E, Denton DA, McKinley MJ, Weller S. A central osmosensitive receptor for renal sodium excretion. J Physiol (1975) 244:497–509.
83. Morris M, McCann SM, Orias R. Evidence for hormonal participation in the natriuretic and kaliuretic responses to intraventricular hypertonic saline and norepinephrine. Proc Soc Exp Biol Med (1976) 152:95–8. doi:10.3181/00379727-152-39336
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
84. McKinley MJ, Denton DA, Coghlan JP, Harvey RB, McDougall JG, Rundgren M, et al. Cerebral osmoregulation of renal sodium excretion-a response analogous to thirst and vasopressin release. Can J Physiol Pharmacol (1987) 65:1724–9. doi:10.1139/y87-271
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
85. Thornborough JR, Passo SS, Rothballer AB. Receptors in cerebral circulation affecting sodium excretion in the cat. Am J Physiol (1973) 225(1):138–41.
86. Andersson B, Jobin M, Olsson K. A study of thirst and other effects of an increased sodium concentration in the 3rd brain ventricle. Acta Physiol Scand (1967) 69(1):29–36. doi:10.1111/j.1748-1716.1967.tb03488.x
87. Mouw DR, Vander AJ. Evidence for brain Na receptors controlling renal Na excretion and plasma renin activity. Am J Physiol (1970) 219:822–32.
88. Ulfendahl HR, Goransson A, Hansell P, Karlsson M, Sjoquist M. Natriuresis obtained by stimulation of the cerebroventricular system with sodium ions indicates a blood bourne natriuretic factor. Acta Physiol Scand (1986) 127:269–71. doi:10.1111/j.1748-1716.1986.tb07904.x
89. Saville MA, Geer PG, Wang BC, Leadley RJ, Goetz KL. A high-salt meal produces natriuresis in humans without elevating plasma atriopeptin. Proc Soc Exp Biol Med (1988) 188:387–93. doi:10.3181/00379727-188-3-RC2
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
90. Hansell P, Goransson A, Leppaluoto J, Arjamaa O, Vakkurri O, Ulfendahl HR. CNS-induced natriuresis is not mediated by the atrial natriuretic factor. Acta Physiol Scand (1987) 129:221–7. doi:10.1111/j.1748-1716.1987.tb08062.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
91. Hansell P. Natriuretic factors. Influence on glomerular filtration, renal blood flow and electrolyte excretion in the rat. Acta Univ Ups (1988) 154:1–29.
92. Ulfendahl HR, Ahlsson A, Hansell P, Hoglund U, Jacobsson E, Lee SL, et al. Studies on the mechanisms underlying CNS-induced natriuresis. Acta Physiol Scand (1989) 136(Suppl 583):75–8.
93. Leksell LG, Congiu M, Denton DA, Fei DT, McKinley MJ, Tarjan E, et al. Influence of mannitol-induced reduction in CSF Na on nervous and endocrine mechanisms involved in the control of fluid balance. Acta Physiol Scand (1981) 112(1):33–40. doi:10.1111/j.1748-1716.1981.tb06779.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
94. Leksell LG, Denton DA, Fei DT, McKinley MJ, Müller AF, Weisinger RS, et al. On the importance of CSF Na in the regulation of renal sodium excretion and renin release. Acta Physiol Scand (1982) 115(1):141–6. doi:10.1111/j.1748-1716.1982.tb07056.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
95. McKinley MJ, Denton DA, Fryday HW, Weisinger RS. Cerebral mechanisms influencing renal sodium excretion in dehydrated sheep. Clin Exp Pharmacol Physiol (1983) 10:521–6. doi:10.1111/j.1440-1681.1983.tb00220.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
96. McKinley MJ, Evered MD, Mathai ML. Renal Na excretion in dehydrated and rehydrated adrenalectomized sheep maintained with aldosterone. Am J Physiol Regul Integr Comp Physiol (2000) 279(1):R17–24.
97. Cox PS, Denton DA, Mouw DR, Tarjan E. Natriuresis induced by localized perfusion within the third cerebral ventricle of sheep. Am J Physiol (1987) 252:R1–6.
98. Cort JH, Lichardus B. The natriuretic activity of jugular vein blood during carotid occlusion. Physiol Bohemoslov (1963) 12:497–501.
99. Andersson B, Lishajko F, Leskell LG. Perturbations in fluid balance induced by medially placed forebrain lesions. Brain Res (1975) 99:261–75. doi:10.1016/0006-8993(75)90028-1
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
100. Keeler R. Effect of chronic preoptic lesions on the renal excretion of sodium in rats. Am J Physiol (1975) 228:1725–8.
101. Johnson AK, Buggy J. Periventricular preoptic-hypothalamus is vital for thirst and normal water economy. Am J Physiol (1978) 234:R122–9.
102. Kaloyanides GJ, Balabanian MB, Bowman RL. Evidence that the brain participates in the humoral natriuretic mechanism of blood volume expansion in the dog. J Clin Invest (1978) 62:1288–95. doi:10.1172/JCI109249
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
103. Bealer SL, Haywoood JR, Gruber KA, Buckalew VM Jr, Fink GD, Brody MJ. Preoptic-hypothalamic periventricular lesions reduce natriuresis to volume expansion. Am J Physiol (1983) 244:R51–7.
104. Mangiapane ML, Thrasher TN, Keil LC, Simpson JB, Ganong WF. Deficits in drinking and vasopressin secretion after lesions of the nucleus medianus. Neuroendocrinology (1983) 37:73–7. doi:10.1159/000123518
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
105. McKinley MJ, Congiu M, Denton DA, Park RG, Penschow J, Simpson JB, et al. The anterior wall of the third cerebral ventricle and homeostatic responses to dehydration. J Physiol (1984) 79:421–7.
106. McKinley MJ, Lichardus B, McDougall JG, Weisinger RS. Periventricular lesions block natriuresis to hypertonic but not isotonic NaCl loads. Am J Physiol (1992) 262(1 Pt 2):F98–107.
107. Peters JP, Welt LG, Simms EAH. A salt wasting syndrome associated with cerebral disease. Trans Assoc Am Physicians (1950) 63:57–64.
109. Nelson PB, Seif SM, Maroon JC, Robinson AG. Hyponatremia in intracranial disease: perhaps not the syndrome of inappropriate secretion of antidiuretic hormone (SIADH). J Neurosurg (1981) 55:938–41. doi:10.3171/jns.1981.55.6.0938
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
110. Nelson PB, Seif SM, Gutal J, Robinson AG. Hyponatremia and natriuresis following subarachnoid hemorrhage in a monkey model. J Neurosurg (1984) 60:233–7. doi:10.3171/jns.1984.60.2.0233
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
111. Diringer M, Ladenson PW, Borel C, Hart GK, Kirsch JR, Hanley DF. Sodium and water regulation in a patient with cerebral salt wasting. Arch Neurol (1989) 46:928–30. doi:10.1001/archneur.1989.00520440124031
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
112. McKinley MJ, Denton DA, Park RG, Weisinger RS. Cerebral involvement in dehydration-induced natriuresis. Brain Res (1983) 263:340–3. doi:10.1016/0006-8993(83)90326-8
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
113. Mouw DR, Vander AJ, Bourgoignie JJ, Kutschinski SS, Mathias NP. Nonpressor mechanisms in CNS-induced natriuresis. Am J Physiol (1979) 237:F157–66.
114. Luke RG. Natriuresis and chloruresis during hydropenia in the rat. Am J Physiol (1973) 224:13–20.
115. Pierce ET, Grekin RJ, Mouw DR. Efferent role of ADH in CNS-induced natriuresis. Am J Physiol (1984) 246:F32–8.
116. Mouw DR, Vander AJ, Landis C, Kutschinski S, Mathias N, Zimmerman D. Dose-response relation of CSF sodium and renal sodium excretion, and its absence in homozygous Brattleboro rats. Neuroendocrinology (1980) 30(4):206–12. doi:10.1159/000123002
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
117. Beasley D, Malvin RL, Mouw DR. CNS-induced natriuresis and renal hemodynamics in conscious rats. Am J Physiol (1983) 245:F763–71.
118. Humphreys MH, Friedler RM, Earley LE. Natriuresis produced by vasopressin or hemorrhage during water diuresis in the dog. Am J Physiol (1970) 219:658–64.
119. Fejes-Toth G, Magyar A, Walter J. Renal response to vasopressin after inhibition of prostaglandin synthesis. Am J Physiol (1977) 232:F416–23.
120. Balment RJ, Brimble MJ, Forsling ML, Musabayane CJ. Natriuretic response of the rat to plasma concentrations of arginine vasopressin within the physiological range. J Physiol (1984) 352:517–26.
121. Thrasher TN, Wade CE, Keil LC, Ramsay DJ. Sodium balance and aldosterone during dehydration and rehydration in the dog. Am J Physiol (1984) 247:R76–83.
122. Lote CJ, McVicar AJ, Smyth DG. Effects of vasopressin-glycine and vasopressin-glycine-lysine-arginine on renal function in the rat. J Endocrinol (1986) 108:255–60. doi:10.1677/joe.0.1080255
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
123. Lote CJ, Thewles A, Wood JA. Vasopressin-induced natriuresis in the conscious rat: role of blood pressure, renal prostaglandin synthesis and the peptide. Am J Physiol (1989) 411:481–91.
124. Park RG, Congiu M, Denton DA, McKinley MJ. Natriuresis induced by arginine vasopressin infusion in sheep. Am J Physiol (1985) 249:F799–805.
125. Merrill DC, Skelton MM, Cowley AW. Humoral control of water and electrolyte excretion during water restriction in sodium-deprived dogs. Kidney Int (1986) 29:1152–61. doi:10.1038/ki.1986.121
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
126. Beasley D, Malvin RL. Role of natriuretic factor in central nervous system (CNS)-induced natriuresis. Proc Soc Exp Biol Med (1985) 178:575–9. doi:10.3181/00379727-178-42044
127. Balment RJ, Brimble MJ, Forsling ML, Kelly LP, Musabayane CT. A synergistic effect of oxytocin and vasopressin on sodium excretion in the neurohypopysectomized rat. J Physiol (1986) 381:453–64.
128. Haller H, Bähr V, Bock A, Distler A, Philipp T. Vasopressin is increased in mineralocorticoid-induced blood pressure increase in man. J Hypertens (1987) 5(5):111–3.
129. Pennington GL, McKinley MJ. Reduction of cerebral NaCl concentration can abolish mineralocorticoid escape. Am J Physiol (1990) 259(5 Pt 2):F839–46.
130. Brooks DP, Share L, Crofton JT, Guthe C, Ling WD, Bohr DF. Increased sensitivity of the osmotic control of vasopressin in sheep with deoxycorticosterone acetate-induced hypertension. J Endocrinol (1985) 107(3):309–15. doi:10.1677/joe.0.1070309
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
131. Brooks FP, Pickford M. The effect of posterior pituitary hormones on the excretion of electrolytes in dogs. J Physiol (1958) 142:468–93.
132. Sawyer WH. Posterior pituitary extracts and excretion of electrolytes by the rat. Am J Physiol (1952) 169:583–7.
133. Chan WY, Sawyer WH. Saluretic action of neurohypophysial peptides in conscious dogs. Am J Physiol (1961) 201:799–803.
134. Bartter FC, Mills IH. The effect of oxytocin on sodium excretion. J Endocrinol (1969) 45(1):v–vi.
135. Kleinman LI, Banks RO. Natriuretic effect of oxytocin in saline-expanded neonatal dogs. Am J Physiol (1980) 239:F589–94.
136. Diez J, Colina I, Guarner F, Quiroga J, Corzo J, Purroy A, et al. Intracerebroventricular infusion of sodium chloride-rich artificial cerebrospinal fluid in rats induces natriuresis and releases an inhibitor of prostaglandin synthesis. Clin Sci (1984) 66:621–4.
137. Buckalew VM Jr, Dimond KA. Effect of vasopressin on sodium excretion and plasma antinatriferic activity in the dog. Am J Physiol (1976) 231:28–33.
138. Schwartzman M, Ferreri NR, Carroll MA, Songu-Mize E, McGiff JC. Renal cytochrome P450-related arachidonate metabolite inhibits Na,K-ATPase. Nature (1985) 314:620–2. doi:10.1038/314620a0
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
139. Gilmore JP, Nemeh MN. Salt depletion inhibits cerebral-induced natriuresis in the dog. Am J Physiol (1984) 247:F725–8.
140. Grim CE, Scoggins BA. The rapid adjustment of renal sodium excretion to changes in dietary sodium intake in sheep. Life Sci (1986) 39:215–22. doi:10.1016/0024-3205(86)90533-3
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
141. Lichardus B, Ponec J. On the role of the hypophysis in the mechanism of body fluid volume regulation in acutely hypophysectomized rats. Endokrinologie (1973) 61:403–12.
142. Clarkson EM, Koutsaimanis KG, Davidman M, DuBois M, Penn WP, deWardener HE. The effects of brain extracts on urinary sodium excretion of the rat and the intracellular sodium concentration of renal tubule fragments. Clin Sci Mol Med (1974) 41:210–3.
143. Sedlakova E, Prusik Z, Skopkova J, Barth T, Kluh I, Cort JH. Isolation of a tridecapeptide from natriuretic fractions of bovine posterior pituitary. Eur J Clin Invest (1974) 4:285–92. doi:10.1111/j.1365-2362.1974.tb00405.x
144. Goto A, Yamada K, Yagi N, Yoshioka M, Sugimoto T. Physiology and pharmacology of endogenous digitalis-like factors. Pharmacol Rev (1992) 44(3):377–99.
145. Schoner W. Endogenous digitalis-like factors. Clin Exp Hypertens A (1992) 14(5):767–814. doi:10.3109/10641969209036220
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
146. de Wardener HE, MacGregor GA, Clarkson EM, Alaghband-Zadeh J, Bitensky L, Chayen J. Effect of sodium intake on ability of human plasma to inhibit renal Na,K-ATPase. Lancet (1981) i:411–2. doi:10.1016/S0140-6736(81)91792-X
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
147. Dikstein S. Stimulability, adenosine triphosphatases, and their control by cellular redox processes. Naturwissenschaften (1971) 58:439–43. doi:10.1007/BF00624617
148. Fenton S, Clarkson E, MacGregor G, Alaghband-Zadeh J, de Wardener HE. An assay of the capacity of biological fluids to stimulate renal glucose-6-phosphate dehydrogenase activity in vitro as a marker of their ability to inhibit sodium potassium-dependent adenosine triphosphatase activity. J Endocrinol (1982) 94(1):99–110. doi:10.1677/joe.0.0940099
149. Lichardus B, Ponec J. Conditions for biological evidence of a natriuretic hormone in experiments with rat cross circulation. Physiol Bohemoslov (1970) 19:330.
150. Nutbourne DM, Howse JD, Schrier RW, Talner LB, Ventom MG, Verroust PJ, et al. The effect of expanding the blood volume of a dog on the short circuit current across an isolated frog skin incorporated in the dogs circulation. Clin Sci (1970) 38:629–48.
151. Clarkson EM, Talner LB, de Wardener HE. The effect of plasma from blood volume expanded dogs on sodium, potassium and PAH transport of renal tubule fragments. Clin Sci (1970) 38:617–27.
152. Buckalew VM Jr, Lancaster CD Jr. Studies of a humoral sodium transport in normal dogs and dogs with ligation of the inferior vena cava. Circ Res (1971) 28-29(Suppl II):II–44–II–52.
153. Epstein M. Cardiovascular and renal effects of head out water immersion in man. Circ Res (1976) 39:619–28. doi:10.1161/01.RES.39.5.619
154. Poston L, Wilkinson SP, Sewell R, Williams R. Sodium transport during natriuresis of volume expansion. A study using peripheral leucocytes. Clin Sci (1982) 63:243–5.
155. Jandhyala BS, Ansari AF. Elevation of sodium levels in the cerebral ventricles of anesthetized dogs triggers the release of an inhibitor of ouabain-sensitive sodium-potasium-ATPase into the circulation. Clin Sci (1986) 70:103–10.
156. Haddy FJ. Natriuretic hormone – the missing link in low renin hypertension? Biochem Pharmacol (1982) 31:3159–61. doi:10.1016/0006-2952(82)90544-5
157. Ludens JH, Clark MA, Kolbasa KP, Hamlyn JM. Digitalis-like factor and ouabain-like compound in plasma of volume-expanded dogs. J Cardiovasc Pharmacol (1993) 22(Suppl 2):S38–41. doi:10.1097/00005344-199322002-00014
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
158. Graves SW, Markides KE, Hollenberg NK. Application of supercritical fluid chromatography to characterize a labile digitalis-like factor. Hypertension (2000) 36(6):1059–64. doi:10.1161/01.HYP.36.6.1059
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
159. Buckalew VM Jr, Martinez FJ, Green WE. The effect of dialysates and ultrafiltrates of plasma of saline-loaded dogs on toad bladder sodium transport. J Clin Invest (1970) 49:926–35. doi:10.1172/JCI106312
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
160. Buggy J, Huot S, Pamnani M, Haddy F. Periventricular forebrain mechanisms for blood pressure regulation. Fed Proc (1984) 43(1):25–31.
161. Mathews WR, DuCharme DW, Hamlyn JM, Harris DW, Mandel F, Clark MA, et al. Mass spectral characterization of an endogenous digitalislike factor from human plasma. Hypertension (1991) 17(6 Pt 2):930–5. doi:10.1161/01.HYP.17.6.930
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
162. Komiyama Y, Nishimura N, Dong XH, Hirose S, Kosaka C, Masaki H, et al. Liquid chromatography mass spectrometric analysis of ouabainlike factor in biological fluid. Hypertens Res (2000) 23(Suppl):S21–7. doi:10.1291/hypres.23.Supplement_S21
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
163. Goto A, Yamada K, Yagi N, Hui C, Nagoshi H, Sasabe M, et al. Digitalis-like factors from human urine. J Cardiovasc Pharmacol (1993) 22(Suppl 2):S58–9. doi:10.1097/00005344-199322002-00019
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
164. Komiyama Y, Dong XH, Nishimura N, Masaki H, Yoshika M, Masuda M, et al. A novel endogenous digitalis, telocinobufagin, exhibits elevated plasma levels in patients with terminal renal failure. Clin Biochem (2005) 38(1):36–45. doi:10.1016/j.clinbiochem.2004.08.005
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
165. Komiyama Y, Nishimura N, Munakata M, Okuda K, Nishino N, Kosaka C, et al. Increases in plasma ouabainlike immunoreactivity during surgical extirpation of pheochromocytoma. Hypertens Res (1999) 22(2):135–9. doi:10.1291/hypres.22.135
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
166. Ferrandi M, Manunta P, Ferrari P, Bianchi G. The endogenous ouabain: molecular basis of its role in hypertension and cardiovascular complications. Front Biosci (2005) 10:2472–7. doi:10.2741/1711
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
167. Huang BS, Amin MS, Leenen FH. The central role of the brain in salt-sensitive hypertension. Curr Opin Cardiol (2006) 21(4):295–304. doi:10.1097/01.hco.0000231398.64362.94
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
168. Schoner W, Scheiner-Bobis G. Endogenous and exogenous cardiac glycosides: their roles in hypertension, salt metabolism, and cell growth. Am J Physiol Cell Physiol (2007) 293(2):C509–36. doi:10.1152/ajpcell.00098.2007
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
169. Manunta P, Ferrandi M, Bianchi G, Hamlyn JM. Endogenous ouabain in cardiovascular function and disease. J Hypertens (2009) 27(1):9–18. doi:10.1097/HJH.0b013e32831cf2c6
170. Xie Z, Askari A. Na(+)/K(+)-ATPase as a signal transducer. Eur J Biochem (2002) 269(10):2434–9. doi:10.1046/j.1432-1033.2002.02910.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
171. Drake MT, Violin JD, Whalen EJ, Wisler JW, Shenoy SK, Lefkowitz RJ. Beta-arrestin-biased agonism at the beta2-adrenergic receptor. J Biol Chem (2008) 283(9):5669–76. doi:10.1074/jbc.M708118200
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
172. Zulian A, Linde CI, Pulina MV, Baryshnikov SG, Papparella I, Hamlyn JM, et al. Activation of c-SRC underlies the differential effects of ouabain and digoxin on Ca(2+) signaling in arterial smooth muscle cells. Am J Physiol Cell Physiol (2013) 304(4):C324–33. doi:10.1152/ajpcell.00337.2012
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
173. Wilde WS, Howard PJ. Renal tubular action of ouabain on Na and K transport during stop-flow and slow-flow technique. J Pharmacol Exp Ther (1960) 130:232–8.
174. Yates NA, McDougall JG. Effects of direct renal arterial infusion of bufalin and ouabain in conscious sheep. Br J Pharmacol (1993) 108(3):627–30. doi:10.1111/j.1476-5381.1993.tb12852.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
175. Hook JB. A positive correlation between natriuresis and inhibition of renal Na, K-adenosine triphosphatase by ouabain. Proc Soc Exp Biol Med (1969) 13:731–4. doi:10.3181/00379727-131-33963
176. Selden R, Smith TW. Ouabain pharmacokinetics in dog and man: determination by radioimmunoassay. Circulation (1972) 45:1176–82. doi:10.1161/01.CIR.45.6.1176
177. Butler VP Jr, Schmidt DH, Smith TW, Haber E, Raynor BD, Demartini P. Effects of sheep digoxin-specific antibodies and their Fab fragments on digoxin pharmacokinetics in dogs. J Clin Invest (1977) 59(2):345–59. doi:10.1172/JCI108647
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
178. Cao Y, Zhao L, Liang Q, Bi K, Wang Y, Luo G. Study of the determination and pharmacokinetics of bufadienolides in dog’s plasma after administration of Liu-Shen-Wan by high performance liquid chromatography time-of-flight mass spectrometry. J Chromatogr B Analyt Technol Biomed Life Sci (2007) 853(1–2):227–33. doi:10.1016/j.jchromb.2007.03.018
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
179. Zhang Y, Tang X, Liu X, Li F, Lin X. Simultaneous determination of three bufadienolides in rat plasma after intravenous administration of bufadienolides extract by ultra performance liquid chromatography electrospray ionization tandem mass spectrometry. Anal Chim Acta (2008) 610(2):224–31. doi:10.1016/j.aca.2008.01.029
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
180. Patel AR, Kurashina T, Granger JP, Kirchner KA. Acute Na+, K+-ATPase inhibition with bufalin impairs pressure natriuresis in the rat. Hypertension (1996) 27(3 Pt 2):668–71. doi:10.1161/01.HYP.27.3.668
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
181. Blanco G, Mercer RW. Isozymes of the Na-K-ATPase: heterogeneity in structure, diversity in function. Am J Physiol (1998) 275(5 Pt 2):F633–50.
182. Doucet A, Barlet C. Evidence for differences in the sensitivity to ouabain of Na, K-ATPase along the nephrons of rabbit kidney. J Biol Chem (1986) 261:993–5.
183. Pittner J, Rhinehart K, Pallone TL. Ouabain modulation of endothelial calcium signaling in descending vasa recta. Am J Physiol Renal Physiol (2006) 291(4):F761–9. doi:10.1152/ajprenal.00326.2005
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
184. Bagrov AY, Fedorova OV. Cardenolide and bufadienolide ligands of the sodium pump. How they work together in NaCl sensitive hypertension. Front Biosci (2005) 10:2250–6. doi:10.2741/1694
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
185. Loreaux EL, Kaul B, Lorenz JN, Lingrel JB. Ouabain-Sensitive alpha1 Na,K-ATPase enhances natriuretic response to saline load. J Am Soc Nephrol (2008) 19(10):1947–54. doi:10.1681/ASN.2008020174
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
186. Doursout MF, Chelly JE, Liang YY, Buckley JP. The ouabain-dependent Na(+)-K+ pump and the brain renin-angiotensin system. Clin Exp Hypertens A (1992) 14(3):393–411. doi:10.3109/10641969209036197
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
187. Yuan CM, Manunta P, Hamlyn JM, Chen S, Bohen E, Yeun J, et al. Long-term ouabain administration produces hypertension in rats. Hypertension (1993) 22(2):178–87. doi:10.1161/01.HYP.22.2.178
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
188. Manunta P, Rogowski AC, Hamilton BP, Hamlyn JM. Ouabain-induced hypertension in the rat: relationships among plasma and tissue ouabain and blood pressure. J Hypertens (1994) 12(5):549–60. doi:10.1097/00004872-199405000-00008
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
189. Kurashina T, Kirchner KA, Granger JP, Patel AR. Chronic sodium-potassium-ATPase inhibition with ouabain impairs renal haemodynamics and pressure natriuresis in the rat. Clin Sci (Lond) (1996) 91(4):497–502.
190. Huang BS, Huang X, Harmsen E, Leenen FH. Chronic central versus peripheral ouabain, blood pressure, and sympathetic activity in rats. Hypertension (1994) 23(6 Pt 2):1087–90. doi:10.1161/01.HYP.23.6.1087
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
191. Zhang J, Lee MY, Cavalli M, Chen L, Berra-Romani R, Balke CW, et al. Sodium pump alpha2 subunits control myogenic tone and blood pressure in mice. J Physiol (2005) 569(Pt 1):243–56. doi:10.1113/jphysiol.2005.091801
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
192. Cao C, Payne K, Lee-Kwon W, Zhang Z, Lim SW, Hamlyn J, et al. Chronic ouabain treatment induces vasa recta endothelial dysfunction in the rat. Am J Physiol Renal Physiol (2009) 296(1):F98–106. doi:10.1152/ajprenal.90429.2008
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
193. Blaustein MP, Leenen FH, Chen L, Golovina VA, Hamlyn JM, Pallone TL, et al. How NaCl raises blood pressure: a new paradigm for the pathogenesis of salt-dependent hypertension. Am J Physiol Heart Circ Physiol (2012) 302(5):H1031–49. doi:10.1152/ajpheart.00899.2011
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
194. Manunta P, Hamilton BP, Hamlyn JM. Salt intake and depletion increase circulating levels of endogenous ouabain in normal men. Am J Physiol Regul Integr Comp Physiol (2006) 290(3):R553–9. doi:10.1152/ajpregu.00648.2005
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
195. Wang JG, Staessen JA, Messaggio E, Nawrot T, Fagard R, Hamlyn JM, et al. Salt, endogenous ouabain and blood pressure interactions in the general population. J Hypertens (2003) 21(8):1475–81. doi:10.1097/00004872-200308000-00010
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
196. Hamlyn JM, Manunta P. Endogenous ouabain: a link between sodium intake and hypertension. Curr Hypertens Rep (2011) 13(1):14–20. doi:10.1007/s11906-010-0161-z
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
197. Manunta P, Maillard M, Tantardini C, Simonini M, Lanzani C, Citterio L, et al. Relationships among endogenous ouabain, alpha-adducin polymorphisms and renal sodium handling in primary hypertension. J Hypertens (2008) 26(5):914–20. doi:10.1097/HJH.0b013e3282f5315f
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
198. Bauer N, Müller-Ehmsen J, Krämer U, Hambarchian N, Zobel C, Schwinger RH, et al. Ouabain-like compound changes rapidly on physical exercise in humans and dogs: effects of beta-blockade and angiotensin-converting enzyme inhibition. Hypertension (2005) 45(5):1024–8. doi:10.1161/01.HYP.0000165024.47728.f7
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
199. Bignami E, Casamassima N, Frati E, Messagio E, Corno L, Zangrillo A, et al. Endogenous ouabain changes rapidly during cardiac pulmonary by pass. J Steroids Horm Sci (2011) S3:002. doi:10.4172/2157-7536.S3-002
200. Bignami E, Casamassima N, Frati E, Lanzani C, Corno L, Alfieri O, et al. Preoperative endogenous ouabain predicts acute kidney injury in cardiac surgery patients. Crit Care Med (2013) 41(3):744–55. doi:10.1097/CCM.0b013e3182741599
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Keywords: salt, sodium, urine, excretion, sodium pump, ouabain, hormone
Citation: Hamlyn JM (2014) Natriuretic hormones, endogenous ouabain, and related sodium transport inhibitors. Front. Endocrinol. 5:199. doi: 10.3389/fendo.2014.00199
Received: 29 August 2014; Accepted: 10 November 2014;
Published online: 03 December 2014.
Edited by:
Vardaman Buckalew, Wake Forest School of Medicine, USAReviewed by:
Vardaman Buckalew, Wake Forest School of Medicine, USAFrans H. H. Leenen, University of Ottawa Heart Institute, Canada
Copyright: © 2014 Hamlyn. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: John M. Hamlyn, Department of Physiology, University of Maryland School of Medicine, 655 West Baltimore Street, Baltimore, MD 21201, USA e-mail:amhhbWx5bkB1bWFyeWxhbmQuZWR1