 Luis V. Syro1*
Luis V. Syro1* Fabio Rotondo2Alex Ramirez3
Fabio Rotondo2Alex Ramirez3 Antonio Di Ieva4
Antonio Di Ieva4 Murat Aydin Sav5Lina M. Restrepo6Carlos A. Serna7Kalman Kovacs2
Murat Aydin Sav5Lina M. Restrepo6Carlos A. Serna7Kalman Kovacs2
- 1Department of Neurosurgery, Hospital Pablo Tobon Uribe and Clinica Medellin, Medellin, Colombia
- 2Laboratory Medicine, Division of Pathology, St. Michael’s Hospital, University of Toronto, Toronto, ON, Canada
- 3Department of Endocrinology, Universidad Pontificia Bolivariana, Medellin, Colombia
- 4Department of Neurosurgery, Australian School of Advanced Medicine, Macquarie University, Sydney, NSW, Australia
- 5Nisantasi Pathology Group, Istanbul, Turkey
- 6Division of Endocrinology, Clinica Medellin, Medellin, Colombia
- 7Laboratorio de Patologia y Citologia Rodrigo Restrepo, Department of Pathology, Clinica Medellin, Medellin, Colombia
Pituitary adenomas are common neoplasms. Their classification is based upon size, invasion of adjacent structures, sporadic or familial cases, biochemical activity, clinical manifestations, morphological characteristics, response to treatment and recurrence. Although they are considered benign tumors, some of them are difficult to treat due to their tendency to recur despite standardized treatment. Functional tumors present other challenges for normalizing their biochemical activity. Novel approaches for early diagnosis, as well as different perspectives on classification, may help to identify subgroups of patients with similar characteristics, creating opportunities to match each patient with the best personalized treatment option. In this paper, we present the progress in the diagnosis and classification of different subgroups of patients with pituitary tumors that may be managed with specific considerations according to their tumor subtype.
Introduction
Pituitary adenomas are benign tumors representing approximately 15–20% of intracranial neoplasms (1). They can present with pituitary dysfunction, neurological deficits (especially visual impairment), and/or invasion into parasellar compartment and/or sphenoid sinuses. Endocrinologically active tumors present different challenges to normalize their hormonal production (2). Initially considered as sporadic tumors, some of them are associated with familial syndromes (3). Morphologically, pituitary adenomas represent a heterogeneous group of tumors and their meticulous pathological classification is required (4).
Early diagnosis of pituitary tumors is advisable and their proper classification is of paramount importance for treatment and prognostic purposes (5). Pituitary adenomas have been classified according to the clinical, radiological, and endocrinological findings, tumor size, and invasiveness. In morphologic/pathologic studies, they were initially classified on the basis of their tinctorial characteristics with hematoxylin–eosin stain as acidophilic, basophilic, amphophilic, and chromophobic adenomas. Immunohistochemical investigation achieved a major progress identifying hormone production by tumor cells. Electron microscopy added further information by defining the cell type of the tumor and its ultrastructure. Recent molecular/genetic/epigenetic methods are still in their initial phase and more research is required.
Despite the standard protocols of treatment, some pituitary adenomas may have a clinically aggressive course with tendency to recur, becoming giant in size, and/or invading surrounding structures (6). At present, there is no consensus regarding the diagnosis of aggressive adenomas. The early recognition of aggressiveness and the prediction of pituitary tumor behavior remain a challenge (7). There are no specific and universally accepted biomarkers yet. The correct diagnosis of histological subtypes of pituitary adenomas may predict clinical aggressive behavior in some cases. Here, we present a novel progress in the diagnosis of acromegaly and different subgroups of pituitary adenomas which, in the opinion of the authors, deserve special mention due to their characteristic clinical behavior and special considerations in terms of diagnosis, treatment and follow-up.
Progress in Diagnosis
Early Diagnosis in Acromegaly
Despite the characteristic physical manifestations and significant comorbidities, the diagnosis of acromegaly may take some years. Its slow and insidious course and its changes are frequently unnoticed by the patient, family members, friends, and physicians. Although new surgical and medical treatments have emerged along with new developments in laboratory tests, the diagnosis of patients with acromegaly has not changed in many years. Reid et al. (8) studied 324 patients from the same institution during two different periods of time and no significant differences were found in terms of clinical symptoms, size of the tumor, and delay at diagnosis in both groups. The clinical recognition of acromegaly has not improved over the last 25 years. Novel approaches for an early diagnosis of acromegaly have been proposed. Based on photographs, Miller et al. (9) assessed the accuracy of diagnosis of acromegaly between a computer program and general physicians. The diagnostic accuracy of the computer model was 86%, whereas that of the physicians was only 26%. In a similar study, Schneider et al. (10) compared classification accuracy of acromegaly by means of a face analysis software. Their program correctly classified 71.9% of patients versus 63.2 and 42.1% by experts and general internists, respectively. In both studies, computer analysis reached diagnosis better than physicians. By using photographs and special software, it can be possible to detect and diagnose acromegalic changes early, even in cases with low clinical suspicion. This innovative approach opens the possibility that, in the near future, using specialized software, mobile apps, and capillary networks, diagnosis can be made earlier. In a society consisting of more well-informed patients, astonishing technological advances and easy internet access, novel ways of improving early diagnosis of acromegaly should be implemented.
Progress in Classification
Familial Pituitary Tumors
Recent findings have revealed the existence of familial pituitary tumor syndromes (3, 11). These syndromes represent a group of diseases with different genetic background and variable phenotype. The most frequently seen are multiple endocrine neoplasia type 1 (MEN1), familial isolated pituitary adenoma (FIPA), and Carney complex (CNC) (Figure 1). Other uncommon familial syndromes include somatotropinoma/paraganglioma (12), pituitary blastoma (13), and X-linked acrogigantism (X-LAG) syndrome (14, 15). These will not be discussed since they are not in the scope of this review.
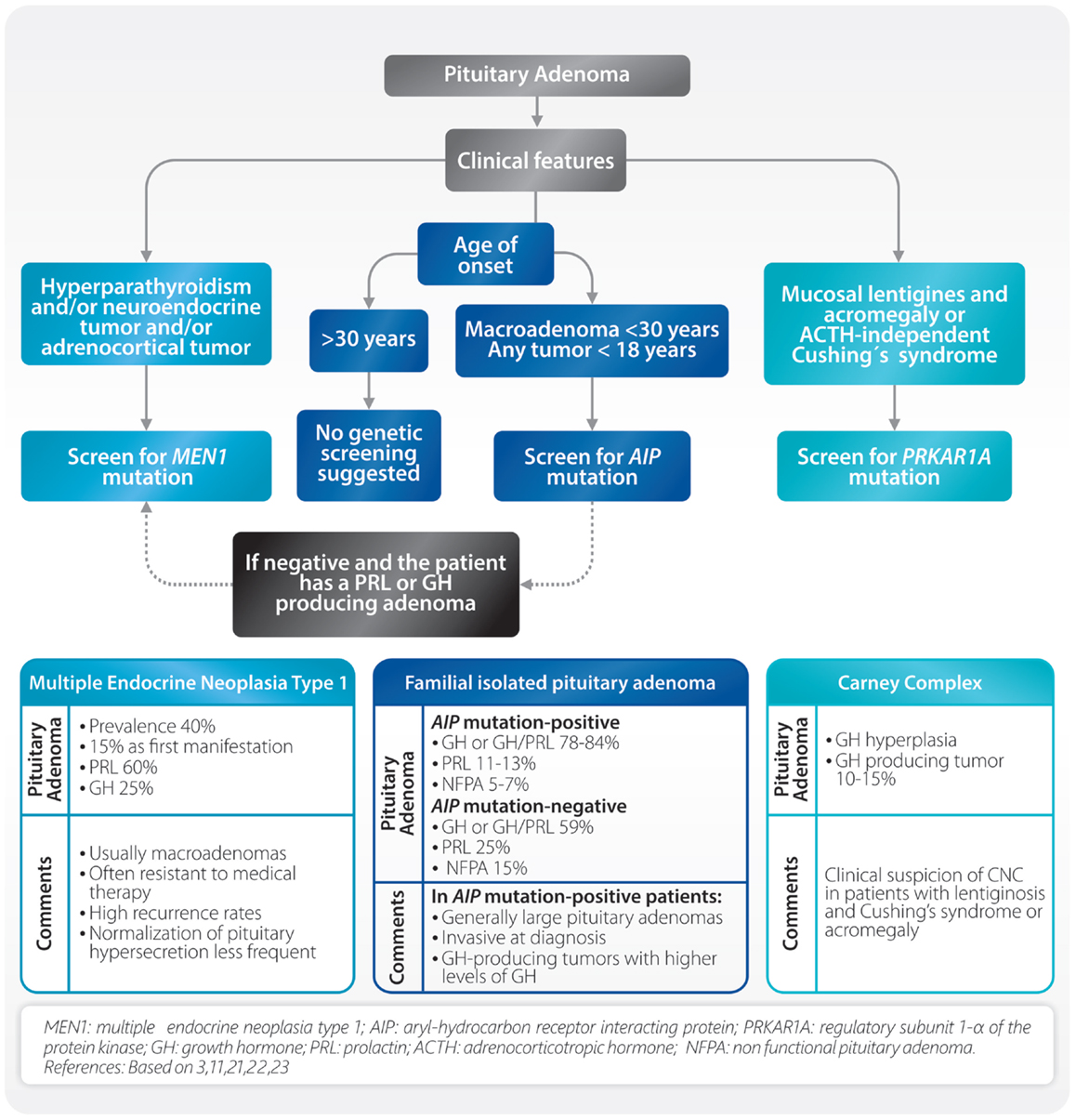
Figure 1. Considerations in apparently sporadic pituitary adenomas and common familial syndromes.
Multiple endocrine neoplasia type 1 (MEN1) is an autosomal-dominant disorder characterized by tumors of the pituitary, parathyroid, endocrine gastrointestinal tract, endocrine pancreas, and adrenal cortex (16). Localized on chromosome 11q13, MEN1 is a tumor suppressor gene encoding menin, a nuclear protein involved in transcriptional regulation, genome stability, cell division, and proliferation. About 5–10% of patients with MEN1 phenotype may not harbor mutations in the coding region of the MEN1 gene. In a small group of patients, mutations in CDKN1B were found. This infrequent MEN1-like syndrome was named MEN4 (17). Patients with MEN1 syndrome usually have a family history of MEN1 and MEN1 gene mutations, which can be identified in 70–95% of patients (16). Clinical diagnosis is established when two of these features are present or when one feature is present together with a first-degree relative with established MEN1. Pituitary tumors in MEN1 patients are present from 10 to 60% and they can be the first clinical manifestation in up to 15% of the cases. Pituitary adenomas associated with MEN1 differ from sporadic ones (18). In MEN1, they are usually diagnosed at earlier ages, frequently macroadenomas, often resistant to medical therapy and with high recurrence rates (18). Although PRL and GH-producing adenomas are the most frequent, almost all types of pituitary adenomas can occur (19). Due to the fact that primary hyperparathyroidism is present in the majority (approximately 90%) of MEN1 patients, parathyroid hyperplasia/adenoma/carcinoma should be ruled out in all cases of apparently sporadic pituitary adenomas. In this setting, examination of the parathyroid gland and its function can help to detect possible MEN1 patients (Figure 1). Characterization of the MEN1 gene product, menin, can also be achieved using immunohistochemistry (IHC) by site-specific MEN1 antibodies. Here, menin can be localized to the nucleus of pituitary adenomas removed from patients with MEN1 (20).
Familial isolated pituitary adenoma is characterized by the presence of pituitary adenomas in two or more members of a family. Prevalence of FIPA is not known but it is probably similar to MEN1 (21). In 20% of these families, a mutation on the aryl-hydrocarbon receptor interacting protein (AIP) gene is found. In patients with apparently sporadic pituitary adenomas under the age of 18 years, AIP mutations are present in approximately 20% (11). FIPA shows an autosomal dominant pattern with incomplete penetrance and a wide variation between families. The affected families can be classified as AIP mutation-positive or AIP mutation-negative, and according to the phenotype as: (a) homogenous, if the same type of pituitary adenoma is present, or (b) heterogeneous, if different types of tumors occur within the same family. There are some differences between AIP mutation-positive and AIP mutation-negative patients (Figure 1). In AIP mutation-positive patients, the mean age of onset is lower (20–24 years), they are predominantly males (63.5%), and they present in 85% of the cases with GH or GH/PRL producing tumors. AIP mutation-positive acromegalic patients have larger and more invasive tumors (22) (Figure 1). At present, published data are inconclusive in regards to the clinical behavior of AIP mutation-negative patients. Diagnosis of AIP-related FIPA relies on the detailed analysis of the pituitary adenoma based on hormone secretion, imaging, and histologic findings. Using IHC techniques, in cases where the patient is AIP mutation-positive, immunostaining for aryl hydrocarbon receptor nuclear translocator (ARNT) and aryl hydrocarbon receptor (AHR) will demonstrate a decrease of ARNT protein expression and an increase of AHR localization in the nucleus (23, 24). Increased expression of nuclear AHR in pituitary adenomas with AIP mutations may indicate a loss of signal in phosphodiesterase 2 enzyme (PDE2A), which normally regulates cyclic adenosine monophosphate (cAMP). As a result, there is an increase in cAMP concentration which may lead to the subsequent formation of a pituitary adenoma (25).
Carney complex, an autosomal dominant syndrome, is characterized by the following features: multiple skin lesions (blue nevus, spotty skin pigmentation, and mucosal lentigines), cardiac myxoma, acromegaly, psammomatous melanocytic schwannoma, thyroid carcinoma, multiple hypoechoic thyroid nodules, and primary pigmented nodular adrenocortical disease (PPNAD) with Cushing’s syndrome (26). It is produced by an inactivating mutation of the regulatory subunit 1-α of the protein kinase A (PRKAR1A). The diagnosis can be established when two of the above mentioned major features occur, or in the presence of one major feature and an inactivating PRKAR1A mutation, or a first-degree relative with CNC. Clinical manifestations are variable even within members of the same family and one-third of the patients present as simplex (sporadic) cases. Lentiginosis is the most common feature of CNC (70%) consisting of small, 2–10 mm brown to black macule on the lips, eyelids, ears, and genitals, and can be present at birth and acquire their clinical characteristics at puberty (26). ACTH independent Cushing’s syndrome due to PPNAD is the main endocrine manifestation (60%) followed by acromegaly due to pituitary adenoma or adenohypophyseal hyperplasia.
In all cases of apparently sporadic pituitary adenomas, an effort for recognizing a familial syndrome must be made because in many instances the clinical behavior and response to treatment are different (18). Primary hyperparathyroidism must be ruled out, because this is present in 90% of MEN1 patients. If other clinical features of MEN1 are present, MEN1 mutation analysis should be performed. In patients with tumors diagnosed before 18 years of age, as well as patients with macroadenomas before 30 years of age, it is advisable to screen AIP and/or MEN1 mutations (27). Clinical suspicion of CNC must arise in patients with lentiginosis and Cushing’s syndrome or acromegaly (Figure 1) (28).
Progress in Morphological Classification
Somatotroph Pituitary Adenomas
GH-producing adenomas constitute 10–15% of all pituitary adenomas. The more common morphological subtypes are densely and sparsely granulated somatotroph adenomas (5, 29). Densely granulated somatotroph adenomas (DGSA) are acidophilic tumors with diffuse and intense GH immunopositivity. Immunostaining demonstrates low molecular weight keratin (LMWK), Cam5.2, in a diffuse perinuclear pattern. Sparsely granulated somatotroph adenomas (SGSA) are composed of chromophobic or mildly acidophilic cells. Immunopositivity for GH is variable, it is usually scarce and of low intensity. The fibrous bodies, initially described by electron microscopy and corresponding to accumulation of paranuclear cytokeratins, are the characteristic feature of this tumor type. With IHC, using LMWK (Cam5.2) the identification of paranuclear “dot” pattern is characteristic of this tumor type and correlates with the electron microscopic finding (30). In recent studies, a correlation between these subtypes and clinical response was confirmed (31, 32). Clinical, histopathological, and radiological characteristics of acromegalic patients were subjected to cluster analysis by Cuevas–Ramos et al. (31). They classified acromegalic patients in three groups based on significant differences in morphology, tumor aggressiveness, treatment responsiveness, expression profile of somatotroph surface receptors, markers of cell senescence, and disease outcomes. Type-1 comprised patients with densely granulated GH tumors, micro or macroadenomas with higher expression of somatostatin receptor 2 (SSTR2), and p21, a cyclin-dependent kinase inhibitor (CDKI) (33, 34). Type-2 comprised patients with mixed densely and sparsely granulated GH tumors and Type-3 comprised patients with sparsely granulated GH tumors. They found that Type-3 adenomas were aggressive macroadenomas, and comprised patients with adverse outcomes, in spite of receiving more treatment modalities. This proposed classification may identify distinctive patterns of disease aggressiveness and outcomes in acromegalic patients and it can be a useful tool for selection criteria in clinical studies. Definite clinical distinction is useful between DGSA and SGSA, because their recognition is helpful as far as prognosis and treatment is concerned (5, 35, 36). Assessment of somatostatin receptors 2 and 5 (SSTR2, SSTR5) stains can be useful to predict the response to medical treatment (37–39).
Crooke’s Cell Adenomas
ACTH-secreting adenomas constitute 10–15% of all pituitary adenomas and present three different morphological subtypes. The densely granulated corticotroph subtype is the most common. This tumor is composed of basophilic or amphophilic, PAS positive cells, strongly immunopositive for ACTH and LMWK (Cam5.2) in a perinuclear pattern. By electron microscopy, numerous secretory granules are noted and bundles of keratin filaments are apparent around the nucleus, consistent with perinuclear LMWK (Cam5.2) immunostaining. Patients with Cushing’s disease and Nelson’s syndrome usually present this histologic subtype. Sparsely granulated corticotroph adenoma, the second subtype is rare. It is immunopositive for LMWK (Cam5.2) and variable for ACTH.
Crooke’s cell adenomas are the third subtype (40). In 1935, Crooke was the first to describe the histologic features of the adenohypophysis in patients with Cushing’s syndrome. He noticed that, in the presence of ACTH secreting adenomas, non-neoplastic corticotrophs often showed accumulation of intracytoplasmic, perinuclear hyaline material. These Crooke’s cells are considered corticotrophs which, in presence of glucocorticoid excess, undergo massive accumulation of perinuclear cytokeratin. Using LMWK (Cam5.2) immunostaining, a strong, ring-like pattern around the nucleus is seen, with displacement of ACTH immunoreactivity under the cell membrane. In some ACTH producing adenomas, for unknown reasons, there is a massive hyaline change in the majority of the cells, the same as the Crooke’s cells seen in the adenohypophysis of patients with glucocorticoid excess (40). The reasons why these cells produce ACTH, and at the same time display Crooke’s hyaline changes are not well understood. Tumors that contain so-called Crooke’s hyaline material in their cytoplasm of more than 50% of the cells are classified as Crooke’s cell adenomas (40, 41). As mentioned, Crooke’s cell tumors may produce ACTH causing Cushing’s disease in 75% of the cases, or may be endocrinologically silent. They frequently exhibit aggressive clinical behavior, with high recurrence rate and invasiveness (41); thus, strict surveillance and eventual multimodal treatment are recommended.
Aggressive Pituitary Adenomas
According to the World Health Organization (WHO), pituitary tumors are classified as typical adenomas, atypical adenomas, and carcinomas (42). The majority are typical adenomas, with monotonous histological features. They are slow growing, well demarcated, non-invasive adenomas, showing no major cellular and nuclear pleomorphism, few mitotic figures, and a Ki-67 nuclear index <3%. Atypical adenomas are tumors that disclose “atypical morphological features suggestive of aggressive behavior” (42), such as invasive growth, elevated mitotic index, a Ki-67 labeling index >3%, and extensive nuclear staining for the p53 protein. Pituitary carcinomas can only be diagnosed if cerebrospinal and/or systemic metastases are documented. They may develop by transformation from adenomas or arise de novo from non-tumorous adenohypophyseal cells. Pituitary carcinomas produce more often PRL or ACTH (43). However, GH, TSH, FSH, LH, or alpha subunit can also be produced or they may be immunohistochemically negative.
The WHO classification of typical and atypical adenomas does not correlate with clinical behavior. Neither all typical adenomas have a benign clinical evolution nor do all atypical adenomas have the tendency to recur or invade surrounding structures. Some tumors exhibit high rate of recurrence, resistance to conventional treatments, and invasion to the parasellar compartment and/or sphenoid sinuses, often requiring multiple surgeries. They seem to represent a distinct entity and may be defined as aggressive pituitary adenomas (6, 44, 45). Their distinction and definition are controversial and elusive. Morphologically, they are pleomorphic, contain mitotic figures, and have a rapid cell proliferative rate. The assessment of the Ki-67 nuclear labeling, using the MIB-1 antibody, can be the most useful tool. If the Ki-67 nuclear labeling index is more than 10%, the tumor could be classified as aggressive adenoma, although there is no agreement on this (7, 43). A recent clinicopathological classification, which takes into account proliferation markers, invasion to cavernous and sphenoid sinus and size, has been proposed (46). For the criteria of invasiveness, the authors considered histological and/or radiological signs of cavernous sinus or sphenoid sinus invasion. For the assessment of cell proliferation, at least two of the three following criteria had to be present: Ki67 ≥3%, 2 or more mitoses per 10 high-power fields, and p53 immunopositivity. Pituitary adenomas were classified in five groups according to the invasion, presence of proliferation, and metastasis. After an 8-year follow-up, invasive and proliferative tumors had an increased probability of tumor persistence or progression. Furthermore, based on the fact that six out of the eight carcinomas in their series were classified as invasive and proliferative at the first surgery, the authors postulated that they are possibly malignant tumors without metastasis and proposed that the term tumor suspected of malignancy be used for them (47). Other authors have suggested the term in situ carcinoma or a premetastatic pituitary carcinoma in the sellar phase (43, 48).
Aggressive adenomas can produce GH, PRL, ACTH, TSH, FSH, LH, alpha subunit, or they can be immunonegative. Some aggressive adenomas are silent, not over-producing hormones, and unassociated clinically with hormonal excess. The question arises whether aggressive pituitary adenomas have malignant potential. It may well be that some of them are actually carcinomas without any accompanying solid evidence of metastases. Some carcinomas may have their malignant nature already before any metastases can be detected and may metastasize afterward. These are important questions; their resolution is of crucial significance. Obviously, new biomarkers have to be investigated to resolve the biologic behavior of aggressive pituitary adenomas and carcinomas. There may be two alternative possibilities that carcinomas may develop: either de novo from normal non-tumorous adenohypophyseal cells or transform gradually from adenoma to aggressive adenoma and carcinoma (1, 49). Further studies are needed to conclusively recognize and identify aggressive pituitary tumors.
Pituitary Transcription Factors
During development, the process of cell differentiation is coordinated by specific transcription factors. They also have some estimated roles in determining the cytodifferentiation and hormone production of pituitary adenomas, and can help in their classification. Pituitary tumors are monoclonal benign adenomas, and secrete specific hormones reflective of their differentiated cell of origin. Pituitary-specific transcription factor 1 (Pit-1) defines cells that can produce GH, PRL, and/or TSH. T-box transcription factor TBX19 (Tpit) identifies corticotrophs. Estrogen receptor alpha (ER-α) cooperates with Pit-1 to enhance PRL secretion; therefore, coexpression of Pit-1 and ER-α is seen in lactotrophs. Guanine-Adenine-Thymine-Adenine binding protein 2 (GATA-2) appears to be an important contributor to thyrotroph development and is coexpressed with Pit-1 in thyrotrophs. Expression of steroidogenic factor 1 (SF1), ER-α, and GATA-2 identify gonadotrophs. Immunohistochemical demonstration of transcription factors helps to accurately classify pituitary adenomas (Figure 2), especially in cases of low or absent of identifiable hormone content (4, 50).
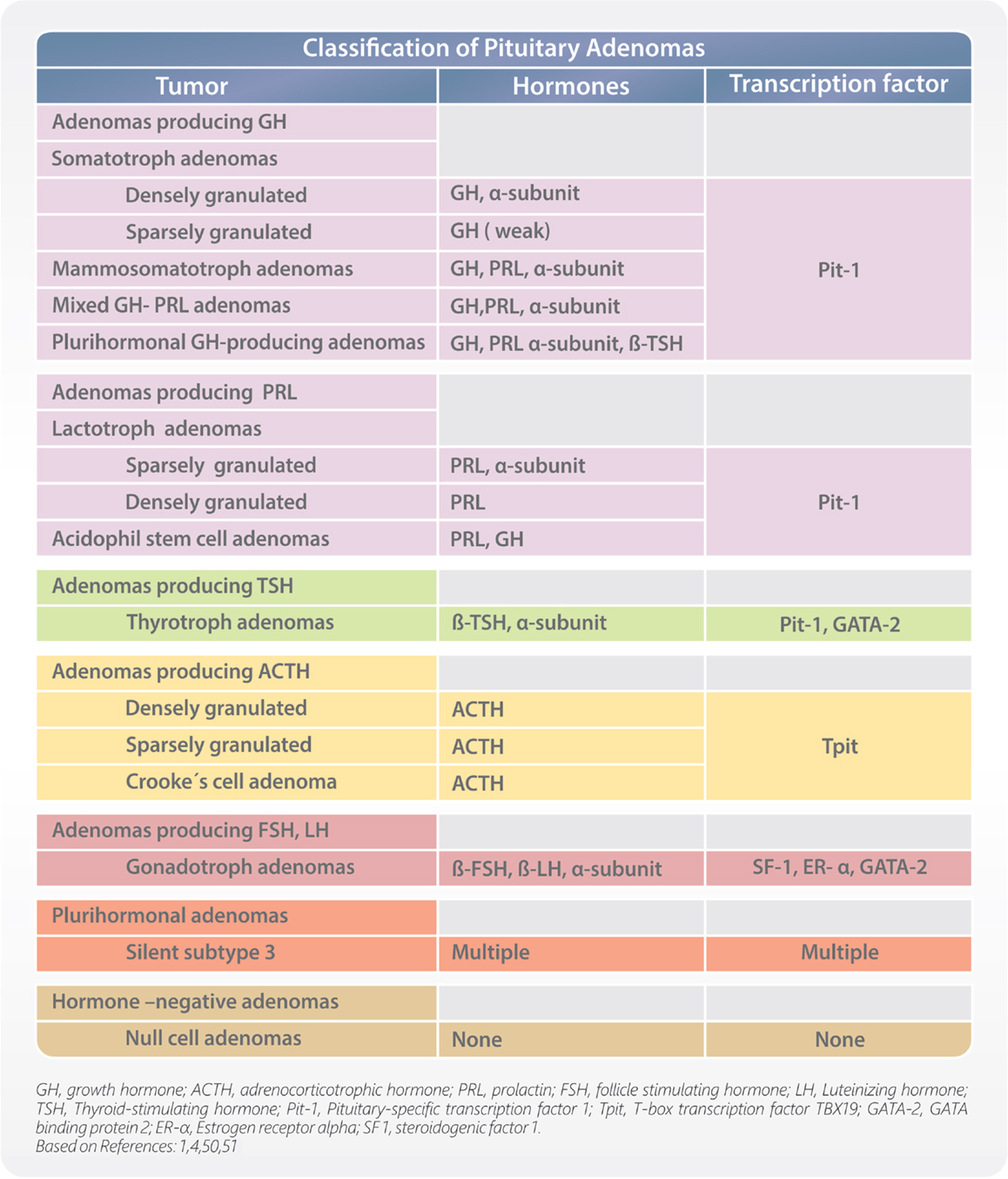
Figure 2. Classification of pituitary adenomas.
The discovery of adenohypophyseal cell plasticity has changed our thinking in the classification of pituitary tumors (51). Earlier, it was universally accepted that pituitary cells can produce only one hormone, cannot change their morphology and hormonal production, and cannot transform to another cell type. This concept has been proven to be inaccurate and the findings of adenohypophyseal cell plasticity changed our understanding completely. At present, conclusive evidence indicates that adenohypophyseal cells can transform to another cell type, they can produce more than one pituitary hormone, and can change their morphology and phenotype. Adenohypophyseal cell plasticity also alters our views in regards to pituitary tumors. The factors that affect pituitary cell transformation are not well known and more work is needed to confirm the mechanism behind cell plasticity. Adenohypophyseal cell plasticity can have a major influence in the proper classification of pituitary tumors. It is possible that pituitary adenoma cells can change their morphology and phenotype and transform to another cell type, known as transdifferentiation. Moreover, some reports have also showed that benign, slow-growing adenomas can transform to aggressive adenomas or carcinomas (52).
Markers of Transformation of Pituitary Adenomas
The search for new, reliable markers to predict the behavior of pituitary tumors is ongoing (1). Some markers have shown promise; however, their utility to conclusively indicate pituitary tumor transformation is debatable. Early studies of markers such as galectin-3 and cyclooxygenase II (Cox-2) have shown inconsistent results. Others, such as matrix metalloproteinase’s (MMPs), p27, p21, vascular endothelial growth factor (VEGF), CD34, hypoxia-inducible factor 1 alpha and pituitary tumor transforming gene (PTTG), show promising roles as markers for indicating tumor behavior but more work is needed to elucidate their role and prognostic value.
Conclusion
Proper classification of pituitary tumors is an important area of research. Novel approaches for early diagnosis as well as different perspectives on their classification may help to identify subgroups of patients that share similar characteristics (31). The recognition of these subgroups creates opportunities to match each patient with the best personalized treatment option. In the subgroups of patients discussed here, some specific characteristics may predict their clinical behavior and their response to treatment. New techniques may help us to reach a more accurate classification and personalized and precise treatment options for patients harboring pituitary adenomas (53).
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
Authors are grateful to the Jarislowsky and Lloyd Carr-Harris Foundations for their support.
References
1. Melmed S. Pathogenesis of pituitary tumors. Nat Rev Endocrinol (2011) 7(5):257–66. doi: 10.1038/nrendo.2011.40
2. Melmed S. Medical progress: acromegaly. N Engl J Med (2006) 355(24):2558–73. doi:10.1056/NEJMra062453
3. Gadelha MR, Trivellin G, Hernandez Ramirez LC, Korbonits M. Genetics of pituitary adenomas. Front Horm Res (2013) 41:111–40. doi:10.1159/000345673
4. Mete O, Asa SL. Therapeutic implications of accurate classification of pituitary adenomas. Semin Diagn Pathol (2013) 30(3):158–64. doi:10.1053/j.semdp.2013.06.002
5. Beatriz M, Lopes S. Growth hormone-secreting adenomas: pathology and cell biology. Neurosurg Focus (2010) 29(4):E2. doi:10.3171/2010.7.FOCUS10169
6. Colao A, Grasso LF, Pivonello R, Lombardi G. Therapy of aggressive pituitary tumors. Expert Opin Pharmacother (2011) 12(10):1561–70. doi:10.1517/14656566.2011.568478
7. Heaney A. Management of aggressive pituitary adenomas and pituitary carcinomas. J Neurooncol (2014) 117(3):459–68. doi:10.1007/s11060-014-1413-6
8. Reid TJ, Post KD, Bruce JN, Nabi Kanibir M, Reyes-Vidal CM, Freda PU. Features at diagnosis of 324 patients with acromegaly did not change from 1981 to 2006: acromegaly remains under-recognized and under-diagnosed. Clin Endocrinol (2010) 72(2):203–8. doi:10.1111/j.1365-2265.2009.03626.x
9. Miller RE, Learned-Miller EG, Trainer P, Paisley A, Blanz V. Early diagnosis of acromegaly: computers vs clinicians. Clin Endocrinol (2011) 75(2):226–31. doi:10.1111/j.1365-2265.2011.04020.x
10. Schneider HJ, Kosilek RP, Gunther M, Roemmler J, Stalla GK, Sievers C, et al. A novel approach to the detection of acromegaly: accuracy of diagnosis by automatic face classification. J Clin Endocrinol Metab (2011) 96(7):2074–80. doi:10.1210/jc.2011-0237
11. Lecoq AL, Kamenicky P, Guiochon-Mantel A, Chanson P. Genetic mutations in sporadic pituitary adenomas-what to screen for? Nat Rev Endocrinol (2015) 11(1):43–54. doi:10.1038/nrendo.2014.181
12. Xekouki P, Szarek E, Bullova P, Giubellino A, Quezado M, Mastroyannis SA, et al. Pituitary adenoma with paraganglioma/pheochromocytoma (3PAs) and succinate dehydrogenase defects in human and mice. J Clin Endocrinol Metab (2015) 100(5):E710–9. doi:10.1210/jc.2014-4297
13. de Kock L, Sabbaghian N, Plourde F, Srivastava A, Weber E, Bouron-Dal Soglio D, et al. Pituitary blastoma: a pathognomonic feature of germ-line DICER1 mutations. Acta Neuropathol (2014) 128(1):111–22. doi:10.1007/s00401-014-1285-z
14. Beckers A, Lodish M, Giampaolo T, Rostomyan L, Lee M, Faucz FR, et al. X-linked acrogigantism (X-LAG) syndrome: clinical profile and therapeutic responses. Endocr Relat Cancer (2015) 22(3):353–67. doi:10.1530/ERC-15-0038
15. Trivellin G, Daly AF, Faucz FR, Yuan B, Rostomyan L, Larco DO, et al. Gigantism and acromegaly due to Xq26 microduplications and GPR101 mutation. N Engl J Med (2014) 371(25):2363–74. doi:10.1056/NEJMoa1408028
16. Thakker RV. Multiple endocrine neoplasia type 1 (MEN1). Best Pract Res Clin Endocrinol Metab (2010) 24(3):355–70. doi:10.1016/j.beem.2010.07.003
17. Schernthaner-Reiter MH, Trivellin G, Stratakis CA. MEN1, MEN4, and carney complex: pathology and molecular genetics. Neuroendocrinology (2015). doi:10.1159/000371819
18. Toledo SP, Lourenco DM Jr, Toledo RA. A differential diagnosis of inherited endocrine tumors and their tumor counterparts. Clinics (Sao Paulo) (2013) 68(7):1039–56. doi:10.6061/clinics/2013(07)24
19. Trouillas J, Labat-Moleur F, Sturm N, Kujas M, Heymann MF, Figarella-Branger D, et al. Pituitary tumors and hyperplasia in multiple endocrine neoplasia type 1 syndrome (MEN1): a case-control study in a series of 77 patients versus 2509 non-MEN1 patients. Am J Surg Pathol (2008) 32(4):534–43. doi:10.1097/PAS.0b013e31815ade45
20. Theodoropoulou M, Cavallari I, Barzon L, D’Agostino DM, Ferro T, Arzberger T, et al. Differential expression of menin in sporadic pituitary adenomas. Endocr Relat Cancer (2004) 11(2):333–44. doi:10.1677/erc.0.0110333
21. Martucci F, Trivellin G, Korbonits M. Familial isolated pituitary adenomas: an emerging clinical entity. J Endocrinol Invest (2012) 35(11):1003–14. doi:10.1007/BF03346742
22. Daly AF, Tichomirowa MA, Petrossians P, Heliovaara E, Jaffrain-Rea ML, Barlier A, et al. Clinical characteristics and therapeutic responses in patients with germ-line AIP mutations and pituitary adenomas: an international collaborative study. J Clin Endocrinol Metab (2010) 95(11):E373–83. doi:10.1210/jc.2009-2556
23. Leontiou CA, Gueorguiev M, van der Spuy J, Quinton R, Lolli F, Hassan S, et al. The role of the aryl hydrocarbon receptor-interacting protein gene in familial and sporadic pituitary adenomas. J Clin Endocrinol Metab (2008) 93(6):2390–401. doi:10.1210/jc.2007-2611
24. Boikos SA, Stratakis CA. Molecular genetics of the cAMP-dependent protein kinase pathway and of sporadic pituitary tumorigenesis. Hum Mol Genet (2007) 16(Spec No 1):R80–7. doi:10.1093/hmg/ddm019
25. Bender AT, Beavo JA. Cyclic nucleotide phosphodiesterases: molecular regulation to clinical use. Pharmacol Rev (2006) 58(3):488–520. doi:10.1124/pr.58.3.5
26. Salpea P, Stratakis CA. Carney complex and McCune Albright syndrome: an overview of clinical manifestations and human molecular genetics. Mol Cell Endocrinol (2014) 386(1–2):85–91. doi:10.1016/j.mce.2013.08.022
27. Beckers A, Aaltonen LA, Daly AF, Karhu A. Familial isolated pituitary adenomas (FIPA) and the pituitary adenoma predisposition due to mutations in the aryl hydrocarbon receptor interacting protein (AIP) gene. Endocr Rev (2013) 34(2):239–77. doi:10.1210/er.2012-1013
28. Syro LV, Builes CE, Di Ieva A, Sav A, Rotondo F, Kovacs K. Improving differential diagnosis of pituitary adenomas. Expert Rev Endocrinol Metab (2014) 9(4):377–86. doi:10.1586/17446651.2014.922412
29. Yamada S, Aiba T, Sano T, Kovacs K, Shishiba Y, Sawano S, et al. Growth hormone-producing pituitary adenomas: correlations between clinical characteristics and morphology. Neurosurgery (1993) 33(1):20–7. doi:10.1227/00006123-199307000-00003
30. Obari A, Sano T, Ohyama K, Kudo E, Qian ZR, Yoneda A, et al. Clinicopathological features of growth hormone-producing pituitary adenomas: difference among various types defined by cytokeratin distribution pattern including a transitional form. Endocr Pathol (2008) 19(2):82–91. doi:10.1007/s12022-008-9029-z
31. Cuevas-Ramos D, Carmichael JD, Cooper O, Bonert VS, Gertych A, Mamelak AN, et al. A structural and functional acromegaly classification. J Clin Endocrinol Metab (2015) 100(1):122–31. doi:10.1210/jc.2014-2468
32. Kiseljak-Vassiliades K, Carlson N, Borges M, Kleinschmidt-DeMasters BK, Lillehei K, Kerr J, et al. Growth hormone tumor histological subtypes predict response to surgical and medical therapy. Endocrine (2014) 49(1):1–11. doi:10.1007/s12020-014-0383-y
33. Chesnokova V, Melmed S. Pituitary senescence: the evolving role of Pttg. Mol Cell Endocrinol (2010) 326(1–2):55–9. doi:10.1016/j.mce.2010.02.012
34. Chesnokova V, Zonis S, Kovacs K, Ben-Shlomo A, Wawrowsky K, Bannykh S, et al. p21(Cip1) restrains pituitary tumor growth. Proc Natl Acad Sci U S A (2008) 105(45):17498–503. doi:10.1073/pnas.0804810105
35. Fougner SL, Casar-Borota O, Heck A, Berg JP, Bollerslev J. Adenoma granulation pattern correlates with clinical variables and effect of somatostatin analogue treatment in a large series of patients with acromegaly. Clin Endocrinol (2012) 76(1):96–102. doi:10.1111/j.1365-2265.2011.04163.x
36. Mori R, Inoshita N, Takahashi-Fujigasaki J, Joki T, Nishioka H, Abe T, et al. Clinicopathological features of growth hormone-producing pituitary adenomas in 242 acromegaly patients: classification according to hormone production and cytokeratin distribution. ISRN Endocrinol (2013) 2013:723432. doi:10.1155/2013/723432
37. Theodoropoulou M, Stalla GK. Somatostatin receptors: from signaling to clinical practice. Front Neuroendocrinol (2013) 34(3):228–52. doi:10.1016/j.yfrne.2013.07.005
38. Chinezu L, Vasiljevic A, Jouanneau E, Francois P, Borda A, Trouillas J, et al. Expression of somatostatin receptors, SSTR2A and SSTR5, in 108 endocrine pituitary tumors using immunohistochemical detection with new specific monoclonal antibodies. Hum Pathol (2014) 45(1):71–7. doi:10.1016/j.humpath.2013.08.007
39. Cuevas-Ramos D, Fleseriu M. Somatostatin receptor ligands and resistance to treatment in pituitary adenomas. J Mol Endocrinol (2014) 52(3):R223–40. doi:10.1530/JME-14-0011
40. Felix IA, Horvath E, Kovacs K. Massive Crooke’s hyalinization in corticotroph cell adenomas of the human pituitary. A histological, immunocytological, and electron microscopic study of three cases. Acta Neurochir (Wien) (1981) 58(3–4):235–43.
41. Di Ieva A, Davidson JM, Syro LV, Rotondo F, Montoya JF, Horvath E, et al. Crooke’s cell tumors of the pituitary. Neurosurgery (2015) 76(5):616–22. doi:10.1227/NEU.0000000000000657
42. Lloyd RV, Kovacs K, Young WF Jr, Farrell W, Asa SL, Trouillas J, et al. Pituitary tumors: introduction. In: DeLlelis RA, Lloyd RV, Heitz PU, Eng C, editors. Pathology and Genetics of Tumours of Endocrine Organs. Lyon: IARC Press (2004). p. 10–3.
43. Heaney AP. Pituitary carcinoma: difficult diagnosis and treatment. J Clin Endocrinol Metab (2011) 96(12):3649–60. doi:10.1210/jc.2011-2031
44. Di Ieva A, Rotondo F, Syro LV, Cusimano MD, Kovacs K. Aggressive pituitary adenomas – diagnosis and emerging treatments. Nat Rev Endocrinol (2014) 10(7):423–35. doi:10.1038/nrendo.2014.64
45. Buchfelder M. Management of aggressive pituitary adenomas: current treatment strategies. Pituitary (2009) 12(3):256–60. doi:10.1007/s11102-008-0153-z
46. Trouillas J, Roy P, Sturm N, Dantony E, Cortet-Rudelli C, Viennet G, et al. A new prognostic clinicopathological classification of pituitary adenomas: a multicentric case-control study of 410 patients with 8 years post-operative follow-up. Acta Neuropathol (2013) 126(1):123–35. doi:10.1007/s00401-013-1084-y
47. Trouillas J. In search of a prognostic classification of endocrine pituitary tumors. Endocr Pathol (2014) 25(2):124–32. doi:10.1007/s12022-014-9322-y
48. Pasquel FJ, Vincentelli C, Brat DJ, Oyesiku NM, Ioachimescu AG. Pituitary carcinoma in situ. Endocr Pract (2013) 19(3):e69–73. doi:10.4158/EP12351.CR
49. Sav A, Rotondo F, Syro LV, Di Ieva A, Cusimano MD, Kovacs K. Invasive, atypical and aggressive pituitary adenomas and carcinomas. Endocrinol Metab Clin North Am (2015) 44(1):99–104. doi:10.1016/j.ecl.2014.10.008
50. Al-Brahim NY, Asa SL. My approach to pathology of the pituitary gland. J Clin Pathol (2006) 59(12):1245–53. doi:10.1136/jcp.2005.031187
51. Asa SL, Ezzat S. The pathogenesis of pituitary tumors. Annu Rev Pathol (2009) 4:97–126. doi:10.1146/annurev.pathol.4.110807.092259
52. Scheithauer BW, Gaffey TA, Lloyd RV, Sebo TJ, Kovacs KT, Horvath E, et al. Pathobiology of pituitary adenomas and carcinomas. Neurosurgery (2006) 59(2):341–53. doi:10.1227/01.NEU.0000223437.51435.6E
Keywords: diagnosis, genetics, pathology, acromegaly, multiple endocrine neoplasia type 1, pituitary adenoma, familial isolated, Carney complex
Citation: Syro LV, Rotondo F, Ramirez A, Di Ieva A, Sav MA, Restrepo LM, Serna CA and Kovacs K (2015) Progress in the diagnosis and classification of pituitary adenomas. Front. Endocrinol. 6:97. doi: 10.3389/fendo.2015.00097
Received: 25 February 2015; Accepted: 26 May 2015;
Published: 12 June 2015
Edited by:
Marcello D. Bronstein, University of São Paulo Medical School, BrazilReviewed by:
Gianluca Tamagno, Mater Misericordiae University Hospital, IrelandLeandro Kasuki, Federal University of Rio de Janeiro, Brazil
Copyright: © 2015 Syro, Rotondo, Ramirez, Di Ieva, Sav, Restrepo, Serna and Kovacs. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Luis V. Syro, Department of Neurosurgery, Hospital Pablo Tobon Uribe and Clinica Medellin, Calle 54 # 46-27, Cons 501, Medellin 050012, Colombia,bHZzeXJvQHVuZS5uZXQuY28=