 Megan Elizabeth Rich
Megan Elizabeth Rich Heather Kingsley Caldwell
Heather Kingsley Caldwell- Laboratory of Neuroendocrinology and Behavior, Department of Biological Sciences, The School of Biomedical Sciences, Kent State University, Kent, OH, USA
Schizophrenia is a chronic debilitating neuropsychiatric disorder estimated to affect 51 million people worldwide. Several symptom domains characterize schizophrenia, including negative symptoms, such as social withdrawal and anhedonia, cognitive impairments, such as disorganized thinking and impaired memory, and positive symptoms, such as hallucinations and delusions. While schizophrenia is a complex neuropsychiatric disorder with no single “cause,” there is evidence that the oxytocin (Oxt) system may be dysregulated in some individuals. Further, treatment with intranasal Oxt reduces some of the heterogeneous symptoms associated with schizophrenia. Since Oxt is known for its modulatory effects on a variety of social and non-social behaviors, it is perhaps not surprising that it may contribute to some aspects of schizophrenia and could also be a useful therapeutic agent. In this review, we highlight what is known about Oxt’s contributions to schizophrenia and schizophrenia-related behaviors and discuss its potential as a therapeutic agent.
Introduction
Schizophrenia, a chronic and debilitating neuropsychiatric disorder, affects 1% of the population worldwide (1). According to the fifth edition of the Diagnostic and Statistical Manual of Mental Disorders, schizophrenia is characterized by a combination of negative symptoms, cognitive dysfunction, and positive symptoms (2). Negative symptoms of schizophrenia include deficits in social behaviors such as social withdrawal, anhedonia, and flattened affect. Cognitive impairments include disorganized thinking and impaired executive function, working memory, and attention (3, 4). Lastly, the positive symptoms of schizophrenia include hallucinations, paranoid delusions, and disorganized speech. Unfortunately, while current antipsychotic medications are effective at ameliorating the positive symptoms, they are not very effective at treating the negative symptoms and cognitive dysfunction associated with schizophrenia, which tend to be more pervasive and persistent (5–7).
Current antipsychotic therapies are based on the dopamine hypothesis of schizophrenia, which proposes that increases in dopamine transmission in the mesolimbic dopamine pathway, and decreases in its activity in the prefrontal cortex contribute to many of the observed symptoms (8–10). As such, typical antipsychotics are dopamine 2 (D2) receptor antagonists, which only reduce positive symptom severity. Atypical antipsychotics on the other hand are reported to alleviate the positive symptoms as well as some of the negative symptoms associated with schizophrenia. These medications inhibit the serotonin 2A receptor (5-HT2A), and to a lesser extent D2 receptors and other neurotransmitter systems associated with schizophrenia, such as the adrenergic and cholinergic systems (11). However, two large clinical studies, the Clinical Antipsychotic Trials of Intervention and Effectiveness (CATIE) and the Cost Utility of the Latest Antipsychotic Drugs in Schizophrenia Study (CUtLASS) found no significant difference between the ability of typical and atypical antipsychotics to reduce the negative symptoms and cognitive dysfunction of schizophrenic patients (12–15). Thus, it is important to better understand the neurochemistry of the negative symptoms and cognitive dysfunction as they often precede the onset of the positive symptoms and act as better predictors of therapeutic outcome (16, 17). Due to the various combinations of symptoms and the wide range of symptom severity, diagnosis and treatment of schizophrenia are difficult; making it extremely important to elucidate which neurological factors may contribute to schizophrenia as well as identify treatments that can effectively lessen symptom severity.
Schizophrenia
Schizophrenia is a heterogeneous group of disorders, and as such no single gene can explain its pathophysiology. Hence, it is not surprising that several neurotransmitter and neuropeptide systems, beyond dopamine, have been implicated in its symptomology (Figure 1) [for review, see Ref. (8, 18–20)]. In addition to the dopamine hypothesis, there is the glutamate hypothesis, which supposes that it is the hypofunctioning of N-methyl-d-aspartate (NMDA) receptors that contribute to the negative symptoms and cognitive impairments associated with schizophrenia (19). Researchers studying the cholinergic and gamma aminobutyric acid (GABA) systems have found that these neurotransmitter systems may also play a role in both the psychotic and cognitive deficits found in schizophrenia patients (20, 21); while serotonin (5-HT) is mainly implicated in only the cognitive dysfunction associated with schizophrenia (22–24). Cannabinoids and monoamine oxidase, which modulate some of these neurotransmitter systems, also appear to also play a role in the negative symptoms and cognitive deficits (25, 26). Since many neuropeptides are often co-released with these neurotransmitters, they likely have a role to play as well. Some of these neuropeptides are neurotensin, cholecystokinin, corticotropin-releasing factor, neuropeptide y, and orexin (18). One neuropeptide that interacts with several of the aforementioned neurotransmitter and neuropeptide systems is the nonapeptide oxytocin (Oxt). Further, there is evidence that Oxt may be important to the etiology, symptom severity, and potential treatment of schizophrenia. First, in schizophrenic patients, there are reports of disruptions in the Oxt system that are affected by treatment with antipsychotics (27, 28). Second, treatment with Oxt as an adjunctive therapy is known to lessen symptom severity in some (29, 30). Third, animal models of schizophrenia suggest that Oxt may be involved in all three symptom domains (31–35).
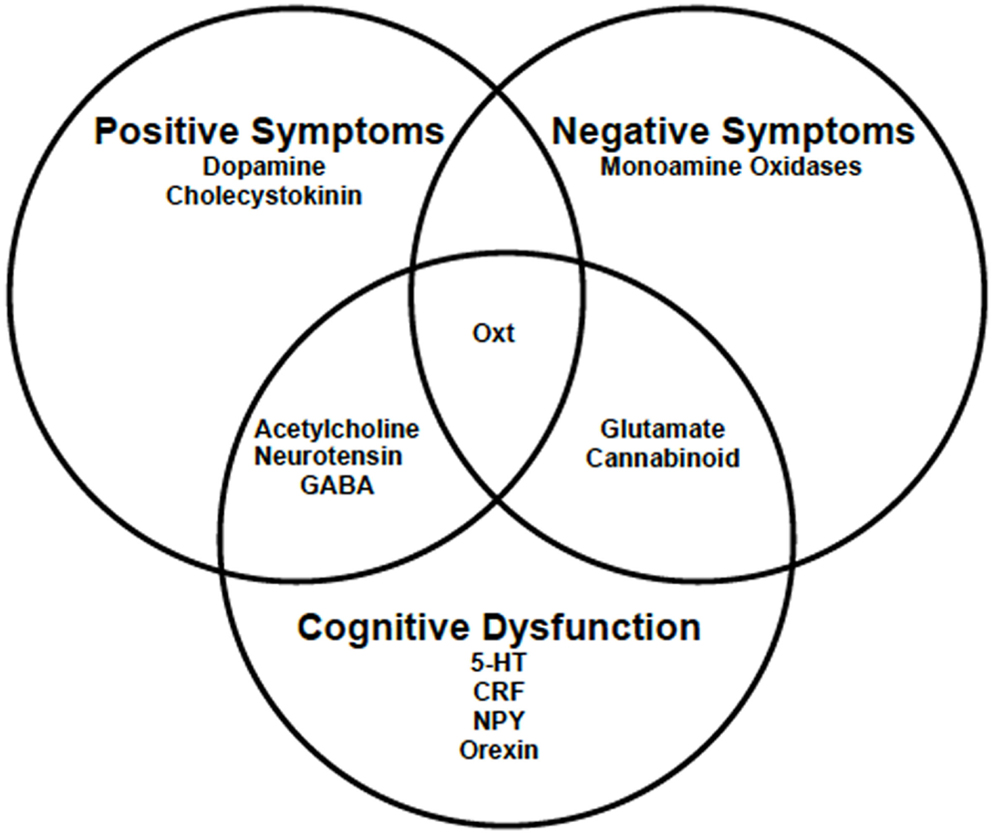
Figure 1. The symptom domains of schizophrenia and the neurotransmitter and neuropeptide systems known to play a role. Research suggests that Oxt may play a role in all three symptom domains associated with schizophrenia. 5-HT, serotonin; CRF, corticotropin-releasing factor; GABA, gamma aminobutyric acid; NPY, neuropeptide y; Oxt, oxytocin.
Oxytocin
Oxt is a nine amino acid peptide hormone, synthesized primarily in neurons of the hypothalamic supraoptic (SON) and paraventricular (PVN) nuclei. To date, a single seven-transmembrane G-protein coupled receptor, known as the Oxt receptor (Oxtr), is thought to mediate the actions of Oxt; although Oxt can also bind to the vasopressin (Avp) 1a and 1b receptors [for review, see Ref. (36)]. The Oxt system is involved in regulating a variety of behaviors [for review, see Ref. (37)] and implicated in aspects of learning and memory, such as spatial and non-spatial memory (38–41). However, more commonly, Oxt is known for its importance to the neuromodulation of social behaviors such as social memory and social recognition, affiliative behaviors, and aggression [for review, see Ref. (37, 42)]. Social behaviors are evolutionarily important because they reduce stress and anxiety (43, 44) and in humans, Oxt facilitates prosocial behaviors and increases feelings of trust and empathy (45–47). Given the effects of Oxt on social behaviors, it is perhaps not surprising that research has focused on the role of Oxt in neuropsychiatric disorders that are characterized by disruptions in social functioning.
Abnormalities in the Oxytocin System
Due to the negative symptoms associated with schizophrenia, and the effects of Oxt on prosocial behaviors, researchers hypothesize that Oxt dysregulation may contribute to the etiology and symptom severity of schizophrenia (29, 30). This hypothesis is supported by studies indicating that disruptions in the Oxt system are linked to the pathophysiology of schizophrenia (28, 48, 49). Altered levels of Oxt are reported in patients with schizophrenia (50, 51). However, the data are conflicting with some studies reporting an increase in Oxt and the Oxt carrier protein neurophysin I (50, 51) and another reporting no change in Oxt levels in cerebral spinal fluid (CSF) (52). However, patients with higher plasma levels of Oxt have less severe positive symptoms and exhibit fewer social deficits (53, 54).
Recently it has been reported that single nucleotide polymorphisms (SNPs) of the OXT and OXTR genes may contribute to symptom severity and treatment efficacy in schizophrenic patients (55–57). SNPs of the OXTR gene are associated with the severity of symptoms and the improvement of the positive symptoms of schizophrenia following treatment with antipsychotics (27, 28). Additionally, post-mortem analysis of brain tissue from unmedicated schizophrenia patients found altered neurophysin immunoreactivity (ir) in the PVN, internal palladium, and substantia nigra (58). Most recently, in patients with schizophrenia and polydipsia, decreases in plasma Oxt were found to correlate with the ability to correctly identify facial emotions (48) as well as malformations in brain areas that mediate neuroendocrine responses such as the anterior lateral hippocampus and amygdala (Amg) (59). Together, these data suggest that alterations in the function of the Oxt system may underlie all three symptom domains associated with schizophrenia. Given the dysregulation of the Oxt system in patients with schizophrenia, Oxt has been studied as a candidate for use as a therapeutic.
Human studies suggest that Oxt may have antipsychotic properties [for review, see Ref. (60, 61)]. Previous work found that injections of Oxt reduce the symptoms of psychosis and anhedonia in patients with schizophrenia (62, 63). Due to the ease of delivery, researchers are now utilizing intranasal administration of Oxt. It should be noted that there is an ongoing debate in the field on whether or not intranasal administration of Oxt is able to cross the blood–brain barrier, but there is evidence that intranasal administration increases Oxt concentrations in CSF in humans and animal models (64–66). In healthy patients, intranasal Oxt increases holistic processing, divergent thinking, and creative cognition (67), and studies in patients diagnosed with schizophrenia report that intranasal Oxt can be beneficial. Specifically, intranasal Oxt can facilitate social cognition (30, 68–70) and alleviate some of the cognitive deficits and positive symptoms in patients with schizophrenia (69). Yet, intranasal Oxt may be most effective as an adjunctive therapy to already prescribed antipsychotics, where chronic treatment is able to ameliorate some of the negative symptoms and the cognitive deficits, as well as the positive symptoms (30, 71, 72). While this research suggests that Oxt treatment has the potential to improve symptoms in all three domains, where in the brain and how these effects are mediated remains unknown. Animal models for schizophrenia are being used to determine where and how Oxt treatment may improve symptoms associated with schizophrenia.
Oxytocin in Humans and Animal Models
There are inherent challenges when studying a multifaceted disorder such as schizophrenia. Therefore, reliable animal models are necessary to understand and develop viable treatments. A good animal model must have phenotypic overlaps with either a behavior or a molecular characteristic of the disease. In humans, schizophrenia is characterized by several endophenotypes, including impairments in social behaviors such as emotion processing, social perception, attributional bias, and theory of mind [for review, see Ref. (73)]. Schizophrenic patients also have deficits in sensorimotor gating, as measured by prepulse inhibition (PPI) of the acoustic startle reflex [for review, see Ref. (74)], and cognitive deficits in verbal and visual memory, and impaired cognitive flexibility [for review, see Ref. (75)]. There are also neuromotor abnormalities such as dysmetria, eye tracking dysfunctions, and saccadic eye movements, which are typically associated with the positive symptoms of schizophrenia [for review, see Ref. (76–78)], as well as structural abnormalities in total brain volume and the volume of specific brain regions including, but not limited to, the hippocampus, the lateral ventricles, and the prefrontal cortex [for review, see Ref. (77, 79)] Co-morbid anxiety disorders are found in 38% of schizophrenia patients, and studies have reported increases in violent behaviors in schizophrenic patients (80–82). While changes in anxiety-like behavior and aggression have not been proposed as animal models for schizophrenia, several existing animal models of schizophrenia result in altered anxiety-like and aggressive behavior (83–91). Further, atypical antipsychotics have been found to reduce anxiety and reverse psychosis-induced aggression in patients with schizophrenia (92–96). Therefore, the examination of anxiety-like and aggressive behaviors seems warranted.
Currently, over 20 animal models are being used to assess the heterogeneous symptoms associated with schizophrenia (97). To study the specific contributions of the Oxt system, several models have been developed. The first utilizes perinatal stress, since research in humans suggests that exposure to adverse environmental conditions during perinatal development increases the risk for schizophrenia (98). Stress during the perinatal period is known to induce the behavioral and molecular characteristics of schizophrenia and is commonly used to model the negative symptoms of schizophrenia (85, 86, 99). The second employs the pharmacological disruption of the dopaminergic and glutamatergic systems, since the pathophysiology of schizophrenia suggests that there is dysfunction in both of these systems. Treatment with amphetamine (AMP), an indirect dopamine agonist, or phencyclidine (PCP), an NMDA receptor antagonist, induces hyperlocomotor activity, which corresponds with the positive symptoms of schizophrenia (100–103). Further, PCP treatment induces both negative symptoms and cognitive dysfunction, such as social withdrawal (104–106), impaired PPI (107), and cognitive deficits (108). The third uses gene knockout, since schizophrenia is a genetic disorder with high levels of heritability. Recently, it has been reported that genetic mutations in Oxt genes are associated with schizophrenia (55). It is for this reason that mice with genetic disruptions of their Oxt systems, such as Oxt and Oxtr knockout mice (Oxt−/− and Oxtr−/−, respectively) have been used to determine their potential contributions to the symptoms associated with schizophrenia. While no single model is sufficient to encompass all of the heterogeneous symptoms of schizophrenia, together these models can help us to better understand the role that Oxt may play in schizophrenia. It should be noted that several of these models are not specific to schizophrenia, and the data are relevent for other neuropsychiatric disorders (109). Currently, all of the aforementioned models are being used to study the relationship of Oxt to the negative symptoms of schizophrenia, and while some have been used to study the cognitive deficits and positive symptoms, more research is needed.
Deficits in Social Behaviors
Oxt has a well-characterized role in the neural regulation of social behaviors [for reviews, see Ref. (37, 42, 63, 110, 111)]. It is therefore not surprising that Oxt is studied for its potential contributions to the modulation of the negative symptoms of schizophrenia (Table 1). This section is broken up according to the approaches described in the previous section, as there is far more data on the contributions of Oxt to deficits in social behaviors than there are for the other symptoms associated with schizophrenia.
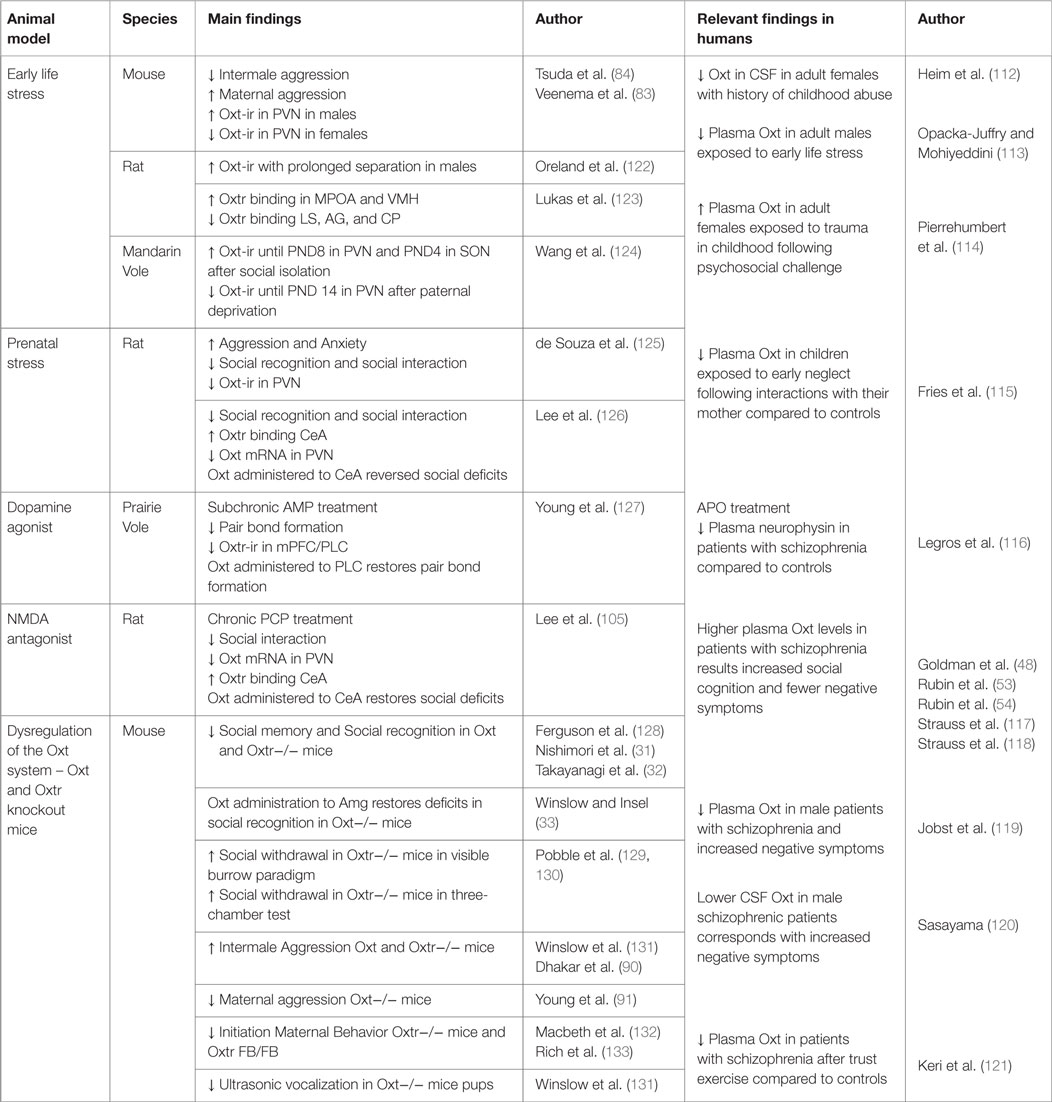
Table 1. Oxt and social deficits associated with schizophrenia.
Perinatal Stress
Research in humans has demonstrated that there is a positive correlation between perinatal exposure to a stressful environment and increased risk of schizophrenia (98). In rodents, maternal separation modifies aggressive behavior, and decreases social recognition, anxiety-like, and depression-like behaviors (85, 86, 99); with the effects of early life stress on aggression and Oxtr distribution being sex specific in both mice and rats. Following maternal separation, male mice exhibit decreases in aggression (83, 84) and increases in Oxt-ir in the PVN (84). However, in female mice, maternal separation results in increases in maternal aggression and decreases in Oxt-ir cells in the PVN (83). Similar to mice, in male Long Evans rats, early life stress results in decreases in intermale aggression, and in male Wistar rats, prolonged maternal separation results in increases in Oxt-ir in the Amg (122), increases in Oxtr binding in the medial pre-optic area (MPOA) and ventromedial hypothalamus (VMH), and decreases in Oxtr binding in the lateral septum (LS), agranular cortex, and caudate putamen (CP) in adulthood (123). Early life stress in female Wistar rats results in increases in aggression (134, 135). Data from another rodent species, mandarin voles, have shown that neonatal social isolation results in increases in Oxt-ir in the PVN until post natal day (PND) 8 and the SON until PND4 in both sexes (124). Further, in vole pups that have been isolated from their fathers there is a downregulation of Oxt-ir neurons until PND14, but these decreases do not persist (124).
In addition to maternal separation, prenatal stress can also cause behavioral effects in rodents that are reflective of symptoms of schizophrenia. Adult male rats subjected to prenatal stress and reared by stressed mothers display lower levels of aggression and social behaviors, and increases in anxiety-like behaviors (125, 126, 136). However, when non-stressed mothers rear pups that are exposed to stress during the prenatal period, the deficits in aggressive behaviors and increases in anxiety do not persist (125). Further, these effects appear to be due to Oxt, as an injection of Oxt into the central amygdala (CeA) is able to restore the social deficits exhibited by male rats subjected to prenatal stress (126). Male offspring raised by their prenatally stressed mothers also have reductions in Oxtr mRNA, fewer Oxt positive magnocellular neurons in the PVN, and increases in Oxtr binding in the CeA (125, 126). These morphological changes in Oxt system are not found when non-stressed dams raise the pups.
The behavioral differences observed between species, strain, and sex that result from stress during the perinatal period appear to be a result of alterations in the Oxt system. Many of the changes in the Oxt system are found within the neuronal network that mediates aggression: the MPOA, LS, anterior hypothalamus, VMH, medial amygdala (MeA), and bed nucleus of the stria terminalis (BNST) (137). There are also changes found in the Oxt system in the PVN, and it is known that stress can modulate aggression via the PVN (137). In males, perinatal stress results in decreases in aggression and increases in Oxt-ir and Oxtr binding (83, 84, 122, 134). However, in females, increases in aggression coincided with decreases in Oxt signaling (83, 134, 135). These sex differences in aggression and Oxt may be a result of estrogen-mediated sex differences in Oxtr regulation (138, 139).
Low levels of licking/grooming (LG) maternal behavior are associated with decreases in estrogen receptor-alpha (ERα) and Oxtr levels in the MPOA in female offspring (138, 139). Further, the interactions of estrogens and the Oxt system may result in changes to the dopamine system, as females reared by low LG dams have fewer dopamine neurons in the VTA (140). Research using dopamine agonists to model schizophrenia suggest that there are important interactions between the Oxt and dopaminergic systems to social cognition. Taken together, the data from perinatal stress models suggest that there can be long-lasting disruptions of Oxt neurochemistry, which may lead to impairments in behaviors that are similar to the negative symptoms of schizophrenia.
Pharmacological Disruption
Pathophysiological studies utilizing dopamine agonists and NMDA receptor antagonists have reaffirmed the importance of Oxt to social cognition in patients with schizophrenia. A study on drug addiction and social behaviors provides insight into the role of Oxt, dopamine, and social behaviors (127). Specifically, in prairie voles, repeated subchronic AMP exposure inhibits pair bond formation (127), decreases Oxtr-ir in the mPFC, and reduces Oxtr activation in the PLC; which is important for partner preference formation (127, 141). Additionally, Oxt direct infusion into the PLC is able to restore AMP-induced impairment in partner preference and alter dopamine levels in the nucleus accumbens (NAcc) (127). Administration of PCP induces social dysfunctions in animals that mimics the negative symptoms associated with schizophrenia [for review, Ref. see (142, 143)]. Oxt mRNA expression is reduced in the PVN of rats and Oxtr binding is increased in the CeA following chronic PCP treatment (105). Further, PCP-induced deficits in social interactions are increased by bilateral infusions of Oxt to the CeA (105). While these data suggest that the interaction of Oxt with both dopamine and glutamate is important for social behavior, the specific mechanisms that mediate these effects remain unclear.
Research on sex behavior in rats suggests that the dopaminergic and Oxt systems can modulate each other (144–146), and the Oxtr is located throughout the mesolimbic dopamine pathway (147, 148). Thus, researchers have hypothesized that Oxt and dopamine may work together to affect on how an individual perceives the salience of social cues [for review, see Ref. (149)]. However, the connection between these two systems and their role in schizophrenia remains murky. Likewise, the link between the Oxt and glutamate systems is also poorly understood. In rat SON preparations, application of both Oxt and Avp inhibits glutamate release (150). However, in cultured rat olfactory bulb neurons glutamate transmission is facilitated (151). More recently, it has been found that in the CeA, Oxt and glutamate are co-released from Oxt neurons (152). More research is still needed to determine how and where Oxt may interact with these neurotransmitter systems to affect social cognition in patients with schizophrenia.
Genetic Disruptions
The use of genetic tools, including Oxt−/− and Oxtr−/− mice have significantly contributed to our understanding of the role of Oxt in the social deficits observed in patients with schizophrenia. Male Oxt−/− and Oxtr−/− mice fail to develop social recognition memory, in essence having social amnesia (31, 32, 128, 153) (Figure 2). Further, an injection of Oxt into the MeA of Oxt−/− mice is able to restore social recognition (33, 154). These deficits in social memory are not specific to males, as female Oxt−/− mice do not show a normal Bruce effect (155, 156). Oxtr−/− mice also display behaviors similar to the negative symptoms of schizophrenia across multiple testing scenarios. In a visible burrow system, which provides a more natural habitat for rodents, Oxtr−/− mice have reductions in social interaction behaviors, spending more time alone and self-grooming than controls (129, 130). In a three-chamber test for sociability Oxtr−/− mice display increases in social withdrawal (129, 130) and in a social proximity test they display reductions in the frequency of nose-to-nose and nose-to-anogenital behaviors (129, 130). These data suggest that a functional Oxt system is necessary for normal social interactions, and that dysregulation of Oxt in schizophrenia could contribute to some of the negative symptoms.
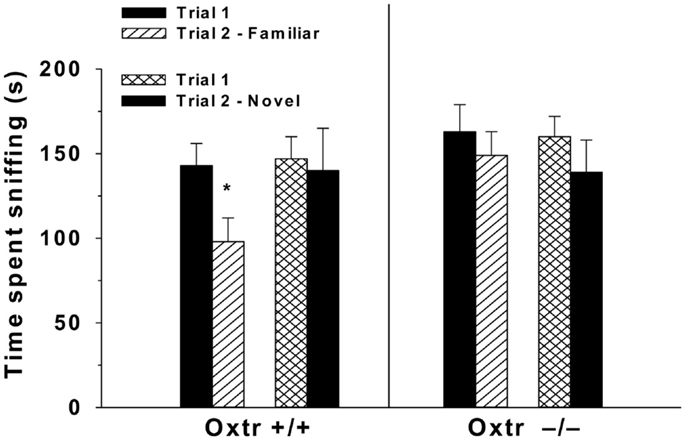
Figure 2. Oxtr−/− mice have impaired social recognition. In a two-trial discrimination task performed over 2 weeks, Oxtr+/+ (n = 8) and Oxtr−/− males (n = 8) were exposed to overiectomized BALB/C female mice during trial 1, and then 30 min later during trial 2 they were exposed to a familiar female on week 1. During week 2 of testing after trial 1, mice were exposed to a novel female during trial 2. Oxtr−/− mice fail to discriminate between the familiar and novel female spending approximately equal amounts of time sniffing both; compared to Oxtr+/+ mice that spend more time sniffing the novel female. Reprinted with permission from Endocrinology, Lee et al. (153).
Research also suggests that Oxt is important for other social behaviors, such as aggression and maternal behavior. Some studies have reported increases in violent behaviors in schizophrenic patients; however, it remains unclear whether this is a symptom of schizophrenia or rather co-morbid disorders (80, 81). Oxt−/− and Oxtr−/− mice have increases in aggressive behavior, and given the dysregulation of the Oxt system in schizophrenia, a functional Oxt system could be important for normal aggressive behavior (32, 90, 131, 153, 157, 158). Specifically, male Oxt−/− mice have heightened aggression when born to null mutant dams, but not when they are born to heterozygous dams (131, 157). Oxtr−/− mice also have heightened intermale aggression, but Oxtr FB/FB do not (32, 90, 153, 158). These data suggest that Oxt exposure during development may have persistent effects on aggressive behavior. Therefore, it could be that developmental Oxt contributes to the etiology of schizophrenia; however, more research is needed before such a claim can be made.
While there are no reported deficits in maternal behavior in patients with schizophrenia, the cognitive impairments associated with schizophrenia may lead to reductions in the ability to acquire necessary parenting skills (159–161). In animal models of schizophrenia, evidence suggests that decreases in maternal behaviors result in the development of behaviors similar to those found in other animal models of schizophrenia (83, 125, 126, 136, 162). Oxtr−/− and Oxtr FB/FB display deficits in the initiation of maternal behavior (32, 132, 133) and Oxt−/− mice pups emit fewer ultrasonic vocalizations when separated from nest; all of which suggest that Oxt contributes to social behavior in rodents (33, 131).
Impaired Cognition
The Oxt system may also be important to the cognitive dysfunctions associated with schizophrenia. One endophenotype of schizophrenia is impaired sensorimotor gating, i.e., the inability to “filter or gate” information (163, 164). Across species, sensorimotor gating can be measured using PPI of the startle reflex. The startle reflex is a defensive response to an abrupt, relatively intense stimuli (165). The neural circuitry that underlies PPI is known as the cortico-striato-pallido-pontine (CSPP) circuit (166). In humans, PPI is measured using electromyographic recordings from eye blink responses (167). In rodents, it is measured using the whole body flinch reflex of an animal to the startle stimulus (168). Patients with schizophrenia not only have reduced PPI but also show less habituation of the startle reflex compared to controls (169). In Brown Norway rats, which have a naturally low PPI, Oxt but not its structural analog carbetocin, is able to significantly increase PPI (170).
Stress during the perinatal period may contribute to deficits in PPI, though there have been contradicting reports, with one group reporting deficits in PPI and another group finding no changes in PPI, some data suggests that early life stress reduces PPI levels in adulthood (162, 171). Changes in the Oxt system have been reported following early life stress, and may have an impact on perinatal stress-induced reductions in PPI, though more research is necessary (83, 122, 123). Further, in models that pharmacologically disrupt PPI, exogenous Oxt is known to reverse these deficits (170). Specifically, in rats, subcutaneous Oxt injections are able to restore deficits in PPI induced by AMP, an indirect dopamine agonist, and dizocipline (MK-801), a specific NMDA receptor antagonist, but not apomorphine (APO), a direct dopamine agonist (172). Finally, genetic disruptions in the Oxt system suggest that a lack of endogenous Oxt appears to be important in the regulation of PPI, as Oxt−/− mice have increased PCP-induced deficits in PPI (173) (Figure 3). This further suggests that the effects of endogenous Oxt on PPI may be specific to the glutamatergic system.
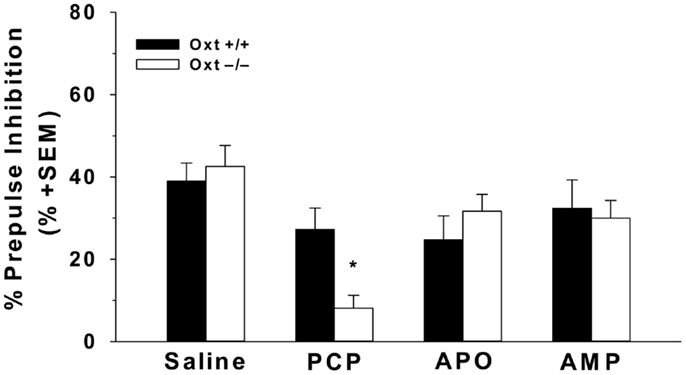
Figure 3. Oxt−/− mice have greater PCP-induced deficits in sensorimotor gating. The acoustic startle of male Oxt+/+ (n = 12) and Oxt−/− mice (n = 8) was measured using the whole body reflex flinch in reaction to a startle tone using startle chambers (SR-LAB; San Diego Instruments, San Diego, CA, USA). Mice were administered either an i.p. injection of 10 mg/kg AMP and APO, a subcutaneous injection 6 mg/kg PCP, or an equivalent volume of 0.9% saline as a control 15 min prior to testing. Testing session consisted of 60 trials, including no stimulus trials, pulse-alone trials, and prepulse + pulse trials. The testing sessions began and ended with the presentation of five 120 db pulse-alone tones. The middle 50 trials consisted of: 10 no pulse tones trials, 30 prepulse + pulse trials at 3, 6, and 12 db above background, and 10 pulse-alone tones at 120 db. A repeated measures design was used with each animal receiving 0.9% saline, AMP, APO, and PCP, with a minimum of 3 days between each trial. Oxt−/− mice display greater reductions in the average percent PPI across three prepulse levels (3, 6, and 12 db above background) following an injection of PCP compared to Oxt+/+ mice. There were no genotypic differences in PPI following injection of AMP or APO. Adapted and reprinted with permission from Macmillan Publishers Ltd: Molecular Psychiatry, Caldwell et al. (173).
Oxt is likely to also contribute to the cognitive deficits associated with schizophrenia, such as impaired spatial memory and cognitive flexibility (3, 174). Similar to the cognitive deficits found in schizophrenia, Oxtr−/− mice display reduced cognitive flexibility, as measured by an inability to alter their behavior during the reversal phase of a t-maze task (175). Since the Oxtr is abundant in the hippocampus of mice, it may be important for memory (176). However, there are divergent reports of Oxt’s effects on spatial memory, suggesting that Oxt may have brain region-specific effects (38, 177). In vitro, hippocampal slices treated with Oxt are able to maintain long-term potentiation longer than untreated slices (38). In mouse dams, a central injection of Oxt is able to improve reference memory on a radial arm maze, but does not affect their short-term memory during acquisition, suggesting that Oxt only improves long-term spatial memory (38). As Oxt can improve anxiety in virgin mice when administered to the Amg or VMH, the effects of Oxt on reference memory may be due to its actions in these brain regions. However, there was no effect on their open-field activity, which suggests direct action on hippocampal neurons (38). Further, dams that receive an intracerebroventricular (i.c.v.) injection of an Oxt antagonist have reductions in reference memory compared to controls (38). But, in rats, Oxt injections into the nucleus basalis of Meynert (NBM) impair spatial memory, as measured by a Morris water maze, while an Oxtr antagonist injected into the NBM facilitates spatial memory (177). Given that disruptions in Oxt signaling appear to contribute to multiple aspects of cognition, and that Oxt may affect memory formation, it is plausible that Oxt may play a role in the cognitive deficits associated with schizophrenia.
The effects of Oxt dysregulation and Oxt treatment on the cognitive dysfunction found in patients with schizophrenia are poorly understood. Studies in both humans and animal models suggest that a functional Oxt system is required for normal sensorimotor gating and cognitive flexibility. The effects Oxt on sensorimotor gating may be specific to the glutamatergic system (172, 173), with mice lacking the obligatory NMDA receptor 1 subunit having impaired PPI (178). Unfortunately, as previously discussed, how these two systems interact remains unclear. The Oxt system is coupled to phospholipase c-β1 (PLC-β1) and glutamate is known to regulate PLC-β1 (36, 179–182). Abnormal expression patterns of PLC-β1 are found in patients with schizophrenia (183, 184). Further, studies using PLC-β1 knockout (PLCβ1−/−) mice find impaired PPI and deficits in working memory (185, 186). Therefore, the PLC-β1 may reflect a point of convergence for the Oxt and glutamate systems in the regulation of sensorimotor gating.
The effects of Oxt treatment on spatial learning are also ambiguous. While research suggests that Oxt in the hippocampus facilitates learning, it impairs memory when injected to the NBM (38, 177). However, while neuronal deficits in the hippocampus have been found, no reductions in neuronal density have been observed in the NBM in patients with schizophrenia (187). So, it is not clear whether or not this brain region is important to the pathophysiology of schizophrenia. In addition to Oxt’s effects on the cognitive deficits, it may also play a role in the positive symptoms associated with schizophrenia.
Neuromotor Abnormalities
In animal models, psychotic symptoms similar to the positive symptoms of schizophrenia can be manifested in rodents by treatment with dopamine agonists and NMDA receptor antagonists, which cause hyperlocomotor activity. While hyperlocomotor activity does not have direct face validity for the positive symptoms of schizophrenia, it does have construct validity as psychotomimetics cause similar neurotransmitter activity in animal models as is found in human schizophrenic patients. However, the behavioral effects are not necessarily similar; though some suggest that hyperlocomotor activity is comparable to some positive symptoms such as grossly disorganized behavior and psychomotor agitation (188, 189). Further, established antipsychotics, which reduce positive symptoms of schizophrenia, consistently reduce the hyperactivity associated with pharmacological agents such as AMP, cocaine, ketamine, and PCP. The antipsychotic efficacy of Oxt is supported by pharmacological manipulations that induce aspects of schizophrenia. During studies on addiction, Oxt decreases drug-induced hyperlocomotor activity (34, 35) while pretreatment with Oxt is able to attenuate the hyperlocomotor activity caused by cocaine, an indirect dopamine agonist (35) (Figure 4). In another study, which examined the effects of Oxt on addiction, i.c.v. injections of Oxt reduce methamphetamine-induced increases in locomotor activity (34). Other research also suggests that Oxt and the glutamatergic system may interact to affect the positive symptoms associated with schizophrenia. In addition to its behavioral effects, PCP induces the excessive release of glutamate within the medial prefrontal cortex (mPFC), which when blocked, suppresses hyperlocomotion (190, 191). Oxt has been found to reduce the PCP-induced symptoms associated with psychosis (173), as well as suppress glutamate release within the mPFC (192). Therefore, Oxt could suppress the hypofunction of glutamate specifically within the mPFC to protect against PCP-induced symptoms of psychosis. Genetic disruptions of the Oxt system also provide evidence that endogenous Oxt may affect locomotor activity, as there is hyperlocomotor activity in infant Oxtr−/− mice; however, this effect is not persistent (32).
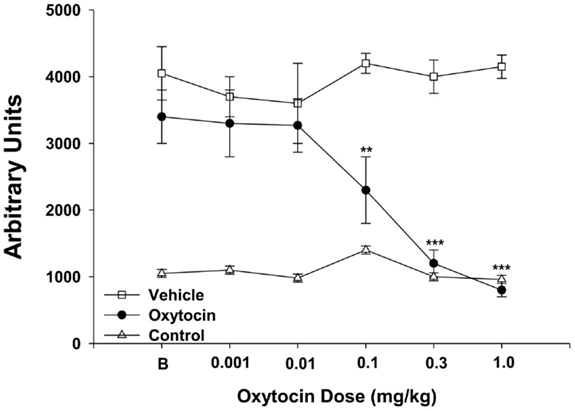
Figure 4. Oxt dose-dependently decreases locomotor activity in self-administering methamphetamine rats. Oxt (n = 5) was administered IP in ascending doses (0.001, 0.01, 0.1, 03, 1 mg/kg) over five consecutive days and equivalent amounts of vehicle (n = 5) were administered. Only the animals treated with Oxt or vehicle self-administered methamphetamine, and the control (n = 8) was used to determine baseline levels of locomotor activity. B = baseline day before oxytocin testing began. **p < 0.01 and ***p < 0.001. There was no difference between rats treated with Oxt compared to the control group at 0.3 and 1 mg/kg Oxt dose. All other comparisons between Oxt treatment and the control group and the vehicle treatment and control groups were significant. Data are shown as mean ± SEM. Adapted and reprinted with permission from Elsevier: Carson et al. (34).
Oxytocin and the Pharmacology of Schizophrenia
Oxt is known to interact with several other neurotransmitter systems that are important in the etiology and treatment of schizophrenia, such as GABA and 5-HT (193). During parturition, Oxt has been found to modulate GABAergic inhibition in rodent models for autism spectrum disorder (ASD) (194). Given that ASDs and schizophrenia share similar endophenotypes, Oxt may also modulate GABA signaling in schizophrenic patients as well. However, further research is necessary to elucidate the role of the interactions of the Oxt system and GABAergic system to the symptomology of schizophrenia. Oxt and 5-HT are known to modulate one another, and both are important for numerous social behaviors and mood (195–197). Specifically, Oxt may exert anxiolytic effects via Oxtr activation in 5-HT neurons (195). Current atypical antipsychotics may provide further evidence for the interactions between the Oxt and 5-HT systems and schizophrenia.
Some of the currently used atypical antipsychotics are known to interact with the Oxt system. The atypical antipsychotics, amperozide and clozapine, increase plasma levels of Oxt, but the typical antipsychotic haloperidol does not (198). Amperozide and clozapine are both a 5-HT2A antagonists, and to lesser extent D2 antagonists, that are reported to decrease both the negative and positive symptoms associated with schizophrenia (199–202). Whereas, the D2 specific antagonist, haloperidol, only appears to alleviate positive symptoms of schizophrenia (203, 204). Further, some atypical antipsychotics cause activation of Oxt cells as measured by cFos ir. Clozapine increases cFos activation in Oxt cells in the PVN, but again, haloperidol treatment does not (205). Similar to the effects of Oxt, in rodents, clozapine attenuates the reduction of cognitive flexibility caused by the sub-chronic PCP treatment (206), and is able to restore normal levels of PPI to brown Norway rats (207). This evidence further supports a role of the Oxt in the cognitive deficits found in schizophrenic patients. In humans, clozapine attenuates both the negative symptoms and cognitive dysfunctions found in patients with schizophrenia (208–212). Therefore, the ability to reduce the social and cognitive deficits may be associated with the ability of clozapine to increase Oxt levels. Further, the specific interactions between the Oxt and serotonergic systems may be important to the social and cognitive deficits found in patients with schizophrenia. However, additional research is necessary to assess how Oxt may affect the symptom domains associated with schizophrenia through its interactions with other neurotransmitters systems.
Conclusion
Given the importance of the Oxt system to the modulation of social behaviors, it is not surprising that across animal models of schizophrenia, Oxt has been implicated in the negative symptoms and deficits in social cognition. Data suggests that developmental, drug induced, and genetic disruptions in the Oxt system lead to the symptoms associated with the negative symptoms observed in schizophrenic patients. However, further research is needed to elucidate the specific mechanisms whereby Oxt exerts these effects. Human and animal models also suggest that research is needed to determine if Oxt can work as a therapeutic agent to improve the social behavior deficits observed in patients with schizophrenia. Oxt also appears to be a contributor to the cognitive and positive symptom domains of schizophrenia; though much more work in this area is needed. While Oxt does not “cause” schizophrenia, its putative impact to all three symptom domains suggests that it may be an important player to the etiology, and perhaps even an effective treatment, of schizophrenia. Using animal models, future research will need to focus on elucidating of the mechanisms of Oxt dysregulation and the interactions between Oxt and other neurotransmitter systems that may contribute to the symptoms associated with schizophrenia.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1. Regier DA, Narrow WE, Rae DS, Manderscheid RW, Locke BZ, Goodwin FK. The de facto US mental and addictive disorders service system. Epidemiologic catchment area prospective 1-year prevalence rates of disorders and services. Arch Gen Psychiatry (1993) 50:85–94. doi: 10.1001/archpsyc.1993.01820140007001
2. American Psychiatric Association. Diagnositc and Statistical Manual of Mental Disorders. Arlington, VA: American Psychiatric Association (2013).
3. Gogtay N, Rapoport J. Clozapine use in children and adolescents. Expert Opin Pharmacother (2008) 9:459–65. doi:10.1517/14656566.9.3.459
4. N.I.O.M. Health. Schizophrenia. Washington, DC: U.S. Department of Health and Human Services (2009).
5. Fenton WS, McGlashan TH. Natural history of schizophrenia subtypes. II. Positive and negative symptoms and long-term course. Arch Gen Psychiatry (1991) 48:978–86. doi:10.1001/archpsyc.1991.01810350018003
6. Mueser KT, McGurk SR. Schizophrenia. Lancet (2004) 363:2063–72. doi:10.1016/S0140-6736(04)16458-1
7. Andreasen NC. Symptoms, signs, and diagnosis of schizophrenia. Lancet (1995) 346:477–81. doi:10.1016/S0140-6736(95)91325-4
8. da Silva Alves F, Figee M, van Amelsvoort T, Veltman D, de Haan L. The revised dopamine hypothesis of schizophrenia: evidence from pharmacological MRI studies with atypical antipsychotic medication. Psychopharmacol Bull (2008) 41:121–32. doi:10.1016/S0920-9964(08)70291-3
9. Walter H, Kammerer H, Frasch K, Spitzer M, Abler B. Altered reward functions in patients on atypical antipsychotic medication in line with the revised dopamine hypothesis of schizophrenia. Psychopharmacology (2009) 206:121–32. doi:10.1007/s00213-009-1586-4
10. Pogarell O, Koch W, Karch S, Dehning S, Muller N, Tatsch K, et al. Dopaminergic neurotransmission in patients with schizophrenia in relation to positive and negative symptoms. Pharmacopsychiatry (2012) 45(Suppl 1):S36–41. doi:10.1055/s-0032-1306313
11. Schotte A, Janssen PF, Gommeren W, Luyten WH, Van Gompel P, Lesage AS, et al. Risperidone compared with new and reference antipsychotic drugs: in vitro and in vivo receptor binding. Psychopharmacology (1996) 124:57–73. doi:10.1007/BF02245606
12. Stroup TS, McEvoy JP, Swartz MS, Byerly MJ, Glick ID, Canive JM, et al. The National Institute of Mental Health Clinical Antipsychotic Trials of Intervention Effectiveness (CATIE) project: schizophrenia trial design and protocol development. Schizophr Bull (2003) 29:15–31. doi:10.1093/oxfordjournals.schbul.a006986
13. Naber D, Lambert M. The CATIE and CUtLASS studies in schizophrenia: results and implications for clinicians. CNS Drugs (2009) 23:649–59. doi:10.2165/00023210-200923080-00002
14. Jones PB, Barnes TR, Davies L, Dunn G, Lloyd H, Hayhurst KP, et al. Randomized controlled trial of the effect on quality of life of second- vs. first-generation antipsychotic drugs in schizophrenia: cost utility of the latest antipsychotic drugs in schizophrenia study (CUtLASS 1). Arch Gen Psychiatry (2006) 63:1079–87. doi:10.1001/archpsyc.63.10.1079
15. Lewis S, Lieberman J. CATIE and CUtLASS: can we handle the truth? Br J Psychiatry (2008) 192:161–3. doi:10.1192/bjp.bp.107.037218
16. Davidson M, Reichenberg A, Rabinowitz J, Weiser M, Kaplan Z, Mark M. Behavioral and intellectual markers for schizophrenia in apparently healthy male adolescents. Am J Psychiatry (1999) 156:1328–35.
17. Cornblatt BA, Erlenmeyer-Kimling L. Global attentional deviance as a marker of risk for schizophrenia: specificity and predictive validity. J Abnorm Psychol (1985) 94:470–86. doi:10.1037/0021-843X.94.4.470
18. LaCrosse AL, Olive MF. Neuropeptide systems and schizophrenia. CNS Neurol Disord Drug Targets (2013) 12:619–32. doi:10.2174/1871527311312050010
19. Coyle JT. Glutamate and schizophrenia: beyond the dopamine hypothesis. Cell Mol Neurobiol (2006) 26:365–84. doi:10.1007/s10571-006-9062-8
20. Foster DJ, Choi DL, Conn PJ, Rook JM. Activation of M1 and M4 muscarinic receptors as potential treatments for Alzheimer’s disease and schizophrenia. Neuropsychiatr Dis Treat (2014) 10:183–91. doi:10.2147/NDT.S55104
21. Deidda G, Bozarth IF, Cancedda L. Modulation of GABAergic transmission in development and neurodevelopmental disorders: investigating physiology and pathology to gain therapeutic perspectives. Front Cell Neurosci (2014) 8:119. doi:10.3389/fncel.2014.00119
22. Meneses A. Memory formation and memory alterations: 5-HT6 and 5-HT7 receptors, novel alternative. Rev Neurosci (2014) 25:325–56. doi:10.1515/revneuro-2014-0001
23. Di Pilato P, Niso M, Adriani W, Romano E, Travaglini D, Berardi F, et al. Selective agonists for serotonin 7 (5-HT7) receptor and their applications in preclinical models: an overview. Rev Neurosci (2014) 25:401–15. doi:10.1515/revneuro-2014-0009
24. Nikiforuk A. The procognitive effects of 5-HT6 receptor ligands in animal models of schizophrenia. Rev Neurosci (2014) 25:367–82. doi:10.1515/revneuro-2014-0005
25. Tan H, Ahmad T, Loureiro M, Zunder J, Laviolette SR. The role of cannabinoid transmission in emotional memory formation: implications for addiction and schizophrenia. Front Psychiatry (2014) 5:73. doi:10.3389/fpsyt.2014.00073
26. Kim SK, Park HJ, Seok H, Jeon HS, Chung JH, Kang WS, et al. Association study between monoamine oxidase A (MAOA) gene polymorphisms and schizophrenia: lack of association with schizophrenia and possible association with affective disturbances of schizophrenia. Mol Biol Rep (2014) 41:3457–64. doi:10.1007/s11033-014-3207-5
27. Souza RP, Ismail P, Meltzer HY, Kennedy JL. Variants in the oxytocin gene and risk for schizophrenia. Schizophr Res (2010) 121:279–80. doi:10.1016/j.schres.2010.04.019
28. Souza RP, de Luca V, Meltzer HY, Lieberman JA, Kennedy JL. Schizophrenia severity and clozapine treatment outcome association with oxytocinergic genes. Int J Neuropsychopharmacol (2010) 13:793–8. doi:10.1017/S1461145710000167
29. Bujanow W. Letter: is oxytocin an anti-schizophrenic hormone? Can Psychiatr Assoc J (1974) 19:323.
30. Feifel D, Macdonald K, Nguyen A, Cobb P, Warlan H, Galangue B, et al. Adjunctive intranasal oxytocin reduces symptoms in schizophrenia patients. Biol Psychiatry (2010) 68:678–80. doi:10.1016/j.biopsych.2010.04.039
31. Nishimori K, Takayanagi Y, Yoshida M, Kasahara Y, Young LJ, Kawamata M. New aspects of oxytocin receptor function revealed by knockout mice: sociosexual behaviour and control of energy balance. Prog Brain Res (2008) 170:79–90. doi:10.1016/S0079-6123(08)00408-1
32. Takayanagi Y, Yoshida M, Bielsky IF, Ross HE, Kawamata M, Onaka T, et al. Pervasive social deficits, but normal parturition, in oxytocin receptor-deficient mice. Proc Natl Acad Sci USA (2005) 102:16096–101. doi:10.1073/pnas.0505312102
33. Winslow JT, Insel TR. The social deficits of the oxytocin knockout mouse. Neuropeptides (2002) 36:221–9. doi:10.1054/npep.2002.0909
34. Carson DS, Cornish JL, Guastella AJ, Hunt GE, McGregor IS. Oxytocin decreases methamphetamine self-administration, methamphetamine hyperactivity, and relapse to methamphetamine-seeking behaviour in rats. Neuropharmacology (2010) 58:38–43. doi:10.1016/j.neuropharm.2009.06.018
35. Sarnyai Z, Szabo G, Kovacs GL, Telegdy G. Oxytocin attenuates the cocaine-induced exploratory hyperactivity in mice. Neuroreport (1990) 1:200–2. doi:10.1097/00001756-199011000-00006
36. Gimpl G, Fahrenholz F. The oxytocin receptor system: structure, function, and regulation. Physiol Rev (2001) 81:629–83.
37. Lee HJ, Macbeth AH, Pagani JH, Young WS III. Oxytocin: the great facilitator of life. Prog Neurobiol (2009) 88:127–51. doi:10.1016/j.pneurobio.2009.04.001
38. Tomizawa K, Iga N, Lu YF, Moriwaki A, Matsushita M, Li ST, et al. Oxytocin improves long-lasting spatial memory during motherhood through MAP kinase cascade. Nat Neurosci (2003) 6:384–90. doi:10.1038/nn1023
39. Bohus B, Urban I, van Wimersma Greidanus TB, de Wied D. Opposite effects of oxytocin and vasopressin on avoidance behaviour and hippocampal theta rhythm in the rat. Neuropharmacology (1978) 17:239–47. doi:10.1016/0028-3908(78)90107-7
40. De Wied D. Long term effect of vasopressin on the maintenance of a conditioned avoidance response in rats. Nature (1971) 232:58–60. doi:10.1038/232058a0
41. Kovacs GL, Vecsei L, Telegdy G. Opposite action of oxytocin to vasopressin in passive avoidance behavior in rats. Physiol Behav (1978) 20:801–2. doi:10.1016/0031-9384(78)90309-8
42. Caldwell HK. Neurobiology of sociability. Adv Exp Med Biol (2012) 739:187–205. doi:10.1007/978-1-4614-1704-0_12
43. Carter CS. Neuroendocrine perspectives on social attachment and love. Psychoneuroendocrinology (1998) 23:779–818. doi:10.1016/S0306-4530(98)00055-9
44. vnas-Moberg KU. Physiological and endocrine effects of social contact. Ann N Y Acad Sci (1997) 807:146–63. doi:10.1111/j.1749-6632.1997.tb51917.x
45. Zak PJ, Stanton AA, Ahmadi S. Oxytocin increases generosity in humans. PLoS One (2007) 2:e1128. doi:10.1371/journal.pone.0001128
46. Feldman R, Weller A, Zagoory-Sharon O, Levine A. Evidence for a neuroendocrinological foundation of human affiliation: plasma oxytocin levels across pregnancy and the postpartum period predict mother-infant bonding. Psychol Sci (2007) 18:965–70. doi:10.1111/j.1467-9280.2007.02010.x
47. Barraza JA, Zak PJ. Empathy toward strangers triggers oxytocin release and subsequent generosity. Ann N Y Acad Sci (2009) 1167:182–9. doi:10.1111/j.1749-6632.2009.04504.x
48. Goldman M, Marlow-O’Connor M, Torres I, Carter CS. Diminished plasma oxytocin in schizophrenic patients with neuroendocrine dysfunction and emotional deficits. Schizophr Res (2008) 98:247–55. doi:10.1016/j.schres.2007.09.019
49. Rosenfeld AJ, Lieberman JA, Jarskog LF. Oxytocin, dopamine, and the amygdala: a neurofunctional model of social cognitive deficits in schizophrenia. Schizophr Bull (2011) 37:1077–87. doi:10.1093/schbul/sbq015
50. Beckmann H, Lang RE, Gattaz WF. Vasopressin – oxytocin in cerebrospinal fluid of schizophrenic patients and normal controls. Psychoneuroendocrinology (1985) 10:187–91. doi:10.1016/0306-4530(85)90056-3
51. Linkowski P, Geenen V, Kerkhofs M, Mendlewicz J, Legros JJ. Cerebrospinal fluid neurophysins in affective illness and in schizophrenia. Eur Arch Psychiatry Neurol Sci (1984) 234:162–5. doi:10.1007/BF00461555
52. Glovinsky D, Kalogeras KT, Kirch DG, Suddath R, Wyatt RJ. Cerebrospinal fluid oxytocin concentration in schizophrenic patients does not differ from control subjects and is not changed by neuroleptic medication. Schizophr Res (1994) 11:273–6. doi:10.1016/0920-9964(94)90021-3
53. Rubin LH, Carter CS, Drogos L, Pournajafi-Nazarloo H, Sweeney JA, Maki PM. Peripheral oxytocin is associated with reduced symptom severity in schizophrenia. Schizophr Res (2010) 124:13–21. doi:10.1016/j.schres.2010.09.014
54. Rubin LH, Carter CS, Drogos L, Jamadar R, Pournajafi-Nazarloo H, Sweeney JA, et al. Sex-specific associations between peripheral oxytocin and emotion perception in schizophrenia. Schizophr Res (2011) 130:266–70. doi:10.1016/j.schres.2011.06.002
55. Teltsh O, Kanyas-Sarner K, Rigbi A, Greenbaum L, Lerer B, Kohn Y. Oxytocin and vasopressin genes are significantly associated with schizophrenia in a large Arab-Israeli pedigree. Int J Neuropsychopharmacol (2012) 15:309–19. doi:10.1017/S1461145711001374
56. Watanabe Y, Kaneko N, Nunokawa A, Shibuya M, Egawa J, Someya T. Oxytocin receptor (OXTR) gene and risk of schizophrenia: case-control and family-based analyses and meta-analysis in a Japanese population. Psychiatry Clin Neurosci (2012) 66:622. doi:10.1111/j.1440-1819.2012.02396.x
57. Montag C, Brockmann EM, Bayerl M, Rujescu D, Muller DJ, Gallinat J. Oxytocin and oxytocin receptor gene polymorphisms and risk for schizophrenia: a case-control study. World J Biol Psychiatry (2013) 14:500–8. doi:10.3109/15622975.2012.677547
58. Mai JK, Berger K, Sofroniew MV. Morphometric evaluation of neurophysin-immunoreactivity in the human brain: pronounced inter-individual variability and evidence for altered staining patterns in schizophrenia. J Hirnforsch (1993) 34:133–54.
59. Goldman MB, Gomes AM, Carter CS, Lee R. Divergent effects of two different doses of intranasal oxytocin on facial affect discrimination in schizophrenic patients with and without polydipsia. Psychopharmacology (2011) 216:101–10. doi:10.1007/s00213-011-2193-8
60. Macdonald K, Feifel D. Oxytocin in schizophrenia: a review of evidence for its therapeutic effects. Acta Neuropsychiatr (2012) 24:130–46. doi:10.1111/j.1601-5215.2011.00634.x
61. Bakermans-Kranenburg MJ, van IJMH. Sniffing around oxytocin: review and meta-analyses of trials in healthy and clinical groups with implications for pharmacotherapy. Transl Psychiatry (2013) 3:e258. doi:10.1038/tp.2013.34
62. McEwen BB. Brain-fluid barriers: relevance for theoretical controversies regarding vasopressin and oxytocin memory research. Adv Pharmacol (2004) 50:531–92. doi:10.1016/S1054-3589(04)50016-9
63. Churchland PS, Winkielman P. Modulating social behavior with oxytocin: how does it work? What does it mean? Horm Behav (2012) 61:392–9. doi:10.1016/j.yhbeh.2011.12.003
64. Dal Monte O, Noble PL, Turchi J, Cummins A, Averbeck BB. CSF and blood oxytocin concentration changes following intranasal delivery in Macaque. PLoS One (2014) 9:e103677. doi:10.1371/journal.pone.0103677
65. Striepens N, Kendrick KM, Hanking V, Landgraf R, Wullner U, Maier W, et al. Elevated cerebrospinal fluid and blood concentrations of oxytocin following its intranasal administration in humans. Sci Rep (2013) 3:3440. doi:10.1038/srep03440
66. Neumann ID, Maloumby R, Beiderbeck DI, Lukas M, Landgraf R. Increased brain and plasma oxytocin after nasal and peripheral administration in rats and mice. Psychoneuroendocrinology (2013) 38:1985–93. doi:10.1016/j.psyneuen.2013.03.003
67. De Dreu CK, Baas M, Roskes M, Sligte DJ, Ebstein RP, Chew SH, et al. Oxytonergic circuitry sustains and enables creative cognition in humans. Soc Cogn Affect Neurosci (2014) 9(8):1159–65. doi:10.1093/scan/nst094
68. Davis MC, Lee J, Horan WP, Clarke AD, McGee MR, Green MF, et al. Effects of single dose intranasal oxytocin on social cognition in schizophrenia. Schizophr Res (2013) 147:393–7. doi:10.1016/j.schres.2013.04.023
69. Pedersen CA, Gibson CM, Rau SW, Salimi K, Smedley KL, Casey RL, et al. Intranasal oxytocin reduces psychotic symptoms and improves Theory of Mind and social perception in schizophrenia. Schizophr Res (2011) 132:50–3. doi:10.1016/j.schres.2011.07.027
70. Averbeck BB, Bobin T, Evans S, Shergill SS. Emotion recognition and oxytocin in patients with schizophrenia. Psychol Med (2012) 42:259–66. doi:10.1017/S0033291711001413
71. Feifel D, Macdonald K, Cobb P, Minassian A. Adjunctive intranasal oxytocin improves verbal memory in people with schizophrenia. Schizophr Res (2012) 139:207–10. doi:10.1016/j.schres.2012.05.018
72. Modabbernia A, Rezaei F, Salehi B, Jafarinia M, Ashrafi M, Tabrizi M, et al. Intranasal oxytocin as an adjunct to risperidone in patients with schizophrenia: an 8-week, randomized, double-blind, placebo-controlled study. CNS Drugs (2013) 27:57–65. doi:10.1007/s40263-012-0022-1
73. Green MF, Horan WP. Social cognition in schizophrenia. Curr Dir Psychol Sci (2010) 19:243–8. doi:10.1177/0963721410377600
74. Velasques B, Machado S, Paes F, Cunha M, Sanfim A, Budde H, et al. Sensorimotor integration and psychopathology: motor control abnormalities related to psychiatric disorders. World J Biol Psychiatry (2011) 12:560–73. doi:10.3109/15622975.2010.551405
75. Lepage M, Bodnar M, Bowie CR. Neurocognition: clinical and functional outcomes in schizophrenia. Can J Psychiatry (2014) 59:5–12.
76. Levy DL, Sereno AB, Gooding DC, O’Driscoll GA. Eye tracking dysfunction in schizophrenia: characterization and pathophysiology. Curr Top Behav Neurosci (2010) 4:311–47. doi:10.1007/7854_2010_60
77. Allen AJ, Griss ME, Folley BS, Hawkins KA, Pearlson GD. Endophenotypes in schizophrenia: a selective review. Schizophr Res (2009) 109:24–37. doi:10.1016/j.schres.2009.01.016
78. Isohanni M, Isohanni I, Koponen H, Koskinen J, Laine P, Lauronen E, et al. Developmental precursors of psychosis. Curr Psychiatry Rep (2004) 6:168–75. doi:10.1007/s11920-004-0061-5
79. Haijma SV, Van Haren N, Cahn W, Koolschijn PC, Hulshoff Pol HE, Kahn RS. Brain volumes in schizophrenia: a meta-analysis in over 18 000 subjects. Schizophr Bull (2013) 39:1129–38. doi:10.1093/schbul/sbs118
80. Fazel S, Gulati G, Linsell L, Geddes JR, Grann M. Schizophrenia and violence: systematic review and meta-analysis. PLoS Med (2009) 6:e1000120. doi:10.1371/journal.pmed.1000120
81. Witt K, van Dorn R, Fazel S. Risk factors for violence in psychosis: systematic review and meta-regression analysis of 110 studies. PLoS One (2013) 8:e55942. doi:10.1371/journal.pone.0055942
82. Braga RJ, Reynolds GP, Siris SG. Anxiety comorbidity in schizophrenia. Psychiatry Res (2013) 210:1–7. doi:10.1016/j.psychres.2013.07.030
83. Veenema AH, Bredewold R, Neumann ID. Opposite effects of maternal separation on intermale and maternal aggression in C57BL/6 mice: link to hypothalamic vasopressin and oxytocin immunoreactivity. Psychoneuroendocrinology (2007) 32:437–50. doi:10.1016/j.psyneuen.2007.02.008
84. Tsuda MC, Yamaguchi N, Ogawa S. Early life stress disrupts peripubertal development of aggression in male mice. Neuroreport (2011) 22:259–63. doi:10.1097/WNR.0b013e328344495a
85. Lapiz MD, Fulford A, Muchimapura S, Mason R, Parker T, Marsden CA. Influence of postweaning social isolation in the rat on brain development, conditioned behavior, and neurotransmission. Neurosci Behav Physiol (2003) 33:13–29. doi:10.1023/A:1021171129766
86. Fone KC, Porkess MV. Behavioural and neurochemical effects of post-weaning social isolation in rodents-relevance to developmental neuropsychiatric disorders. Neurosci Biobehav Rev (2008) 32:1087–102. doi:10.1016/j.neubiorev.2008.03.003
87. Lipska BK, Jaskiw GE, Weinberger DR. Postpubertal emergence of hyperresponsiveness to stress and to amphetamine after neonatal excitotoxic hippocampal damage: a potential animal model of schizophrenia. Neuropsychopharmacology (1993) 9:67–75. doi:10.1038/npp.1993.44
88. Tseng KY, Chambers RA, Lipska BK. The neonatal ventral hippocampal lesion as a heuristic neurodevelopmental model of schizophrenia. Behav Brain Res (2009) 204:295–305. doi:10.1016/j.bbr.2008.11.039
89. Mei L, Xiong WC. Neuregulin 1 in neural development, synaptic plasticity and schizophrenia. Nat Rev Neurosci (2008) 9:437–52. doi:10.1038/nrn2392
90. Dhakar MB, Rich ME, Reno EL, Lee HJ, Caldwell HK. Heightened aggressive behavior in mice with lifelong versus postweaning knockout of the oxytocin receptor. Horm Behav (2012) 62:86–92. doi:10.1016/j.yhbeh.2012.05.007
91. Young WS III, Shepard E, DeVries AC, Zimmer A, LaMarca ME, Ginns EI, et al. Targeted reduction of oxytocin expression provides insights into its physiological roles. Adv Exp Med Biol (1998) 449:231–40. doi:10.1007/978-1-4615-4871-3_30
92. Huf G, Alexander J, Allen MH, Raveendran NS. Haloperidol plus promethazine for psychosis-induced aggression. Cochrane Database Syst Rev (2009) 3:CD005146. doi:10.1002/14651858.CD005146.pub2
93. Huf G, Alexander J, Allen MH. Haloperidol plus promethazine for psychosis induced aggression. Cochrane Database Syst Rev (2005) 4:CD005146.10.1002/14651858.CD005146.pub2
94. Hershenberg R, Gros DF, Brawman-Mintzer O. Role of atypical antipsychotics in the treatment of generalized anxiety disorder. CNS Drugs (2014) 28:519–33. doi:10.1007/s40263-014-0162-6
95. Buckley PF. Efficacy of quetiapine for the treatment of schizophrenia: a combined analysis of three placebo-controlled trials. Curr Med Res Opin (2004) 20:1357–63. doi:10.1185/030079904125004510
96. Mullen J, Jibson MD, Sweitzer D. A comparison of the relative safety, efficacy, and tolerability of quetiapine and risperidone in outpatients with schizophrenia and other psychotic disorders: the quetiapine experience with safety and tolerability (QUEST) study. Clin Ther (2001) 23:1839–54. doi:10.1016/S0149-2918(00)89080-3
97. Carpenter WT, Koenig JI. The evolution of drug development in schizophrenia: past issues and future opportunities. Neuropsychopharmacology (2008) 33:2061–79. doi:10.1038/sj.npp.1301639
98. Schmitt A, Malchow B, Hasan A, Falkai P. The impact of environmental factors in severe psychiatric disorders. Front Neurosci (2014) 8:19. doi:10.3389/fnins.2014.00019
99. Jones CA, Watson DJ, Fone KC. Animal models of schizophrenia. Br J Pharmacol (2011) 164:1162–94. doi:10.1111/j.1476-5381.2011.01386.x
100. Robinson TE, Becker JB. Enduring changes in brain and behavior produced by chronic amphetamine administration: a review and evaluation of animal models of amphetamine psychosis. Brain Res (1986) 396:157–98. doi:10.1016/0165-0173(86)90002-0
101. Featherstone RE, Rizos Z, Kapur S, Fletcher PJ. A sensitizing regimen of amphetamine that disrupts attentional set-shifting does not disrupt working or long-term memory. Behav Brain Res (2008) 189:170–9. doi:10.1016/j.bbr.2007.12.032
102. Featherstone RE, Kapur S, Fletcher PJ. The amphetamine-induced sensitized state as a model of schizophrenia. Prog Neuropsychopharmacol Biol Psychiatry (2007) 31:1556–71. doi:10.1016/j.pnpbp.2007.08.025
103. Kalinichev M, Robbins MJ, Hartfield EM, Maycox PR, Moore SH, Savage KM, et al. Comparison between intraperitoneal and subcutaneous phencyclidine administration in Sprague-Dawley rats: a locomotor activity and gene induction study. Prog Neuropsychopharmacol Biol Psychiatry (2008) 32:414–22. doi:10.1016/j.pnpbp.2007.09.008
104. Sams-Dodd F. Distinct effects of d-amphetamine and phencyclidine on the social behaviour of rats. Behav Pharmacol (1995) 6:55–65. doi:10.1097/00008877-199501000-00009
105. Lee PR, Brady DL, Shapiro RA, Dorsa DM, Koenig JI. Social interaction deficits caused by chronic phencyclidine administration are reversed by oxytocin. Neuropsychopharmacology (2005) 30:1883–94. doi:10.1038/sj.npp.1300722
106. Qiao H, Noda Y, Kamei H, Nagai T, Furukawa H, Miura H, et al. Clozapine, but not haloperidol, reverses social behavior deficit in mice during withdrawal from chronic phencyclidine treatment. Neuroreport (2001) 12:11–5. doi:10.1097/00001756-200101220-00010
107. Mansbach RS, Geyer MA. Effects of phencyclidine and phencyclidine biologs on sensorimotor gating in the rat. Neuropsychopharmacology (1989) 2:299–308. doi:10.1016/0893-133X(89)90035-3
108. Egerton A, Reid L, McKerchar CE, Morris BJ, Pratt JA. Impairment in perceptual attentional set-shifting following PCP administration: a rodent model of set-shifting deficits in schizophrenia. Psychopharmacology (2005) 179:77–84. doi:10.1007/s00213-004-2109-y
109. Nestler EJ, Hyman SE. Animal models of neuropsychiatric disorders. Nat Neurosci (2010) 13:1161–9. doi:10.1038/nn.2647
110. Olff M, Frijling JL, Kubzansky LD, Bradley B, Ellenbogen MA, Cardoso C, et al. The role of oxytocin in social bonding, stress regulation and mental health: an update on the moderating effects of context and interindividual differences. Psychoneuroendocrinology (2013) 38:1883–94. doi:10.1016/j.psyneuen.2013.06.019
111. Bartz JA, Zaki J, Bolger N, Ochsner KN. Social effects of oxytocin in humans: context and person matter. Trends Cogn Sci (2011) 15:301–9. doi:10.1016/j.tics.2011.05.002
112. Heim C, Young LJ, Newport DJ, Mletzko T, Miller AH, Nemeroff CB. Lower CSF oxytocin concentrations in women with a history of childhood abuse. Mol Psychiatry (2009) 14:954–8. doi:10.1038/mp.2008.112
113. Opacka-Juffry J, Mohiyeddini C. Experience of stress in childhood negatively correlates with plasma oxytocin concentration in adult men. Stress (2012) 15:1–10. doi:10.3109/10253890.2011.560309
114. Pierrehumbert B, Torrisi R, Laufer D, Halfon O, Ansermet F, Beck Popovic M. Oxytocin response to an experimental psychosocial challenge in adults exposed to traumatic experiences during childhood or adolescence. Neuroscience (2010) 166:168–77. doi:10.1016/j.neuroscience.2009.12.016
115. Fries ABW, Ziegler TE, Kurian JR, Jacoris S, Pollak SD. Early experience in humans is associated with changes in neuropeptides critical for regulating social behavior. Proc Natl Acad Sci USA (2005) 102:17237–40. doi:10.1073/pnas.0504767102
116. Legros JJ, Gazzotti C, Carvelli T, Franchimont P, Timsit-Berthier M, von Frenckell R, et al. Apomorphine stimulation of vasopressin- and oxytocin-neurophysins. Evidence for increased oxytocinergic and decreased vasopressinergic function in schizophrenics. Psychoneuroendocrinology (1992) 17:611–7. doi:10.1016/0306-4530(92)90019-4
117. Strauss GP, Keller WR, Koenig JI, Gold JM, Frost KH, Buchanan RW. Plasma oxytocin levels predict social cue recognition in individuals with schizophrenia. Schizophr Res (2015) 162:47–51. doi:10.1016/j.schres.2015.01.034
118. Strauss GP, Keller WR, Koenig JI, Gold JM, Ossenfort KL, Buchanan RW. Plasma oxytocin levels predict olfactory identification and negative symptoms in individuals with schizophrenia. Schizophr Res (2015) 162:57–61. doi:10.1016/j.schres.2014.12.023
119. Jobst A, Dehning S, Ruf S, Notz T, Buchheim A, Henning-Fast K, et al. Oxytocin and vasopressin levels are decreased in the plasma of male schizophrenia patients. Acta Neuropsychiatr (2014) 26:347–55. doi:10.1017/neu.2014.20
120. Sasayama D, Hattori K, Teraishi T, Hori H, Ota M, Yoshida S, et al. Negative correlation between cerebrospinal fluid oxytocin levels and negative symptoms of male patients with schizophrenia. Schizophr Res (2012) 139:201–6. doi:10.1016/j.schres.2012.06.016
121. Keri S, Kiss I, Kelemen O. Sharing secrets: oxytocin and trust in schizophrenia. Soc Neurosci (2009) 4:287–93. doi:10.1080/17470910802319710
122. Oreland S, Gustafsson-Ericson L, Nylander I. Short- and long-term consequences of different early environmental conditions on central immunoreactive oxytocin and arginine vasopressin levels in male rats. Neuropeptides (2010) 44:391–8. doi:10.1016/j.npep.2010.06.001
123. Lukas M, Bredewold R, Neumann ID, Veenema AH. Maternal separation interferes with developmental changes in brain vasopressin and oxytocin receptor binding in male rats. Neuropharmacology (2010) 58:78–87. doi:10.1016/j.neuropharm.2009.06.020
124. Wang L, Zhang W, Wu R, Kong L, Feng W, Cao Y, et al. Neuroendocrine responses to social isolation and paternal deprivation at different postnatal ages in mandarin voles. Dev Psychobiol (2014) 56:1214–28. doi:10.1002/dev.21202
125. de Souza MA, Centenaro LA, Menegotto PR, Henriques TP, Bonini J, Achaval M, et al. Prenatal stress produces social behavior deficits and alters the number of oxytocin and vasopressin neurons in adult rats. Neurochem Res (2013) 38:1479–89. doi:10.1007/s11064-013-1049-5
126. Lee PR, Brady DL, Shapiro RA, Dorsa DM, Koenig JI. Prenatal stress generates deficits in rat social behavior: Reversal by oxytocin. Brain Res (2007) 1156:152–67. doi:10.1016/j.brainres.2007.04.042
127. Young KA, Liu Y, Gobrogge KL, Wang H, Wang Z. Oxytocin reverses amphetamine-induced deficits in social bonding: evidence for an interaction with nucleus accumbens dopamine. J Neurosci (2014) 34:8499–506. doi:10.1523/JNEUROSCI.4275-13.2014
128. Ferguson JN, Young LJ, Hearn EF, Matzuk MM, Insel TR, Winslow JT. Social amnesia in mice lacking the oxytocin gene. Nat Genet (2000) 25:284–8. doi:10.1038/77040
129. Pobbe RL, Pearson BL, Blanchard DC, Blanchard RJ. Oxytocin receptor and Mecp2 308/Y knockout mice exhibit altered expression of autism-related social behaviors. Physiol Behav (2012) 107:641–8. doi:10.1016/j.physbeh.2012.02.024
130. Pobbe RL, Pearson BL, Defensor EB, Bolivar VJ, Young WS III, Lee HJ, et al. Oxytocin receptor knockout mice display deficits in the expression of autism-related behaviors. Horm Behav (2012) 61:436–44. doi:10.1016/j.yhbeh.2011.10.010
131. Winslow JT, Hearn EF, Ferguson J, Young LJ, Matzuk MM, Insel TR. Infant vocalization, adult aggression, and fear behavior of an oxytocin null mutant mouse. Horm Behav (2000) 37:145–55. doi:10.1006/hbeh.1999.1566
132. Macbeth AH, Stepp JE, Lee HJ, Young WS III, Caldwell HK. Normal maternal behavior, but increased pup mortality, in conditional oxytocin receptor knockout females. Behav Neurosci (2010) 124:677–85. doi:10.1037/a0020799
133. Rich ME, deCardenas EJ, Lee HJ, Caldwell HK. Impairments in the initiation of maternal behavior in oxytocin receptor knockout mice. PLoS One (2014) 9:e98839. doi:10.1371/journal.pone.0098839
134. Boccia ML, Pedersen CA. Brief vs. long maternal separations in infancy: contrasting relationships with adult maternal behavior and lactation levels of aggression and anxiety. Psychoneuroendocrinology (2001) 26:657–72. doi:10.1016/S0306-4530(01)00019-1
135. Padoin MJ, Cadore LP, Gomes CM, Barros HM, Lucion AB. Long-lasting effects of neonatal stimulation on the behavior of rats. Behav Neurosci (2001) 115:1332–40. doi:10.1037/0735-7044.115.6.1332
136. Patin V, Lordi B, Vincent A, Caston J. Effects of prenatal stress on anxiety and social interactions in adult rats. Brain Res Dev Brain Res (2005) 160:265–74. doi:10.1016/j.devbrainres.2005.09.010
137. Nelson RJ, Trainor BC. Neural mechanisms of aggression. Nat Rev Neurosci (2007) 8:536–46. doi:10.1038/nrn2174
138. Champagne F, Diorio J, Sharma S, Meaney MJ. Naturally occurring variations in maternal behavior in the rat are associated with differences in estrogen-inducible central oxytocin receptors. Proc Natl Acad Sci USA (2001) 98:12736–41. doi:10.1073/pnas.221224598
139. Francis DD, Champagne FC, Meaney MJ. Variations in maternal behaviour are associated with differences in oxytocin receptor levels in the rat. J Neuroendocrinol (2000) 12:1145–8. doi:10.1046/j.1365-2826.2000.00599.x
140. Pena CJ, Neugut YD, Calarco CA, Champagne FA. Effects of maternal care on the development of midbrain dopamine pathways and reward-directed behavior in female offspring. Eur J Neurosci (2014) 39:946–56. doi:10.1111/ejn.12479
141. Young LJ, Lim MM, Gingrich B, Insel TR. Cellular mechanisms of social attachment. Horm Behav (2001) 40:133–8. doi:10.1006/hbeh.2001.1691
142. Jentsch JD, Roth RH. The neuropsychopharmacology of phencyclidine: from NMDA receptor hypofunction to the dopamine hypothesis of schizophrenia. Neuropsychopharmacology (1999) 20:201–25. doi:10.1016/S0893-133X(98)00060-8
143. Moghaddam B, Jackson ME. Glutamatergic animal models of schizophrenia. Ann N Y Acad Sci (2003) 1003:131–7. doi:10.1196/annals.1300.065
144. Melis MR, Melis T, Cocco C, Succu S, Sanna F, Pillolla G, et al. Oxytocin injected into the ventral tegmental area induces penile erection and increases extracellular dopamine in the nucleus accumbens and paraventricular nucleus of the hypothalamus of male rats. Eur J Neurosci (2007) 26:1026–35. doi:10.1111/j.1460-9568.2007.05721.x
145. Succu S, Sanna F, Cocco C, Melis T, Boi A, Ferri GL, et al. Oxytocin induces penile erection when injected into the ventral tegmental area of male rats: role of nitric oxide and cyclic GMP. Eur J Neurosci (2008) 28:813–21. doi:10.1111/j.1460-9568.2008.06385.x
146. Baskerville TA, Allard J, Wayman C, Douglas AJ. Dopamine-oxytocin interactions in penile erection. Eur J Neurosci (2009) 30:2151–64. doi:10.1111/j.1460-9568.2009.06999.x
147. Freund-Mercier MJ, Stoeckel ME, Klein MJ. Oxytocin receptors on oxytocin neurones: histoautoradiographic detection in the lactating rat. J Physiol (1994) 480(Pt 1):155–61. doi:10.1113/jphysiol.1994.sp020349
148. Yoshimura R, Kiyama H, Kimura T, Araki T, Maeno H, Tanizawa O, et al. Localization of oxytocin receptor messenger ribonucleic acid in the rat brain. Endocrinology (1993) 133:1239–46. doi:10.1210/endo.133.3.8396014
149. Love TM. Oxytocin, motivation and the role of dopamine. Pharmacol Biochem Behav (2014) 119:49–60. doi:10.1016/j.pbb.2013.06.011
150. Curras-Collazo MC, Gillard ER, Jin J, Pandika J. Vasopressin and oxytocin decrease excitatory amino acid release in adult rat supraoptic nucleus. J Neuroendocrinol (2003) 15:182–90. doi:10.1046/j.1365-2826.2003.00976.x
151. Osako Y, Otsuka T, Taniguchi M, Oka T, Kaba H. Oxytocin enhances presynaptic and postsynaptic glutamatergic transmission between rat olfactory bulb neurones in culture. Neurosci Lett (2001) 299:65–8. doi:10.1016/S0304-3940(00)01779-1
152. Knobloch HS, Charlet A, Hoffmann LC, Eliava M, Khrulev S, Cetin AH, et al. Evoked axonal oxytocin release in the central amygdala attenuates fear response. Neuron (2012) 73:553–66. doi:10.1016/j.neuron.2011.11.030
153. Lee HJ, Caldwell HK, Macbeth AH, Tolu SG, Young WS III. A conditional knockout mouse line of the oxytocin receptor. Endocrinology (2008) 149:3256–63. doi:10.1210/en.2007-1710
154. Ferguson JN, Aldag JM, Insel TR, Young LJ. Oxytocin in the medial amygdala is essential for social recognition in the mouse. J Neurosci (2001) 21:8278–85.
155. Bruce HM, Parrott DM. Role of olfactory sense in pregnancy block by strange males. Science (1960) 131:1526. doi:10.1126/science.131.3412.1526
156. Bruce HM. A block to pregnancy in the mouse caused by proximity of strange males. J Reprod Fertil (1960) 1:96–103. doi:10.1530/jrf.0.0010096
157. DeVries AC, Young WS III, Nelson RJ. Reduced aggressive behaviour in mice with targeted disruption of the oxytocin gene. J Neuroendocrinol (1997) 9:363–8. doi:10.1046/j.1365-2826.1997.t01-1-00589.x
158. Macbeth AH, Lee HJ, Edds J, Young WS III. Oxytocin and the oxytocin receptor underlie intrastrain, but not interstrain, social recognition. Genes Brain Behav (2009) 8:558–67. doi:10.1111/j.1601-183X.2009.00506.x
159. Park JM, Solomon P, Mandell DS. Involvement in the child welfare system among mothers with serious mental illness. Psychiatr Serv (2006) 57:493–7. doi:10.1176/appi.ps.57.4.493
160. Reupert A, Maybery D. Families affected by parental mental illness: a multiperspective account of issues and interventions. Am J Orthopsychiatry (2007) 77:362–9. doi:10.1037/0002-9432.77.3.362
161. Lagan M, Knights K, Barton J, Boyce PM. Advocacy for mothers with psychiatric illness: a clinical perspective. Int J Ment Health Nurs (2009) 18:53–61. doi:10.1111/j.1447-0349.2008.00576.x
162. Millstein RA, Ralph RJ, Yang RJ, Holmes A. Effects of repeated maternal separation on prepulse inhibition of startle across inbred mouse strains. Genes Brain Behav (2006) 5:346–54. doi:10.1111/j.1601-183X.2005.00172.x
163. Braff D. Psychophysiological and information processing approaches to schizophrenia. In: Charney DS, Bunney BS, editors. Foundations of Mental Illness. Oxford, NY: Oxford University Press (1999). p. 258–71.
164. Perry W, Feifel D, Minassian A, Bhattacharjie I, Braff DL. Information processing deficits in acutely psychotic schizophrenia patients medicated and unmedicated at the time of admission. Am J Psychiatry (2002) 159:1375–81. doi:10.1176/appi.ajp.159.8.1375
166. Swerdlow NR, Geyer MA, Braff DL. Neural circuit regulation of prepulse inhibition of startle in the rat: current knowledge and future challenges. Psychopharmacology (Berl) (2001) 156:194–215. doi:10.1007/s002130100799
167. Braff DL, Grillon C, Geyer MA. Gating and habituation of the startle reflex in schizophrenic patients. Arch Gen Psychiatry (1992) 49:206–15. doi:10.1001/archpsyc.1992.01820030038005
168. Swerdlow NR, Martinez ZA, Hanlon FM, Platten A, Farid M, Auerbach P, et al. Toward understanding the biology of a complex phenotype: rat strain and substrain differences in the sensorimotor gating-disruptive effects of dopamine agonists. J Neurosci (2000) 20:4325–36.
169. Geyer MA, Braff DL. Habituation of the Blink reflex in normals and schizophrenic patients. Psychophysiology (1982) 19:1–6. doi:10.1111/j.1469-8986.1982.tb02589.x
170. Feifel D, Shilling PD, Belcher AM. The effects of oxytocin and its analog, carbetocin, on genetic deficits in sensorimotor gating. Eur Neuropsychopharmacol (2012) 22:374–8. doi:10.1016/j.euroneuro.2011.09.004
171. Ellenbroek BA, van den Kroonenberg PTJM, Cools AR. The effects of an early stressful life event on sensorimotor gating in adult rats. Schizophr Res (1998) 30:251–60. doi:10.1016/S0920-9964(97)00149-7
172. Feifel D, Reza T. Oxytocin modulates psychotomimetic-induced deficits in sensorimotor gating. Psychopharmacology (1999) 141:93–8. doi:10.1007/s002130050811
173. Caldwell HK, Stephens SL, Young WS III. Oxytocin as a natural antipsychotic: a study using oxytocin knockout mice. Mol Psychiatry (2009) 14:190–6. doi:10.1038/sj.mp.4002150
174. Glahn DC, Therman S, Manninen M, Huttunen M, Kaprio J, Lonnqvist J, et al. Spatial working memory as an endophenotype for schizophrenia. Biol Psychiatry (2003) 53:624–6. doi:10.1016/S0006-3223(02)01641-4
175. Sala M, Braida D, Lentini D, Busnelli M, Bulgheroni E, Capurro V, et al. Pharmacologic rescue of impaired cognitive flexibility, social deficits, increased aggression, and seizure susceptibility in oxytocin receptor null mice: a neurobehavioral model of autism. Biol Psychiatry (2011) 69:875–82. doi:10.1016/j.biopsych.2010.12.022
176. Insel TR, Winslow JT, Witt DM. Homologous regulation of brain oxytocin receptors. Endocrinology (1992) 130:2602–8. doi:10.1210/en.130.5.2602
177. Wu W, Yu LC. Roles of oxytocin in spatial learning and memory in the nucleus basalis of Meynert in rats. Regul Pept (2004) 120:119–25. doi:10.1016/j.regpep.2004.02.017
178. Duncan GE, Moy SS, Perez A, Eddy DM, Zinzow WM, Lieberman JA, et al. Deficits in sensorimotor gating and tests of social behavior in a genetic model of reduced NMDA receptor function. Behav Brain Res (2004) 153:507–19. doi:10.1016/j.bbr.2004.01.008
179. Rhee SG, Bae YS. Regulation of phosphoinositide-specific phospholipase C isozymes. J Biol Chem (1997) 272:15045–8. doi:10.1074/jbc.272.24.15045
180. Stoop R. Neuromodulation by oxytocin and vasopressin. Neuron (2012) 76:142–59. doi:10.1016/j.neuron.2012.09.025
181. Kim D, Jun KS, Lee SB, Kang NG, Min DS, Kim YH, et al. Phospholipase C isozymes selectively couple to specific neurotransmitter receptors. Nature (1997) 389:290–3. doi:10.1038/38508
182. Hannan AJ, Kind PC, Blakemore C. Phospholipase C-beta1 expression correlates with neuronal differentiation and synaptic plasticity in rat somatosensory cortex. Neuropharmacology (1998) 37:593–605. doi:10.1016/S0028-3908(98)00056-2
183. Lin XH, Kitamura N, Hashimoto T, Shirakawa O, Maeda K. Opposite changes in phosphoinositide-specific phospholipase C immunoreactivity in the left prefrontal and superior temporal cortex of patients with chronic schizophrenia. Biol Psychiatry (1999) 46:1665–71. doi:10.1016/S0006-3223(99)00036-0
184. Shirakawa O, Kitamura N, Lin XH, Hashimoto T, Maeda K. Abnormal neurochemical asymmetry in the temporal lobe of schizophrenia. Prog Neuropsychopharmacol Biol Psychiatry (2001) 25:867–77. doi:10.1016/S0278-5846(01)00149-X
185. Koh HY, Kim D, Lee J, Lee S, Shin HS. Deficits in social behavior and sensorimotor gating in mice lacking phospholipase Cbeta1. Genes Brain Behav (2008) 7:120–8. doi:10.1111/j.1601-183X.2007.00351.x
186. McOmish CE, Burrows E, Howard M, Scarr E, Kim D, Shin HS, et al. Phospholipase C-beta1 knockout mice exhibit endophenotypes modeling schizophrenia which are rescued by environmental enrichment and clozapine administration. Mol Psychiatry (2008) 13:661–72. doi:10.1038/sj.mp.4002046
187. Arendt T, Bigl V, Arendt A, Tennstedt A. Loss of neurons in the nucleus basalis of Meynert in Alzheimer’s disease, paralysis agitans and Korsakoff’s Disease. Acta Neuropathol (1983) 61:101–8. doi:10.1007/BF00697388
188. Jones DNC, Gartlon JE, Minassian A, Perry W, Geyer MA. Developing new drugs for schizophrenia: from animals to the clinic. In: McArthur R, Borsini R, editors. Animal and Translational Models for CNS Drug Discovery: Psychiatric Disorders. New York, NY: Elsevier (2008). p. 199–262.
189. Powell SB, Zhou X, Geyer MA. Prepulse inhibition and genetic mouse models of schizophrenia. Behav Brain Res (2009) 204:282–94. doi:10.1016/j.bbr.2009.04.021
190. Moghaddam B, Adams BW. Reversal of phencyclidine effects by a group II metabotropic glutamate receptor agonist in rats. Science (1998) 281:1349–52. doi:10.1126/science.281.5381.1349
191. Homayoun H, Jackson ME, Moghaddam B. Activation of metabotropic glutamate 2/3 receptors reverses the effects of NMDA receptor hypofunction on prefrontal cortex unit activity in awake rats. J Neurophysiol (2005) 93:1989–2001. doi:10.1152/jn.00875.2004
192. Ninan I. Oxytocin suppresses basal glutamatergic transmission but facilitates activity-dependent synaptic potentiation in the medial prefrontal cortex. J Neurochem (2011) 119:324–31. doi:10.1111/j.1471-4159.2011.07430.x
193. Emiliano AB, Cruz T, Pannoni V, Fudge JL. The interface of oxytocin-labeled cells and serotonin transporter-containing fibers in the primate hypothalamus: a substrate for SSRIs therapeutic effects? Neuropsychopharmacology (2007) 32:977–88. doi:10.1038/sj.npp.1301206
194. Tyzio R, Nardou R, Ferrari DC, Tsintsadze T, Shahrokhi A, Eftekhari S, et al. Oxytocin-mediated GABA inhibition during delivery attenuates autism pathogenesis in rodent offspring. Science (2014) 343:675–9. doi:10.1126/science.1247190
195. Yoshida M, Takayanagi Y, Inoue K, Kimura T, Young LJ, Onaka T, et al. Evidence that oxytocin exerts anxiolytic effects via oxytocin receptor expressed in serotonergic neurons in mice. J Neurosci (2009) 29:2259–71. doi:10.1523/JNEUROSCI.5593-08.2009
196. Eaton JL, Roache L, Nguyen KN, Cushing BS, Troyer E, Papademetriou E, et al. Organizational effects of oxytocin on serotonin innervation. Dev Psychobiol (2012) 54:92–7. doi:10.1002/dev.20566
197. Jorgensen H, Riis M, Knigge U, Kjaer A, Warberg J. Serotonin receptors involved in vasopressin and oxytocin secretion. J Neuroendocrinol (2003) 15:242–9. doi:10.1046/j.1365-2826.2003.00978.x
198. vnas-Moberg KU, Alster P, Svensson TH. Amperozide and clozapine but not haloperidol or raclopride increase the secretion of oxytocin in rats. Psychopharmacology (1992) 109:473–6. doi:10.1007/BF02247726
199. Bjork A, Olsson NG, Christensson E, Martinsson K, Olsson O. Effects of amperozide on biting behavior and performance in restricted-fed pigs following regrouping. J Anim Sci (1988) 66:669–75.
200. Gonyou HW, Parfet KA, Anderson DB, Olson RD. Effects of amperozide and azaperone on aggression and productivity of growing-finishing pigs. J Anim Sci (1988) 66:2856–64.
201. Egbe PC. Locomotor effects of amperozide. Antagonism of amphetamine-induced locomotor stimulation. Arzneimittelforschung (1989) 39:1223–4.
202. Meltzer HY, Matsubara S, Lee JC. Classification of typical and atypical antipsychotic drugs on the basis of dopamine D-1, D-2 and serotonin2 pKi values. J Pharmacol Exp Ther (1989) 251:238–46.
205. Kiss A, Bundzikova J, Pirnik Z, Mikkelsen JD. Different antipsychotics elicit different effects on magnocellular oxytocinergic and vasopressinergic neurons as revealed by Fos immunohistochemistry. J Neurosci Res (2010) 88:677–85. doi:10.1002/jnr.22226
206. Abdul-Monim Z, Reynolds GP, Neill JC. The effect of atypical and classical antipsychotics on sub-chronic PCP-induced cognitive deficits in a reversal-learning paradigm. Behav Brain Res (2006) 169:263–73. doi:10.1016/j.bbr.2006.01.019
207. Feifel D, Shilling PD, Melendez G. Clozapine and PD149163 elevate prepulse inhibition in Brown Norway rats. Behav Neurosci (2011) 125:268–72. doi:10.1037/a0022691
208. Ciapparelli A, Ducci F, Carmassi C, Carlini M, Paggini R, Catena M, et al. Predictors of response in a sample of treatment-resistant psychotic patients on clozapine. Eur Arch Psychiatry Clin Neurosci (2004) 254:343–6. doi:10.1007/s00406-004-0511-4
209. Kane JM, Honigfeld G, Singer J, Meltzer H. Clozapine in treatment-resistant schizophrenics. Psychopharmacol Bull (1988) 24:62–7.
210. Kaneda Y, Jayathilak K, Meltzer H. Determinants of work outcome in neuroleptic-resistant schizophrenia and schizoaffective disorder: cognitive impairment and clozapine treatment. Psychiatry Res (2010) 178:57–62. doi:10.1016/j.psychres.2009.04.001
211. Tiihonen J, Leucht S. Clozapine resistance – augmentation strategies. Eur Neuropsychopharmacol (2013) 23:338. doi:10.1016/j.euroneuro.2012.04.020
Keywords: dopamine, early life stress, glutamate, social cognition, sensorimotor gating
Citation: Rich ME and Caldwell HK (2015) A role for oxytocin in the etiology and treatment of schizophrenia. Front. Endocrinol. 6:90. doi: 10.3389/fendo.2015.00090
Received: 01 December 2014; Accepted: 15 May 2015;
Published: 03 June 2015
Edited by:
Hubert Vaudry, University of Rouen, FranceReviewed by:
Jacques Epelbaum, Université Paris Descartes, FranceKatsuhiko Nishimori, Tohoku University, Japan
Copyright: © 2015 Rich and Caldwell. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Heather Kingsley Caldwell, Laboratory of Neuroendocrinology and Behavior, Department of Biological Sciences, The School of Biomedical Sciences, Kent State University, PO Box 5190, 121 Cunningham Hall, Kent, OH 44242, USA,aGNhbGR3ZWxAa2VudC5lZHU=