 Biagio Arcidiacono1
Biagio Arcidiacono1 Stefania Iiritano1
Stefania Iiritano1 Eusebio Chiefari1
Eusebio Chiefari1 Francesco S. Brunetti2
Francesco S. Brunetti2 Guoqiang Gu3
Guoqiang Gu3 Daniela Patrizia Foti1
Daniela Patrizia Foti1 Antonio Brunetti1*
Antonio Brunetti1*- 1Department of Health Sciences, University “Magna Græcia” of Catanzaro, Catanzaro, Italy
- 2Department of Medical and Surgical Sciences, University “Magna Græcia” of Catanzaro, Catanzaro, Italy
- 3Department of Cell and Developmental Biology, Center of Stem Cell Biology, Vanderbilt Medical Center, Nashville, TN, USA
The high-mobility group AT-hook 1 (HMGA1) protein is a nuclear architectural factor that can organize chromatin structures. It regulates gene expression by controlling the formation of stereospecific multiprotein complexes called “enhanceosomes” on the AT-rich regions of target gene promoters. Previously, we reported that defects in HMGA1 caused decreased insulin receptor expression and increased susceptibility to type 2 diabetes mellitus in humans and mice. Interestingly, mice with disrupted HMGA1 gene had significantly smaller islets and decreased insulin content in their pancreata, suggesting that HMGA1 may have a direct role in insulin transcription and secretion. Herein, we investigate the regulatory roles of HMGA1 in insulin transcription. We provide evidence that HMGA1 physically interacts with PDX-1 and MafA, two critical transcription factors for insulin gene expression and beta-cell function, both in vitro and in vivo. We then show that the overexpression of HMGA1 significantly improves the transactivating activity of PDX-1 and MafA on human and mouse insulin promoters, while HMGA1 knockdown considerably decreased this transactivating activity. Lastly, we demonstrate that high glucose stimulus significantly increases the binding of HMGA1 to the insulin (INS) gene promoter, suggesting that HMGA1 may act as a glucose-sensitive element controlling the transcription of the INS gene. Together, our findings provide evidence that HMGA1, by regulating PDX-1- and MafA-induced transactivation of the INS gene promoter, plays a critical role in pancreatic beta-cell function and insulin production.
Introduction
Production and secretion of insulin from beta cells of the pancreas maintain glucose homeostasis in vertebrates (from fish to human). Absence or dysfunction of beta cells results in diabetes mellitus, a set of genetically heterogeneous disorders featured by high blood sugar and related complications. The majority of diabetes is type 2 diabetes, which is featured by defects in both pancreatic insulin secretion (beta-cell dysfunction) and peripheral insulin action (insulin resistance) (1, 2). Therefore, understanding beta-cell generation and insulin production/secretion holds keys to understand and potentially cure diabetes.
Insulin production in beta cells is controlled at both transcriptional and translational levels (3). High glucose upregulates both insulin transcription and preproinsulin translation. To this end, glucose regulates proinsulin translation by altering the phosphorylation status of eukaryotic initiation factor 2α (4), while it promotes insulin transcription by activation of several beta-cell specific transcription factors (5). Yet, direct connections between high glucose and activation of beta-cell transcription factors have not been established.
Regulation of insulin (INS) gene expression has been characterized by several groups. A 500-bp stretch of DNA upstream of the INS transcription start site controls its transcription. Several cis-acting regulatory sequences reside in this region, with highly conserved A3, C1, E1, and CRE cis-acting regulatory elements (6, 7). These promoter regions bind ubiquitous factors and a pool of beta-cell specific transcription factors (8–14). Among these, the pancreatic and duodenal homeobox factor-1 (PDX-1) and MafA (V-maf musculoaponeurotic fibrosarcoma oncogene homolog A) are the most notable factors. PDX-1 forms multiprotein complexes with several beta-cell transcription factors to synergistically activate the INS promoter (7). It also mediates a role of high glucose in the up-regulation of INS transcription in humans and rodents (15–22). Consequently, targeted disruption of PDX-1 gene in beta cells leads to beta-cell dysfunction and overt diabetes in mice (23), whereas mutations in PDX-1 have been linked to pancreatic agenesis and diabetes in humans (24, 25). Similarly, MafA activates INS transcription through the E1 and C1 elements of the INS gene promoter (26–31). Its inactivation results in immature beta cells with lowered insulin expression and secretion and severe glucose intolerance (32, 33).
High-mobility group AT-hook 1 (HMGA1) is an architectural transcription factor that binds to the minor groove of AT-rich regions of DNA (34). This binding alters the DNA conformation and recruits transcription factors to the transcription initiation site to assemble stereospecific DNA–protein complexes (so-called enhanceosomes) that drive gene transcription (35, 36). The fact that HMGA1 is expressed at high levels in most cell types during embryonic development suggests its wide-spread roles in gene expression and function (37). We have previously demonstrated that loss of function in HMGA1 in both human and mice compromised pancreatic endocrine function and resulted in diabetes (38). We further showed that binding of PDX-1 to the INS gene promoter was reduced in nuclear extracts from HMGA1-deficient mice, in which PDX-1 protein expression was unaffected compared with wild-type animals (38). Furthermore, perturbation of HMGA1 in the insulin-secreting beta-cell line INS-1 transfected with an antisense expression vector specific for HMGA1 impaired glucose-stimulated insulin secretion, suggesting that HMGA1 may have a direct role in insulin production and pancreatic islet development through PDX-1 and/or other nuclear molecular partners. In this context, it is noteworthy that Ins gene transcription is reduced in Hmga1-deficient mice, in which pancreatic islets are up to 80% smaller compared to those of wild-type mice (38), and reports of others have suggested that HMGA1 binds to the A3/4 region of the INS gene promoter (14).
In this study, we investigated whether HMGA1 can directly activate the INS gene by synergizing with PDX-1 and MafA. Additionally, the effect of glucose on HMGA1 affinity to the INS gene promoter was also investigated.
Materials and Methods
Glutathione S-Transferase Pull-Down Assay and Co-Immunoprecipitation
Human 35S-labeled PDX-1 and MafA proteins were synthesized in vitro using the TNT-T7 quick-coupled transcription/translation system (Promega), as previously reported (36). Glutathione S-transferase (GST)-tagged human HMGA1 was obtained using the pcDNA1-GST/HMGA1 expression vector, a kind gift from D. Thanos (Biomedical Research Foundation, Academy of Athens, Athens, Greece). For direct coupling of antibody to protein A-Sepharose beads (GE Healthcare), an anti-HMGA1 polyclonal antibody was mixed with beads and bound for 1 h with rotation at room temperature, as described previously (36). Antibody-coupled protein A beads were washed twice in phosphate-buffered saline and used in immunoprecipitation studies. Briefly, aliquots of INS-1 cell nuclear extract, HMGA1, PDX-1, or MafA together were incubated for 3 h with rotation at 4°C with 10 μl of antibody-coupled protein A beads. Beads were recovered by gentle centrifugation and washed three times with 500 μl of NETN wash buffer [0.1% NP-40, 150 mM NaCl, 1 mM EDTA, 50 mM Tris-HCl (pH 8.0)]. Protein was removed from the beads by boiling in sample buffer for 5 min and analyzed by SDS-PAGE and immunoblotting (39). Antibodies used for these studies were as follows: anti-HMGA1 (39), anti-PDX-1 (40), and anti-MafA (Abcam).
Cell Cultures and Nuclear Extracts
Human embryonic kidney (HEK) 293 cells and human epithelial carcinoma (HeLa) cells were cultured in DMEM (Invitrogen) supplemented with 10% fetal bovine serum (FBS), 2 mM glutamine, penicillin (100 U/ml), and streptomycin (100 μg/ml) in a humidified 5% CO2 atmosphere at 37°C. INS-1 rat insulinoma cells were cultured in RPMI-1640 medium supplemented with 10% FBS, 2 mM glutamine, penicillin (100 U/ml), streptomycin (100 μg/ml), 50 μM beta-mercaptoethanol, and 100 mM HEPES buffer (Sigma). Nuclear extracts were prepared from cultured cells as described previously (41, 42). For each extract, an equal number of nuclei were homogenized, and the final protein concentration in the extracts was determined using the colorimetric assay of Bradford (Bio-Rad).
Plasmid Vectors, Small Interfering RNA, and Transient Transfection
The promoter regions of human and mouse INS genes were amplified from genomic DNA, using modified specific primers (Table 1). Sequence-verified promoters were then cloned into the pGL3-basic reporter vector (Promega) to generate the human (phINS-Luc) and mouse (pmINSII-Luc) luciferase (Luc) reporter plasmids. The expression plasmids were as follows: pcDNA3/HA-HMGA1 (a generous gift from G. Manfioletti, University of Trieste, Italy); pcDNA3-MafA (a generous gift from P. Gold, Gold Biotechnology, Inc., St. Louis, MO, USA); pSG5-ATG-hPDX-1 (a generous gift from C. V. Wright, Vanderbilt University Medical Center, Nashville, TN, USA). For gene silencing experiments, mouse and human anti-HMGA1 small interfering RNAs (siRNAs) (Santa Cruz Biotech) were used. In all knockdown experiments, cells were transfected with 100 pmol of HMGA1 siRNA in 6 well plates, and incubated without further treatment for 72 h before being used in subsequent analyses. Transient transfections of cultured cells were carried out using the Lipofectamine 2000 method (Invitrogen), and Luc activity was assayed 48–96 h later, using the dual-luciferase reporter assay system (Promega) (43).
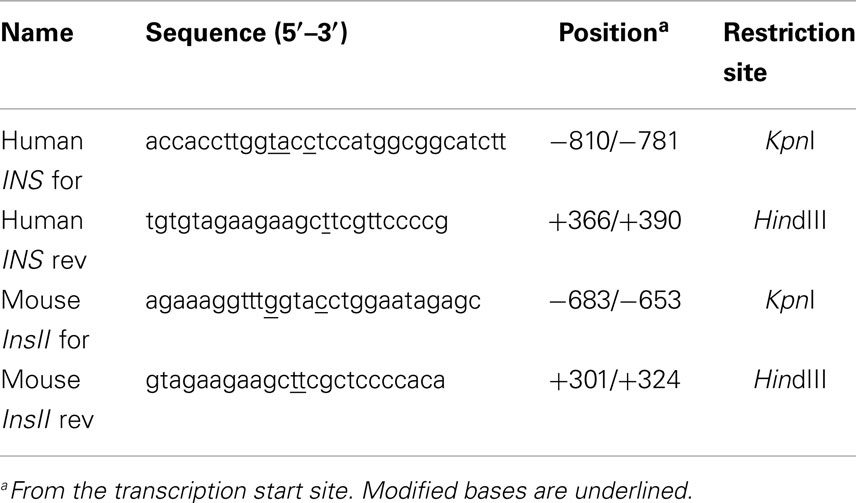
Table 1. Primers used for plasmid construction of human and mouse INS promoter-containing vectors.
Glucose Stimulation of Insulin Release
INS-1 cells (ATCC) were cultured in RPMI-1640 medium (GIBCO) supplemented with 10% FBS, 2 mM glutamine, penicillin (100 U/ml), streptomycin (100 μg/ml), 50 μM beta-mercaptoethanol, 100 mM HEPES, in a humidified 5% CO2 atmosphere at 37°C. For insulin secretion, INS-1 cells were transiently transfected at 60% confluence with HMGA1 siRNA or negative control siRNA as above. Seventy-two hours later the tissue culture medium was switched to fresh medium containing 5 mmol/l glucose and the cells were grown for additional 18 h. After this, the cells were washed with Hanks’ balanced salt solution (HBSS) containing 140 mM NaCl, 4.7 mM KCl, 1.2 mM KH2PO4, 1.16 mM MgSO4, 20 mM HEPES, 2.5 mM CaCl2, 25.5 mM NaHCO3, 0.2% BSA, and further incubated for 2 h in either low (3 mM) or high (15 mM) glucose condition. Then, the insulin concentration in the medium was measured by a commercial rat insulin ELISA kit (Mercodia Inc.).
Chromatin Immunoprecipitation
Chromatin immunoprecipitation (ChIP) was performed in INS-1 cells as described previously (38). As soon as the cells reached confluence, they were incubated in HBSS buffer with 3 or 15 mM glucose for 30 min at 37°C. Then, the cells were washed with 1× PBS and fresh RPMI was added. DNA–protein complexes were cross-linked by adding formaldehyde diluted to 1% final concentration for 10 min at room temperature, followed by blocking with glycine for 2 min. Cells were washed twice with cold PBS and lysed on ice using SDS lysis buffer (1% SDS, 10 mM EDTA, 50 mM Tris pH 8). Chromatin samples were sonicated on ice and the formaldehyde-fixed DNA–protein complexes were immunoprecipitated with anti-HMGA1 antibody (Santa Cruz Biotech) and the following sequence-specific primers for the rat InsI gene promoter were used for PCR amplification of immunoprecipitated DNA, using PCR ready-to-go beads (GE Healthcare). Rat InsI (NC_005100.3): for 5′-CTGGGAAATGAGGTGGAAAA-3′ (−328/−308 from the trasncription start site); rev 5′-AGGAGGGGTAGGTAGGCAGA-3′ (−108/−88 from the trasncription start site). PCR products were electrophoretically resolved on 1.5% agarose gel and stained with ethidium bromide staining solution. PCR products were confirmed by sequence analysis.
Real-Time PCR
For qRT-PCR, total cellular RNA was extracted from INS-1 cells using the RNAqueous-4PCR kit and subjected to DNase treatment (Ambion). RNA levels were normalized against 18S ribosomal RNA in each sample, and cDNAs were synthesized from 2 μg of total RNA using the RETROscript first strand synthesis kit (Ambion). Primers for rat InsI, HMGA1, PDX-1, MafA, and Rps9 were designed according to sequences from the GenBank database (Table 2). A real-time thermocycler (Eppendorf Mastercycler ep realplex ES) was used to perform quantitative PCR. SYBR Green fluorescence was measured, and relative quantification was made against the Rps9 cDNA used as an internal standard (44). All PCR reactions were done in triplicate.
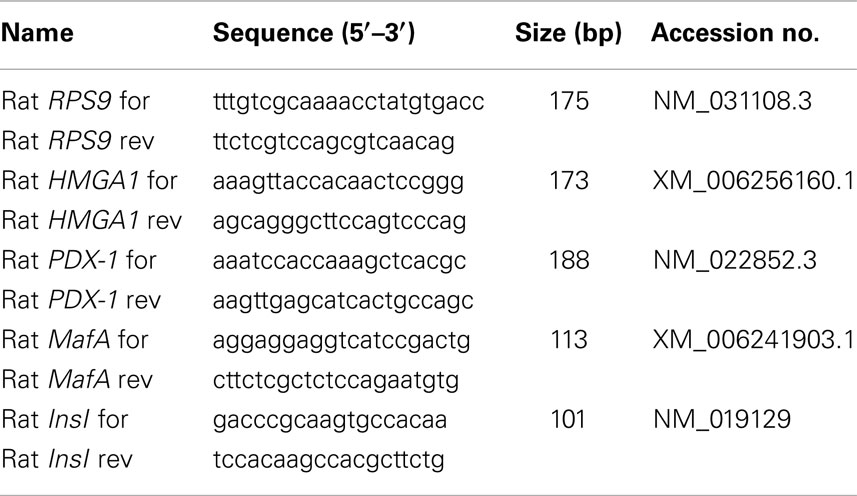
Table 2. Gene-specific primers for Real-Time PCR.
Statistical Analysis
All calculations were performed with SPSS 20.0 statistical software (SPSS Inc.). Mean values were compared with t-tests. A p-value <0.05 (two-tailed) was considered significant.
Results
HMGA1 Physically Interacts with PDX-1 and MafA in vitro and in vivo
Figure 1 shows a schematic representation of the promoter region of human, mouse, and rat INS genes. Binding sites for HMGA1, PDX-1, and MafA were identified within the promoter of the rat Ins gene by sequence analysis with MatINspector (version 8.1, Genomatix, http://www.genomatix.de/). Bioinformatics analysis of the human and mouse INS promoters predicted no HMGA1 binding sites, although the existence of non-recognizable DNA motifs cannot be completely excluded. Instead, binding sites for PDX-1 and MafA were predicted in these promoters as for the rat Ins gene. Consistent with this DNA promoter analysis, the existence of HMGA1 in a cocomplex with PDX-1 was demonstrated previously by us and others in supershift experiments in electrophoretic mobility shift assays using anti-HMGA1 and anti-PDX-1 antibodies, in the presence of the rat InsI E2A3/4 mini-enhancer DNA (14, 38). In both investigations, binding of PDX-1 to this promoter element was increased in the presence of HMGA1, suggesting a functional cooperation between HMGA1 and PDX-1 on the INS promoter.
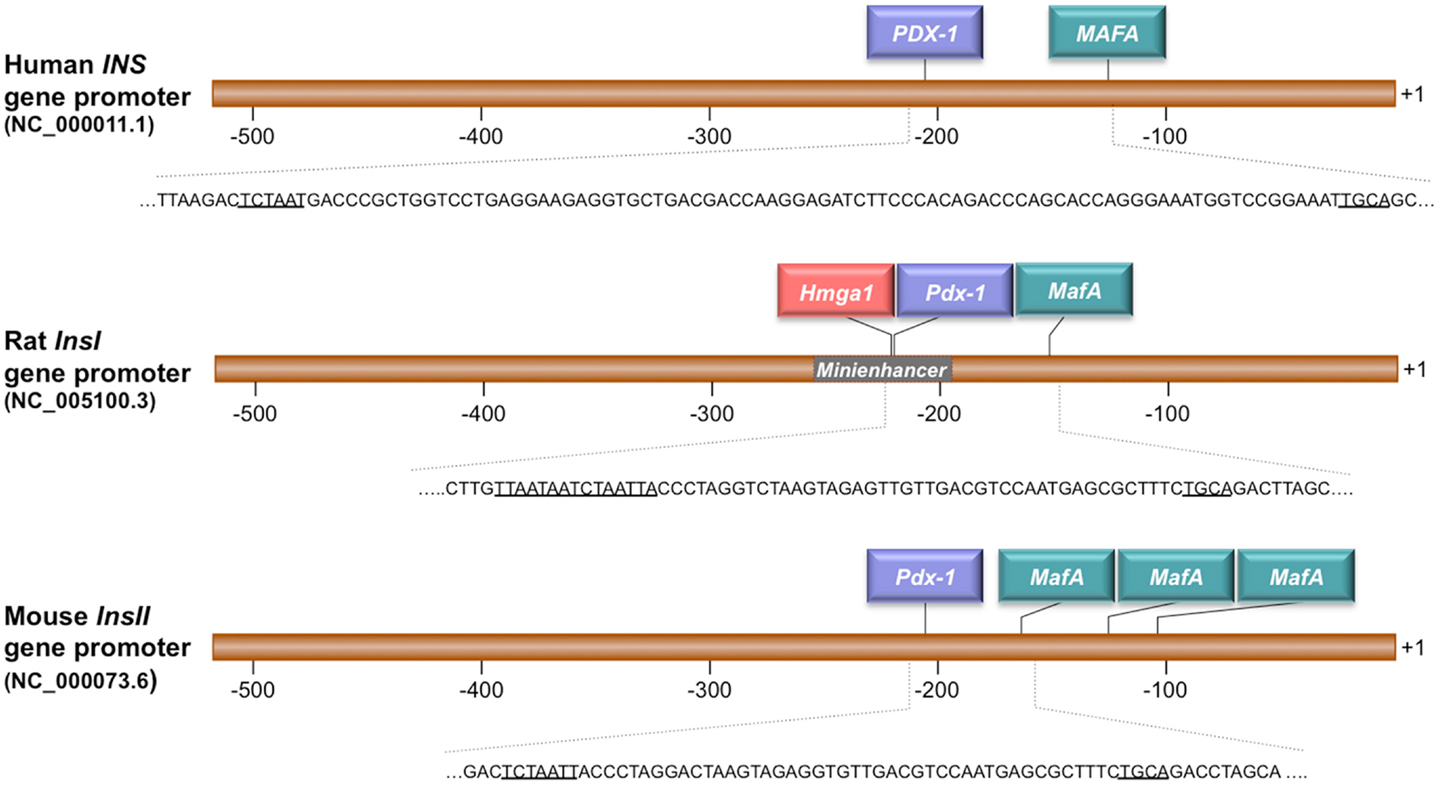
Figure 1. Schematic representation of the human (INS), rat (InsI), and mouse (InsII) promoter regions. HMGA1, PDX-1, and MafA binding sites are underlined in each gene sequence. The numbers indicate positions in base pairs relative to the transcriptional start site (+1). The rat insulin mini-enhancer element E2A3/4 is indicated.
In an attempt to determine whether and/or how HMGA1 regulates INS gene expression, we first analyzed whether HMGA1 interacts with PDX-1 and MafA without DNA. In vitro-translated 35S-labeled proteins PDX-1 and MafA were analyzed for their ability to be specifically retained by a GST-HMGA1 affinity resin. As shown in Figure 2A, PDX-1 and MafA were both retained by GST-HMGA1, but not GST alone, indicating that both these factors physically interact with HMGA1 in vitro, in the absence of DNA. The high specificity of the interaction of HMGA1 with PDX-1 and MafA was highlighted by the observation that no protein–protein contact was detected between HMGA1 and Neuro D (Figure 2A), a transactivator that is critical for INS gene expression in both human and mouse beta cells (45). To verify whether this protein–protein interaction in vitro could occur also in vivo, we performed co-immunoprecipitation studies using an anti-HMGA1 antibody immobilized on protein A-Sepaharose beads. As shown in Figure 2B, immunoprecipitation of HMGA1 from INS-1 nuclear extracts specifically pulled down PDX-1 and MafA, as shown by Western Blot analysis with well-characterized anti-PDX-1 or anti-MafA antibodies. These findings unequivocally demonstrate that HMGA1 physically interacts with PDX-1 and MafA both in vitro and in vivo, and suggest that physical interactions between these nuclear factors may constitute a fundamental prerequisite for functional cooperation between HMGA1, PDX-1, and MafA.
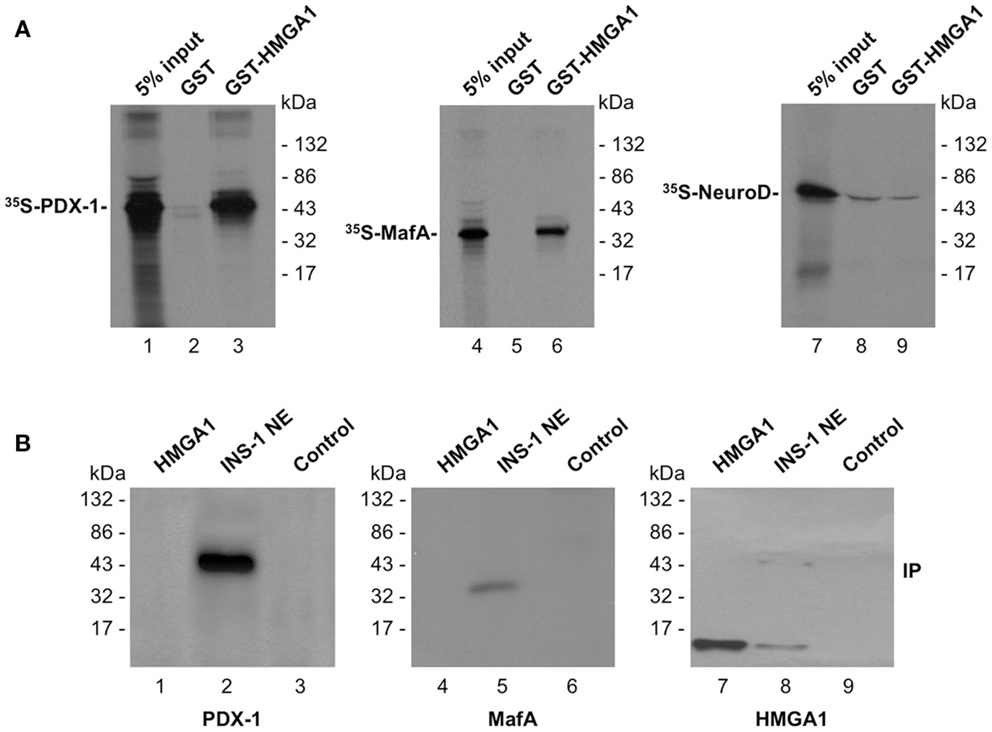
Figure 2. Physical association between HMGA1, PDX-1, and MafA. (A) SDS-PAGE of 35S-PDX-1, 35S-MafA, and 35S-NeuroD bound to GST or GST-HMGA1 resin (lanes 1–9). In lanes 1, 4, and 7 labeled protein was added directly onto the gel without binding to and elution from GST protein resin. (B) Immunoprecipitation (IP) of HMGA1, PDX-1, and MafA by using the anti-HMGA1 antibody followed by immunoblotting with the anti-PDX-1 antibody (lanes 1–3), the anti-MafA antibody (lanes 4–6), or the anti-HMGA1 antibody (lanes 7–9) after reprobing the same transfer. Lanes: 1, 4, and 7, 10 ng of pure HMGA1, 2, 5, and 8, INS-1 nuclear extract (NE; 500 μg). In lanes 1, 4, and 7, protein was directly applied to the gel without binding to and elution from protein A beads. To prove specificity, pure Sp1 (15 ng) was used for immunoprecipitation by the anti-HMGA1 antibody (lanes 3, 6, and 9, control). The faint band in lane 8 at 43 kDa represents non-specific signal. A representative of three separate assays is shown for each condition.
HMGA1 Potentiates the Activities of PDX-1 and MafA on INS Gene Transcription
We next determined whether HMGA1, PDX-1, and MafA functionally cooperate to activate INS transcription. HEK-293 cells were cotransfected transiently with Luc reporter plasmids containing human or mouse INS gene promoter and various effector vectors. HEK-293 cells were ideally suited for studying the effects of these proteins on transcription since they express only low levels of HMGA1 and no PDX-1 or MafA. As shown in Figure 3A, forced expression of single transcription factors HMGA1, PDX-1, and MafA in HEK-293 cells activated the human INS promoter to levels of two to threefold above the background with the recombinant reporter vector (phINS-Luc) alone. An ulterior increase in promoter activity was found in the presence of a combination of two of these transcription factors, while a further additive effect was observed in the promoter activity of phINS-Luc in cells overexpressing all three proteins (HMGA1, PDX-1, and MafA) together. Comparable or better results were obtained in HEK-293 cells when the mouse Ins promoter (pmInsII-Luc) was utilized for transactivation assays (Figure 3B). Differences in the activity of human and mouse INS gene promoters in HEK-293 cells may reflect the differences between humans and mice in terms of transcriptional activation of genes associated with pancreatic function (38). The greater increase in promoter activity of the pmInsII-Luc compared to the phINS-Luc in cells overexpressing MafA is likely attributable to the greater number of MafA binding sites within the mouse InsII promoter. Given that full sequence analysis with MatINspector predicted no HMGA1 binding sites in the human and mouse INS promoters, these results indicate that HMGA1 acts in concert with PDX-1 and MafA to regulate the expression of human and mouse INS genes through a mechanism that does not require its direct binding to DNA. Similar findings, indicating that HMGA1 acts to regulate the expression of estrogen responsive genes through a mechanism that does not require direct binding to DNA, have been reported elsewhere (46).
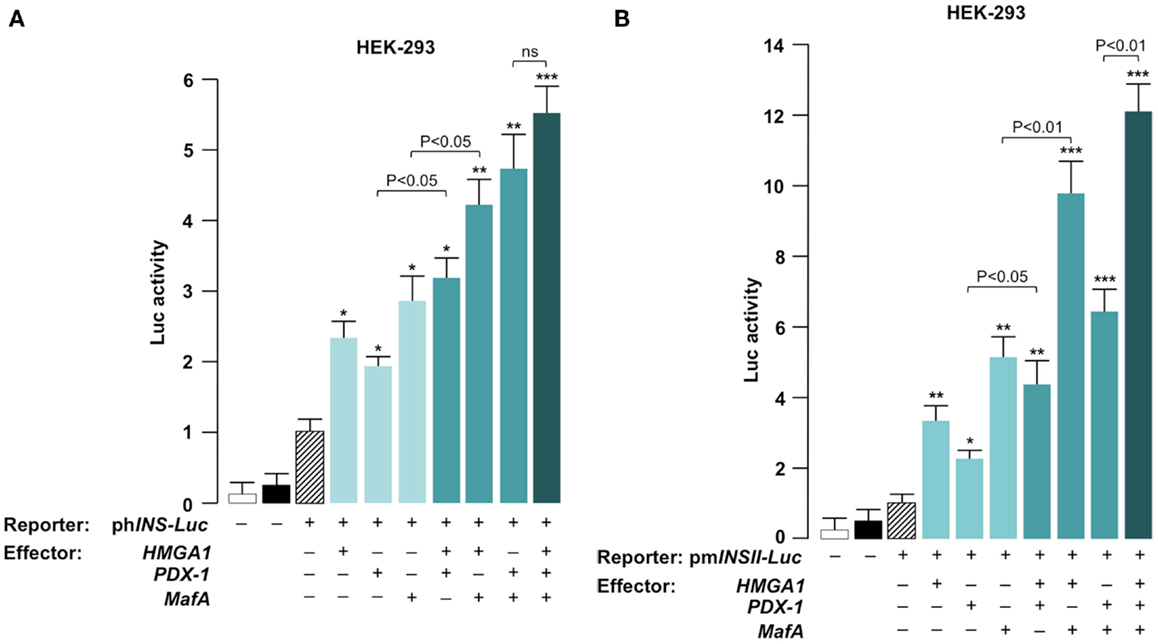
Figure 3. Functional significance of HMGA1, PDX-1, and MafA for INS gene transcription. HEK-293 cells were cotransfected with 1 μg of human (phINS-Luc) (A) or mouse (pmINSII-Luc) (B) Luc reporter plasmids, in the absence or presence of effector vectors for HMGA1, PDX-1, and MafA (0.5 μg each), either alone or in double or triple combinations. Data represent the means ± SE for three separate experiments; values are expressed as factors by which Luc activity increased as compared with the level of Luc activity obtained in transfections with reporter vector alone (slashed bar), which is assigned an arbitrary value of 1. Open bar, mock (no DNA); black bar, pGL3-basic (vector without an insert). *p < 0.05; **p < 0.01; ***p < 0.001 versus control (slashed bar).
We next tested whether HMGA1 is required for high transactivation activity of PDX-1 and MafA on INS gene in beta cells. For this purpose, we performed HMGA1 knockdown in INS-1 cells with siRNA approaches. As shown in Figure 4A, INS-luc activity failed to increase in INS-1 cells with overexpression of HMGA1, PDX-1, and MafA separately or in combination, probably because of the saturating levels of endogenous HMGA1, PDX-1, and MafA in this insulin-producing cell line. However, the promoter activity of pmInsII-Luc was significantly blunted in INS-1 cells with HMGA1 knockdown (Figure 4A), indicating that HMGA1 is required for full activation of INS transcription in vivo. A similar reduction in INS promoter activity was observed in HeLa cells that naturally express HMGA1 but not PDX-1 and MafA. In this case, whereas ectopic expression of PDX-1 and MafA, together, only weakly activated the expression of the reporter vector phINS-Luc, knockdown of endogenous HMGA1 significantly prevented this activation (Figure 4B). Consistently with the results reported above, and in agreement with our previous findings that pancreatic insulin mRNA transcripts were reduced in HMGA1-knockout mice (38), INS mRNA levels were significantly lower in INS-1 cells in the presence of siRNA directed against HMGA1, whereas mRNA abundances of PDX-1 and MafA were not altered by siRNA-induced down-regulation of HMGA1 (Figure 4C).
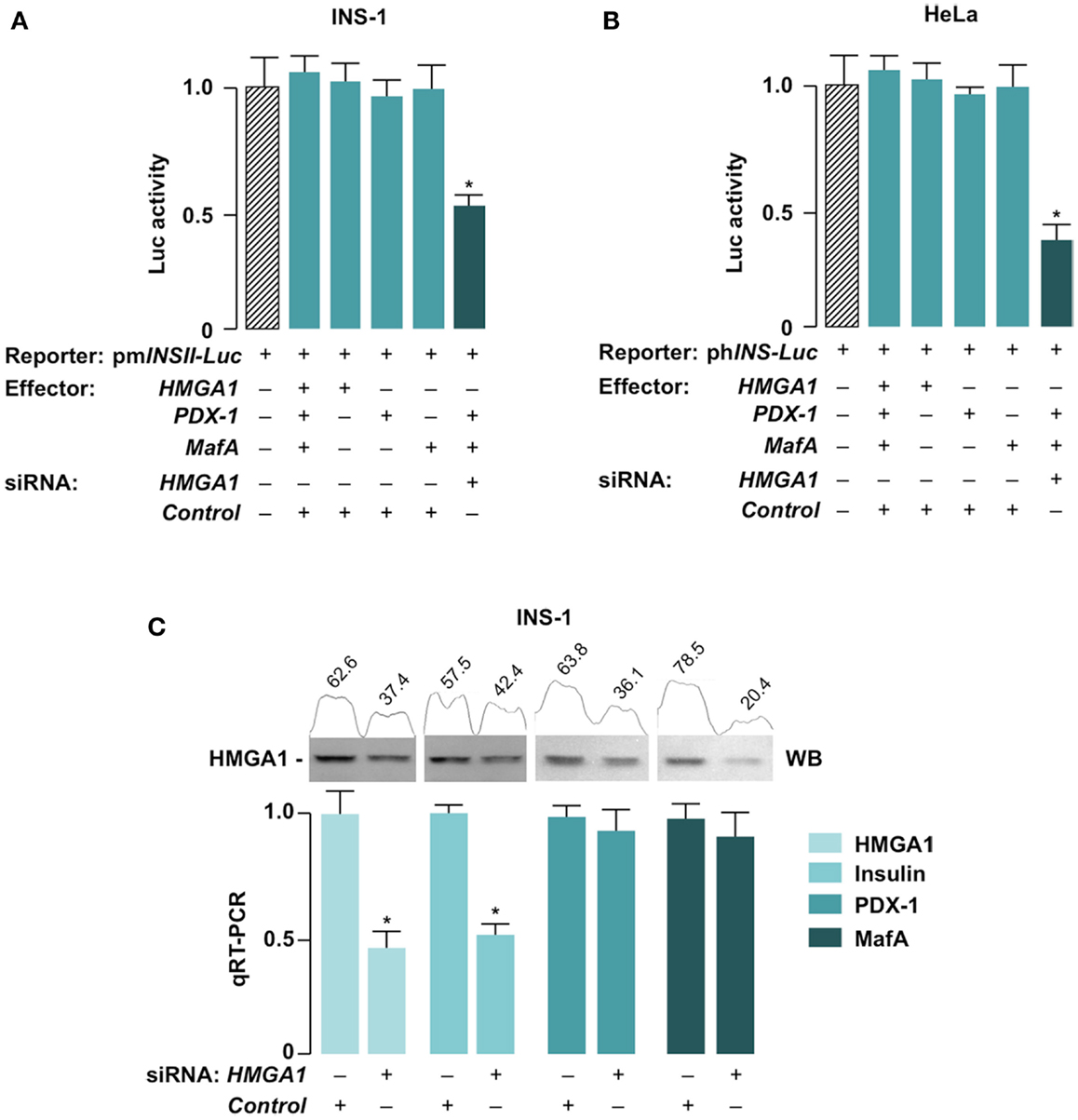
Figure 4. Suppression of INS promoter and mRNA by siRNA to HMGA1. INS-1 (A) and HeLa (B) cells were incubated without or with siRNA targeting HMGA1 (100 pmol) and transfected thereafter with 1 μg of mouse (pmINSII-Luc) or human (phINS-Luc) Luc reporter plasmid, respectively, either in the absence or presence of effector vectors (0.5 μg each) for HMGA1, PDX-1, and MafA. Values are expressed relative to the Luc activity obtained in transfections with the Luc reporter plasmid alone (slashed bar) that is assigned an arbitrary value of 1. Results are the means ± SE of triplicates from three independent transfections. *p < 0.05 versus control (slashed bar). (C) INS-1 cells were transfected with siRNA against HMGA1 or a non-targeting control siRNA, and mRNA levels for HMGA1, Insulin, PDX-1, and MafA were measured by quantitative RT-PCR (qRT-PCR). Results are the mean ± SEM of at least three separate transfections, each in triplicate. *p < 0.05 versus the relative control. Western blots (WB) of HMGA1, either before or after HMGA1 siRNA treatment, are shown in the autoradiograms and are from four independent experiments. Densitometric slot blot analysis, using the ImageJ software program, is shown. Numbers on the peaks are the size of the corresponding slot as a percentage of the total size of the two slots in each condition.
Taken together, these data confirm and extend previous observations and provide strong evidence that HMGA1 is required for full transactivation of the INS gene in beta cells.
Effects of HMGA1 on Glucose-Induced INS Gene Expression and Insulin Secretion
High glucose levels not only trigger a rapid secretion of insulin stored in the beta-cell but also stimulate the synthesis of new insulin molecules by inducing INS gene transcription. As reported previously, one of the mechanisms whereby glucose stimulates INS gene transcription in pancreatic beta cells is via activation and nuclear translocation of the transcription factor PDX-1 (25, 47), in addition to the up-regulation of MafA transcription (48). In the light of the data presented above, indicating the cooperative interactions between HMGA1, PDX-1, and MafA, we investigated whether HMGA1 plays a role in glucose-induced insulin transcription. To this end, we first carried out ChIP experiments in INS-1 cells at basal (3 mM) and stimulatory (15 mM) glucose concentrations. Results showed that binding of HMGA1 to the A3/A4 region of the INS gene promoter was low in INS-1 cells incubated in low glucose condition, while this binding was significantly increased under high glucose conditions (Figure 5A). These findings demonstrate that binding of HMGA1 to the INS promoter is stimulated by high glucose and indicate that HMGA1 could mediate the INS transactivation ability of high glucose. To further support this conclusion, HMGA1 siRNA-treated INS-1 cells were used to test the effects of glucose on insulin release into the culture medium. As shown in Figure 5B, a significant increase in insulin secretion was obtained when the medium glucose content was increased from 3 to 15 mM. No further increase in insulin secretion was observed by transfection with HMGA1 expression vector, most probably due to the endogenous HMGA1 present in INS-1 cells. However, pretreatment of INS-1 cells with HMGA1 siRNA resulted in a significant reduction in glucose-induced insulin secretion into the incubation medium (Figure 5B), confirming the central importance of HMGA1 in this scenario. Taken together, these results provide new insights into the molecular mechanisms regulating INS gene expression and emphasize the role of HMGA1 as an essential molecule necessary for proper beta-cell insulin secretion.
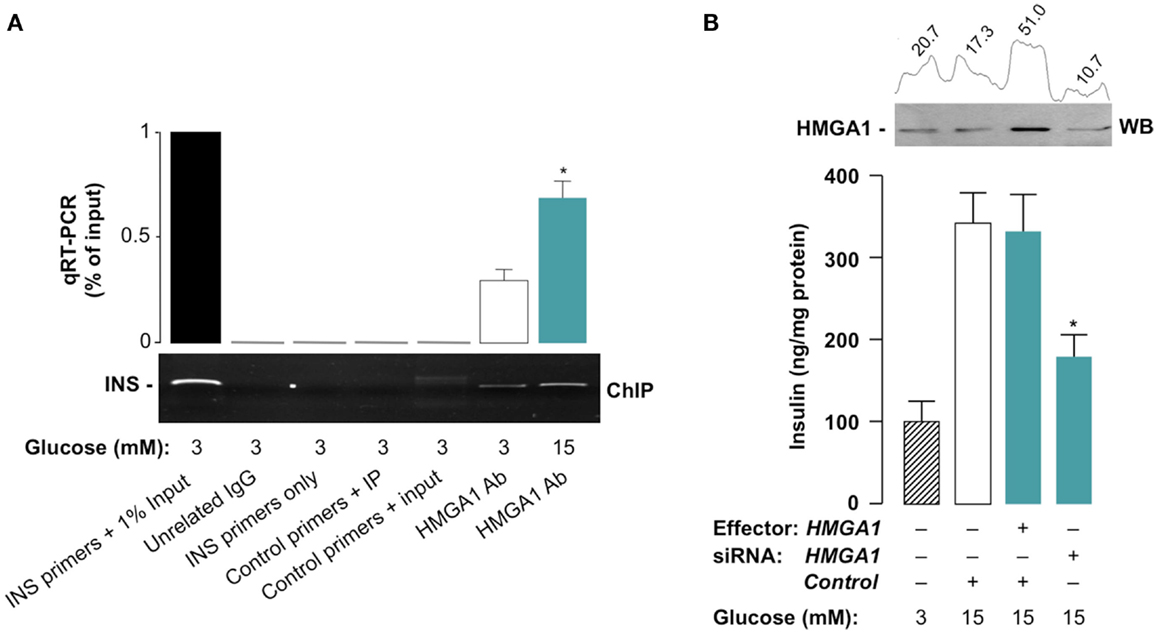
Figure 5. Functional significance of HMGA1 for glucose-induced INS gene expression and insulin secretion. (A) ChIP of the rat Ins promoter gene in INS-1 cells treated with either basal (3 mM) or stimulatory (15 mM) glucose concentrations, using an anti-HMGA1 specific antibody (Ab). A representative assay is shown, together with qRT-PCR of ChIP-ed samples. p < 0.05 versus control (white bar). (B) Insulin secretion from HMGA1 siRNA-treated INS-1 cells. Results are the mean ± SEM for three independent experiments conducted in triplicate. p < 0.05 versus control (white bar). A representative WB of HMGA1 is shown in the autoradiogram, together with densitometric slot blot analysis in each condition.
Discussion
Functional defects in the pancreatic beta-cell result in type 2 diabetes mellitus, a metabolic disorder in which, for various reasons, beta-cell insulin secretion may be profoundly affected at the multiple stages of the natural history of the disease. At the initial stage, peripheral insulin resistance increases demands of insulin secretion to stabilize blood sugar, which requires increased insulin transcription and translation (49). Yet sustained high glucose and insulin secretion eventually induce stress responses in beta cells to attenuate insulin production and secretion. Supporting the complexity and the importance of insulin production and secretion, genome-wide association studies have shown that most of the multiple susceptibility genes that facilitate diabetes development are involved in controlling beta-cell mass and function (1, 2). Thus, understanding factors that regulate insulin transcription and translation is critical to prevent or delay the development of type 2 diabetes.
By binding to AT-rich regions in the minor groove of DNA, HMGA1 contributes importantly to the transcriptional activation of many mammalian genes. By itself, HMGA1 has no intrinsic transcriptional activity; rather, it binds DNA to modulate its conformation so that other transcription factors can be recruited and assembled at the transcription initiation site to regulate transcription (34–37). These findings suggest that HMGA1 acts as a general nuclear factor to regulate gene expression. Indeed, promoters of genes that are expressed in a tissue-specific fashion are often regulated by the combination of tissue-specific and ubiquitous nuclear proteins, where the ubiquitous factor can support and facilitate the action of tissue-specific transcription factors (36, 50). As a part of an investigation into the genetic basis of insulin resistance syndromes and diabetes, we previously reported that loss of HMGA1 expression considerably decreased beta-cell insulin expression and severely reduced insulin secretion, causing a phenotype with hypoinsulinemia and glucose intolerance (38). Interestingly, pancreatic islets from Hmga1-knockout mice were nearly 80% smaller compared with wild-type islets, indicating that decreased insulin secretion in this mutant mouse model was dependent, at least in part, on reduced beta-cell mass, and suggesting that HMGA1 may play a direct role in pancreatic islet development and insulin production.
In this study, we provide direct evidence that HMGA1 is required for proper transcription of the INS gene in culture cells. We show that functional integrity of HMGA1 was necessary for full transactivation of the INS promoter by PDX-1 and MafA in cells that do not normally produce insulin. In contrast, repression of endogenous HMGA1 protein function adversely affected both PDX-1- and MafA-induced transactivation of the INS gene in insulin-producing cells. These findings demonstrate that HMGA1 plays significant roles in the transcriptional activities of these nuclear factors in the context of the INS gene. Underscoring the importance of this biochemical activity, our ChIP-qRT-PCR-based analysis showed that glucose upregulates HMGA1’s activity to the endogenous INS chromosomal locus. It is likely also that after a glucose challenge a deficit in HMGA1 may dampen INS gene transcriptional rate, thereby reducing insulin production.
Functional cooperation between HMGA1, PDX-1, and MafA in the transactivation of the INS promoter could be mediated by HMGA1-induced changes in DNA structure, thereby triggering a chain of molecular events that enhances the affinity of PDX-1 and MafA for their target DNA sequence and perhaps for other DNA-binding proteins that bind in the immediate vicinity, thus promoting the formation of an active transcription complex to which coactivators are recruited. While the biological relevance of these findings with regard to type 1 and type 2 diabetes remains to be clarified, it is tempting to hypothesize that a putative defect in HMGA1, by adversely affecting binding of PDX-1 and MafA to the INS gene, under either basal or glucose-stimulated conditions, may impair INS gene transcription, leading to decreased insulin synthesis and secretion. Consistent with the high sequence homology of the human and mouse INS genes (7), our data on in vitro INS gene transactivation demonstrate a striking similarity between the two species. This is particularly noteworthy in the light of several observations in the literature, indicating that substantial differences exist in pancreatic islet morphogenesis and function between humans and mice (38, 51, 52). Thus, caution is required when generalizing these results, as well as additional confirmation in future studies.
In conclusion, our data in the present work consistently suggest that HMGA1 may act as a glucose sensor that facilitates activation of the INS gene and insulin release. In addition, they further support the notion that HMGA1 is an architectural element tightly linked to glucose metabolism and metabolic disorders (38, 44, 53–60). Future studies on the interplays among HMGA1, PDX-1, and MafA might be useful in understanding the molecular basis of clinical phenotypes in certain clinical conditions where insulin secretion becomes compromised (i.e., diabetes mellitus and other categories of glucose intolerance). Understanding these mechanisms should augment our capacity to identify novel therapeutic targets for the prevention and treatment of these diseases.
Author Contributions
Performed and designed the experiments (Biagio Arcidiacono, Stefania Iiritano); drafting of the manuscript (Eusebio Chiefari, Daniela Patrizia Foti, Antonio Brunetti); critical revision and editing of the manuscript (Antonio Brunetti, Guoqiang Gu, Francesco S. Brunetti). All authors have approved the submitted version.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
The authors would like to thank the following individuals for providing materials used in this study: P. Gold, G. Manfioletti, D. Thanos, and C. V. Wright. This work was supported by Telethon-Italy (grant GGP04245) and MIUR (protocol 2004062059-002 Italy) to Antonio Brunetti. Dr. Biagio Arcidiacono is supported as a postdoctoral researcher by a grant from Regione Calabria (PO Calabria FSE 2007-2013).
References
1. Brunetti A, Chiefar E, Foti D. Recent advances in the molecular genetics of type 2 diabetes mellitus. World J Diabetes (2014) 5(2):128–40. doi:10.4239/wjd.v5.i2.128
2. Brunetti A, Chiefari E, Foti D. Perspectives on the contribution of genetics to the pathogenesis of type 2 diabetes mellitus. Recenti Prog Med (2011) 102(12):468–75. doi:10.1701/998.10858
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
3. Fu Z, Gilbert ER, Liu D. Regulation of insulin synthesis and secretion and pancreatic beta-cell dysfunction in diabetes. Curr Diabetes Rev (2013) 9(1):25–53. doi:10.2174/15733998130104
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
4. Vander Mierde D, Scheuner D, Quintens R, Patel R, Song B, Tsukamoto K, et al. Glucose activates a protein phosphatase-1-mediated signaling pathway to enhance overall translation in pancreatic beta-cells. Endocrinology (2007) 148(2):609–17. doi:10.1210/en.2006-1012
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
5. Sharma A, Stein R. Glucose-induced transcription of the insulin gene is mediated by factors required for beta-cell-type-specific expression. Mol Cell Biol (1994) 14(2):871–9.
6. German M, Ashcroft S, Docherty K, Edlund H, Edlund T, Goodison S, et al. The insulin gene promoter. A simplified nomenclature. Diabetes (1995) 44(8):1002–4. doi:10.2337/diab.44.8.1002
7. Hay CW, Docherty K. Comparative analysis of insulin gene promoters: implications for diabetes research. Diabetes (2006) 55(12):3201–13. doi:10.2337/db06-0788
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
8. Walker MD, Edlund T, Boulet AM, Rutter WJ. Cell-specific expression controlled by the 5’-flanking region of insulin and chymotrypsin genes. Nature (1983) 306(5943):557–61. doi:10.1038/306557a0
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
9. Melloul D, Ben-Neriah Y, Cerasi E. Glucose modulates the binding of an islet-specific factor to a conserved sequence within the rat I and the human insulin promoters. Proc Natl Acad Sci U S A (1993) 90(9):3865–9. doi:10.1073/pnas.90.9.3865
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
10. Melloul D, Marshak S, Cerasi E. Regulation of insulin gene transcription. Diabetologia (2002) 45(3):309–26. doi:10.1007/s00125-001-0728-y
11. German MS, Wang J. The insulin gene contains multiple transcriptional elements that respond to glucose. Mol Cell Biol (1994) 14(6):4067–75.
12. Petersen HV, Serup P, Leonard J, Michelsen BK, Madsen OD. Transcriptional regulation of the human insulin gene is dependent on the homeodomain protein STF1/IPF1 acting through the CT boxes. Proc Natl Acad Sci U S A (1994) 91(22):10465–9. doi:10.1073/pnas.91.22.10465
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
13. Sander M, German MS. The beta cell transcription factors and development of the pancreas. J Mol Med (1997) 75(5):327–40. doi:10.1007/s001090050118
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
14. Ohneda K, Mirmira RG, Wang J, Johnson JD, German MS. The homeodomain of PDX-1 mediates multiple protein-protein interactions in the formation of a transcriptional activation complex on the insulin promoter. Mol Cell Biol (2000) 20(3):900–11. doi:10.1128/MCB.20.3.900-911.2000
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
15. Boam DS, Docherty K. A tissue-specific nuclear factor binds to multiple sites in the human insulin-gene enhancer. Biochem J (1989) 264(1):233–9.
16. Leonard J, Peers B, Johnson T, Ferreri K, Lee S, Montminy MR. Characterization of somatostatin transactivating factor-1, a novel homeobox factor that stimulates somatostatin expression in pancreatic islet cells. Mol Endocrinol (1993) 7(10):1275–83. doi:10.1210/me.7.10.1275
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
17. Ohlsson H, Karlsson K, Edlund T. IPF1, a homeodomain-containing transactivator of the insulin gene. EMBO J (1993) 12(11):4251–9.
18. Miller CP, McGehee RE Jr, Habener JF. IDX-1: a new homeodomain transcription factor expressed in rat pancreatic islets and duodenum that transactivates the somatostatin gene. EMBO J (1994) 13(5):1145–56.
19. Jonsson J, Carlsson L, Edlund T, Edlund H. Insulin-promoter-factor 1 is required for pancreas development in mice. Nature (1994) 371(6498):606–9. doi:10.1038/371606a0
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
20. Marshak S, Totary H, Cerasi E, Melloul D. Purification of the beta-cell glucose-sensitive factor that transactivates the insulin gene differentially in normal and transformed islet cells. Proc Natl Acad Sci U S A (1996) 93(26):15057–62. doi:10.1073/pnas.93.26.15057
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
21. McKinnon CM, Docherty K. Pancreatic duodenal homeobox-1, PDX-1, a major regulator of beta cell identity and function. Diabetologia (2001) 44(10):1203–14. doi:10.1007/s001250100628
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
22. Kaneto H, Miyatsuka T, Kawamori D, Yamamoto K, Kato K, Shiraiwa T, et al. PDX-1 and MafA play a crucial role in pancreatic beta-cell differentiation and maintenance of mature beta-cell function. Endocr J (2008) 55(2):235–52. doi:10.1507/endocrj.K07E-041
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
23. Ahlgren U, Jonsson J, Jonsson L, Simu K, Edlund H. Beta-cell specific inactivation of the mouse Ipf1/Pdx1 gene results in loss of the beta-cell phenotype and maturity onset diabetes. Genes Dev (1998) 12(12):1763–8. doi:10.1101/gad.12.12.1763
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
24. Stoffers DA, Ferrer J, Clarke WL, Habener JF. Early-onset type-II diabetes mellitus (MODY4) linked to IPF1. Nat Genet (1997) 17(2):138–9. doi:10.1038/ng1097-138
25. Macfarlane WM, Frayling TM, Ellard S, Evans JC, Allen LI, Bulman MP, et al. Missense mutations in the insulin promoter factor-1 gene predispose to type 2 diabetes. J Clin Invest (1999) 104(9):R33–9. doi:10.1172/JCI7449
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
26. Olbrot M, Rud J, Moss LG, Sharma A. Identification of beta-cell-specific insulin gene transcription factor RIPE3b1 as mammalian MafA. Proc Natl Acad Sci U S A (2002) 99(10):6737–42. doi:10.1073/pnas.102168499
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
27. Kataoka K, Shioda S, Ando K, Sakagami K, Handa H, Yasuda K. Differentially expressed Maf family transcription factors, c-Maf and MafA, activate glucagon and insulin gene expression in pancreatic islet alpha- and beta-cells. J Mol Endocrinol (2004) 32(1):9–20. doi:10.1677/jme.0.0320009
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
28. Matsuoka TA, Zhao L, Artner I, Jarrett HW, Friedman D, Means A, et al. Members of the large Maf transcription family regulate insulin gene transcription in islet beta cells. Mol Cell Biol (2003) 23(17):6049–62. doi:10.1128/MCB.23.17.6049-6062.2003
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
29. Kaneto H, Matsuoka TA, Kawashima S, Yamamoto K, Kato K, Miyatsuka T, et al. Role of MafA in pancreatic beta-cells. Adv Drug Deliv Rev (2009) 61(7–8):489–96. doi:10.1016/j.addr.2008.12.015
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
30. Hang Y, Stein R. MafA and MafB activity in pancreatic β cells. Trends Endocrinol Metab (2011) 22(9):364–73. doi:10.1016/j.tem.2011.05.003
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
31. Zhao X, Mohan R, Özcan S, Tang X. MicroRNA-30d induces insulin transcription factor MafA and insulin production by targeting mitogen-activated protein 4 kinase 4 (MAP4K4) in pancreatic β-cells. J Biol Chem (2012) 287(37):31155–64. doi:10.1074/jbc.M112.362632
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
32. Guo S, Dai C, Guo M, Taylor B, Harmon JS, Sander M, et al. Inactivation of specific β cell transcription factors in type 2 diabetes. J Clin Invest (2013) 123(8):3305–16. doi:10.1172/JCI65390
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
33. Artner I, Hang Y, Mazur M, Yamamoto T, Guo M, Lindner J, et al. MafA and MafB regulate genes critical to beta-cells in a unique temporal manner. Diabetes (2010) 59(10):2530–9. doi:10.2337/db10-0190
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
34. Bustin M, Reeves R. High-mobility-group chromosomal proteins: architectural components that facilitate chromatin function. Prog Nucleic Acid Res Mol Biol (1996) 54:35–100. doi:10.1016/S0079-6603(08)60360-8
35. Munshi N, Agalioti T, Lomvardas S, Merika M, Chen G, Thanos D. Coordination of a transcriptional switch by HMGI(Y) acetylation. Science (2001) 293(5532):1133–6.
36. Foti D, Iuliano R, Chiefari E, Brunetti A. A nucleoprotein complex containing Sp1, C/EBP beta, and HMGI-Y controls human insulin receptor gene transcription. Mol Cell Biol (2003) 23(8):2720–32. doi:10.1128/MCB.23.8.2720-2732.2003
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
37. Cleynen I, Van de Ven WJ. The HMGA proteins: a myriad of functions. Int J Oncol (2008) 32(2):289–305. doi:10.3892/ijo.32.2.289
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
38. Foti D, Chiefari E, Fedele M, Iuliano R, Brunetti L, Paonessa F, et al. Lack of the architectural factor HMGA1 causes insulin resistance and diabetes in humans and mice. Nat Med (2005) 11(7):765–73. doi:10.1038/nm1254
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
39. Brunetti A, Manfioletti G, Chiefari E, Goldfine ID, Foti D. Transcriptional regulation of the insulin receptor by the high mobility group protein HMGI(Y). FASEB J (2001) 15(2):492–500. doi:10.1096/fj.00-0190com
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
40. Offield MF, Jetton TL, Labosky PA, Ray M, Stein RW, Magnuson MA, et al. PDX-1 is required for pancreatic outgrowth and differentiation of the rostral duodenum. Development (1996) 122(3):983–95.
41. Brunetti A, Foti D, Goldfine ID. Identification of unique nuclear regulatory proteins for the insulin receptor gene, which appear during myocyte and adipocyte differentiation. J Clin Invest (1993) 92(3):1288–95. doi:10.1172/JCI116702
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
42. Brunetti A, Brunetti L, Foti D, Accili D, Goldfine ID. Human diabetes associated with defects in regulatory proteins for the insulin receptor gene. J Clin Invest (1996) 97(1):258–62. doi:10.1172/JCI118400
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
43. Paonessa F, Foti D, Costa V, Chiefari E, Brunetti G, Leone F, et al. Activator protein-2 overexpression accounts for increased insulin receptor expression in human breast cancer. Cancer Res (2006) 66(10):5085–93. doi:10.1158/0008-5472.CAN-05-3678
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
44. Chiefari E, Paonessa F, Iiritano S, Le Pera I, Palmieri D, Brunetti G, et al. The cAMP-HMGA1-RBP4 system: a novel biochemical pathway for modulating glucose homeostasis. BMC Biol (2009) 7:24. doi:10.1186/1741-7007-7-24
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
45. Wang S, Yan J, Anderson DA, Xu Y, Kanal MC, Cao Z, et al. Neurog3 gene dosage regulates allocation of endocrine and exocrine cell fates in the developing mouse pancreas. Dev Biol (2010) 339(1):26–37. doi:10.1016/j.ydbio.2009.12.009
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
46. Massaad-Massade L, Navarro S, Krummrei U, Reeves R, Beaune P, Barouki R. HMGA1 enhances the transcriptional activity and binding of the estrogen receptor to its responsive element. Biochemistry (2002) 41(8):2760–8. doi:10.1021/bi011455j
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
47. Andrali SS, Sampley ML, Vanderford NL, Ozcan S. Glucose regulation of insulin gene expression in pancreatic beta-cells. Biochem J (2008) 415(1):1–10. doi:10.1042/BJ20081029
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
48. Vanderford NL. Regulation of β-cell-specific and glucose-dependent MafA expression. Islets (2011) 3(1):35–7. doi:10.4161/isl.3.1.14032
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
49. Unger RH. Reinventing type 2 diabetes: pathogenesis, treatment, and prevention. JAMA (2008) 299(10):1185–7. doi:10.1001/jama.299.10.1185
50. Milos PM, Zaret KS. A ubiquitous factor is required for C/EBP-related proteins to form stable transcription complexes on an albumin promoter segment in vitro. Genes Dev (1992) 6(6):991–1004. doi:10.1101/gad.6.6.991
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
51. Eizirik DL, Pipeleers DG, Ling Z, Welsh N, Hellerström C, Andersson A. Major species differences between humans and rodents in the susceptibility to pancreatic beta-cell injury. Proc Natl Acad Sci U S A (1994) 91(20):9253–6. doi:10.1073/pnas.91.20.9253
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
52. Kim SK, Hebrok M. Intercellular signals regulating pancreas development and function. Genes Dev (2001) 15(2):111–27. doi:10.1101/gad.859401
53. Semple RK. From bending DNA to diabetes: the curious case of HMGA1. J Biol (2009) 8(7):64. doi:10.1186/jbiol164
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
54. Chiefari E, Iiritano S, Paonessa F, Le Pera I, Arcidiacono B, Filocamo M, et al. Pseudogene-mediated posttranscriptional silencing of HMGA1 can result in insulin resistance and type 2 diabetes. Nat Commun (2010) 1:40. doi:10.1038/ncomms1040
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
55. Chiefari E, Tanyolaç S, Paonessa F, Pullinger CR, Capula C, Iiritano S, et al. Functional variants of the HMGA1 gene and type 2 diabetes mellitus. JAMA (2011) 305(9):903–12. doi:10.1001/jama.2011.207
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
56. Iiritano S, Chiefari E, Ventura V, Arcidiacono B, Possidente K, Nocera A, et al. The HMGA1-IGF-I/IGFBP system: a novel pathway for modulating glucose uptake. Mol Endocrinol (2012) 26(9):1578–89. doi:10.1210/me.2011-1379
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
57. Liu L, Ding H, Wang HR, Xu YJ, Cui GL, Wang PH, et al. Polymorphism of HMGA1 is associated with increased risk of type 2 diabetes among Chinese individuals. Diabetologia (2012) 55(6):1685–8. doi:10.1007/s00125-012-2518-0
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
58. Chiefari E, Nevolo MT, Arcidiacono B, Maurizio E, Nocera A, Iiritano S, et al. HMGA1 is a novel downstream nuclear target of the insulin receptor signaling pathway. Sci Rep (2012) 2:251. doi:10.1038/srep00251
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
59. Chiefari E, Tanyolaç S, Iiritano S, Sciacqua A, Capula C, Arcidiacono B, et al. A polymorphism of HMGA1 is associated with increased risk of metabolic syndrome and related components. Sci Rep (2013) 3:1491. doi:10.1038/srep01491
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
60. Pullinger CR, Goldfine ID, Tanyolaç S, Movsesyan I, Faynboym M, Durlach V, et al. Evidence that an HMGA1 gene variant associates with type 2 diabetes, body mass index, and high-density lipoprotein cholesterol in a Hispanic-American population. Metab Syndr Relat Disord (2014) 12(1):25–30. doi:10.1089/met.2013.0086
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Keywords: HMGA1, PDX-1, MafA, insulin gene, beta-cells, transcription, diabetes
Citation: Arcidiacono B, Iiritano S, Chiefari E, Brunetti FS, Gu G, Foti DP and Brunetti A (2015) Cooperation between HMGA1, PDX-1, and MafA is essential for glucose-induced insulin transcription in pancreatic beta cells. Front. Endocrinol. 5:237. doi: 10.3389/fendo.2014.00237
Received: 23 October 2014; Paper pending published: 05 December 2014;
Accepted: 18 December 2014; Published online: 13 January 2015.
Edited by:
Nicholas Michael Morton, The University of Edinburgh, UKReviewed by:
Niamh Xavier Cawley, National Institutes of Health, USAShareen Forbes, The University of Edinburgh, UK
Copyright: © 2015 Arcidiacono, Iiritano, Chiefari, Brunetti, Gu, Foti and Brunetti. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Antonio Brunetti, Department of Health Sciences, University “Magna Græcia” of Catanzaro, Viale Europa (Località Germaneto), Catanzaro 88100, Italy e-mail:YnJ1bmV0dGlAdW5pY3ouaXQ=