 Mathieu Méquinion
Mathieu Méquinion Christophe Chauveau
Christophe Chauveau Odile Viltart
Odile Viltart- 1INSERM UMR-S1172, Development and Plasticity of Postnatal Brain, Lille, France
- 2Pathophysiology of Inflammatory Bone Diseases, EA 4490, University of the Littoral Opal Coast, Boulogne sur Mer, France
- 3INSERM UMR-S1172, Early stages of Parkinson diseases, University Lille 1, Lille, France
Extensive studies were performed to decipher the mechanisms regulating feeding due to the worldwide obesity pandemy and its complications. The data obtained might be adapted to another disorder related to alteration of food intake, the restrictive anorexia nervosa. This multifactorial disease with a complex and unknown etiology is considered as an awful eating disorder since the chronic refusal to eat leads to severe, and sometimes, irreversible complications for the whole organism, until death. There is an urgent need to better understand the different aspects of the disease to develop novel approaches complementary to the usual psychological therapies. For this purpose, the use of pertinent animal models becomes a necessity. We present here the various rodent models described in the literature that might be used to dissect central and peripheral mechanisms involved in the adaptation to deficient energy supplies and/or the maintenance of physiological alterations on the long term. Data obtained from the spontaneous or engineered genetic models permit to better apprehend the implication of one signaling system (hormone, neuropeptide, neurotransmitter) in the development of several symptoms observed in anorexia nervosa. As example, mutations in the ghrelin, serotonin, dopamine pathways lead to alterations that mimic the phenotype, but compensatory mechanisms often occur rendering necessary the use of more selective gene strategies. Until now, environmental animal models based on one or several inducing factors like diet restriction, stress, or physical activity mimicked more extensively central and peripheral alterations decribed in anorexia nervosa. They bring significant data on feeding behavior, energy expenditure, and central circuit alterations. Animal models are described and criticized on the basis of the criteria of validity for anorexia nervosa.
Introduction
Eating disorders represent a large field of investigation in industrialized societies where food intake behaviors and quality of food become indisputable and incoherent. Research projects are currently focused on obesity, a dramatic consequence of overconsumption of fat and carbohydrates. However, populations of these societies also suffer of other dramatic but underinvestigated eating disorders. These eating disorders are presently defined according to the American Manual of Psychiatry DSM-5 (1) and divided into three main subtypes of eating disorders: anorexia nervosa (AN), bulimia nervosa (BN), and binge eating disorder (BED). The main characteristics of these three subtypes are summarized in Table 1. As usually mentioned by psychiatrists, the subtype determination at the time of diagnosis should be considered carefully since the majority of women with AN crossed over between the other subtypes [BED or BN; (2)].
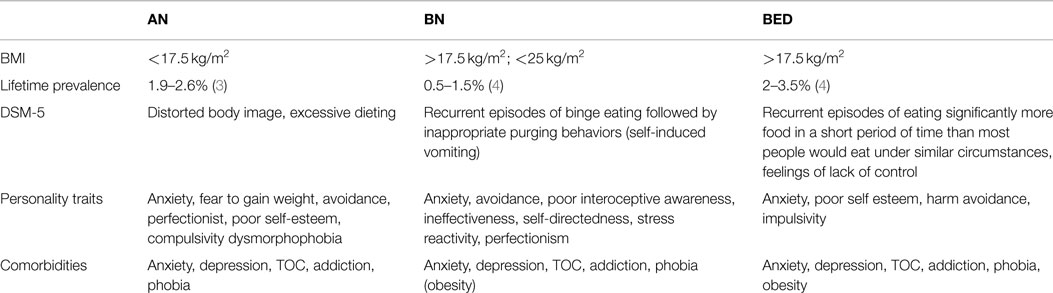
Table 1. Main characteristics of the mean eating disorders: anorexia nervosa (AN), bulimia nervosa (BN), binge eating disorder (BED).
Anorexia is said to be restrictive if during the past 3 months the person has not engaged in recurrent bulimic crises or purging behavior (i.e., self-induced vomiting or abuse of laxatives, diuretics, or enemas). One might consider this restrictive AN (AN-R) subtype as an awful eating disorder as the chronic refusal to eat leads to severe and sometimes irreversible complications for the whole organism, until death. AN-R is considered as a multifactorial disease with a complex etiology. The dramatic physiological and psychological consequences on health generated by the low food intake might lead to central and/or peripheral reprograming that permits the organism to endure in a first step, this reduced energy supply. A better understanding of the different facets of this disease becomes an urgent necessity to find novel therapeutic approaches complementary to the classic psychological therapies.
The objectives of this review are first to present briefly the main pathophysiological alterations observed in AN-R patients, then to introduce the different animal models that are currently used or could be used to better apprehend the physiological, metabolic, and neurobiological dysfunctions associated with AN-R, and finally to discuss the potential contribution of these models for understanding the pathology.
Physiological Alterations in Restrictive Anorexia Nervosa: From Neurobiology to Genetic Polymorphisms
The two faces of anorexia, physiological and psychological, which were first used to describe the disease, then were neglected by the psychiatrists and psychologists for years are now more and more widely accepted by numerous clinicians and practicians to interfere.
Physiopathological Alterations
The recent DSM-5 (2013) suggests diagnosing AN-R by three major criteria. The first criterion is a severe and persistent restriction of energy intake leading to significantly low body weight in context of what is minimally expected for age, sex, developmental trajectory, and physical health. The gradual loss of weight can reach more than 50% of the initial body weight. The second criterion is the intense fear of gaining weight or of becoming fat. The third criterion is a disturbance in the way AN patients experience their body weight or shape (dysmorphophobia), associated with persistent lack of recognition of the seriousness of the current low body weight. Another important criterion, amenorrhea or the absence of at least three menstrual cycles, was removed in the DSM-5. This criterion was deleted since it cannot be applied to patients of different age and gender. Moreover, some data describe individuals who exhibit all other symptoms and signs of AN, but still report some menstrual activity (5, 6).
Anorexia nervosa has one of the highest mortality rates of all psychiatric diseases (7, 8). In a 21-year follow-up study, Löwe et al. (9) showed that 16% of AN patients deceased due to consequences of the illness. Among them, about 50% died because of somatic complications and the other 50% committed suicide. In fact, the course of AN is extremely variable, with approximately 50–60% of individuals with AN that recover, 20–30% that partially recover, and 10–20% that remain chronically ill (9, 10). Among the different clinical studies conducted on AN patients, low ionic plasma concentrations, symptomatic hypoglycemia, and anemia are often associated with lymphopenia that can generate opportunistic infections or hepatic cytolysis in some cases. However, contradictory results were published concerning essential amino acid levels in plasma of AN patients and healthy controls (11–13). Modifications in essential metabolites might be related to the generalized amyotrophy often described in AN patients. Moreover, increase in the metabolic hormone levels (like ghrelin or cortisol) is often observed, and the endocrine function of adipose tissue is modified resulting in increased circulating levels of adiponectin and decreased concentrations of leptin (14, 15). Usually, AN-R patients also showed a nutritionally acquired hepatic resistance to GH with decreased production of IGF-1 and increased GH levels. Such increase is due to (i) a reduction of IGF-1 feedback on pituitary and hypothalamus GH secretion and (ii) high levels of ghrelin, a GH secretagogue (16). Additionally, osteoporosis, another main complication of AN affecting 20–50% of cases, has been observed and is often irreversible (17, 18). Behavioral changes like physical (or intellectual) hyperactivity observed in 31–80% of the cases might also be associated with AN (19). Finally, disordered fluid intake is currently associated with AN-R, 54% of patients drinking excessively, and 28% drinking restrictively (20). This leads to relatively frequent renal complications (21).
Neurobiological Alterations
Anorexia nervosa is often associated with psychiatric comorbidities like depression, anxiety, obsessive–compulsive or personality disorders, and drug abuse (22). It becomes more and more accepted that AN-R resembles an addictive behavior disorder linked to food deprivation, weight loss, or physical activity. In fact, neuroimaging studies have first pointed out morphological changes affecting gray and white matters (23). The systematic review of Phillipou et al. (24) summarizes a number of brain differences, which are reported in AN patients. The neural profile of AN corresponds to a predominant imbalance between the reward (meso-cortico-limbic system) and inhibition (prefrontal cortex) systems of the brain. Recent data of Kullmann et al. (25) suggest that AN patients showed a reduced connectivity in the brain areas involved in the cognitive control and an increased connectivity in regions important for salience processing. The demonstrated altered integrity of the inferior frontal cortex might contribute to the physical hyperactivity developed by AN patients due to its role in the general behavioral inhibition like motor response. Furthermore, dysfunction of the central monoaminergic systems has been related. The review of Bari and Robbins (26) describes the implication of these systems as pathological neural substrates of diseases. They underline that prefrontal noradrenergic neurotransmission is involved in the inhibition of an already initiated response whereas dopaminergic system appears to modulate motor readiness for both inhibition/activation and reward, respectively at the level of the dorsal and ventral striatum. Dopamine has been associated with the expression of an appetitive reward system (27), and probably works in mutual opponency with a system that signals the prediction of punishment instead of reward. Serotonin neuromodulation might contribute to the more affective part of the inhibition behavior and/or the wanting behavior. Serotonin has a critical role in the adaptation of animals to aversive events, in the inhibition of appetite, and in anxious and obsessive behaviors, as well as in depression. Furthermore, harm avoidance is a temperament trait highly observed in AN patients (28), that reflects inhibition and anxiety and involves both dopamine and serotonin (5-HT) neurotransmission (29). AN patients show decreased dopaminergic metabolite levels in the cerebro-spinal fluid as well as increased dopaminergic D2/D3 receptor density (30, 31). Similarly, levels of serotonin markers like blood serotonin contents, plasma tryptophan are lower in AN patients compared to non-eating disordered subjects (32). Brain imaging studies using serotonin-specific radioligands have consistently shown 5-HT1A receptor binding is increased in cortical and limbic structures in ill and recovered AN patients (33, 34), whereas 5-HT2A receptor binding remains normal in ill patients (33). 5-HT transporter activity is also increased in recovery AN patients (35). The basal hyperfunctioning of the serotonergic pathway described in these various studies may be related not only to alteration in the reward process of food intake but also to anxiety, behavioral inhibition, and body image distortions (29, 36, 37).
Finally, one might also consider the involvement of the endocannabinoid neurotransmission in the neurobiological changes observed in AN patients. As reviewed by Monteleone and Maj (38) in a positron emission tomography study, AN patients showed a dysregulated endocannabinoid tone with enhanced plasma anandamide (AEA) levels and an increased number of cannabinoid type 1 receptors (CB1) in the insula and inferior frontal and temporal cortex of underweight AN patients. These data underline or suggest that altered food intake in AN patients may be a consequence of aberrant reward processing combined with an exaggerated cognitive control [see review in Ref. (39)]. Consequently, the current psychopharmacologic strategy in the treatment of AN uses typical and atypical antipsychotics, tetrahydrocannabinol, anticonvulsants, antidepressants, which modulate the synaptic signals of these neuromediators, but which have been illusive for decades [see Ref. (40)]. Thus, dissecting the mechanisms of action of the different neuropeptides/neurotransmitters involved in the regulation of food intake, as well as in the motivational aspects of feeding, becomes a necessity to open new perspectives for an efficient therapy of this disease complementary to the psychological approaches.
Genetics
As clearly summarized by Scherag et al. (41), formal genetic studies suggested a substantial genetic influence in eating disorders and particularly in AN. The possible involvement of genetic components was strengthened by several twin and family studies concluding that AN presents genetic etiological components for 33–84% of the patients (42–45). Beside this, genome-wide linkage screens have been performed in order to identify unknown genes involved in AN. In the following paragraphs, presented data without reference to publication where cited in Scherag et al. (41).
Investigation on the genes directly involved in the regulation of feeding and energy expenditure was performed. The leptinergic–melanocortinergic system includes several key factors of the regulation of food intake and body weight. Surprisingly, despite the anorexigenic role of the leptin hormone, critically involved in the regulation of energy balance and adaptation of organism to semi-starvation, mutation analysis of the leptin gene and of the leptin receptor gene did not show any association with AN (46, 47). Agouti related peptide (AgRP), an orexigenic peptide, acts downstream of leptin through inhibition of central melanocortin receptors (MC receptors). Several studies concluded that the Ala67Thr AgRP polymorphism is significantly associated with AN. However, the involvement of this polymorphism in AN patients remains to be determined. This mutation would cause a lower inhibition the MC4R, a decrease in food intake, and would increase the risk of developing anorexia (48, 49). Brain-derived neutrophic factor (BDNF) is indirectly involved in the negative control of food intake. Low plasma levels of BDNF were determined in acute patients with AN. Several studies found that variants of BDNF and BDNF receptors (TrkB) are associated with AN. Moreover, AN patients often display high plasma levels of adiponectin, an adipocyte hormone known to play a role in the regulation of food intake and energy expenditure. Recently, a German study showed that several single nucleotide polymorphisms within the adiponectin (AdipoQ) locus were associated with adiponectin serum levels or eating behavior (50), but there is no convincing published study on the linkage between adiponectin gene polymorphism and AN.
Among the neurotransmitters suspected, genes involved in the serotonergic and dopaminergic systems have been pointed out. An overexpression of serotonin was suggested in AN. An association was shown with AN for serotonin transporter, serotonin receptors, and tryptophan hydroxylase 2 expressions. Moreover, positive but non-significant associations were also observed for dopamine D2 and D4 receptors and catechol-O-methyltransferase genes. The norepinephrine system was also investigated as low norepinephrine serum levels were always measured in recovery AN patients. Variants of the norepinephrine transporter gene that could lead to a lower norepinephrine reuptake have been associated with AN.
The endocannabinoid system is particularly involved in the regulation of appetite, food intake, and energy balance. Cannabinoids stimulate food intake through activation CB1. A study on 52 families showed that an allele of CB1 gene was more often transmitted in the restricted AN group. Moreover, in the Japanese population, Ando et al. (51) showed an association of a polymorphism of fatty acid amide hydrolase, which role is to inhibit the activity of the main CB1 ligand (N-arachidonoyl-ethanolamide), with AN.
As a first general comment on these data, it is to note that for several genes there is no evidence to suggest that any of the polymorphisms identified has a functional consequence on the biological activity or expression of the resulting protein. This may lead us to ponder these data when we try to establish linkages between polymorphisms and physiology or etiology. A second conclusion is that most of the polymorphisms that were shown to be associated with AN are related to the central nervous system, and particularly factors involved in the regulation of energy balance.
Inputs of Animal Models of AN
Development of appropriate animal model of AN appears to be something difficult given the complex etiology. Although psychological factors play a pivotal role in the development of AN, a better understanding of the biological basis of this eating disorder can help to improve current treatments additional to therapies currently used by psychologists and psychiatrists. However, due to obvious ethic reasons, all the aspects of AN remain difficult to assess rendering necessary to develop relevant animal models. Thus, in rodents, different genetic and environmental models have been developed with varying degrees of success.
Genetic Models
Two categories of genetic models are commonly used: models presenting spontaneous mutations and genetically engineered models that can be constitutive or conditional.
Spontaneous Mutations
Anx/anx mice. This model has been extensively studied and described (52). The mutant mice anx/anx emerged spontaneously in the Jackson Laboratories (Bar Harbor, USA) in 1976. These mice are characterized by an emaciated appearance, a reduction in food intake, and early death 3–5 weeks after the birth (53). Moreover, serotonergic hyperinnervation and decrease in the striatal dopamine concentration and its metabolites may contribute to alterations in the locomotor and reward systems (54, 55). The anx/anx phenotype is associated with an approximative 50% downregulation of the gene Ndufaf1 in the hypothalamus. It encodes a protein required for assembly of mitochondrial complex I (56). These mice exhibited several deviations in the hypothalamic neurotransmitter and neuropeptidergic systems involved in the regulation of food intake and energy metabolism, with a down-regulation of anorexigenic peptides POMC and CART and variations in the expression of the orexigenic NPY and AgRP peptides in the arcuate nucleus (54, 57–59). The reduction of the leptin peak, usually observed around postnatal day 8, could alter the arcuate neuronal development (60). These data are associated with mitochondrial dysfunction and neurodegeneration/neuroinflammation processes (52, 56, 61, 62). All these data suggest that this natural genetic model of anorexia represents an excellent model of anorexia–cachexia syndrome characterized by an inflammatory response that might be useful to dissect mechanisms that lead to physiological dysfunctions observed in AN. Here, the main limitations of this genetic anorexia model are: (i) the premature death of the mice before reaching puberty and (ii) effects observed on both male and female mice.
Lou/C rats. Lou/C rat is a rat substrain obtained from a Wistar rat selection at the Louvain University (Belgium). Lou/C rats are mainly characterized by a long life span until 35 months in male and 40 months in female (63, 64). These rats present the particularity to be resistant to diet-induced obesity and age-induced obesity since they exhibited a spontaneous food restriction, by eating fewer calories per day than Wistar rats in standard chow diet (63). The decreased food intake level is associated with a lower body weight (65, 66) itself associated with high energy expenditure and high sympathetic tone in the white and brown adipose tissues (67). Interestingly, Lou/C rats develop also an osteoporosis related to age associated with increased bone marrow adiposity (68). Lou/C rats mimic leptin, insulin, ghrelin, GH, and IGF-1 alterations observed in AN patients (66, 69–71). At central level, Lou/C rats present an upregulation of the hypothalamic AgRP, NPY, and orexin mRNA, and a down-regulation of leptin and ghrelin receptors in the arcuate and ventromedial hypothalamic nuclei (70).
Even if this rat strain presents various common alterations observed in AN patients, it is a more suitable model of healthy aging (64, 72).
Genetically Engineered Mice
Beside spontaneous mutation models, various genetically engineered models have been developed. In humans, genomic association studies have shown that various gene polymorphisms seem particularly linked to AN (see “Genetics”). In view of these data, we have summarized results from studies on animal models with modified genes encoding molecules involved in neuropeptidergic circuits and monoaminergic systems. For a more complete overview, animal models based on genetic alterations of peripheral factors are also presented (Table 2). It is noteworthy that the interest of all these models is discussed independently of the purpose of the original studies and thus of their intrinsic interest.
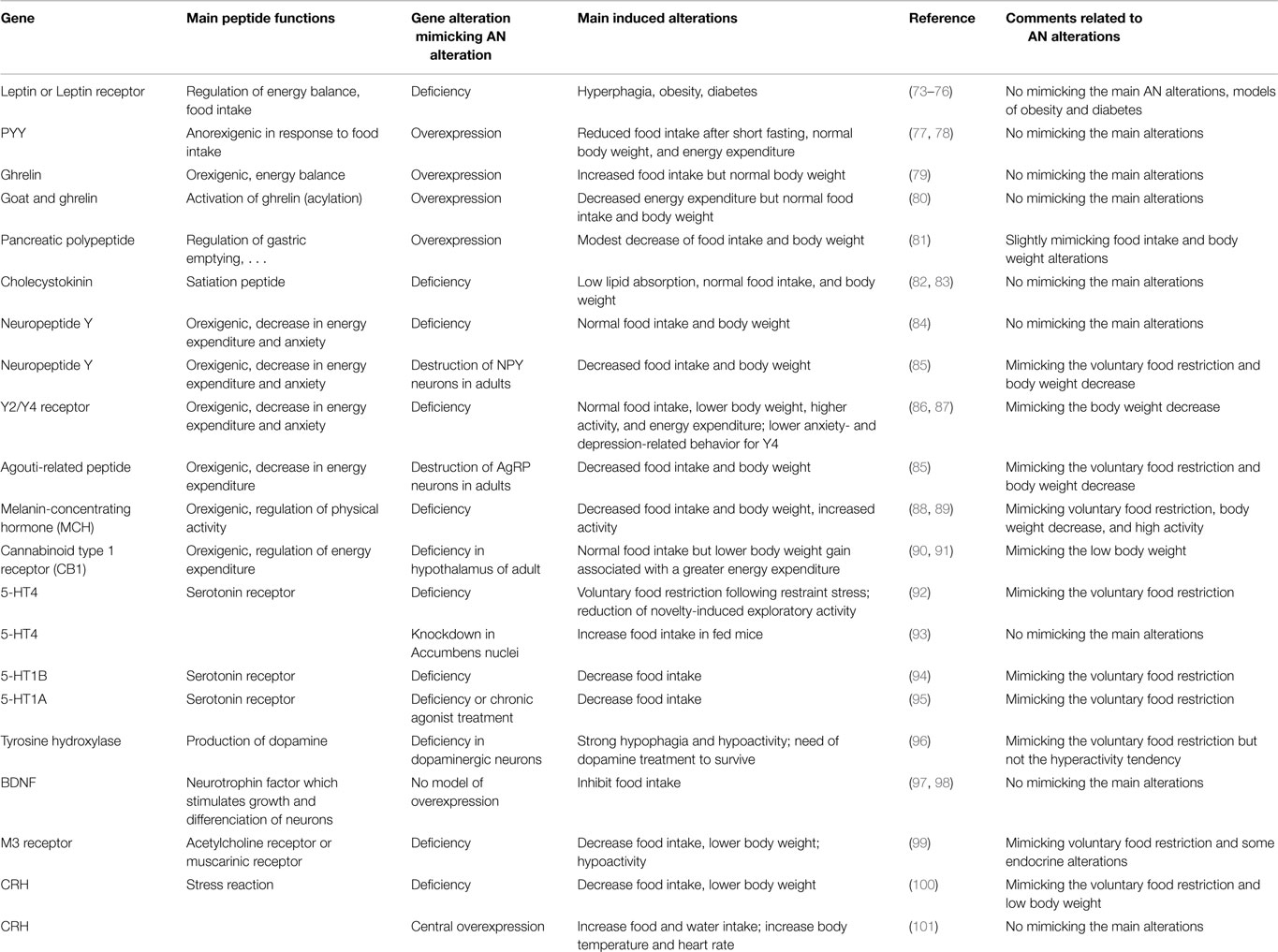
Table 2. Presentation of the most pertinent model to decipher subtle peripheral and central mechanisms that might be involved in anorexia nervosa.
Peripheral factors: hormones involved in the regulation of the energy metabolism. As a consequence of leptin anorexigenic function, leptin-deficient (ob/ob) or leptin receptor-deficient (db/db) mice display a phenotype of hyperphagia and obesity [see reviews in Ref. (76, 102)]. Even if the plasma levels of leptin are low in AN, these genetic models did not mimic pathology alterations. Mice overexpressing the ghrelin peptide in their stomach show higher plasma levels of bioactive (acyl) and total (acyl and non-acyl) ghrelin. They display a slight increase in food intake but not in body weight (79). To increase acyl-ghrelin plasma levels, it might be necessary to also increase the expression of GOAT (ghrelin O-acyltransferase), enzyme involved in the ghrelin acylation. Contradictory results were obtained for GOAT expression levels in stomach after 12–36 h of fasting, whereas chronic and severe food restrictions (21 days, 70% restriction) increase GOAT expression in rat (103). Mice overexpressing GOAT display higher concentrations of acyl-ghrelin without any changes in body weight or food intake (80). Thus, engineering genetic alterations of the ghrelin system in mice did not succeed in mimicking AN alterations despite the essential role of this hormone in the maintenance of glucose homeostasis on food restriction condition (104–106). The anorexigenic peptide PYY is physiologically released in response to food intake and its plasma levels increased in patients with AN. Mice overexpressing PYY display normal weight gain and food intake (77). These observations could suggest that this model should be excluded from the list of AN models, but a recent study (78) showed that when PYY overexpression begins in adult mice, it induces a reduced food intake after 24-h fasting. However, these mice display no significant difference of body weight or energy expenditure when compared to wild type mice. The pancreatic polypeptide (PP) produced in pancreas after food intake inhibits gastric emptying, and contributes to the important satiety effect of cholecystokinin (CCK). Baseline PP concentrations were similar between AN patients and healthy controls (107) or higher in AN patients (108), but these concentrations increased much more in AN patients than in controls after a meal test (107, 108). Transgenic mice over-expressing PP display a slightly lowered body weight associated with a modest reduction of food intake (81). CCK is a gut hormone stimulated by fatty meals and inducing satiety. It is also involved in the control of gastrointestinal motility and in anxiety behaviors. The response of CCK to a meal test was four-fold lower in AN patients than in healthy control group (107). Interestingly, CCK deficient mice display a normal food intake and a normal body weight when fed a basal diet (82, 83). Thus, once again, this model does not mimic the main alterations observed in patients with AN.
Neuropeptidergic systems. Modifications in the expression of neuropeptides permit to generate central alterations that might explain mechanisms giving rise to some of the symptoms described in AN patients.
In the arcuate hypothalamic nucleus, the two populations of orexigenic and anorexigenic neurons and their receptors, respectively the AgRP/NPY and αMSH/CART (α-melanocyte stimulating hormone/cocaine amphetamine related peptides) neurons, have been the focus of numerous studies in an attempt to better understand the finely tuned regulation of food intake. During fasting, NPY and AgRP gene expressions are up-regulated, and αMSH and CART gene expressions are down-regulated in hypothalamus. Moreover, various experiments suggest that NPY/AgRP inhibits directly the activity of αMSH neurons through a corelease of GABA, as well as an action on MC4R-bearing cells. Inactivation of genes encoding NPY, AgRP, or both has little effect on energy balance (109). Mice KO for NPY present significant changes neither in their body weight nor in their food intake, but become hyperphagic following food deprivation (110, 111). Surprsingly, mice KO for both Y2 and Y4 receptors exhibited a reduction in adiposity and an increase in lean mass, but without significant changes in food intake. Energy expenditure and physical activity were significantly increased in Y4-KO and particularly in Y2-KO/Y4-KO (87). Such models might be valuable to study the involvment of NPY and its receptors in the modulation of body composition and energy metabolism that are dramatically disturbed in AN. Contrary to the Y2 and Y4 receptors, the Y1-KO and Y5-KO mice develop the late-onset obesity with an increase in food intake and adiposity (112–114). This implies compensatory mechanism in feeding behavior in these KO mice and underlines the complexity of the NPY-food intake regulation system. Selective acute deletion of AgRP neurons in the adult mouse inhibits feeding and can lead to starvation not observed when the ablation is performed in neonatal mice before AgRP neurons are mature (85). Wu et al. (115) show in Ay/a mice no discernable effect on the anorexia phenotype caused by AgRP neuron ablation, suggesting that excessive activation of the melanocortin signaling is not responsible for starvation. Compensatory mechanisms may occur and hide the potential role of certain peptides (116, 117). Unfortunately, in these models, physiological data are rarely presented, their use are of interest to better understand the dialog existing between these populations of neurons by deciphering the involvement of their receptors in specific conditions. These approaches can highlight the main homeostatic pathway disturbed in AN.
The lateral hypothalamus contains MCH (melanin concentrating hormone) orexigenic neurons, described to be essential in the control of food intake and physical activity (88). In his review, Macneil (118) points out the various mouse models where disruption of MCH signaling results in altered energy homeostasis. Indeed, targeted inactivation of the MCH gene in mice induces reduced body weight and leanness due to hypophagia associated with an increased metabolic rate, despite reduced amount of both leptin and arcuate nucleus proopiomelanocortin mRNA (88). KO MCH mice also display an increased running-wheel activity during dark period (89). In the Promch/ataxin-3 mouse, 60–70% of MCH-expressing neurons degenerate in the first few weeks of life. Thus, at the age of 7-week, mice developed reduced body weight due to hypophagia and increased energy expenditure, body length, fat mass, lean mass, and leptin levels (119). Similarly, the Mchr1−/− mice were less susceptible to diet-induced obesity, and the leanness was a consequence of hyperactivity and altered metabolism. The manipulation of the MCH system remains one of the most interesting to reproduce many of the symptoms described in AN. The progressive degeneration of an orexigenic neuronal population induces a voluntary food restriction that impacts the overall physiology of the animal.
The lateral hypothalamic area also comprises another population of orexigenic neurons: the orexin/hypocretin (Hcrt) neurons which are implicated in various functions altered in AN. Indeed, in a neuron-ablated strategy, the orexin/ataxin-3 transgenic mice severely reduced the formation of food anticipatory activity (FAA) under food restriction conditions (120). Furthermore, in a recent study, Ramanathan and Siegel (121) report gender differences in Hcrt KO mice. Hcrt KO females had increased body weight associated with increases in various components of the body composition, despite a decreased food and water intake not observed so drastically in the males. This promising model remains complex to interpret in the case of AN, because of the multiple roles in which orexin is involved.
Among the other neuropeptidergic systems involved in AN, the cannabinoid system must be pointed out. Mice invalidated for CB1 in hypothalamus showed a significant weight loss associated with greater energy expenditure despite a normocaloric food intake in standard diet (90). The mechanisms involved in such adaptations need to be more investigated since pharmacological manipulation of the endocannabinoid system is currently discussed as potential strategy for the treatment of anxiety disorders, depression, and AN (122, 123). Similarly, the opioïd system is known to play a role in the control of homeostatic and hedonic pathways. Thus, mice knockout for the opioid receptors like the μ-receptor display no significant difference in body weight, food intake, locomotor activity, or dark respiratory quotient when fed with regular chow diet compared to wild type mice, but they are resistant to diet-induced obesity and display more important weight loss during food deprivation (124–126). They also show a decrease in food motivation as demonstrated in an operant paradigm for chow diet or sucrose pellets, and a reduction of FAA in a daily scheduled food access compared to wild type mice (127, 128). These models might be of interest more specially to dissect the complex mechanisms that regulate the non-homeostatic aspects of the feeding in AN patients.
Neurotransmitters: dopamine and serotonin. As mentioned above, in AN patients, neuroimaging studies as well as dosages in the cerebro-spinal fluid report alterations in the serotonergic and dopaminergic systems.
Concerning the serotonergic system, pharmacological treatments that increase serotonin disponibility lower consumption of food in humans and rodents (129, 130). The model of mice genetically modified for 5-HT4 receptors has been extensively studied as a model of anorexia (131). Briefly, these mice were characterized by a voluntary food restriction, only following restrained stress, and by an attenuation of novelty-induced exploratory activity (92). Conversely, the knockdown of 5-HT4 receptor in nucleus accumbens increases food intake only in fed mice (93). Likewise, mice lacking 5-HT1B receptor food restricted (20%, 3 days) eat less than the wild type mice when standard food ration is given. They also show an increased locomotion (94). Mice lacking 5-HT1A receptor or wild type mice chronically treated subcutaneously with a 5-HT1A receptor agonist display a decrease of their food intake (95). The interpretation of data obtained from manipulation of the serotonergic system is rendered difficult due to the large number of receptors and the various effects they have depending of their location at the synaptic level and in the brain. Thus, to better elucidate the role of serotonin in the feeding behavior, it is preferable to use conditioned deletion or the cre-lox technology to avoid large effects that might be more the result of compensatory mechanisms than a true action of the neurotransmitter.
Concerning the dopaminergic system, Szczypka et al. (96) used initially a gene-targeting strategy to inactivate specifically the tyrosine hydroxylase (TH) gene in dopaminergic neurons, sparing the production of dopamine as a precursor for adrenaline and noradrenaline. These mice, called “dopamine deficient mice,” became hypophagic and died from starvation at 34 days because they showed locomotor deficiencies. Routine treatment with l-DOPA restored a food intake similar to wild type mice. Using viral strategy (96, 132–134), the involvement of dorsal striatum and accumbens nucleus has been demonstrated in locomotion and motivation, respectively, underlining the importance of dopamine to execute behaviors necessary to seek and ingest properly food. In AN, dopamine deficiencies might contribute to alterations in the accomplishment of these behaviors. Moreover, motivation aspects of feeding are also under the influence of medial prefrontal cortex and amygdala as recently demonstrated and involved D1 and D2 receptors (135, 136). These recent data emphasize the complexity of the regulation of feeding motivation, complete brain imaging data obtained in humans in these brain regions, and point out the need of more targeted pharmacological treatments (137).
Other genes. Other genes are also studied in the case of AN and are potential targets involved in the maintenance and/or evolution of the disease, like BDNF (brain-derived neurotrophic factor), CRH (corticotropin-releasing hormone), the glutamate receptors, the muscarinic type receptors even if they are also involved in a variety of functions (138, 139) rendering difficult to dissect precisely their actual role in the regulation of hunger/feeding. BDNF, a neurotrophic factor, is also a central regulator of energy balance, since BDNF suppresses food intake by acting on hypothalamic neurons (97, 98). Unfortunately, to our knowledge, no studies on hypothalamic overexpression of BDNF and feeding behavior are described in the literature. Investigating the CRH system in the case of AN is rendered difficult, even if the link is obvious, since AN patients often present stress-related disorders like anxiety and depression. Among the genetically modified models, the CRH-KO mice model described by Jacobson (100), the mice fed with chow diet present a decreased food intake associated with a lowered body weight loss than mice fed with restricted protein diet. In the opposite, mice who overexpress central CRH display changes in autonomic variables, like increased body temperature and heart rate, as well as increased food and water consumption, when compared with wild type mice (101). Thus, as detailed along the review, the HPA axis plays a key role in the regulation of the homeostatic and non-homeostatic aspects of the AN altered feeding, but the precise role remains to be determined in this case due to numerous brain areas involved. Muscarinic receptors (M1 to M5) are involved in acetylcholine signaling and in various functions at peripheral and central level (140). The M3 receptor has been associated with alterations that are observed in AN. Indeed, M3 KO mice are hypoactive and display a voluntary food restriction associated with lower body weight compared to wild type mice. These transgenic mice also present lower fat deposits associated with reduction of plasma leptin and insulin concentrations. Moreover, M3 KO mice present an up-regulation of AgRP and down-regulation of POMC and MCH in hypothalamus compared to control (99). Due to the large distribution of these receptors in the CNS, targeted strategies of gene deletion must be chosen to assess precisely their involvement in the regulation of food intake (141).
Conclusion. The main results obtained on mouse models in which one gene expression was modified to follow the alteration of the corresponding protein levels described in AN patients was summarized on the Table 2. These lead us to mention that most of these models are more relevant for obesity or display no specific phenotype related to AN. Interestingly, this table points out that most of the alterations related to these genes induce phenotypes very different of the pathologic ones. This could be linked to the fact that alterations of factors in AN patients appear often to be opposite to the physiological and behavioral alterations obtained in these genetic models. As examples, the plasma levels of leptin and ghrelin, respectively, low and high in AN patients, might normally lead to an increase in food intake, which is not the case in the disease, reflecting a physiological adaptation that is not well-perceived at the central and/or peripheral levels.
Thus, even if these genetic models gave comprehensive informations about some mechanisms related to the processes regulating homeostatic and non-homeostatic regulation of food intake, these models are most often used on short term protocols and do not allow to follow the physiological and neurobiological evolutions of the phenotype while restrictive AN is usually a chronic disease. Furthermore, they focus on certain aspects of the disease such as hypophagia, hyperactivity, or motivational disturbances without taking into account a general view of the whole body functioning. To circumvent these drawbacks, the use of “environmental” model allows us to reconsider some of these aspects.
Environmental Models
Various environmental animal models have been proposed to mimic various symptoms of AN. These models are usually based on qualitative or quantitative modifications in the pattern of distribution of the meal, including period of quantitative food restriction or limited time of food access as well as exposure to chronic or acute stress.
Animal Model Based on One Inducing Factor
Dietary restriction models. Various studies have focused on adaptations induced by dietary restriction to determine contribution of energy imbalance or nutriment deficiency in changes observed in AN patients. Some studies focused on life span, cancer prevalence, or metabolic syndrome have brought data useful for understanding AN-related alterations [see review in Ref. (142)]. Altogether, the different feeding paradigms lead to various but complementary results.
Food restriction (FR). Most of the studies using chronic FR used mild restriction protocols. Restricted animals were fed usually 30–40% less than ad libitum control ones. However, it must be noted that in animal facilities, rodents are usually overfeed of about 30% compared to their physiological needs resulting in a significant weight gain over the time and leading to the use of overweight animals as reference (143). In FR protocols, body weight changes are age and gender dependent. Breeding weaned mice onto 30% FR lead to gain weight, even less rapidly than control ones (144). On the contrary, feeding adult mice (10 weeks of age) with 30% FR induces a loss of 20% of their body weight in 1 week (145). Thus, such FR models should be considered as valuable models of balanced feeding as shown by the induced longer lifespan (146).
In the quantitative food restriction models, the severity of the restriction generates various levels of weight loss associated with modifications of energy expenditure and respiratory quotient (145, 147–150). Indeed, long-term 30% FR in mice leads to a significant shift to carbohydrate metabolism during the meal (145). In rats, a 30% FR applied during 48 h or 14 days induced a significant body weight loss associated with decrease in plasma leptin concentrations, but only acute food deprivation leads to a decrease in glycemia and plasma insulin concentrations. At central level, both protocols induce up-regulation of hypothalamic AgRP and NPY mRNA associated with down-regulation of POMC mRNA (151). A 30% FR applied for 9 weeks in 3–week-old mice impacts bone mineral content more rapidly than when it is applied in older mice (9–14 weeks old) (144, 152, 153). Food restriction is associated with emotional impairments (154). C57Bl/6 mice subjected to a 20% caloric restriction for 8–12 days exhibit an anxiety-like behavior (155). Moreover, in 20–30% FR rats for 7–10 days, a decrease in dopamine levels in the nucleus accumbens occurs associated with an impairment of the expression of genes related to the dopamine (156). These alterations could be involved in reward sensitivity and emotional and motivation related behaviors observed in AN patients.
Alternate feeding experiments. Alternate feeding experiments with animal fed 1 day every two days appeared to induce alterations close to that observed on 40% FR models. Mice under alternate feeding from 12 to 65 weeks of age displayed a 20% increase in their body weight while this increase reached 60% for control mice (157).
Severe food restriction. Severe food restriction studies (50–70% restriction) are much less common (158, 159). Because of their severity, these studies are often shorter while numerous changes need several weeks to develop (160, 161). However, in a 50% FR on a long term protocol, mice show a decrease in energy expenditure after a meal associated with a decrease in lipid oxidation (150). Severe FR on 5-week protocol induced emotional impairments on rats. They showed increased anxiety like behavior, decreased serotonin turnover in the hippocampus and hypothalamus, and a decreased expression of 5-HT reuptake transporter in the raphe nucleus (162). Alterations of dopamine and DOPAC levels in septum and hypothalamus are associated with conditioning fear and control in food intake (163–167). The dopaminergic signaling was also shown to be modified (168) in the mesolimbic circuitry, strongly involved in the modulation of the motivational aspects of the food intake. Altogether, these protocols mimic various AN symptoms such as body weight loss associated with alterations in reproductive function, metabolic, endocrine, and neuro-endocrine systems (Table 3). Moreover, these models bring very interesting informations about the potential mechanisms sustaining physiological alterations observed in AN, and due to chronic caloric restriction, but they do not take into account two other major components widely described in AN, namely stress and physical activity. Other models have been developed to determine the role and involvement of both of these factors.
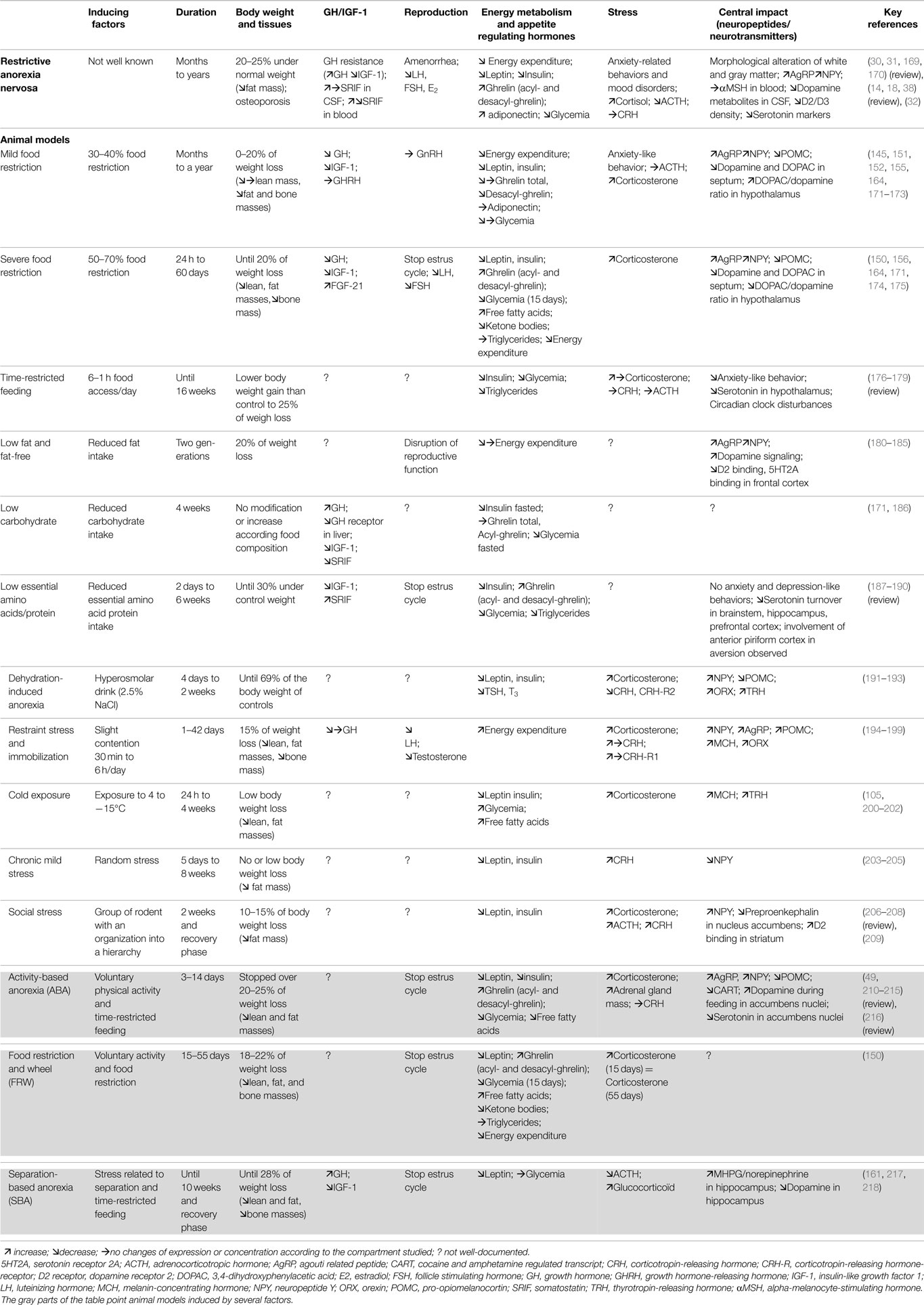
Table 3. Environmental models: main physiological and neurobiological changes observed in rodent models manipulated for one or several factors.
Time-restricted feeding (TR). Time-restricted feeding consists in ad libitum energy intake, but within few hours each day. Recently, Rothschild et al. (179) wrote a comprehensive review on the links between TR and metabolic diseases in animal models and human. Sherman et al. (219) showed that a 3-h food access each day for 16 weeks induces a food intake 15% lower and a body weight increase a half lower in adult male mice compared to their ad libitum control mice. But in these experiments, restricted animal are fed during the light period. Longer durations of daily food access were also studied, but they had a lower impact on food intake and body weight. Most of the time-restricted studies demonstrated slight or no changes in body weight gain when compared to control group, but an improvement of markers of metabolic disease risks. They also pointed out the link between disruption of the molecular circadian clock and metabolic disorders even under high fat diet (219, 220). These models mimic neither severe food restriction nor body weight decrease described in AN. TR feeding also leads to a reduction in the anxiety-like behavior and alteration of the serotonin system of rats (176, 221). The authors suggest that the decrease in the essential amino acid tryptophan in the hypothalamus may be the consequence of plasma tryptophan decreases, and thus contribute to the decrease in the serotonin synthesis. The related hypothalamic variations are suggested to provoke a compensatory upregulation of postsynaptic 5-HT receptors to precipitate AN.
Low fat diet. Animal models based on low fat diet (4–5% of fat/g) could take into account the fact that patients with AN not only reduce their food intake, but also select their foods. But two main difficulties limit the use of these models to study AN. First, foods with 4% of fat are commonly used as low fat diet, even if this is the fat level suggested for standard rodent food, while 10% fat diets usually lead to overweight with time and age. Second, almost all studies focused on comparisons between high-fat and low-fat diet consequences or focused on the effects of low-fat diet on obese mice.
Fat-free diet. The first studies conducted on rats submitted to fat-free diet during from 60 days to 6 weeks display a decrease of body weight (80% compared to control), a lower growth with emaciation appearance associated with increase of water intake, no difference in food intake compared to control rats (180, 181). It was also described impairment of reproductive function in male and female rats (181, 222). Respiratory quotient measured in rats under fat-free diets (1 month) but submitted to carbohydrate access following 14 h of fasting evidenced a shift to lipid metabolism (182). Variations of plasma lipid induced by low-fat diet and fat-free diet are sensed by neurons of ventromedial hypothalamus (223–225). However, to our knowledge, only the study of Staszkiewicz et al. (185) showed an upregulation of AgRP and NPY expression in low-fat diet group. In parallel, a lower dopamine signaling is described in rats submitted during two generations of α-linolenic acid deficient diet compared to normal chow diet as well as a lower 5-HT2 binding was observed till in the frontal cortex, even if no significant difference was observed concerning body weight between groups (183, 184, 226, 227). Moreover, although these neurotransmitters are known to be related to anxiety- and depression-like behaviors, no behavioral test was conducted in these studies.
Low carbohydrate diet. Patients with AN also select food with low carbohydrate content in the aim to reduce their calorie intake. But in rats, low carbohydrate diets moderately impact body weight (171), and mice on a zero-carbohydrate diet significantly gain more weight than animals consuming standard chow, despite similar caloric intake. These zero-carbohydrate fed mice also exhibited metabolic disruptions, while low carbohydrate diets in humans induce greater weight loss than isocaloric food (228). These results do not lead to consider low carbohydrate diet fed mice as relevant models for AN. Finally, studies on high/low fat or high/low carbohydrate diets revealed great differences in the use of fat and carbohydrate between mice and humans.
Indispensable amino acid deficient diet (IAA). Indispensable or essential amino acids are neither synthesized nor stored in organisms. In AN patients, one might consider that severe food restriction may alter the concentrations of plasma essential amino acids and might have drastic nutritional consequences (229). Several studies examining plasma amino acid levels display conflicting results in AN with higher, lower, or no significant differences compared to controls (11–13). However, a decrease in plasma tryptophan and a decrease in the tryptophan/large neutral amino acid ratio in acutely underweight AN patients are usually observed (32, 230–233). Thus, animal models based on essential amino acid restriction do not appear to be suitable models for AN. However, they could mimic some induced alterations, because essential amino acid restriction induces an adaptive behavior of food deprivation or because they are related to tryptophan. Various protocols have been developed using more commonly threonine, leucine, or valine deficient diets (189). Interpretation of changes observed should be taken with caution, since some alterations are related to energy deficit and others are related to the amino acid deficiency itself. In particular, valine deficient diet induced food restriction, a greater weight loss than for other IAA diets (approximatively 20% of their initial body weight), and increased of plasma acylghrelin and des-acylghrelin concentrations after 6 days of protocol (188). However, this valine deficient diet must be taken with caution since it leads to neurotoxicity not observed with an isoleucine deficient diet for example (189, 234–236). In another series of experiments using a combination of IAA deficient diets, Narita et al. (189) showed after 15 days of protocol, a decrease of glycemia, plasma triglycerides, leptin, insulin, and IGF-1 levels as well as a blockage of the estrous cycle in diestrus stage. Chronic tryptophan deficient diets (until 6 weeks) in rodents also lead to a progressive decrease of body weight (237–239). In contrast to acute deficient diet, no anxiety- or depressive-like behavior was observed in rodent despite a decrease in tryptophan concentration and serotonin turnover in brainstem, hippocampus, and prefrontal cortex (238–241). Indeed, no sucrose preference was observed in acute deficient rats while an increase of sucrose consumption was observed in mice after 5 weeks of tryptophan deficiency (239, 241). Unfortunately, to our knowledge, no studies determine the alterations of brain circuits regulating energy homeostasis. Rodents also develop different strategies to overcome the amino acid unbalance, including stopping the ingestion of food, change in the choice of food; they also develop a foraging behavior (to find complementary food); they establish an aversion with a learning phase and memorization of taste and smell to avoid the consumption of deficiency food in future (190, 242–244).
Dehydration-induced anorexia. As highlighted in part I, AN patients present relatively frequent osmoregulation impairment and renal complications due to their drink intake behaviors (21, 245, 246). Gutman and Krausz (247) pointed out a drastic decrease of food intake after acute subcutaneous injection of a hypertonic solution in rat. A “dehydration-induced anorexia” (DIA) model was developed by Watts (248). It consisted in a scheduled consumption of a hyperosmolar solution of NaCl (2.5%). This protocol has been tested for 4–14 days (192, 248). It provokes reduced food intake with a negative energy balance that is similar to those seen in pair-fed food-restricted animals: weight up to 69% under the body weight of control rodents, increased corticosterone, lowered leptin and insulin plasma levels (191). The food restriction is due a change in the pattern of food intake, a reduction of meal duration, and an inhibitory effect on gastric motility (249, 250). At the central level, DIA and pair-fed groups share up-regulation of NPY and down-regulation of POMC mRNA in the arcuate nucleus, and up-regulation of orexin mRNA in the lateral hypothalamic area only in pair-fed groups (191, 193). Beside, a down-regulation of CRH mRNA expression in the paraventricular nucleus and higher plasma corticosterone levels are observed only in DIA group (192). This model displays some common alterations also observed in AN. However, the drastic changes in osmolarity, which are not always observed in patients, might limit the use of DIA to decipher the central and peripheral mechanisms that can lead to chronic renal failure.
Stress models. A growing body of literature associated stress and anxiety as critical factors in the development of eating disorders like AN (251). Several animal models have been developed to evaluate mechanisms linking response to stressful events and alterations of food intake. In this section, we will not discuss data related to anorexia induced by the administration of lipopolysaccharides or endotoxemia. The most extensive studies concern restraint stress, cold exposure, or chronic mild stress (CMS).
Restraint stress. In rodents, limiting movements for a determined period (30 min to 6 h) every day generates a stress and a body weight decrease depending on the duration and type of immobilization (195). Indeed, animals are immobilized in a plastic tube or by attaching the four limbs to metal mounts with adhesive tape. Body weight loss up to 15% impacts both lean and fat masses, and is associated with a voluntary food restriction after an acute stress session (like 2 h), or when the stress is repeated (195, 252–254). Moreover, this low body weight is maintained even after a recovery period (199). Repetition of the restraint stress induces long lasting increased plasma corticosterone, ACTH and ghrelin concentrations, and decreased plasma leptin and insulin concentrations (255–258). In long duration experiments, bone physiology alterations are also observed (259). Rats exhibit an increase of energy expenditure and body temperature during the stress followed by return to control values (199, 260). At central level, noticable changes in the activation and/or expression of genes involved in the control of food intake are described. Acute restraint stress increases the number of activated neurons in several brain areas compared to controls, while repeated stress effects are lowered probably because of habituation (261–263). In these studies, modifications in the activation of the HPA axis are the most documented. Restraint stress protocols increase plasma corticosterone concentrations, which are associated with an increased activation and expression of CRH in the paraventricular nucleus (198, 264). However, such increases are not anymore observed when the stress is repeated (198, 199, 265). Considering the anorexigenic effects of intracerebroventricular injections of CRH, this peptide has been suspected to be responsible for the voluntary food restriction observed in this type of protocol (265). Both acute and repeated restraint stress in rats induce decreased number of neurons immunoreactive for Fos and AgRP in arcuate nucleus, while the number of neurons immunoreactive for Fos and MC4R increases in the lateral hypothalamic area but decreases in the arcuate nucleus on the long term (263). Such reduction in MC4R cell activation may signify a desensitization of feeding regulatory pathways in the arcuate nucleus after repeated stress exposure that may be indicative of a shift toward more orexigenic behaviors, as signals promoting feeding become more prominent. In another study where a 2-week chronic restraint stress is applied on mice, inhibition of food intake occurs until the end of the first week and is associated with also an up-regulation of POMC mRNA in the arcuate nucleus (258). The data obtained with NPY are less clear since acute restraint stress increases NPY mRNA expression in the arcuate nucleus. This expression is normal in the case of chronic stress (266). Thus, the relative balance between orexigenic and anorexigenic pathway activation appears to be dependent on whether the stress is acute or repeated. Finally, stress induces a very rapid degradation of GH (267) and thus a decrease in plasma GH concentrations (268, 269). The release of somatostatin, a major inhibitor of the GHRH release, increases in the median eminence level following acute restraint stress, and thus might be a major factor in this GH drop (270).
These results suggest that food intake may be increased or decreased as a consequence of stress, and may play a role in eating disorders from anorexia to binge-eating leading to obesity and other stress-associated metabolic disorders. Once again, this psychological stress impacts differentially the brain areas involved in the regulation of food intake rendering difficult to use such protocol to study precisely the mechanisms involved in AN.
Cold exposure. One hypothesis on the origins of hyperactivity often observed in AN is that it would be a form of thermoregulatory behavior. Studies on the effects of ambient temperature or heat treatment on AN patients displaying hyperactivity strengthen this hypothesis (271, 272). Cold exposure is a physiological stress used to determine mechanisms involved in control of body temperature. The protocol used temperature exposure from 4°C to -15°C and for a duration ranging from 24 h to 4 weeks. Usually, relatively low body weight loss is observed and is not always associated with a decrease in food intake (200–202, 273–276). This body weight loss is associated with a decrease in both lean and fat masses and an increase in brown adipose tissue mass (202, 277–279). Short term exposure (1–24 h) or long term exposure (8 days) to cold stress (at 4°C) increases blood glucose, plasma adrenaline, and corticosterone concentrations, and decreases plasma leptin and insulin concentrations (200, 276, 279, 280). Cold exposure also leads to increased glucose uptake by peripheral tissues associated with increased liver glycogen, lipolysis in white and brown adipose tissues, and concomitant to lipogenesis in these tissues (200, 279, 281). Activation of lipolysis in the different fat depots involves the sympathetic nervous system as suggested by an increased noradrenergic turnover (276, 277). Lower temperatures (under 0°C) during 2 weeks induced in mice, a more important body weight loss associated with higher food intake and lower body temperature (202). Cold exposure leads to activation of numerous brain areas involved in thermoregulation located in the hindbrain (282, 283), in the hypothalamus, and in the forebrain (280, 284). Cold exposure during 4 days (4°C) leads also to increase of MCH expression in the hypothalamus (201), suggesting the involvment of this neuropeptide directly or indirectly in such variations. However, the origin of these variations is unclear but they are probably due to the role of MCH in control of energy expenditure (201, 285).
Chronic mild stress. Depression is another sign classically observed in AN. The most valuable animal model of depression like behavior was developed by Willner et al. (203). This model called CMS consists to expose rodents to mild stress applied randomly and daily during 3 to 9 weeks. In this kind of protocol conducted on rodents, the body weight is slightly diminished but, a notable reduction of sucrose consumption, sign of anhedonia, is described (203, 204, 286, 287). The body weight loss concerns decreased subcutaneaous and visceral fat mass associated with decreased plasma leptin and insulin concentrations. However, these changes are not specific to CMS protocol because they are also observed in the “weight match” control group (205). At central level, CMS animals exhibit up-regulation of CRH in paraventricular nucleus while its expression is reduced in the “weight match” group (205). Other peptide expressions also are altered, with especially a down-regulation of NPY in the arcuate nucleus (204). The CMS is described to have anxiogenic effects through a stronger neuronal activation in various brain areas, as well as a decreased neurogenesis in the hippocampus (288). Recent reviews (289–291) pointed out a role of ghrelin in depression and anxiety, even if it is again still a subject of debate. Its receptor is present in structures known to be involved in mood disorders like hippocampus and amygdala. The model presents the advantages to mimic alterations of the stress axis and anhedonia for palatable food associated with a slight body weight loss. However, the complexity of the stress procedure and a rapid recovery limit the interest of CMS to mirror AN.
Social stress. Another kind of acute/chronic stress is related to rodent social interactions. The main models are based on social defeat stress and the visible burrow system (VBS). The social defeat stress was first used as a model of anxiety and depression (292). A rodent (intruder) is placed in the home cage of another rodent (resident). The interactions between the two animals are usually rapid and lead to aggressive behaviors, with a dominant and a subordinate. The defeat social stress leads to a markedly decrease of body weight in animal following 1 h session of stress (206). The repetition of this stress induces also a higher reactivity to an acute restraint stress with increased plasma corticosterone and ACTH concentrations, but with a normalization of values after stress (293). A decrease in locomotor activity is also observed associated with reduced social interaction in the presence of a non-aggressive rodent (206, 293). An increased nocturnal food intake is noticed and not observed in the case of VBS protocol (209, 293). The VBS protocol induces a more complex social defeat stress since it is based on the establishment of a hierarchy in a group of male rats leading to dominance hierarchies with offensive and defensive behaviors (294). At the end of the confrontation period, a dominant male rat (DOM) takes the ascendancy over other rats qualified like subordinate males (SUB). VBS protocol induces decrease of body weight associated with a decrease of food intake only in SUB male rats (208, 209, 295). The body weight reduction is associated only with a decrease in subcutaneous fat mass, whereas lean mass is unchanged and visceral fat mass is increased (208). The pattern of food intake is modified with a decrease of meal duration (209). SUB rats display also endocrine changes with a decrease of plasma leptin and insulin concentrations compared to DOM and control rats (296, 297). Studies related to alterations at the central level have mainly focused on the HPA axis, particularly affected in the SUB rats, with increased plasma corticosterone concentrations correlated with increased expression of CRH in the paraventricular nucleus and amygdala (208, 295, 296, 298). The chronically elevated corticosterone levels may create an orexigenic drive through upregulation of NPY and AgRP in the SUB rats as well as the loss of fat mass seen in both DOM and SUB, which indicates a negative energy balance, and may also create an orexigenic drive through similar mechanisms. Such observations are validated by the behavior observed in a recovery phase where the rodents become hyperphagic and increase drastically their fat mass (299). These observations render the model inadequate for studying the recovery period after food restriction even if the model generates transiently a food restriction during the protocol. It should be underlined that a noticable decrease of palatable food is observed as in the CMS protocol in the recovery phase (207, 208, 300). These changes have been attributed to alterations in dopamine transporter binding and dopamine receptor (D2) binding which are reduced or increased respectively in the striatum and accumbens nucleus in SUB group (207). The VBS protocol also leads to changes in the SUB serotonergic system in various brain areas involved in the modulation of stress (294, 301). This model is interesting to study the impact of chronic social stress on food intake and its homeostatic and non-homeostatic regulation. But the main drawbacks are: the absence in human of such notion of subordinate and dominant; the recovery period which shows a binge-eating behavior that is rare in recovered AN patients; the short term duration (around 15 days) excluding the development of long term alteration like osteoporosis. Finally, there is currently no or few data about the regulation of energy balance. The VBS model presents an important limit that reduces its use to study AN: it is applicable only on males.
Animal Model Based on Several Inducing Factors
Separation-based anorexia. Separation-based anorexia model is another model of chronic social stress not often used until now. This model is based on stress produced by a physical separation of mice belonging to the same group and associated with a food restriction or a time-restricted feeding (TR) (217). This study was initially conducted on Sabra female mice. Only few studies were published on this strain of mouse with high body weight. Food was provided during the light phase for 1 h a day. Control mice with the same feeding schedule lost 10% of their day 0 body weight within 18 days, and daily ate 2.84 g of food. Separated and time-restricted mice lost 28% of their initial body weight, and daily ate 2.33 g of food. In this group, 21% of mice died before reaching the targeted body weight loss of 33–35%. Separated and time-restricted mice ate 65% of the daily requirements and reach the same level of body weight loss than mice fed 40% of the daily requirements without being separated. These data suggested that separation of mice increases metabolic demands. This first study was followed by two studies on the same model and conducted by the same team. Both of them dealt with the effects of tyrosine treatments on central nervous system functions. Hao et al. (218) showed that SBA mice display an increase in 3-methoxy-4-hydroxyphenyglycol/norepinephrine ratio, an up-regulation of the cholinergic signaling, and a decrease in the dopamine concentration in hippocampus. In 2002, the effects of tyrosine treatments on HPA axis were studied on this model (302). This second central study pointed out a specific pattern of central alterations in SBA mice when compared to FR and active mice despite similar body weight loss. To allow studies on long term metabolic and central adaptations on a usual mouse strain, we recently adapted this model to C57Bl/6 young adult female mice. Food access was progressively reduced from 6 to 2 h a day within 2 weeks and then maintained at 2 h a day for up to 8 weeks (161). We have shown that this protocol induces significant weight loss with a reduction from 20 to 25% of initial body weight. Interestingly, the body weight loss observed in SBA group is not attributable to the timed food access as SBA mice eat only 10% less than ad libitum group. Moreover, such a difference in body weight is not observed in the TR group without separation. We suspect that this difference is partly due to rising energy costs both through the separation-induced stress and higher thermogenesis needs caused by the separation. Body weight loss is related to a decrease in lean mass and visceral and subcutaneous fat masses. In parallel, SBA mice present a blocking of their reproductive function and bone mass gain. Like in AN patients, various endocrine changes are observed. Thus, SBA mice display lower plasma leptin concentrations. Furthermore, disruption of the GH/IGF-1 associated with alteration in bone physiology was observed at 2 and 10 weeks. At metabolic level, protocol induces an up-regulation of several genes (UCP1, PGC1a, Prdm16) especially in the subcutaneous adipose tissue of SBA mice, suggesting the emergence of beige/brite adipocytes in this specific fat depot. Moreover, after 10 weeks of SBA, protocol mice were submitted to a 10-week recovery period with free food access in normal cage. During this recovery period, mice correct their various alterations including body weight, food intake, reproductive function, body composition, endocrine factors, and adipose tissue metabolism. However, SBA mice maintain low plasma leptin concentrations and low leptin expression in visceral fat tissue despite a full normalization of fat mass (161).
This long term model appears interesting as it mimics numerous central and peripheral alterations described or suggested in AN, and allows a recovery study. However, the increased energy expenditure related to chronic stress and high needs of thermogenesis does not match the decrease usually described in patients.
Activity models. In 1967, Routtenberg and Kuznesof developed a protocol, where rats isolated in a cage were allowed to have a timed food access, 1 h per day, combined to a voluntary activity. This model later named activity-based anorexia (ABA)/self-starvation/semistarvation-induced hyperactivity/food restriction-induced hyperactivity/wheel-induced feeding suppression model produces a rapid weight loss, close to 25% of their initial weight within days and food intake, physical hyperactivity, hypothermia, impaired estrous cycle in females, and increases in HPA axis activity (215, 303, 304). Moreover, rats eat less than inactive rats fed with the same schedule. This procedure led rapidly to a “self-starvation” or self-deprivation behavior resembling to that observed in restrictive AN patients and leading rapidly to the death of animals due to the voluntary privation of food (around 7 days). It is currently the most well-known animal model of anorexia (216, 305) and has been adapted to mice (306, 307). Recently, Lewis and Brett (308) reduced progressively the food access duration to maintain mice longer than 7 days. Following this new protocol, Jésus et al. (309) demonstrated alterations of intestinal permeability. In many aspects, all these models mimic numerous physiological alterations observed in AN. However, as specified in the review of Klenotich and Dulawa (310), the ABA paradigm is strongly dependent on the rodent strain, on age and gender (307, 311), on temperature [increasing the temperature to 32°C strongly reduces the ABA behavior, (312)], and on the time of the day the animals receive food. In fact, Boakes and Juraskova (313) and Boakes (210) demonstrated that the “self-starvation” observed in ABA rats might reflect both the reduced palatability of the dry chow for a dehydrated animal and satiety signals from a stomach full of water. Finally, in all these protocols, rodents were isolated in their cage to permit individual metabolic and physiological measures, but isolation creates a social stress adding on the physiological stress of food deprivation, rendering the protocol more drastic. Thus, all these studies present limitations that maintain a distance with AN. Recently, we have developed a modified ABA model on female mice, named here Food Restriction and Wheel (FRW) model that aims: (i) to prevent the social stress by using two mice per cage and (ii) to follow on the long term (up to 10 weeks) physiological alterations induced by a combination of physical activity and a food restriction of 50% (150). All of these activity models present metabolic, endocrine, and neurobiological alterations that might be the basis to study adequately some of physiological mechanisms altered in AN patients. Finally, they all exhibited a FAA, which occurs between 2 and 5 h before food intake distribution, and which is also described in AN patients (314).
The body weight loss observed both in ABA and FRW rodents is related to decrease of lean and subcutaneous/visceral fat masses after 7–14 days of protocol (150, 315). Physical activity at short term exacerbates decreased fat mass and has no protective effect on bone composition and lean mass (150, 211, 213, 316). When the protocol is maintained on the long term (55 days) like in FRW protocol, physical activity participates to body weight stabilization and to a significant slight body weight regain compared to pair-fed group (150). The long term protocol induces alterations in the bone mineral content leading in AN patients to osteoporosis. Indeed, in FRW mice, physical activity, currently described to stimulate bone formation, did not prevent on long term protocol the termination of bone mass acquisition induced by food restriction. Similar data were also described in SBA female mice subjected to a protocol of chronic stress associated with caloric restriction as previously mentioned. Such data confirmed the absence of protective effect of activity on bone mineral content in AN. In the ABA model, Pardo et al. (315) underline a differential tissue-specific expression pattern of ghrelin and leptin receptor at peripheral level reflecting tissue specific mechanisms to control energy homeostasis. The study of intestinal barrier indicates that the ABA protocol generates an increased colonic permeability associated with altered tight junction expression (309). These recent data open new windows to decipher the impact of gut microbiota in the deregulation of energy metabolism as well as the hepatic injury occurring in AN patients.
Besides alterations in various peripheral tissues, numerous endocrine changes are similar to that described in AN patients. Overall, ABA mice present lower plasma leptin and insulin concentrations and higher total plasma ghrelin and corticosterone concentrations (212, 215, 317). Moreover, energy metabolic factors are also changed in ABA/FRW mice with, in particular, an increase of free fatty acid and a decrease of glycemia (150, 213). On the long term, most of the endocrine alterations persist like lower plasma leptin concentrations, higher plasma total ghrelin concentrations still associated to lower glycemia, plasma ketone bodies, and higher free fatty acid in FRW mice (150). As highlighted previously, food restriction might induce shift in the energy metabolism regulations. Combination of food restriction and voluntary physical activity leads to a higher carbohydrate metabolism and a lower fat oxidation during the light period like the ad libitum control groups whereas at long term, FRW mice adopt a similar profile than the pair-fed group with a lipid metabolism more prominent. These changes point out the complexity of the peripheral regulation of nutrient and energy supplies, engaging probably hormones like leptin or ghrelin, which act on adipose tissues, muscles, or liver, might contribute to the changes/reduction in energy expenditure observed in FRW and pair-fed controls both at short and long term.
Central alterations are also observed in ABA protocols with an up-regulation of AgRP and NPY mRNA expression associated with a down-regulation of POMC and CART expression in the arcuate nucleus compared to control mice (211, 318–320). Surprisingly, no differences were observed concerning MCH and orexin expression in lateral hypothalamic area or CRH expression in paraventricular nucleus (211). However, until now, there is no study that evaluates potential changes in the expression of ghrelin and leptin receptors in ABA mice. Such information might be of importance since GHSR KO mice or intracerebroventricular injection of GHSR1a antagonist decreased the behavior of FAA and did not modify the food intake (321). Likewise, chronic subcutaneous or intracerebroventricular leptin injections lead to lower running wheel activity associated or not with reduction of food intake (322–324). Such fundamental researches are conducted to aim finding potential treatment using leptin or ghrelin to reduce hyperactivity frequently associated with AN, and leading to its excessive to emaciated phenotype. Indeed, intracerebroventricular injection of αMSH, whose release in hypothalamus is stimulated by leptin, enhances the ABA phenotype (325). Likewise, the specific sites of action of ghrelin and/or leptin in the ABA protocol should also be clarified. As an example, injections of ghrelin agonist in the lateral dorsal tegmental nucleus or its target, the ventral tegmental area, stimulate locomotor activity and food intake (326, 327). ABA mice are also shown to exhibit higher concentrations of noradrenaline, serotonine, but lower dopamine concentrations in the mediobasal hypothalamus compared to pair-fed and control groups (302, 328, 329). Moreover, Verhagen et al. (214) showed in the nucleus accumbens of ABA rats a lower circadian serotonergic activity without any changes for the circadian dopamine activity compared to control. These monoamines were suggested to play a role in voluntary food restriction in ABA rodents and in comorbidities observed in AN patients [i.e., depression or obsessive compulsive disorders; (137)]. It was suggested that reduction of physical activity is due to inhibition of serotonin release via 5HT1A autoreceptors in raphe nucleus (329–333). The opioid and endocannabinoid systems are also modified in the ABA model with increased plasma βendorphin concentration and pituitary βendorphin content in rats (334). This hyperendorphinism in the hypothalamo-pituitary-adrenal axis was linked to the auto-addiction hypothesis of AN. Furthermore, intraperitoneal injections of ABA mice with Δ9-tetrahydrocannabinol, an exogene ligand of cannabinoid receptors, increase their food intake, attenuate the body weight loss, reduce the energy expenditure, but increase the mortality rate compared to ABA mice vehicle-treated (308, 335). Due to the large distribution of endocannabinoid and opioid receptors in the brain, further studies are needed to clarify more precisely the mechanisms involved and the finely tuned interactions between all these homeostatic and non-homeostatic structures.
The ABA/FRW protocols also affect two other main endocrine functions: stress and reproduction. In ABA rodents, like in FRW mice (on the short and long term), a disruption of estrus cycle, vaginal closure, and reduction of ovaries size, and also hormone disturbances including a decrease of plasma testosterone and luteinizing hormone concentrations have been noted (150, 329, 336, 337). Reproduction axis is normalized when rodents are placed in recovery conditions, which reflect that reproductive disturbance is the result of energy unbalance (337). Concerning the HPA axis, ABA protocols induce on the short-term increased plasma corticosterone and ACTH concentrations and adrenal gland hypertrophy, but no significant modification of CRH expression in paraventricular nucleus compared to controls (211, 318, 338). Intracerebroventricular injection of CRH antagonist injection during the protocol leads to blunt the ABA phenotype (318). Furthermore, ABA adrenalectomized rats do not display increased wheel running activity (212). Once again, these data suggest that HPA axis is essential to apparition of ABA phenotype and point out the role of the glucocorticoids in the pathophysiology of AN. Somatotrope axis is another axis disrupted in AN patients, but in our knowledge there is no study using ABA protocol or associated protocols showing such alterations.
As mentioned earlier, one characteristic of the ABA model is the FAA. Several studies have documented the potential factors and neuronal structures leading to this particular behavior that can be generated like a foraging behavior or to increase the internal temperature due to energy deficit (212, 215, 321–323, 339). FAA itself can also influence the pattern of food intake. Indeed, in the FRW protocol, mice display a shift in the meal initiation compared to the pair-fed group (150). One explanation, suggested by Woods (340), considered eating to be a homeostatic stressful event, because the digested nutriments that reached the blood during and after a meal markedly disrupt energy homeostasis. Thus, the combination of both events, activity and feeding, could generate a stressful energy event especially in the short term, leading to increase in corticosterone levels and resulting to delay the meal initiation. Such phenomenon could occur in the ABA model, where the pattern of food intake has never been measured in metabolic cages, as it was done for FRW mice. The “self starvation” observed might thus be due to this delay in the initiation of the meal, which is, as mentioned above, time limited. Concerning the temperature, the ABA protocol induces a decrease of body temperature (341, 342). In addition, even if a negative correlation between FAA and body temperature was observed, no causal link has been demonstrated (325). Nevertheless, it was suggested that the decrease of body temperature is one of the factors contributing to physical activity (342). When ABA rats have access to a warm platform, they decrease their running wheel activity (320, 325, 343), similarly as observed in AN patients whose excessive physical activity vary depending on the ambient temperature (272).
All the data collected with both ABA and FRW models are totally useful to dissect the different mechanisms involved in the maintenance of the AN phenotype. Combining the different approaches on the short and long term will have an indubitable benefit to study the interactions between the various peripheral and central actors whose dialogs seem strongly impaired.
Conclusion
This review aims to depict the different animal models currently used or potentially interesting to study one or several aspects of restrictive AN (Figure 1). The definition of a pertinent animal model of psychiatric disorder remains extremely difficult. In the case of AN, more specially the restrictive subtype, many symptoms can be mimicked in rodents like the body weight loss, the changes in energy expenditure, increased physical activity, several endocrine and neurotransmitters changes that reflects similar physiological and neurobiological mechanisms inherent to the natural and adapted regulation of feeding. In this sense, some of the currently available animal models described here answer to the “face validity,” i.e., they mimic most of the symptoms of the human pathology. However, AN is usually associated with a refusal to eat. In rodents, such behavior is not natural, even if a kind of self-starvation is observed in migratory and hibernating animals. The “self-starvation” induced by some protocols does not reflect the human starvation, which is classically described to be associated with a personality trait involving neuronal inhibitory cognitive circuits. Even if self starvation is observed in some models like the well known ABA model, one may considered that the starvation is essentially due to physiological factors like temperature, dryness of the food, or even the delay in the initiation of the meal due to the intense physical activity observed before feeding. These models give certainly important information’s about the physiological changes occurring at this period, but do not reflect the self-starvation observed in human, which remains to be understood. Is it only driven by cognitive inputs or is it under the influence of factors regulating the feeding homeostasis like ghrelin or leptin which receptors are distributed in numerous “non-homeostatic” brain areas? Brain imaging might help to solve this question and would permit to give more credit to what we obtained in animal models. Even if all of these models do not fully answer to criterion of “construct validity,” i.e., a common etiology or similar conditions of induction, they fulfill the “predictive validity,” as the different pharmacological treatments used to restore body weight and other altered functions give encouraging results. As a conclusion, it is to note that current environmental models based on a combination of several inducing factors appear to be more relevant than the other models but may be to further improve studies on AN, new models coupling genetic and environmental factors remain to create and assess.
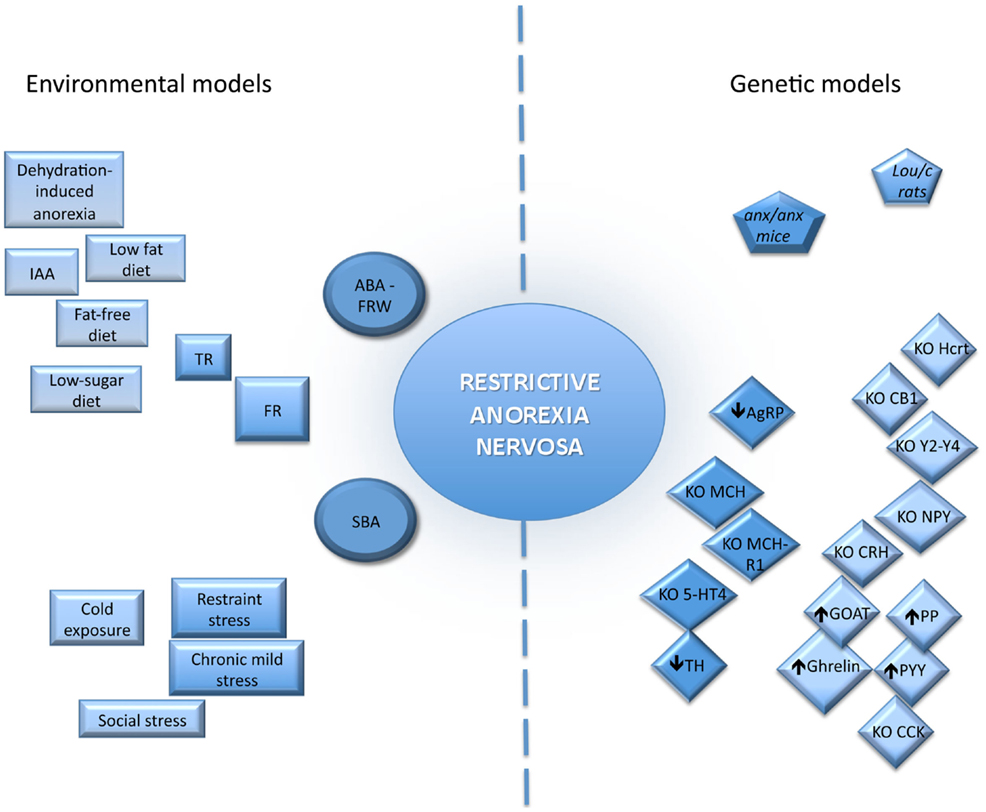
Figure 1. Schematic representation of the relevance of the different animal models for anorexia nervosa, environmental and genetics, described in the review. The more the models mimic restrictive anorexia nervosa symptoms the closer they are to the center of the figure. In the environmental models, the squares correspond to models based on one inducing factors and the circles to models based on several inducing factors. In the genetic models, the pentagons point out genetic models with spontaneous mutations whereas the diamonds show the genetically engineered mice: knock out (KO), transgenic (↑), specific deletion (↓). ABA, activity based anorexia; AgRP, agouti related peptide; CB1, cannabinoid receptor type 1; CCK, cholescystokin; CRH, corticotropin releasing hormone; FR, food restriction; IAA, indispensable amino acid deficient diet; GOAT, ghrelin O-acyltransferase; Hcrt, orexin/hypocretin; MCH, melanocortin concentrating hormone; NPY, neuropeptide Y; PP, polypancreatic peptide; PYY, peptide YY; SBA, separation based anorexia; TH, tyrosine hydroxylase; TR, time-restricted feeding; 5-HT R, serotonin receptor.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1. American Psychiatric Association. Diagnostic and Statistical Manual of Mental Disorders, Fifth Edition, DSM-5. American Psychiatric Association (2013).
2. Eddy KT, Dorer DJ, Franko DL, Tahilani K, Thompson-Brenner H, Herzog DB. Diagnostic crossover in anorexia nervosa and bulimia nervosa: implications for DSM-V. Am J Psychiatry (2008) 165(2):245–50. doi: 10.1176/appi.ajp.2007.07060951
3. Garcia FD, Délavenne H, Déchelotte P. Atypical eating disorders a review. Nutr Diet Suppl (2011) 3:67–75. doi:10.2147/NDS.S10239
4. Hudson JI, Hiripi E, Pope HG Jr, Kessler RC. The prevalence and correlates of eating disorders in the National Comorbidity Survey Replication. Biol Psychiat (2007) 61(3):348–58. Erratum in: Biol Psychiatry (2012) 72(2):164.
5. Dalle Grave R, Calugi S, Marchesini G. Is amenorrhea a clinically useful criterion for the diagnosis of anorexia nervosa? Behav Res Ther (2008) 46(12):1290–4. doi:10.1016/j.brat.2008.08.007
6. Abbate Daga G, Campisi S, Marzola E, Rocca G, Peris C, Campagnoli C, et al. Amenorrhea in eating disorders: poor stability of symptom after a one-year treatment. Eat Weight Disord (2012) 17(2):e78–85.
7. Papadopoulos FC, Ekbom A, Brandt L, Ekselius L. Excess mortality, causes of death and prognostic factors in anorexia nervosa. Br J Psychiatry (2009) 194(1):10–7. doi:10.1192/bjp.bp.108.054742
8. Weiselberg EC, Gonzalez M, Fisher M. Eating disorders in the twenty-first century. Minerva Ginecol (2011) 63(6):531–45.
9. Löwe B, Zipfel S, Buchholz C, Dupont Y, Reas DL, Herzog W. Long-term outcome of anorexia nervosa in a prospective 21-year follow-up study. Psychol Med (2001) 31(5):881–90.
10. Fisher M. The course and outcome of eating disorders in adults and in adolescents: a review. Adolesc Med (2003) 14(1):149–58.
11. Föcker M, Timmesfeld N, Scherag S, Knoll N, Singmann P, Wang-Sattler R, et al. Comparison of metabolic profiles of acutely ill and short-term weight recovered patients with anorexia nervosa reveals alterations of 33 out of 163 metabolites. J Psychiatr Res (2012) 46(12):1600–9. doi:10.1016/j.jpsychires.2012.08.015
12. Palova S, Charvat J, Masopust J, Klapkova E, Kvapil M. Changes in the plasma amino acid profile in anorexia nervosa. J Int Med Res (2007) 35(3):389–94. doi:10.1177/147323000703500314
13. Moyano D, Vilaseca MA, Artuch R, Lambruschini N. Plasma amino acids in anorexia nervosa. Eur J Clin Nutr (1998) 52(9):684–9. doi:10.1038/sj.ejcn.1600625
14. Estour B, Germain N, Diconne E, Frere D, Cottet-Emard JM, Carrot G, et al. Hormonal profile heterogeneity and short-term physical risk in restrictive anorexia nervosa. J Clin Endocrinol Metab (2010) 95(5):2203–10. doi:10.1210/jc.2009-2608
15. Germain N, Galusca B, Grouselle D, Frere D, Billard S, Epelbaum J, et al. Ghrelin and obestatin circadian levels differentiate bingeing-purging from restrictive anorexia nervosa. J Clin Endocrinol Metab (2010) 95(6):3057–62. doi:10.1210/jc.2009-2196
16. Nogueira JP, Valéro R, Maraninchi M, Lorec AM, Samuelian-Massat C, Bégu-Le Corroller A, et al. Growth hormone level at admission and its evolution during refeeding are predictive of short-term outcome in restrictive anorexia nervosa. Br J Nutr (2013) 109(12):2175–81. doi:10.1017/S000711451200431X
17. Misra M, Miller KK, Cord J, Prabhakaran R, Herzog DB, Goldstein M, et al. Relationships between serum adipokines, insulin levels, and bone density in girls with anorexia nervosa. J Clin Endocrinol Metab (2007) 92(6):2046–52. doi:10.1210/jc.2006-2855
18. Legroux-Gérot I, Vignau J, d’Herbomez M, Flipo RM, Cortet B. Predictive factors of change in BMD at 1 and 2 years in women with anorexia nervosa: a study of 146 cases. Osteoporos Int (2012) 23(12):2855–61. doi:10.1007/s00198-012-1919-8
19. Hebebrand J, Casper R, Treasure J, Schweiger U. The need to revise the diagnostic criteria for anorexia nervosa. J Neural Transm (2004) 111(7):827–40. doi:10.1007/s00702-004-0136-9
20. Hart S, Abraham S, Franklin RC, Russell J. The reasons why eating disorder patients drink. Eur Eat Disord Rev (2011) 19(2):121–8. doi:10.1002/erv.1051
21. Stheneur C, Bergeron S, Lapeyraque AL. Renal complications in anorexia nervosa. Eat Weight Disord (2014) 19(4):455–60. doi:10.1007/s40519-014-0138-z
22. Erdur L, Kallenbach-Dermutz B, Lehmann V, Zimmermann-Viehoff F, Köpp W, Weber C, et al. Somatic comorbidity in anorexia nervosa: first results of a 21-year follow-up study on female inpatients. Biopsychosoc Med (2012) 6(1):4. doi:10.1186/1751-0759-6-4
23. Seitz J, Bühren K, von Polier GG, Heussen N, Herpertz-Dahlmann B, Konrad K. Morphological changes in the brain of acutely ill and weight-recovered patients with anorexia nervosa. A meta-analysis and qualitative review. Z Kinder Jugendpsychiatr Psychother (2014) 42(1):7–17. doi:10.1024/1422-4917/a000265
24. Phillipou A, Rossell SL, Castle DJ. The neurobiology of anorexia nervosa: a systematic review. Aust N Z J Psychiatry (2014) 48(2):128–52. doi:10.1177/0004867413509693
25. Kullmann S, Giel KE, Teufel M, Thiel A, Zipfel S, Preissl H. Aberrant network integrity of the inferior frontal cortex in women with anorexia nervosa. Neuroimage Clin (2014) 12(4):615–22. doi:10.1016/j.nicl.2014.04.002
26. Bari A, Robbins TW. Inhibition and impulsivity: behavioral and neural basis of response control. Prog Neurobiol (2013) 108:44–79. doi:10.1016/j.pneurobio.2013.06.005
27. Schultz W. Dopamine neurons and their role in reward mechanisms. Curr Opin Neurobiol (1997) 7(2):191–7. doi:10.1016/S0959-4388(97)80007-4
28. Atiye M, Miettunen J, Raevuori-Helkamaa A. A meta-analysis of temperament in eating disorders. Eur Eat Disord Rev (2014) 2014:29. doi:10.1002/erv.2342
29. Bailer UF, Frank GK, Price JC, Meltzer CC, Becker C, Mathis CA, et al. Interaction between serotonin transporter and dopamine D2/D3 receptor radioligand measures is associated with harm avoidant symptoms in anorexia and bulimia nervosa. Psychiatry Res (2013) 211(2):160–8. doi:10.1016/j.pscychresns.2012.06.010
30. Kaye WH, Frank GK, McConaha C. Altered dopamine activity after recovery from restricting-type anorexia nervosa. Neuropsychopharmacology (1999) 21(4):503–6. doi:10.1016/S0893-133X(99)00053-6
31. Frank GK, Bailer UF, Henry SE, Drevets W, Meltzer CC, Price JC, et al. Increased dopamine D2/D3 receptor binding after recovery from anorexia nervosa measured by positron emission tomography and [11c]raclopride. Biol Psychiatry (2005) 58(11):908–12. doi:10.1016/j.biopsych.2005.05.003
32. Gauthier C, Hassler C, Mattar L, Launay JM, Callebert J, Steiger H, et al. Symptoms of depression and anxiety in anorexia nervosa: links with plasma tryptophan and serotonin metabolism. Psychoneuroendocrinology (2014) 39:170–8. doi:10.1016/j.psyneuen.2013.09.009
33. Bailer UF, Frank GK, Henry SE, Price JC, Meltzer CC, Mathis CA, et al. Exaggerated 5-HT1A but normal 5-HT2A receptor activity in individuals ill with anorexia nervosa. Biol Psychiatry (2007) 61(9):1090–9. doi:10.1016/j.biopsych.2006.07.018
34. Galusca B, Costes N, Zito NG, Peyron R, Bossu C, Lang F, et al. Organic background of restrictive-type anorexia nervosa suggested by increased serotonin 1A receptor binding in right frontotemporal cortex of both lean and recovered patients: [18F]MPPF PET scan study. Biol Psychiatry (2008) 64(11):1009–13. doi:10.1016/j.biopsych.2008.06.006
35. Bailer UF, Frank GK, Henry SE, Price JC, Meltzer CC, Becker C, et al. Serotonin transporter binding after recovery from eating disorders. Psychopharmacology (Berl) (2007) 195(3):315–24. doi:10.1007/s00213-007-0896-7
36. Kaye WH, Fudge JL, Paulus M. New insights into symptoms and neurocircuits function of anorexia nervosa. Nat Rev Neurosci (2009) 10(8):573–84. doi:10.1038/mm2682
37. Bailer UF, Kaye WH. Serotonin: imaging findings in eating disorders. Curr Top Behav Neurosci (2011) 6:59–79. doi:10.1007/7854_2010_78
38. Monteleone P, Maj M. Dysfunctions of leptin, ghrelin, BDNF and endocannabinoids in eating disorders: beyond the homeostatic control of food intake. Psychoneuroendocrinology (2013) 38(3):312–30. doi:10.1016/j.psyneuen.2012.10.021
39. Park RJ, Godier LR, Cowdrey FA. Hungry for reward: how can neuroscience inform the development of treatment for anorexia nervosa? Behav Res Ther (2014) 62:47–59. doi:10.1016/j.brat.2014.07.007
40. Brewerton TD. Antipsychotic agents in the treatment of anorexia nervosa: neuropsychopharmacologic rationale and evidence from controlled trials. Curr Psychiatry Rep (2012) 14(4):398–405. doi:10.1007/s11920-012-0287-6
41. Scherag S, Hebebrand J, Hinney A. Eating disorders: the current status of molecular genetic research. Eur Child Adolesc Psychiatry (2010) 19(3):211–26. doi:10.1007/s00787-009-0085-9
42. Wade TD, Bulik CM, Neale M, Kendler KS. Anorexia nervosa and major depression: shared genetic and environmental risk factors. Am J Psychiatry (2000) 157(3):46971. doi:10.1176/appi.ajp.157.3.469
43. Klump KL, Miller KB, Keel PK, McGue M, Iacono WG. Genetic and environmental influences on anorexia nervosa syndromes in a population-based twin sample. Psychol Med (2001) 31(4):73740. doi:10.1017/S0033291701003725
44. Kortegaard LS, Hoerder K, Joergensen J, Gillberg C, Kyvik KO. A preliminary population-based twin study of self-reported eating disorder. Psychol Med (2001) 31(2):3615. doi:10.1017/S0033291701003087
45. Boraska V, Franklin CS, Floyd JA, Thornton LM, Huckins LM, Southam L, et al. A genome-wide association study of anorexia nervosa. Mol Psychiatry (2014) 19(10):1085–94. doi:10.1038/mp.2013.187
46. Hinney A, Bornscheuer A, Depenbusch M, Mierke B, Tölle A, Middeke K, et al. No evidence for involvement of the leptin gene in anorexia nervosa, bulimia nervosa, underweight or early onset extreme obesity: identification of two novel mutations in the coding sequence and a novel polymorphism in the leptin gene linked upstream region. Mol Psychiatry (1998) 3:539–43. doi:10.1038/sj.mp.4000394
47. Quinton ND, Meechan DW, Brown K, Eastwood H, Blakemore AI. Single nucleotide polymorphisms in the leptin receptor gene: studies in anorexia nervosa. Psychiatr Genet (2004) 14:191–4. doi:10.1097/00041444-200412000-00004
48. Vink T, Hinney A, van Elburg AA, van Goozen SH, Sandkuijl LA, Sinke RJ, et al. Association between an agouti-related protein gene polymorphism and anorexia nervosa. Mol Psychiatry (2001) 6(3):3258. doi:10.1038/sj.mp.4000854
49. de Rijke CE, Jackson PJ, Garner KM, van Rozen RJ, Douglas NR, Kas MJ, et al. Functional analysis of the Ala67Thr polymorphism in agouti related protein associated with anorexia nervosa and leanness. Biochem Pharmacol (2005) 70(2):308–16. doi:10.1016/j.bcp.2005.04.033
50. Rohde K, Keller M, Horstmann A, Liu X, Eichelmann F, Stumvoll M, et al. Role of genetic variants in ADIPOQ in human eating behavior. Genes Nutr (2015) 10(1):449. doi:10.1007/s12263-014-0449-8
51. Ando T, Tamura N, Mera T, Morita C, Takei M, Nakamoto C, et al. Japanese genetic research group for eating disorders (2014). Association of the c.385C >A (p.Pro129Thr) polymorphism of the fatty acid amide hydrolase gene with anorexia nervosa in the Japanese population. Mol Genet Genomic Med (2014) 2(4):313–8. doi:10.1002/mgg3.69
52. Nilsson IA, Lindfors C, Schalling M, Hökfelt T, Johansen JE. Anorexia and hypothalamic degeneration. Vitam Horm (2013) 92:27–60. doi:10.1016/B978-0-12-410473-0.00002-7
53. Maltais LJ, Lane PW, Beamer WG. Anorexia, a recessive mutation causing starvation in preweanling mice. J Hered (1984) 75(6):468–72.
54. Jahng JW, Houpt TA, Kim SJ, Joh TH, Son JH. Neuropeptide Y mRNA and serotonin innervation in the arcuate nucleus of anorexia mutant mice. Brain Res (1998) 790(1–2):67–73. doi:10.1016/S0006-8993(98)00049-3
55. Johansen JE, Teixeira VL, Johansson C, Serrão P, Berggren PO, Soares-Da-Silva P, et al. Altered dopaminergic transmission in the anorexic anx/anx mouse striatum. Neuroreport (2001) 12(12):2737–41. doi:10.1097/00001756-200108280-00029
56. Lindfors C, Nilsson IA, Garcia-Roves PM, Zuberi AR, Karimi M, Donahue LR, et al. Hypothalamic mitochondrial dysfunction associated with anorexia in the anx/anx mouse. Proc Natl Acad Sci U S A (2011) 108(44):18108–13. doi:10.1073/pnas.1114863108
57. Broberger C, Johansen J, Schalling M, Hökfelt T. Hypothalamic neurohistochemistry of the murine anorexia (anx/anx) mutation: altered processing of neuropeptide Y in the arcuate nucleus. J Comp Neurol (1997) 387(1):124–35. doi:10.1002/(SICI)1096-9861(19971013)387:1<124::AID-CNE10>3.0.CO;2-U
58. Broberger C, Johansen J, Brismar H, Johansson C, Schalling M, Hökfelt T. Changes in neuropeptide Y receptors and pro-opiomelanocortin in the anorexia (anx/anx) mouse hypothalamus. J Neurosci (1999) 19(16):7130–9.
59. Mercader JM, Lozano JJ, Sumoy L, Dierssen M, Visa J, Gratacòs M, et al. Hypothalamus transcriptome profile suggests an anorexia-cachexia syndrome in the anx/anx mouse model. Physiol Genomics (2008) 35(3):341–50. doi:10.1152/physiolgenomics.90255.2008
60. Johansen JE, Broberger C, Lavebratt C, Johansson C, Kuhar MJ, Hökfelt T, et al. Hypothalamic CART and serum leptin levels are reduced in the anorectic (anx/anx) mouse. Mol Brain Res (2000) 84(1–2):97–105. doi:10.1016/S0169-328X(00)00228-X
61. Lachuer J, Ouyang L, Legras C, Del Rio J, Barlow C. Gene expression profiling reveals an inflammatory process in the anx/anx mutant mice. Brain Res Mol Brain Res (2005) 139(2):372–6. doi:10.1016/j.molbrainres.2005.06.003
62. Nilsson I, Lindfors C, Fetissov SO, Hökfelt T, Johansen JE. Aberrant agouti-related protein system in the hypothalamus of the anx/anx mouse is associated with activation of microglia. J Comp Neurol (2008) 507(1):1128–40. doi:10.1002/cne.21599
63. Veyrat-Durebex C, Alliot J. Changes in pattern of macronutrient intake during aging in male and female rats. Physiol Behav (1997) 62(6):1273–8. doi:10.1016/S0031-9384(97)00304-1
64. Alliot J, Boghossian S, Jourdan D, Veyrat-Durebex C, Pickering G, Meynial-Denis D, et al. The LOU/c/jall rat as an animal model of healthy aging? J Gerontol A Biol Sci Med Sci (2002) 57(8):B312–20. doi:10.1093/gerona/57.8.B312
65. Héliès JM, Diane A, Langlois A, Larue-Achagiotis C, Fromentin G, Tomé D, et al. Comparison of fat storage between Fischer 344 and obesity-resistant Lou/C rats fed different diets. Obes Res (2005) 13(1):3–10. doi:10.1038/oby.2005.3
66. Veyrat-Durebex C, Montet X, Vinciguerra M, Gjinovci A, Meda P, Foti M, et al. The Lou/C rat: a model of spontaneous food restriction associated with improved insulin sensitivity and decreased lipid storage in adipose tissue. Am J Physiol Endocrinol Metab (2009) 296(5):E1120–32. doi:10.1152/ajpendo.90592.2008
67. Perrin D, Mamet J, Géloën A, Morel G, Dalmaz Y, Pequignot JM. Sympathetic and brain monoaminergic regulation of energy balance in obesity-resistant rats (Lou/C). Auton Neurosci (2003) 109(1–2):1–9. doi:10.1016/j.autneu.2003.08.008
68. Duque G, Rivas D, Li W, Li A, Henderson JE, Ferland G, et al. Age-related bone loss in the LOU/c rat model of healthy ageing. Exp Gerontol (2009) 44(3):183–9. doi:10.1016/j.exger.2008.10.004
69. Kappeler L, Zizzari P, Grouselle D, Epelbaum J, Bluet-Pajot MT. Plasma and hypothalamic peptide-hormone levels regulating somatotroph function and energy balance in fed and fasted states: a comparative study in four strains of rats. J Neuroendocrinol (2004) 16(12):980–8. doi:10.1111/j.1365-2826.2004.01259.x
70. Mitchell SE, Nogueiras R, Rance K, Rayner DV, Wood S, Dieguez C, et al. Circulating hormones and hypothalamic energy balance: regulatory gene expression in the Lou/C and Wistar rats. J Endocrinol (2006) 190(3):571–9. doi:10.1677/joe.1.06576
71. Veyrat-Durebex C, Poher AL, Caillon A, Somm E, Vallet P, Charnay Y, et al. Improved leptin sensitivity as a potential candidate responsible for the spontaneous food restriction of the Lou/C rat. PLoS One (2013) 8(9):e73452. doi:10.1371/journal.pone.0073452
72. Kollen M, Stéphan A, Faivre-Bauman A, Loudes C, Sinet PM, Alliot J, et al. Preserved memory capacities in aged Lou/C/Jall rats. Neurobiol Aging (2010) 31(1):129–42. doi:10.1016/j.neurobiolaging.2008.03.010
73. Ingalls AM, Dickie MM, Snell GD. Obese, a new mutation in the house mouse. J Hered (1950) 41(12):317–8.
75. Weigle DS, Bukowski TR, Foster DC, Holderman S, Kramer JM, Lasser G, et al. Recombinant ob protein reduces feeding and body weight in the ob/ob mouse. J Clin Invest (1995) 96(4):2065–70. doi:10.1172/JCI118254
76. Coleman DL. Obese and diabetes: two mutant genes causing diabetes-obesity syndromes in mice. Diabetologia (1978) 14:141–8. doi:10.1007/BF00429772
77. Boey D, Lin S, Enriquez RF, Lee NJ, Slack K, Couzens M, et al. PYY transgenic mice are protected against diet-induced and genetic obesity. Neuropeptides (2008) 42(1):19–30. doi:10.1016/j.npep.2007.11.003
78. Shi YC, Hämmerle CM, Lee IC, Turner N, Nguyen AD, Riepler SJ, et al. Adult-onset PYY overexpression in mice reduces food intake and increases lipogenic capacity. Neuropeptides (2012) 46(4):173–82. doi:10.1016/j.npep.2012.04.001
79. Bewick GA, Kent A, Campbell D, Patterson M, Ghatei MA, Bloom SR, et al. Mice with hyperghrelinemia are hyperphagic and glucose intolerant and have reduced leptin sensitivity. Diabetes (2009) 58(4):840–6. doi:10.2337/db08-1428
80. Kirchner H, Gutierrez JA, Solenberg PJ, Pfluger PT, Czyzyk TA, Willency JA, et al. GOAT links dietary lipids with the endocrine control of energy balance. Nat Med (2009) 15(7):741–5. doi:10.1038/nm.1997
81. Ueno N, Inui A, Iwamoto M, Kaga T, Asakawa A, Okita M. Decreased food intake and body weight in pancreatic polypeptide-overexpressing mice. Gastroenterology (1999) 117(6):1427–32. doi:10.1016/S0016-5085(99)70293-3
82. Lacourse KA, Swanberg LJ, Gillespie PJ, Rehfeld JF, Saunders TL, Samuelson LC. Pancreatic function in CCK-deficient mice: adaptation to dietary protein does not require CCK. Am J Physiol (1999) 276(5 Pt 1):G1302–9.
83. Lo CM, King A, Samuelson LC, Kindel TL, Rider T, Jandacek RJ, et al. Cholecystokinin knockout mice are resistant to high-fat diet-induced obesity. Gastroenterology (2010) 138(5):1997–2005. doi:10.1053/j.gastro.2010.01.044
84. Palmiter RD, Erickson JC, Hollopeter G, Baraban SC, Schwartz MW. Life without neuropeptide Y. Recent Prog Horm Res (1998) 53:163–99.
85. Luquet S, Perez FA, Hnasko TS, Palmiter RD. NPY/AgRP neurons are essential for feeding in adult mice but can be ablated in neonates. Science (2005) 310(5748):683–5. doi:10.1126/science.1115524
86. Painsipp E, Wultsch T, Edelsbrunner ME, Tasan RO, Singewald N, Herzog H, et al. Reduced anxiety-like and depression-related behavior in neuropeptide Y Y4 receptor knockout mice. Genes Brain Behav (2008) 7(5):532–42. doi:10.1111/j.1601-183X.2008.00389.x
87. Zhang L, Riepler SJ, Turner N, Enriquez RF, Lee IC, Baldock PA. Y2 and Y4 receptor signaling synergistically act on energy expenditure and physical activity. Am J Physiol Regul Integr Comp Physiol (2010) 299(6):R1618–28. doi:10.1152/ajpregu.00345.2010
88. Shimada M, Tritos NA, Lowell BB, Flier JS, Maratos-Flier E. Mice lacking melanin-concentrating hormone are hypophagic and lean. Nature (1998) 396(6712):670–4. doi:10.1038/25341
89. Kokkotou E, Jeon JY, Wang X, Marino FE, Carlson M, Trombly DJ, et al. Mice with MCH ablation resist diet-induced obesity through strain-specific mechanisms. Am J Physiol Regul Integr Comp Physiol (2005) 289(1):R117–24. doi:10.1152/ajpregu.00861.2004
90. Cardinal P, Bellocchio L, Clark S, Cannich A, Klugmann M, Lutz B, et al. Hypothalamic CB1 cannabinoid receptors regulate energy balance in mice. Endocrinology (2012) 153(9):4136–43. doi:10.1210/en.2012-1405
91. Ravinet Trillou C, Delgorge C, Menet C, Arnone M, Soubrié P. CB1 cannabinoid receptor knockout in mice leads to leanness, resistance to diet-induced obesity and enhanced leptin sensitivity. Int J Obes (2004) 28:640–8. doi:10.1038/sj.ijo.0802583
92. Compan V, Zhou M, Grailhe R, Gazzara RA, Martin R, Gingrich J, et al. Attenuated response to stress and novelty and hypersensitivity to seizures in 5-HT4 receptor knock-out mice. J Neurosci (2004) 24(2):412–9. doi:10.1523/JNEUROSCI.2806-03.2004
93. Jean A, Conductier G, Manrique C, Bouras C, Berta P, Hen R, et al. Anorexia induced by activation of serotonin 5-HT4 receptors is mediated by increases in CART in the nucleus accumbens. Proc Natl Acad Sci U S A (2007) 104(41):16335–40. doi:10.1073/pnas.0701471104
94. Jean A, Laurent L, Bockaert J, Charnay Y, Dusticier N, Nieoullon A, et al. The nucleus accumbens 5-HTR4-CART pathway ties anorexia to hyperactivity. Transl Psychiatry (2012) 11(2):e203. doi:10.1038/tp.2012.131
95. Butt I, Hong A, Di J, Aracena S, Banerjee P, Shen CH. The effects of serotonin1A receptor on female mice body weight and food intake are associated with the differential expression of hypothalamic neuropeptides and the GABAA receptor. Neuropeptides (2014) 48(5):313–8. doi:10.1016/j.npep.2014.07.003
96. Szczypka MS, Rainey MA, Kim DS, Alaynick WA, Marck BT, Matsumoto AM, et al. Feeding behavior in dopamine-deficient mice. Proc Natl Acad Sci U S A (1999) 96(21):12138–43. doi:10.1073/pnas.96.21.12138
97. Unger TJ, Calderon GA, Bradley LC, Sena-Esteves M, Rios M. Selective deletion of Bdnf in the ventromedial and dorsomedial hypothalamus of adult mice results in hyperphagic behavior and obesity. J Neurosci (2007) 27(52):14265–74. doi:10.1523/JNEUROSCI.3308-07.2007
98. Toriya M, Maekawa F, Maejima Y, Onaka T, Fujiwara K, Nakagawa T, et al. Long-term infusion of brain-derived neurotrophic factor reduces food intake and body weight via a corticotrophin-releasing hormone pathway in the paraventricular nucleus of the hypothalamus. J Neuroendocrinol (2010) 22(9):987–95. doi:10.1111/j.1365-2826.2010.02039.x
99. Yamada M, Miyakawa T, Duttaroy A, Yamanaka A, Moriguchi T, Makita R, et al. Mice lacking the M3 muscarinic acetylcholine receptor are hypophagic and lean. Nature (2001) 410(6825):207–12. doi:10.1038/35065604
100. Jacobson L. Lower weight loss and food intake in protein-deprived, corticotropin releasing hormone-deficient mice correlate with glucocorticoid insufficiency. Endocrinology (1999) 140(8):3543–51. doi:10.1210/endo.140.8.6910
101. Dirks A, Groenink L, Bouwknecht JA, Hijzen TH, Van Der Gugten J, Ronken E, et al. Overexpression of corticotropin-releasing hormone in transgenic mice and chronic stress-like autonomic and physiological alterations. Eur J Neurosci (2002) 16(9):1751–60. doi:10.1046/j.1460-9568.2002.02245.x
102. Friedman JM. Leptin at 14 y of age: an ongoing story. Am J Clin Nutr (2009) 89(3):973S–9S. doi:10.3945/ajcn.2008.26788B
103. González CR, Vázquez MJ, López M, Diéguez C. Influence of chronic undernutrition and leptin on GOAT mRNA levels in rat stomach mucosa. J Mol Endocrinol (2008) 41(6):415–21. doi:10.1677/JME-08-0102
104. Sun Y, Butte NF, Garcia JM, Smith RG. Characterization of adult ghrelin and ghrelin receptor knockout mice under positive and negative energy balance. Endocrinology (2008) 149(2):843–50. doi:10.1210/en.2007-0271
105. Zhao TJ, Liang G, Li RL, Xie X, Sleeman MW, Murphy AJ, et al. Ghrelin O-acyltransferase (GOAT) is essential for growth hormone-mediated survival of calorie-restricted mice. Proc Natl Acad Sci U S A (2010) 107(16):7467–72. doi:10.1073/pnas.1002271107
106. McFarlane MR, Brown MS, Goldstein JL, Zhao TJ. Induced ablation of ghrelin cells in adult mice does not decrease food intake, body weight, or response to high-fat diet. Cell Metab (2014) 20(1):54–60. doi:10.1016/j.cmet.2014.04.007
107. Tomasik PJ, Sztefko K, Starzyk J, Rogatko I, Szafran Z. Entero-insular axis in children with anorexia nervosa. Psychoneuroendocrinology (2005) 30(4):364–72. doi:10.1016/j.psyneuen.2004.10.003
108. Kinzig KP, Coughlin JW, Redgrave GW, Moran TH, Guarda AS. Insulin, glucose, and pancreatic polypeptide responses to a test meal in restricting type anorexia nervosa before and after weight restoration. Am J Physiol Endocrinol Metab (2007) 292(5):E1441–6. doi:10.1152/ajpendo.00347.2006
109. Qian S, Chen H, Weingarth D, Trumbauer ME, Novi DE, Guan X, et al. Neither agouti-related protein nor neuropeptide Y is critically required for the regulation of energy homeostasis in mice. Mol Cell Biol (2002) 22(14):5027–35. doi:10.1128/MCB.22.14.5027-5035.2002
110. Erickson JC, Clegg KE, Palmiter RD. Sensitivity to leptin and susceptibility to seizures of mice lacking neuropeptide Y. Nature (1996) 381(6581):415–21. doi:10.1038/381415a0
111. Gunapala KM, Gallardo CM, Hsu CT, Steele AD. Single gene deletions of orexin, leptin, neuropeptide Y, and ghrelin do not appreciably alter food anticipatory activity in mice. PLoS One (2011) 6(3):e18377. doi:10.1371/journal.pone.0018377
112. Marsh DJ, Hollopeter G, Kafer KE, Palmiter RD. Role of the Y5 neuropeptide Y receptor in feeding and obesity. Nat Med (1998) 4(6):718–21. doi:10.1038/nm0698-718
113. Pedrazzini T, Seydoux J, Künstner P, Aubert JF, Grouzmann E, Beermann F, et al. Cardiovascular response, feeding behavior and locomotor activity in mice lacking the NPY Y1 receptor. Nat Med (1998) 4(6):722–6. doi:10.1038/nm0698-722
114. Nguyen AD, Mitchell NF, Lin S, Macia L, Yulyaningsih E, Baldock PA, et al. Y1 and Y5 receptors are both required for the regulation of food intake and energy homeostasis in mice. PLoS One (2012) 7(6):e40191. doi:10.1371/journal.pone.0040191
115. Wu Q, Howell MP, Cowley MA, Palmiter RD. Starvation after AgRP neuron ablation is independent of melanocortin signaling. Proc Natl Acad Sci U S A (2008) 105(7):2687–92. doi:10.1073/pnas.0712062105
116. Edelsbrunner ME, Painsipp E, Herzog H, Holzer P. Evidence from knockout mice for distinct implications of neuropeptide-Y Y2 and Y4 receptors in the circadian control of locomotion, exploration, water and food intake. Neuropeptides (2009) 43(6):491–7. doi:10.1016/j.npep.2009.08.007
117. Atalayer D, Robertson KL, Haskell-Luevano C, Andreasen A, Rowland NE. Food demand and meal size in mice with single or combined disruption of melanocortin type 3 and 4 receptors. Am J Physiol Regul Integr Comp Physiol (2009) 298(6):R1667–74. doi:10.1152/ajpregu.00562.2009
118. Macneil DJ. The role of melanin-concentrating hormone and its receptors in energy homeostasis. Front Endocrinol (Lausanne) (2013) 4:49. doi:10.3389/fendo.2013.00049
119. Alon T, Friedman JM. Late-onset leanness in mice with targeted ablation of melanin concentrating hormone neurons. J Neurosci (2006) 26:389–97. doi:10.1523/JNEUROSCI.1203-05.2006
120. Akiyama M, Yuasa T, Hayasaka N, Horikawa K, Sakurai T, Shibata S. Reduced food anticipatory activity in genetically orexin (hypocretin) neuron-ablated mice. Eur J Neurosci (2004) 20(11):3054–62. doi:10.1111/j.1460-9568.2004.03749.x
121. Ramanathan L, Siegel JM. Gender differences between hypocretin/orexin knockout and wild type mice: age, body weight, body composition, metabolic markers, leptin and insulin resistance. J Neurochem (2014) 131(5):615–24. doi:10.1111/jnc.12840
122. Marco EM, García-Gutiérrez MS, Bermúdez-Silva FJ, Moreira FA, Guimarães F, Manzanares J, et al. Endocannabinoid system and psychiatry: in search of a neurobiological basis for detrimental and potential therapeutic effects. Front Behav Neurosci (2011) 5(5):63. doi:10.3389/fnbeh.2011.00063
123. Giacoppo S, Mandolino G, Galuppo M, Bramanti P, Mazzon E. Cannabinoids: new promising agents in the treatment of neurological diseases. Molecules (2014) 19(11):18781–816. doi:10.3390/molecules191118781
124. Tabarin A, Diz-Chaves Y, Carmona Mdel C, Catargi B, Zorrilla EP, Roberts AJ, et al. Resistance to diet-induced obesity in mu-opioid receptor-deficient mice: evidence for a “thrifty gene”. Diabetes (2005) 54(12):3510–6. doi:10.2337/diabetes.54.12.3510
125. Czyzyk TA, Nogueiras R, Lockwood JF, McKinzie JH, Coskun T, Pintar JE, et al. Kappa-opioid receptors control the metabolic response to a high-energy diet in mice. FASEB J (2010) 24(4):1151–9. doi:10.1096/fj.09-143610
126. Czyzyk TA, Romero-Picó A, Pintar J, McKinzie JH, Tschöp MH, Statnick MA, et al. Nogueiras R. Mice lacking δ-opioid receptors resist the development of diet-induced obesity. FASEB J (2012) 26(8):3483–92. doi:10.1096/fj.12-208041
127. Kas MJ, van den Bos R, Baars AM, Lubbers M, Lesscher HM, Hillebrand JJ. Mu-opioid receptor knockout mice show diminished food-anticipatory activity. Eur J Neurosci (2004) 20(6):1624–32. doi:10.1111/j.1460-9568.2004.03581.x
128. Papaleo F, Kieffer BL, Tabarin A, Contarino A. Decreased motivation to eat in mu-opioid receptor-deficient mice. Eur J Neurosci (2007) 25(11):3398–405. doi:10.1111/j.1460-9568.2007.05595.x
129. Compan V. Chapter 8: do limits of neuronal plasticity represent an opportunity for mental diseases, such as addiction to food and illegal drugs? Use and utilities of serotonin receptor knock-out mice. In: Chattopadhyay A, editor. Serotonin Receptors in Neurobiology. Boca Raton, FL: CRC Press (2007). p. 207–39.
130. Lucas JJ, Yamamoto A, Scearce-Levie K, Saudou F, Hen R. Absence of fenfluramine-induced anorexia and reduced c-Fos induction in the hypothalamus and central amygdaloid complex of serotonin 1B receptor knock-out mice. J Neurosci (1998) 18(14):5537–44.
131. Bockaert J, Claeysen S, Compan V, Dumuis A. 5-HT(4) receptors, a place in the sun: act two. Curr Opin Pharmacol (2011) 11(1):87–93. doi:10.1016/j.coph.2011.01.012
132. Zhou QY, Palmiter RD. Dopamine-deficient mice are severely hypoactive, adipsic, and aphagic. Cell (1995) 83(7):1197–209. doi:10.1016/0092-8674(95)90145-0
133. Szczypka MS, Kwok K, Brot MD, Marck BT, Matsumoto AM, Donahue BA. Dopamine production in the caudate putamen restores feeding in dopamine-deficient mice. Neuron (2001) 30(3):819–28. doi:10.1016/S0896-6273(01)00319-1
134. Heusner CL, Hnasko TS, Szczypka MS, Liu Y, During MJ, Palmiter RD. Viral restoration of dopamine to the nucleus accumbens is sufficient to induce a locomotor response to amphetamine. Brain Res (2003) 980(2):266–74. doi:10.1016/S0006-8993(03)02986-X
135. Anderberg RH, Anefors C, Bergquist F, Nissbrandt H, Skibicka KP. Dopamine signaling in the amygdala, increased by food ingestion and GLP-1, regulates feeding behavior. Physiol Behav (2014) 136:135–44. doi:10.1016/j.physbeh.2014.02.026
136. Land BB, Narayanan NS, Liu RJ, Gianessi CA, Brayton CE, Grimaldi DM, et al. Medial prefrontal D1 dopamine neurons control food intake. Nat Neurosci (2014) 17(2):248–53. doi:10.1038/nn.3625
137. van Gestel MA, Kostrzewa E, Adan RA, Janhunen SK. Pharmacological manipulations in animal models of anorexia and binge eating in relation to humans. Br J Pharmacol (2014) 171(20):4767–84. doi:10.1111/bph.12789
138. Rask-Andersen M, Olszewski PK, Levine AS, Schiöth HB. Molecular mechanisms underlying anorexia nervosa: focus on human gene association studies and systems controlling food intake. Brain Res Rev (2009) 62(2):147–64. doi:10.1016/j.brainresrev.2009.10.007
139. Kim SF. Animal models of eating disorders. Neuroscience (2012) 211:2–12. doi:10.1016/j.neuroscience.2012.03.024
140. Matsui M, Yamada S, Oki T, Manabe T, Taketo MM, Ehlert FJ. Functional analysis of muscarinic acetylcholine receptors using knockout mice. Life Sci (2004) 75(25):2971–81. doi:10.1016/j.lfs.2004.05.034
141. Wei J, Walton EA, Milici A, Buccafusco JJ. M1-M5 muscarinic receptor distribution in rat CNS by RT-PCR and HPLC. J Neurochem (1994) 63(3):815–21. doi:10.1046/j.1471-4159.1994.63030815.x
142. Speakman JR, Mitchell SE. Caloric restriction. Mol Aspects Med (2011) 32(3):159–221. doi:10.1016/j.mam.2011.07.001
143. Austad SN. Does caloric restriction in the laboratory simply prevent overfeeding and return house mice to their natural level of food intake? Sci Aging Knowledge Environ (2001) 2001(6):e3. doi:10.1126/sageke.2001.6.pe3
144. Devlin MJ, Cloutier AM, Thomas NA, Panus DA, Lotinun S, Pinz I, et al. Caloric restriction leads to high marrow adiposity and low bone mass in growing mice. J Bone Miner Res (2010) 25(9):2078–88. doi:10.1002/jbmr.82
145. Bruss MD, Khambatta CF, Ruby MA, Aggarwal I, Hellerstein MK. Calorie restriction increases fatty acid synthesis and whole body fat oxidation rates. Am J Physiol Endocrinol Metab (2010) 298(1):E108–16. doi:10.1152/ajpendo.00524.2009
146. Chung KW, Kim DH, Park MH, Choi YJ, Kim ND, Lee J, et al. Recent advances in calorie restriction research on aging. Exp Gerontol (2013) 48(10):1049–53. doi:10.1016/j.exger.2012.11.007
147. Avraham Y, Dagon Y, Magen I, Berry EM. Models of anorexia. Drug Discov Today Dis Models (2005) 2(3):197–204.
148. Dos Santos ZA, Da Silva RJ, Bacurau RFP, Tirapegui J, Ribeiro SML. Effect of food restriction and intense physical training on estrus cyclicity and plasma leptin concentrations in rats. J Nutr Sci Vitaminol (Tokyo) (2010) 57(1):1–8. doi:10.3177/jnsv.57.1
149. Ravussin Y, Gutman R, Diano S, Shanabrough M, Borok E, Sarman B, et al. Effects of chronic weight perturbation on energy homeostasis and brain structure in mice. Am J Physiol Regul Integr Comp Physiol (2011) 300(6):R1352–62. doi:10.1152/ajpregu.00429.2010
150. Méquinion M, Caron E, Zgheib S, Stievenard A, Zizzari P, Tolle V, et al. Physical activity: benefit or weakness in metabolic adaptations in a mouse model of chronic food restriction? Am J Physiol Endocrinol Metab (2015) 308(3):E241–55. doi:10.1152/ajpendo.00340.2014
151. Bi S, Robinson BM, Moran TH. Acute food deprivation and chronic food restriction differentially affect hypothalamic NPY mRNA expression. Am J Physiol Regul Integr Comp Physiol (2003) 285(5):R1030–6. doi:10.1152/ajpregu.00734.2002
152. Hamrick MW, Ding KH, Ponnala S, Ferrari SL, Isales CM. Caloric restriction decreases cortical bone mass but spares trabecular bone in the mouse skeleton: implications for the regulation of bone mass by body weight. J Bone Miner Res (2008) 23(6):870–8. doi:10.1359/jbmr.080213
153. Padovani M, Lavigne JA, Chandramouli GVR, Perkins SN, Barrett JC, Hursting SD. Distinct effects of calorie restriction and exercise on mammary gland gene expression in C57Bl/6 mice. Cancer Prev Res (Phila) (2009) 2(12):1076–87. doi:10.1158/1940-6207.CAPR-09-0034
154. Hart PC, Bergner CL, Smolinsky AN, Dufour BD, Egan RJ, Laporte JL, et al. Experimental models of anxiety for drug discovery and brain research. Methods Mol Biol (2010) 602:299–321. doi:10.1007/978-1-60761-058-8_18
155. Yamamoto Y, Tanahashi T, Kawai T, Chikahisa S, Katsuura S, Nishida K, et al. Changes in behavior and gene expression induced by caloric restriction in C57BL/6 mice. Physiol Genomics (2009) 39(3):227–35. doi:10.1152/physiolgenomics.00082.2009
156. Pothos EN, Creese I, Hoebel BG. Restricted eating with weight loss selectively decreases extracellular dopamine in the nucleus accumbens and alters dopamine response to amphetamine, morphine, and food intake. J Neurosci (1995) 15(10):6640–50.
157. Tatsumi S, Ito M, Asaba Y, Tsutsumi K, Ikeda K. Life-long caloric restriction reveals biphasic and dimorphic effects on bone metabolism in rodents. Endocrinology (2008) 149(2):634–41. doi:10.1210/en.2007-1089
158. Heresi G, Chandra RK. Effects of severe calorie restriction on thymic factor activity and lymphocyte stimulation response in Rats. J Nutr (1980) 110:1888–93.
159. Gairdner SE, Amara CE. Serum leptin is not correlated with body fat in severe food restriction. Appl Physiol Nutr Metab (2012) 37:1063–71. doi:10.1139/h2012-092
160. Dhahbi JM, Kim HJ, Mote PL, Beaver RJ, Spindler SR. Temporal linkage between the phenotypic and genomic responses to caloric restriction. Proc Natl Acad Sci U S A (2004) 101:5524–9. doi:10.1073/pnas.0305300101
161. Zgheib S, Méquinion M, Lucas S, Leterme D, Ghali O, Tolle V, et al. Long-term physiological alterations and recovery in a mouse model of separation associated with time-restricted feeding: a tool to study anorexia nervosa related consequences. PLoS One (2014) 9:e103775. doi:10.1371/journal.pone.0103775
162. Jahng JW, Kim JG, Kim HJ, Kim BT, Kang DW, Lee JH. Chronic food restriction in young rats results in depression- and anxiety-like behaviors with decreased expression of serotonin reuptake transporter. Brain Res (2007) 30(1150):100–7. doi:10.1016/j.brainres.2007.02.080
163. Gillard ER, Dang DQ, Stanley BG. Evidence that neuropeptide Y and dopamine in the perifornical hypothalamus interact antagonistically in the control of food intake. Brain Res (1993) 628(1–2):128–36. doi:10.1016/0006-8993(93)90947-L
164. Avraham Y, Bonne O, Berry EM. Behavioral and neurochemical alterations caused by diet restriction – the effect of tyrosine administration in mice. Brain Res (1996) 732(1–2):133–44. doi:10.1016/0006-8993(96)00514-8
165. Garpenstrand H, Annas P, Ekblom J, Oreland L, Fredrikson M. Human fear conditioning is related to dopaminergic and serotonergic biological markers. Behav Neurosci (2001) 115(2):358–64. doi:10.1037/0735-7044.115.2.358
166. Fetissov SO, Meguid MM, Sato T, Zhang LH. Expression of dopaminergic receptors in the hypothalamus of lean and obese Zucker rats and food intake. Am J Physiol Regul Integr Comp Physiol (2002) 283(4):R905–10. doi:10.1152/ajpregu.00092.2002
167. Calandreau L, Jaffard R, Desmedt A. Dissociated roles for the lateral and medial septum in elemental and contextual fear conditioning. Learn Mem (2007) 14(6):422–9. doi:10.1101/lm.531407
168. Lindblom J, Johansson A, Holmgren A, Grandin E, Nedergård C, Fredriksson R, et al. Increased mRNA levels of tyrosine hydroxylase and dopamine transporter in the VTA of male rats after chronic food restriction. Eur J Neurosci (2006) 23(1):180–6. doi:10.1111/j.1460-9568.2005.04531.x
169. Gwirtsman HE, Kaye WH, George DT, Jimerson DC, Ebert MH, Gold PW. Central and peripheral ACTH and cortisol levels in anorexia nervosa and bulimia. Arch Gen Psychiatry (1989) 46(1):61–9. doi:10.1001/archpsyc.1989.01810010063009
170. Méquinion M, Langlet F, Zgheib S, Dickson S, Dehouck B, Chauveau C, et al. Ghrelin: central and peripheral implications in anorexia nervosa. Front Endocrinol (Lausanne) (2013) 26(4):15. doi:10.3389/fendo.2013.00015
171. Oster MH, Fielder PJ, Levin N, Cronin MJ. Adaptation of the growth hormone and insulin-like growth factor-I axis to chronic and severe calorie or protein malnutrition. J Clin Invest (1995) 95(5):2258–65. doi:10.1172/JCI117916
172. Johansson A, Fredriksson R, Winnergren S, Hulting AL, Schiöth HB, Lindblom J. The relative impact of chronic food restriction and acute food deprivation on plasma hormone levels and hypothalamic neuropeptide expression. Peptides (2008) 29(9):1588–95. doi:10.1016/j.peptides.2008.04.018
173. Reimer RA, Maurer AD, Lau DC, Auer RN. Long-term dietary restriction influences plasma ghrelin and GOAT mRNA level in rats. Physiol Behav (2010) 99(5):605–10. doi:10.1016/j.physbeh.2010.01.034
174. Sisk CL, Bronson FH. Effects of food restriction and restoration on gonadotropin and growth hormone secretion in immature male rats. Biol Reprod (1986) 35(3):554–61. doi:10.1095/biolreprod35.3.554
175. Kubicky RA, Wu S, Kharitonenkov A, De Luca F. Role of fibroblast growth factor 21 (FGF-21) in undernutrition-related attenuation of growth in mice. Endocrinology (2012) 153(5):2287–95. doi:10.1210/en.2011-1909
176. Haider S, Haleem DJ. Decreases of brain serotonin following a food restriction schedule of 4 weeks in male and female rats. Med Sci Monit (2000) 6(6):1061–7.
177. Belda X, Ons S, Carrasco J, Armario A. The effects of chronic food restriction on hypothalamic-pituitary-adrenal activity depend on morning versus evening availability of food. Pharmacol Biochem Behav (2005) 81(1):41–6. doi:10.1016/j.pbb.2005.02.009
178. Verwey M, Amir S. Variable restricted feeding disrupts the daily oscillations of Period2 expression in the limbic forebrain and dorsal striatum in rats. J Mol Neurosci (2012) 46(2):258–64. doi:10.1007/s12031-011-9529-z
179. Rothschild J, Hoddy KK, Jambazian P, Varady KA. Time-restricted feeding and risk of metabolic disease: a review of human and animal studies. Nutr Rev (2014) 72(5):308–18. doi:10.1111/nure.12104
180. McAmis AJ, Anderson WE, Mendel LB. Growth of rats on “fat-free” diets. J Biol Chem (1929) 82:247–62.
181. Burr GO, Burr MM. On the nature and role of the fatty acids essential in nutrition. J Biol Chem (1930) 86:587–621.
182. Wesson LG, Burr GO. The metabolic rate and respiratory quotients of rats on a fat-defi cient diet. J Biol Chem (1931) 91:525–39.
183. Delion S, Chalon S, Guilloteau D, Besnard JC, Durand G. Alpha-linolenic acid dietary deficiency alters age-related changes of dopaminergic and serotoninergic neurotransmission in the rat frontal cortex. J Neurochem (1996) 66(4):1582–91. doi:10.1046/j.1471-4159.1996.66041582.x
184. Zimmer L, Delpal S, Guilloteau D, Aïoun J, Durand G, Chalon S. Chronic n-3 polyunsaturated fatty acid deficiency alters dopamine vesicle density in the rat frontal cortex. Neurosci Lett (2000) 284(1–2):25–8. doi:10.1016/S0304-3940(00)00950-2
185. Staszkiewicz J, Horswell R, Argyropoulos G. Chronic consumption of a low-fat diet leads to increased hypothalamic agouti-related protein and reduced leptin. Nutrition (2007) 23(9):665–71. doi:10.1016/j.nut.2007.06.001
186. Bielohuby M, Sawitzky M, Stoehr BJ, Stock P, Menhofer D, Ebensing S, et al. Lack of dietary carbohydrates induces hepatic growth hormone (GH) resistance in rats. Endocrinology (2011) 152(5):1948–60. doi:10.1210/en.2010-1423
187. Cheng Y, Meng Q, Wang C, Li H, Huang Z, Chen S, et al. Leucine deprivation decreases fat mass by stimulation of lipolysis in white adipose tissue and upregulation of uncoupling protein 1 (UCP1) in brown adipose tissue. Diabetes (2010) 59(1):17–25. doi:10.2337/db09-0929
188. Goto S, Nagao K, Bannai M, Takahashi M, Nakahara K, Kangawa K, et al. Anorexia in rats caused by a valine-deficient diet is not ameliorated by systemic ghrelin treatment. Neuroscience (2010) 166(1):333–40. doi:10.1016/j.neuroscience.2009.12.013
189. Narita K, Nagao K, Bannai M, Ichimaru T, Nakano S, Murata T, et al. Dietary deficiency of essential amino acids rapidly induces cessation of the rat estrous cycle. PLoS One (2011) 6(11):e28136. doi:10.1371/journal.pone.0028136
190. Anthony TG, Gietzen DW. Detection of amino acid deprivation in the central nervous system. Curr Opin Clin Nutr Metab Care (2013) 16(1):96–101. doi:10.1097/MCO.0b013e32835b618b
191. Watts AG, Sanchez-Watts G, Kelly AB. Distinct patterns of neuropeptide gene expression in the lateral hypothalamic area and arcuate nucleus are associated with dehydration-induced anorexia. J Neurosci (1999) 19(14):6111–21.
192. de Gortari P, Mancera K, Cote-Vélez A, Amaya MI, Martínez A, Jaimes-Hoy L, et al. Involvement of CRH-R2 receptor in eating behavior and in the response of the HPT axis in rats subjected to dehydration-induced anorexia. Psychoneuroendocrinology (2009) 34(2):259–72. doi:10.1016/j.psyneuen.2008.09.010
193. García-Luna C, Amaya MI, Alvarez-Salas E, de Gortari P. Prepro-orexin and feeding-related peptide receptor expression in dehydration-induced anorexia. Regul Pept (2010) 159(1–3):54–60. doi:10.1016/j.regpep.2009.09.011
194. Du Ruisseau P, Taché Y, Brazeau P, Collu R. Effects of chronic immobilization stress on pituitary hormone secretion, on hypothalamic factor levels, and on pituitary responsiveness to LHRH and TRH in female rats. Neuroendocrinology (1979) 29(2):90–9. doi:10.1159/000122910
195. Marti O, Marti J, Armario A. Effects of chronic stress on food intake in rats: influence of stressor intensity and duration of daily exposure. Physiol Behav (1994) 55(4):747–53. doi:10.1016/0031-9384(94)90055-8
196. Baubet V, Fèvre-Montange M, Gay N, Debilly G, Bobillier P, Cespuglio R. Effects of an acute immobilization stress upon proopiomelanocortin (POMC) mRNA levels in the mediobasal hypothalamus: a quantitative in situ hybridization study. Brain Res Mol Brain Res (1994) 26(1–2):163–8. doi:10.1016/0169-328X(94)90087-6
197. Rybkin II, Zhou Y, Volaufova J, Smagin GN, Ryan DH, Harris ABS. Effect of restraint stress on food intake and body weight is determined by time of day. Am J Physiol (1997) 273(5 Pt 2):R1612–22.
198. Viau V, Sawchenko PE. Hypophysiotropic neurons of the paraventricular nucleus respond in spatially, temporally, and phenotypically differentiated manners to acute vs. Repeated restraint stress. J Comp Neurol (2002) 445(4):293–307. doi:10.1002/cne.10178
199. Harris RBS, Palmondon J, Leshin S, Flatt WP, Richard D. Chronic disruption of body weight but not of stress peptides or receptors in rats exposed to repeated restraint stress. Horm Behav (2006) 49(5):615–25. doi:10.1016/j.yhbeh.2005.12.001
200. Gasparetti AL, de Souza CT, Pereira-da-Silva M, Oliveira RL, Saad MJ, Carneiro EM, et al. Cold exposure induces tissue-specific modulation of the insulin-signalling pathway in Rattus norvegicus. J Physiol (2003) 552(Pt 1):149–62. doi:10.1113/jphysiol.2003.050369
201. Pereira Da Silva M, Torsoni MA, Nourani HV, Augusto VD, Souza CT, Gasparetti AL, et al. Hypothalamic melanin-concentrating hormone is induced by cold exposure and participates in the control of energy expenditure in rats. Endocrinology (2003) 144(11):4831–40. doi:10.1210/en.2003-0243
202. Zhao ZJ, Chi QS, Cao J, Han YD. The energy budget, thermogenic capacity and behavior in Swiss mice exposed to a consecutive decrease in temperatures. J Exp Biol (2010) 213(Pt 23):3988–97. doi:10.1242/jeb.046821
203. Willner P, Towell A, Sampson D, Sophokleous S, Muscat R. Reduction of sucrose preference by chronic unpredictable mild stress, and its restoration by a tricyclic antidepressant. Psychopharmacology (Berl) (1987) 93(3):358–64. doi:10.1007/BF00187257
204. Kim H, Whang WW, Kim HT, Pyun KH, Cho SY, Hahm DH, et al. Expression of neuropeptide Y and cholecystokinin in the rat brain by chronic mild stress. Brain Res (2003) 983(1–2):201–8. doi:10.1016/S0006-8993(03)03087-7
205. Flak JN, Jankord R, Solomon MB, Krause EG, Herman JP. Opposing effects of chronic stress and weight restriction on cardiovascular, neuroendocrine and metabolic function. Physiol Behav (2011) 104(2):228–34. doi:10.1016/j.physbeh.2011.03.002
206. Meerlo P, Overkamp GJ, Daan S, Van Den Hoofdakker RH, Koolhaas JM. Changes in behaviour and body weight following a single or double social defeat in rats. Stress (1996) 1(1):21–32. doi:10.3109/10253899609001093
207. Lucas LR, Celen Z, Tamashiro KL, Blanchard RJ, Blanchard DC, Markham C, et al. Repeated exposure to social stress has long-term effects on indirect markers of dopaminergic activity in brain regions associated with motivated behavior. Neuroscience (2004) 124(2):449–57. doi:10.1016/j.neuroscience.2003.12.009
208. Tamashiro KLK, Hegeman MA, Nguyen MNN, Melhorn SJ, Ma LY, Woods SC, et al. Dynamic body weight and body composition changes in response to subordination stress. Physiol Behav (2007) 91(4):440–8. doi:10.1016/j.physbeh.2007.04.004
209. Melhorn SJ, Krause EG, Scott KA, Mooney MR, Johnson JD, Woods SC, et al. Meal patterns and hypothalamic NPY expression during chronic social stress and recovery. Am J Physiol Regul Integr Comp Physiol (2010) 299(3):R813–22. doi:10.1152/ajpregu.00820.2009
210. Boakes RA. Self-starvation in the rat: running versus eating. Span J Psychol (2007) 10(2):251–7. doi:10.1017/S113874160000651X
211. de Rijke CE, Hillebrand JJ, Verhagen LA, Roeling TA, Adan RA. Hypothalamic neuropeptide expression following chronic food restriction in sedentary and wheel-running rats. J Mol Endocrinol (2005) 35(2):381–90. doi:10.1677/jme.1.01808
212. Duclos M, Gatti C, Bessière B, Mormède P. Tonic and phasic effects of corticosterone on food restriction-induced hyperactivity in rats. Psychoneuroendocrinology (2009) 34(3):436–45. doi:10.1016/j.psyneuen.2008.10.008
213. Filaire E, Rouveix M, Massart A, Gladine C, Davicco MJ, Durand D. Lipid peroxidation and antioxidant status in rat: effect of food restriction and wheel running. Eur J Appl Physiol (2009) 107(2):243–50. doi:10.1007/s00421-009-1121-7
214. Verhagen LAW, Luijendijk MCM, Korte-Bouws GAH, Korte SM, Adan RAH. Dopamine and serotonin release in the nucleus accumbens during starvation-induced hyperactivity. Eur Neuropsychopharmacol (2009) 19:309–16. doi:10.1016/j.euroneuro.2008.12.008
215. Adan RAH, Hillebrand JJG, Danner UN, Cardona Cano S, Kas MJH, Verhagen LAW. Neurobiology driving hyperactivity in activity-based anorexia. Curr Top Behav Neurosci (2010) 6:229–50. doi:10.1007/7854_2010_77
216. Gutierrez E. A rat in the labyrinth of anorexia nervosa: contributions of the activity-based anorexia rodent model to the understanding of anorexia nervosa. Int J Eat Disord (2013) 46(4):289–301. doi:10.1002/eat.22095
217. van Leeuwen SD, Bonne OB, Avraham Y, Berry EM. Separation as a new animal model for self-induced weight loss. Physiol Behav (1997) 62(1):77–81. doi:10.1016/S0031-9384(97)00144-3
218. Hao S, Avraham Y, Bonne O, Berry EM. Separation-induced body weight loss, impairment in alternation behavior, and autonomic tone: effects of tyrosine. Pharmacol Biochem Behav (2001) 68:273–81. doi:10.1016/S0091-3057(00)00448-2
219. Sherman H, Genzer Y, Cohen R, Chapnik N, Madar Z, Froy O. Timed high-fat diet resets circadian metabolism and prevents obesity. FASEB J (2012) 26(8):3493–502. doi:10.1096/fj.12-208868
220. Hatori M, Vollmers C, Zarrinpar A, DiTacchio L, Bushong EA, Gill S, et al. Time-restricted feeding without reducing caloric intake prevents metabolic diseases in mice fed a high-fat diet. Cell Metab (2012) 15(6):848–60. doi:10.1016/j.cmet.2012.04.019
221. Inoue K, Zorrilla EP, Tabarin A, Valdez GR, Iwasaki S, Kiriike N, et al. Reduction of anxiety after restricted feeding in the rat: implication for eating disorders. Biol Psychiatry (2004) 55(11):1075–81. doi:10.1016/j.biopsych.2004.01.026
222. Funch JP, Jart A, Dam H. The effects of diets with no fat or with hydrogenated or unhydrogenated fat on growth and tissue pathology of rats. Br J Nutr (1960) 14:171–81. doi:10.1079/BJN19600023
223. Obici S, Feng Z, Morgan K, Stein D, Karkanias G, Rossetti L. Central administration of oleic acid inhibits glucose production and food intake. Diabetes (2002) 51(2):271–5. doi:10.2337/diabetes.51.2.271
224. Le Foll C, Dunn-Meynell A, Musatov S, Magnan C, Levin BE. FAT/CD36: a major regulator of neuronal fatty acid sensing and energy homeostasis in rats and mice. Diabetes (2013) 62(8):2709–16. doi:10.2337/db12-1689
225. Le Foll C, Dunn-Meynell AA, Miziorko HM, Levin BE. Regulation of hypothalamic neuronal sensing and food intake by ketone bodies and fatty acids. Diabetes (2014) 63(4):1259–69. doi:10.2337/db13-1090
226. Delion S, Chalon S, Hérault J, Guilloteau D, Besnard JC, Durand G. Chronic dietary alpha-linolenic acid deficiency alters dopaminergic and serotoninergic neurotransmission in rats. J Nutr (1994) 124(12):2466–76.
227. Zimmer L, Hembert S, Durand G, Breton P, Guilloteau D, Besnard JC, et al. Chronic n-3 polyunsaturated fatty acid diet-deficiency acts on dopamine metabolism in the rat frontal cortex: a microdialysis study. Neurosci Lett (1998) 240(3):177–81. doi:10.1016/S0304-3940(97)00938-5
228. Borghjid S, Feinman RD. Response of C57Bl/6 mice to a carbohydrate-free diet. Nutr Metab (Lond) (2012) 9(1):69. doi:10.1186/1743-7075-9-69
229. Prousky JE. Pellagra may be a rare secondary complication of anorexia nervosa: a systematic review of the literature. Altern Med Rev (2003) 8(2):180–5.
230. Schreiber W, Schweiger U, Werner D, Brunner G, Tuschl RJ, Laessle RG, et al. Circadian pattern of large neutral amino acids, glucose, insulin, and food intake in anorexia nervosa and bulimia nervosa. Metabolism (1991) 40:503–7. doi:10.1016/0026-0495(91)90231-K
231. Askenazy F, Candito M, Caci H, Myquel M, Chambon P, Darcourt G, et al. Whole blood serotonin content, tryptophan concentrations, and impulsivity in anorexia nervosa. Biol Psychiatry (1998) 43:188–95. doi:10.1016/S0006-3223(97)00299-0
232. Ehrlich S, Franke L, Schneider N, Salbach-Andrae H, Schott R, Craciun EM, et al. Aromatic amino acids in weight-recovered females with anorexia nervosa. Int J Eat Disord (2009) 42:166–72. doi:10.1002/eat.20575
233. Comai S, Bertazzo A, Carretti N, Podfigurna-Stopa A, Luisi S, Costa CVL. Serum levels of tryptophan, 5-hydroxytryptophan and serotonin in patients affected with different forms of amenorrhea. Int J Tryptophan Res (2010) 3:69–75. doi:10.4137/IJTR.S3804
234. Cusick PK, Koehler KM, Ferrier B, Haskell BE. The neurotoxicity of valine deficiency in rats. J Nutr (1978) 108(7):1200–6.
235. Cusick PK, Lowrie PM, Mehta T, Haskell BE. The ultrastructure of red nuclei neurons in the valine-deficient rat. Peptides (1979) 19(3):527–35.
236. Nagao K, Bannai M, Nakahara K, Murakami N. Functions of dietary valine as revealed by dietary valine-deficient. Amino Acids (2012) 42(4):1397–404. doi:10.1007/s00726-011-0836-z
237. D’Souza DN, Zhang Y, Garcia F, Battaglia G, Van de Kar LD. Fluoxetine-induced changes in body weight and 5-HT1A receptor-mediated hormone secretion in rats on a tryptophan-deficient diet. Am J Physiol Regul Integr Comp Physiol (2004) 286(2):R390–7. doi:10.1152/ajpregu.00335.2003
238. Browne CA, Clarke G, Dinan TG, Cryan JF. An effective dietary method for chronic tryptophan depletion in two mouse strains illuminates a role for 5-HT in nesting behaviour. Neuropharmacology (2012) 62(5–6):1903–15. doi:10.1016/j.neuropharm.2011.12.009
239. Lee H, Ohno M, Ohta S, Mikami T. Regular moderate or intense exercise prevents depression-like behavior without change of hippocampal tryptophan content in chronically tryptophan-deficient and stressed mice. PLoS One (2013) 8(7):e66996. doi:10.1371/journal.pone.0066996
240. Ardis TC, Cahir M, Elliott JJ, Bell R, Reynolds GP, Cooper SJ. Effect of acute tryptophan depletion on noradrenaline and dopamine in the rat brain. J Psychopharmacol (2009) 23(1):51–5. doi:10.1177/0269881108089597
241. Blokland A, Lieben C, Deutz NE. Anxiogenic and depressive-like effects, but no cognitive deficits, after repeated moderate tryptophan depletion in the rat. J Psychopharmacol (2002) 16(1):39–49. doi:10.1177/026988110201600112
242. Leung PM, Rogers QR, Harper AE. Effect of amino acid imbalance on dietary choice in the rat. J Nutr (1968) 95(3):483–92.
243. Leung PM, Rogers QR. Food intake: regulation by plasma amino acid pattern. Life Sci (1969) 8(2):1–9. doi:10.1016/0024-3205(69)90110-6
244. Feurté S, Nicolaidis S, Berridge KC. Conditioned taste aversion in rats for a threonine-deficient diet: demonstration by the taste reactivity test. Physiol Behav (2000) 68(3):423–9. doi:10.1016/S0031-9384(99)00202-4
245. Caregaro L, Di Pascoli L, Favaro A, Nardi M, Santonastaso P. Sodium depletion and hemoconcentration: overlooked complications in patients with anorexia nervosa? Nutrition (2005) 21(4):438–45. doi:10.1016/j.nut.2004.08.022
246. Kanbur N, Katzman DK. Impaired osmoregulation in anorexia nervosa: review of the literature. Pediatr Endocrinol Rev (2011) 8(3):218–21.
247. Gutman Y, Krausz M. Regulation of food and water intake in rats as related to plasma osmolarity and volume. Physiol Behav (1969) 4(3):311–3. doi:10.1016/0031-9384(69)90181-4
248. Watts AG. Dehydration-associated anorexia: development and rapid reversal. Physiol Behav (1999) 65(4–5):871–8. doi:10.1016/S0031-9384(98)00244-3
249. Callahan JB, Rinaman L. The postnatal emergence of dehydration anorexia in rats is temporally associated with the emergence of dehydration-induced inhibition of gastric emptying. Physiol Behav (1998) 64(5):683–7. doi:10.1016/S0031-9384(98)00139-5
250. Boyle CN, Lorenzen SM, Compton D, Watts AG. Dehydration-anorexia derives from a reduction in meal size, but not meal number. Physiol Behav (2012) 105(2):305–14. doi:10.1016/j.physbeh.2011.08.005
251. Hardaway JA, Crowley NA, Bulik CM, Kash TL. Integrated circuits and molecular components for stress and feeding: implications for eating disorders. Genes Brain Behav (2015) 14(1):85–97. doi:10.1111/gbb.12185
252. Shimizu N, Oomura Y, Kai Y. Stress-induced anorexia in rats mediated by serotonergic mechanisms in the hypothalamus. Physiol Behav (1989) 46(5):835–41. doi:10.1016/0031-9384(89)90045-0
253. Harris RB, Zhou J, Mitchell T, Hebert S, Ryan DH. Rats fed only during the light period are resistant to stress-induced weight loss. Physiol Behav (2002) 76(4–5):543–50. doi:10.1016/S0031-9384(02)00754-0
254. Chiba S, Numakawa T, Ninomiya M, Richards MC, Wakabayashi C, Kunugi H. Chronic restraint stress causes anxiety- and depression-like behaviors, downregulates glucocorticoid receptor expression, and attenuates glutamate release induced by brain-derived neurotrophic factor in the prefrontal cortex. Prog Neuropsychopharmacol Biol Psychiatry (2012) 39(1):112–9. doi:10.1016/j.pnpbp.2012.05.018
255. Harris RB, Mitchell TD, Simpson J, Redmann SM Jr, Youngblood BD, Ryan DH. Weight loss in rats exposed to repeated acute restraint stress is independent of energy or leptin status. Am J Physiol Regul Integr Comp Physiol (2002) 282(1):R77–88.
256. Zheng J, Dobner A, Babygirija R, Ludwig K, Takahashi T. Effects of repeated restraint stress on gastric motility in rats. Am J Physiol Regul Integr Comp Physiol (2009) 296(5):R1358–65. doi:10.1152/ajpregu.90928.2008
257. Calvez J, Fromentin G, Nadkarni N, Darcel N, Even P, Tomé D, et al. Inhibition of food intake induced by acute stress in rats is due to satiation effects. Physiol Behav (2011) 104(5):675–83. doi:10.1016/j.physbeh.2011.07.012
258. Jeong JY, Lee DH, Kang SS. Effects of chronic restraint stress on body weight, food intake, and hypothalamic gene expressions in mice. Endocrinol Metab (Seoul) (2013) 28(4):288–96. doi:10.3803/EnM.2013.28.4.288
259. Patterson-Buckendahl P, Rusnák M, Fukuhara K, Kvetnanský R. Repeated immobilization stress reduces rat vertebral bone growth and osteocalcin. Am J Physiol Regul Integr Comp Physiol (2001) 280(1):R79–86.
260. Gao B, Kikuchi-Utsumi K, Ohinata H, Hashimoto M, Kuroshima A. Repeated immobilization stress increases uncoupling protein 1 expression and activity in Wistar rats. Jpn J Physiol (2003) 53(3):205–13. doi:10.2170/jjphysiol.53.205
261. Girotti M, Pace TW, Gaylord RI, Rubin BA, Herman JP, Spencer RL. Habituation to repeated restraint stress is associated with lack of stress-induced c-Fos expression in primary sensory processing areas of the rat brain. Neuroscience (2006) 138(4):1067–81. doi:10.1016/j.neuroscience.2005.12.002
262. Liu J, Garza JC, Truong HV, Henschel J, Zhang W, Lu XY. The melanocortinergic pathway is rapidly recruited by emotional stress and contributes to stress-induced anorexia and anxiety-like behavior. Endocrinology (2007) 148(11):5531–40. doi:10.1210/en.2007-0745
263. Chagra SL, Zavala JK, Hall MV, Gosselink KL. Acute and repeated restraint differentially activate orexigenic pathways in the rat hypothalamus. Regul Pept (2011) 167(1):70–8. doi:10.1016/j.regpep.2010.11.006
264. Larsen PJ, Mau SE. Effect of acute stress on the expression of hypothalamic messenger ribonucleic acids encoding the endogenous opioid precursors preproenkephalin A and proopiomelanocortin. Peptides (1994) 15(5):783–90. doi:10.1016/0196-9781(94)90030-2
265. Krahn DD, Gosnell BA, Majchrzak MJ. The anorectic effects of CRH and restraint stress decrease with repeated exposures. Biol Psychiatry (1990) 27(10):1094–102. doi:10.1016/0006-3223(90)90046-5
266. Sweerts BW, Jarrott B, Lawrence AJ. The effect of acute and chronic restraint on the central expression of prepro-neuropeptide Y mRNA in normotensive and hypertensive rats. J Neuroendocrinol (2001) 13(7):608–17. doi:10.1046/j.1365-2826.2001.00674.x
267. Xu J, Bekaer AJM, Dupont J, Rouve S, Annesi-Maesano I, De Magalhaes Filho CD, et al. Exploring endocrine GH pattern in mice using rank plot analysis and random blood samples. J Endocrinol (2010) 208(2):119–29. doi:10.1677/JOE-10-0317
268. Armario A, Marti O, Gavald A, Giralt M, Jolfn T. Effects of chronic immobilization stress on GH and TSH secretion in the rat: response to hypothalamic regulatory factors. Psychoneuroendocrinology (1993) 18(5–6):405–13. doi:10.1016/0306-4530(93)90015-D
269. Yoshizato H, Fujikawa T, Soya H, Tanaka M, Nakashima K. The growth hormone (GH) gene is expressed in the lateral hypothalamus: enhancement by GH-releasing hormone and repression by restraint stress. Endocrinology (1998) 139(5):2545–51. doi:10.1210/endo.139.5.6009
270. Benyassi A, Gavalà A, Armario A, Arancibia S. Role of somatostatin in the acute immobilization stress-induced GH decrease in rat. Life Sci (1992) 52(4):361–70. doi:10.1016/0024-3205(93)90149-W
271. Gutierrez E, Vazquez R. Heat in the treatment of patients with anorexia nervosa. Eat Weight Disord (2001) 6(1):49–52. doi:10.1007/BF03339752
272. Carrera O, Adan RA, Gutierrez E, Danner UN, Hoek HW, van Elburg AA, et al. Hyperactivity in anorexia nervosa: warming up not just burning-off calorie. PLoS One (2012) 7(7):e41851. doi:10.1371/journal.pone.0041851
273. Nakatsuka H, Shoji Y, Tsuda T. Effects of cold exposure on gaseous metabolism and body composition in the rat. Comp Biochem Physiol A Comp Physiol (1983) 75(1):21–5. doi:10.1016/0300-9629(83)90038-5
274. Rowland N. Effects of chronic cold exposure on wheel running, food intake and fatty acid synthesis in Syrian hamsters. Physiol Behav (1984) 33(2):253–6. doi:10.1016/0031-9384(84)90107-0
275. Ohtani N, Sugano T, Ohta M. Alterations in monoamines and GABA in the ventromedial and paraventricular nuclei of the hypothalamus following cold exposure: a reduction in noradrenaline induces hyperphagia. Brain Res (1999) 842(1):6–14. doi:10.1016/S0006-8993(99)01796-5
276. Brito NA, Brito MN, Bartness TJ. Differential sympathetic drive to adipose tissues after food deprivation, cold exposure or glucoprivation. Am J Physiol Regul Integr Comp Physiol (2008) 294(5):R1445–52. doi:10.1152/ajpregu.00068.2008
277. Young JB, Saville E, Rothwell NJ, Stock MJ, Landsberg L, Dana CA. Effect of diet and cold exposure on norepinephrine turnover in brown adipose tissue of the rat. J Clin Invest (1982) 69(5):1061–71. doi:10.1172/JCI110541
278. McAllister TA, Thompson JR, Samuels SE. Skeletal and cardiac muscle protein turnover during cold acclimation in young rats. Am J Physiol Regul Integr Comp Physiol (2000) 278(3):R705–11.
279. Manfredi LH, Zanon NM, Garófalo MA, Navegantes LC, Kettelhut IC. Effect of short-term cold exposure on skeletal muscle protein breakdown in rats. J Appl Physiol (1985) (2013) 115(10):1496–505. doi:10.1152/japplphysiol.00474.2013
280. Cabral A, Valdivia S, Reynaldo M, Cyr NE, Nillni EA, Perello M. Short-term cold exposure activates TRH neurons exclusively in the hypothalamic paraventricular nucleus and raphe pallidus. Neurosci Lett (2012) 518(2):86–91. doi:10.1016/j.neulet.2012.04.059
281. Yu XX, Lewin DA, Forrest W, Adams SH. Cold elicits the simultaneous induction of fatty acid synthesis and oxidation in murine brown adipose tissue: prediction from differential gene expression and confirmation in vivo. FASEB J (2002) 16(2):155–68. doi:10.1096/fj.01-0568com
282. Rogers RC, Barnes MJ, Hermann GE. Leptin “gates” thermogenic action of thyrotropin-releasing hormone in the hindbrain. Brain Res (2009) 27(1295):135–41. doi:10.1016/j.brainres.2009.07.063
283. Barnes MJ, Rogers RC, Van Meter MJ, Hermann GE. Co-localization of TRHR1 and LepRb receptors on neurons in the hindbrain of the rat. Brain Res (2010) 8(1355):70–85. doi:10.1016/j.brainres.2010.07.094
284. Cano G, Passerin AM, Schiltz JC, Card JP, Morrison SF, Sved AF. Anatomical substrates for the central control of sympathetic outflow to interscapular adipose tissue during cold exposure. J Comp Neurol (2003) 460(3):303–26. doi:10.1002/cne.10643
285. Glick M, Segal-Lieberman G, Cohen R, Kronfeld-Schor N. Chronic MCH infusion causes a decrease in energy expenditure and body temperature, and an increase in serum IGF-1 levels in mice. Endocrine (2009) 36(3):479–85. doi:10.1007/s12020-009-9252-5
286. Bielajew C, Konkle AT, Merali Z. The effects of chronic mild stress on male Sprague-Dawley and Long Evans rats: I. Biochemical and physiological analyses. Behav Brain Res (2002) 136(2):583–92. doi:10.1016/S0166-4328(02)00222-X
287. Cong WN, Golden E, Pantaleo N, White CM, Maudsley S, Martin B. Ghrelin receptor signaling: a promising therapeutic target for metabolic syndrome and cognitive dysfunction. CNS Neurol Disord Drug Targets (2010) 9(5):557–63. doi:10.2174/187152710793361513
288. de Andrade JS, Céspedes IC, Abrão RO, Dos Santos TB, Diniz L, Britto LR, et al. Chronic unpredictable mild stress alters an anxiety-related defensive response, Fos immunoreactivity and hippocampal adult neurogenesis. Behav Brain Res (2013) 1(250):81–90. doi:10.1016/j.bbr.2013.04.031
289. Sominsky L, Spencer SJ. Eating behavior and stress: a pathway to obesity. Front Psychol (2014) 5:434. doi:10.3389/fpsyg.2014.00434
290. Labarthe A, Fiquet O, Hassouna R, Zizzari P, Lanfumey L, Ramoz N, et al. Ghrelin-derived peptides: a link between appetite/reward, GH axis, and psychiatric disorders? Front Endocrinol (Lausanne) (2014) 5:163. doi:10.3389/fendo.2014.00163
291. Wittekind DA, Kluge M. Ghrelin in psychiatric disorders – A review. Psychoneuroendocrinology (2015) 52C:176–94. doi:10.1016/j.psyneuen.2014.11.013
292. Miczek KA, O’Donnell JM. Intruder-evoked aggression in isolated and nonisolated mice: effects of psychomotor stimulants and l-dopa. Psychopharmacology (Berl) (1978) 57(1):47–55. doi:10.1007/BF00426957
293. Bhatnagar S, Vining C, Iyer V, Kinni V. Changes in hypothalamic-pituitary-adrenal function, body temperature, body weight and food intake with repeated social stress exposure in rats. J Neuroendocrinol (2006) 18(1):13–24. doi:10.1111/j.1365-2826.2005.01375.x
294. Blanchard DC, Cholvanich P, Blanchard RJ, Clow DW, Hammer RPJ, Rowlett JK, et al. Serotonin, but not dopamine, metabolites are increased in selected brain regions of subordinate male rats in a colony environment. Brain Res (1991) 568(1–2):61–6. doi:10.1016/0006-8993(91)91379-F
295. Blanchard C, Spencer R, Weiss SM, Blanchard RJ, McEwen B, Sakai RR. Visible burrow system as a model of chronic social stress: behavioral and neuroendocrine correlatesd. Psychoneuroendocrinology (1995) 20(2):117–34. doi:10.1016/0306-4530(94)E0045-B
296. Tamashiro KLK, Nguyen MNN, Fujikawa T, Xu T, Ma LY, Woods SC, et al. Metabolic and endocrine consequences of social stress in a visible burrow system. Physiol Behav (2004) 80(5):683–93. doi:10.1016/j.physbeh.2003.12.002
297. Tamashiro KLK, Hegeman MA, Sakai RR. Chronic social stress in a changing dietary environment. Physiol Behav (2006) 89(4):536–42. doi:10.1016/j.physbeh.2006.05.026
298. Albeck DS, McKittrick CR, Blanchard DC, Blanchard RJ, Nikulina J, McEwen BS, et al. Chronic social stress alters levels of corticotropin-releasing factor and arginine vasopressin mRNA in rat brain. J Neurosci (1997) 17(12):4895–903.
299. Smeltzer M, Scott K, Melhorn S, Krause E, Sakai R. Amylin blunts hyperphagia and reduces weight and fat gain during recovery in socially stressed rats. Am J Physiol Regul Integr Comp Physiol (2012) 303(6):R676–82. doi:10.1152/ajpregu.00090.2012
300. Kumar J, Chuang JC, Na ES, Kuperman A, Gillman AG, Mukherjee S, et al. Differential effects of chronic social stress and fluoxetine on meal patterns in mice. Appetite (2013) 64:81–8. doi:10.1016/j.appet.2012.12.023
301. McKittrick CR, Blanchard DC, Blanchard RJ, McEwen BS, Sakai RR. Serotonin receptor binding in a colony model of chronic social stress. Biol Psychiatry (1995) 37(6):383–93. doi:10.1016/0006-3223(94)00152-S
302. Avraham Y, Hao S, Mendelson S, Berry EM. Tyrosine improves appetite, cognition, and exercise tolerance in activity anorexia. Med Sci Sports Exerc (2001) 33(12):2104–10. doi:10.1097/00005768-200112000-00020
303. Hall JF, Hanford PV. Activity as a function of a restricted feeding schedule. J Comp Physiol Psychol (1954) 47(5):362–3. doi:10.1037/h0060276
304. Routtenberg A, Kuznesof AW. Self-starvation of rats living in activity wheels on a restricted feeding schedule. J Comp Physiol Psychol (1967) 64(3):414–21. doi:10.1037/h0025205
305. Carrera O, Fraga A, Pellón R, Gutiérrez E. Rodent model of activity-based anorexia. Curr Protoc Neurosci (2014) 10:67:9.47.1–9.47.11. doi:10.1002/0471142301.ns0947s67
306. Siegfried Z, Berry EM, Hao S, Avraham Y. Animal models in the investigation of anorexia. Physiol Behav (2003) 79(1):39–45. doi:10.1016/S0031-9384(03)00103-3
307. Gelegen C, Collier DA, Campbell IC, Oppelaar H, van den Heuvel J, Adan RA, et al. Difference in susceptibility to activity-based anorexia in two inbred strains of mice. Eur Neuropsychopharmacol (2007) 17(3):199–205. doi:10.1016/j.euroneuro.2006.04.007
308. Lewis DY, Brett RR. Activity-based anorexia in C57/BL6 mice: effects of the phytocannabinoid, Delta9-tetrahydrocannabinol (THC) and the anandamide analogue, OMDM-2. Eur Neuropsychopharmacol (2010) 20(9):622–31. doi:10.1016/j.euroneuro.2010.04.002
309. Jésus P, Ouelaa W, François M, Riachy L, Guérin C, Aziz M, et al. Alteration of intestinal barrier function during activity-based anorexia in mice. Clin Nutr (2014) 33(6):1046–53. doi:10.1016/j.clnu.2013.11.006
310. Klenotich SJ, Dulawa SC. The activity-based anorexia mouse model. Methods Mol Biol (2012) 829:377–93. doi:10.1007/978-1-61779-458-2_25
311. Pjetri E, de Haas R, de Jong S, Gelegen C, Oppelaar H, Verhagen LA, et al. Identifying predictors of activity based anorexia susceptibility in diverse genetic rodent populations. PLoS One (2012) 7(11):e50453. doi:10.1371/journal.pone.0050453
312. Cerrato M, Carrera O, Vazquez R, Echevarría E, Gutierrez E. Heat makes a difference in activity-based anorexia: a translational approach to treatment development in anorexia nervosa. Int J Eat Disord (2012) 45(1):26–35. doi:10.1002/eat.20884
313. Boakes RA, Juraskova I. The role of drinking in the suppression of food intake by recent activity. Behav Neurosci (2001) 115:718–30. doi:10.1037/0735-7044.115.3.718
314. Scheurink AJ, Boersma GJ, Nergårdh R, Södersten P. Neurobiology of hyperactivity and reward: agreeable restlessness in anorexia nervosa. Physiol Behav (2010) 100(5):490–5. doi:10.1016/j.physbeh.2010.03.016
315. Pardo M, Roca-Rivada A, Al-Massadi O, Seoane LM, Camiña JP, Casanueva FF. Peripheral leptin and ghrelin receptors are regulated in a tissue-specific manner in activity-based anorexia. Peptides (2010) 31(10):1912–9. doi:10.1016/j.peptides.2010.06.022
316. Duclos M, Ouerdani A, Mormède P, Konsman JP. Food restriction-induced hyperactivity: addiction or adaptation to famine? Psychoneuroendocrinology (2013) 38(6):884–97. doi:10.1016/j.psyneuen.2012.09.012
317. Aravich PF, Stanley EZ, Doerries LE. Exercise in food-restricted rats produces 2DG feeding and metabolic abnormalities similar to anorexia nervosa. Physiol Behav (1995) 57(1):147–53. doi:10.1016/0031-9384(94)00277-C
318. Kawaguchi M, Scott KA, Moran TH, Bi S. Dorsomedial hypothalamic corticotropin-releasing factor mediation of exercise-induced anorexia. Am J Physiol Regul Integr Comp Physiol (2005) 288(6):R1800–5. doi:10.1152/ajpregu.00805.2004
319. Nergårdh R, Ammar A, Brodin U, Bergström J, Scheurink A, Södersten P. Neuropeptide Y facilitates activity-based-anorexia. Psychoneuroendocrinology (2007) 32(5):493–502. doi:10.1016/j.psyneuen.2007.03.002
320. Gutierrez JA, Solenberg PJ, Perkins DR, Willency JA, Knierman MD, Jin Z, et al. Ghrelin octanoylation mediated by an orphan lipid transferase. Proc Natl Acad Sci U S A (2008) 105(17):6320–5. doi:10.1073/pnas.0800708105
321. Verhagen LA, Egecioglu E, Luijendijk MC, Hillebrand JJ, Adan RA, Dickson SL. Acute and chronic suppression of the central ghrelin signaling system reveals a role in food anticipatory activity. Eur Neuropsychopharmacol (2012) 21(5):384–92. doi:10.1016/j.euroneuro.2010.06.005
322. Exner C, Hebebrand J, Remschmidt H, Wewetzer C, Ziegler A, Herpertz S. Leptin suppresses semi-starvation induced hyperactivity in rats: implications for anorexia nervosa. Mol Psychiatry (2000) 5(5):476–81. doi:10.1038/sj.mp.4000771
323. Hillebrand JJG, Koeners MP, de Rijke CE, Martien JH, Kas MJH, Adan RAH. Leptin treatment in activity-based anorexia. Biol Psychiatry (2005) 58(2):165–71. doi:10.1016/j.biopsych.2005.03.011
324. Verhagen LA, Luijendijk MC, Adan RA. Leptin reduces hyperactivity in an animal model for anorexia nervosa via the ventral tegmental area. Eur Neuropsychopharmacol (2011) 21(3):274–81. doi:10.1016/j.euroneuro.2010.11.006
325. Hillebrand JJ, de Rijke CE, Brakkee JH, Kas MJ, Adan RA. Voluntary access to a warm plate reduces hyperactivity in activity-based anorexia. Physiol Behav (2005) 85(2):151–7. doi:10.1016/j.physbeh.2005.03.017
326. Jerlhag E, Egecioglu E, Dickson SL, Andersson M, Svensson L, Engel JA. Ghrelin stimulates locomotor activity and accumbal dopamine-overflow via central cholinergic systems in mice: implications for its involvement in brain reward. Addict Biol (2006) 11(1):45–54. doi:10.1111/j.1369-1600.2006.00002.x
327. Zigman JM, Nakano Y, Coppari R, Balthasar N, Marcus JN, Lee CE, et al. Mice lacking ghrelin receptors resist the development of diet-induced obesity. J Clin Invest (2005) 115(12):3564–72. doi:10.1172/JCI26002
328. Broocks A, Liu J, Pirke KM. Semistarvation-induced hyperactivity compensates for decreased norepinephrine and dopamine turnover in the mediobasal hypothalamus of the rat. J Neural Transm Gen Sect (1990) 79(1–2):113–24. doi:10.1007/BF01251006
329. Pirke KM, Broocks A, Wilckens T, Marquard R, Schweiger U. Starvation-induced hyperactivity in the rat: the role of endocrine and neurotransmitter changes. Neurosci Biobehav Rev (1993) 17(3):287–94. doi:10.1016/S0149-7634(05)80012-0
330. Wilckens T, Schweiger U, Pirke M. Activation of 5-HT1c-receptors suppresses excessive wheel running induced by semi-starvation in the rat. Psychopharmacology (Berl) (1992) 109(1–2):77–84. doi:10.1007/BF02245483
331. Altemus M, Glowa JR, Galliven E, Leong YM, Murphy DL. Effects of serotonergic agents on food-restriction-induced hyperactivity. Pharmacol Biochem Behav (1996) 53(1):123–31. doi:10.1016/0091-3057(95)02003-9
332. Atchley DPD, Eckel LA. Fenfluramine treatment in female rats accelerates the weight loss associated with activity-based anorexia. Pharmacol Biochem Behav (2005) 83(4):547–53. doi:10.1016/j.pbb.2006.03.016
333. Atchley DPD, Eckel LA. Treatment with 8-OH-DPAT attenuates the weight loss associated with activity-based anorexia in female rats. Pharmacol Biochem Behav (2006) 83(4):547–53. doi:10.1016/j.pbb.2006.03.016
334. Aravich PF, Rieg TS, Lauterio TJ, Doerries LE. β-Endorphin and dynorphin abnormalities in rats subjected to exercise and restricted feeding: relationship to anorexia nervosa? Brain Res (1993) 622(1–2):1–8. doi:10.1016/0006-8993(93)90794-N
335. Verty ANA, Evetts MJ, Crouch GJ, McGregor IS, Stefanidis A, Oldfield BJ. The cannabinoid receptor agonist THC attenuates weight loss in a rodent model of activity-based anorexia. Neuropsychopharmacology (2011) 36:1349–58. doi:10.1038/npp.2011.19
336. Watanabe K, Hara C, Ogawa N. Feeding conditions and estrous cycle of female rats under the activity-stress procedure from aspects of anorexia nervosa. Physiol Behav (1992) 51(4):827–32. doi:10.1016/0031-9384(92)90122-I
337. Dixon DP, Ackert AM, Eckel LA. Development of, and recovery from, activity-based anorexia in female rats. Physiol Behav (2003) 80(2–3):273–9. doi:10.1016/j.physbeh.2003.08.008
338. Hillebrand JJ, Heinsbroek AC, Kas MJ, Adan RA. The appetite suppressant d-fenfluramine reduces water intake, but not food intake, in activity-based anorexia. J Mol Endocrinol (2006) 36(1):153–62. doi:10.1677/jme.1.01887
339. Hillebrand JJG, Kas MJH, Roger AH. α-MSH enhances activity-based anorexia. Peptides (2005) 26(10):1690–6. doi:10.1016/j.peptides.2004.11.027
340. Woods SC. The eating paradox: how we tolerate food. Psychol Rev (1991) 98(4):488–505. doi:10.1037/0033-295X.98.4.488
341. Morrow NS, Schall M, Grijalva CV, Geiselman PJ, Garrick T, Nuccion S, et al. Body temperature and wheel running predict survival times in rats exposed to activity-stress. Physiol Behav (1997) 62(4):815–25. doi:10.1016/S0031-9384(97)00243-6
342. Gutiérrez E, Vásquez R, Boakes RA. Activity-based anorexia: ambient temperature has been a neglected factor. Psychon Bull Rev (2002) 9(2):239–49. doi:10.3758/BF03196278
Keywords: genetic models, environmental models, anorexia nervosa, acute stress, social stress, food restriction, activity/hyperactivity
Citation: Méquinion M, Chauveau C and Viltart O (2015) The use of animal models to decipher physiological and neurobiological alterations of anorexia nervosa patients. Front. Endocrinol. 6:68. doi: 10.3389/fendo.2015.00068
Received: 12 February 2015; Accepted: 15 April 2015;
Published: 19 May 2015
Edited by:
Hubert Vaudry, University of Rouen, FranceReviewed by:
Herbert Herzog, Garvan Institute, AustraliaHelene Volkoff, Memorial University, Canada
Copyright: © 2015 Méquinion, Chauveau and Viltart. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Odile Viltart, INSERM UMR-S1172, «Stades précoces de la maladie de Parkinson», University Lille 1, Centre JPARC, Bâtiment Biserte, Place de Verdun, Lille Cedex 59045, France,b2RpbGUudmlsdGFydEB1bml2LWxpbGxlMS5mcg==