 Maria Magdalena Montt-Guevara1†
Maria Magdalena Montt-Guevara1† Maria Silvia Giretti1†Eleonora Russo1Andrea Giannini1Paolo Mannella1Andrea Riccardo Genazzani1
Maria Silvia Giretti1†Eleonora Russo1Andrea Giannini1Paolo Mannella1Andrea Riccardo Genazzani1 Alessandro David Genazzani2
Alessandro David Genazzani2 Tommaso Simoncini1*
Tommaso Simoncini1*
- 1Molecular and Cellular Gynecological Endocrinology Laboratory (MCGEL), Department of Clinical and Experimental Medicine, University of Pisa, Pisa, Italy
- 2Department of Obstetrics and Gynecology, University of Modena and Reggio Emilia, Modena, Italy
Estetrol (E4) is a natural human estrogen that is present at high concentrations during pregnancy. E4 has been reported to act as an endogenous estrogen receptor modulator, exerting estrogenic actions on the endometrium or the central nervous system but presenting antagonistic effects on the breast. Due to these characteristics, E4 is currently being developed for a number of clinical applications, including contraception and menopausal hormone therapy. Endothelial nitric oxide (NO) is a key player for vascular function and disease during pregnancy and throughout aging in women. Endothelial NO is an established target of estrogens that enhance its formation in human endothelial cells. We here addressed the effects of E4 on the activity and expression of the endothelial nitric oxide synthase (eNOS) in cultured human umbilical vein endothelial cells (HUVEC). E4 stimulated the activation of eNOS and NO secretion in HUVEC. E4 was significantly less effective compared to E2, and a peculiar concentration-dependent effect was found, with higher amounts of E4 being less effective than lower concentrations. When E2 was combined with E4, an interesting pattern was noted. E4 antagonized NO synthesis induced by pregnancy-like E2 concentrations. However, E4 did not impede the modest induction of NO synthesis associated with postmenopausal-like E2 levels. These results support the hypothesis that E4 may be a regulator of NO synthesis in endothelial cells and raise questions on its peculiar signaling in this context. Our results may be useful to interpret the role of E4 during human pregnancy and possibly to help develop this interesting steroid for clinical use.
Introduction
Estetrol [estra-1,3,5(10)-triene-3,15α, 16α, 17β-tetrol] is an estrogenic hormone discovered in 1965 by Diczfalusy and co-workers at the Karolinska Institute in Stockholm (1, 2). E4 is produced exclusively by the human fetal liver during pregnancy. It is present in fetal blood and reaches maternal circulation through the placenta and can be detected in amniotic fluid and maternal urine (3–6). E4 concentrations increase exponentially during pregnancy and peak at term with fetal levels about 10–20 times higher than in the mother. After delivery, blood levels of E4 become rapidly undetectable (7–9).
The pharmacological properties of E4 have been intensely investigated, but yet the physiological function of E4 has not been clearly understood. For example, in contrast with E2, E4 does not stimulate nor binds to sex hormone-binding globulin (SHBG) (10) and also does not inhibit the activity of cytochrome P450 liver enzymes; it is only minimally metabolized and completely excreted in the urine unaltered (11). E4 has a long half-life in humans, with an average of 28 h, which is about twofold longer than E2 (12).
E4 has a low to moderate affinity for both human estrogen receptor alpha (ER alpha) and ER beta with a four/fivefold preference for ER alpha (11). Based on this relatively low receptor binding affinity compared to E2, E4 was originally thought to be a weak estrogen (13). Subsequently, it was found that in rats, E4 acts as an estrogen on bone, brain, vagina, and endometrium (14, 15). However, it surprisingly acts as an estrogen antagonist on the rat breast DMBA model, where it prevents the development of new breast tumors and stimulates the regression of pre-existing ones (16, 17). The estrogen-antagonistic effect of E4 in the breast has been further supported by the observation of the ability of this steroid to decrease the stimulatory effect of E2 on cytoskeletal rearrangement, horizontal migration, and matrix invasion of ER+ T47-D human breast cancer cells (18).
The role of estrogens on the cardiovascular system has been largely studied. Estrogens promote vasodilatation, endothelial remodeling, and repair and counteract atherosclerosis (19). A key mechanism of action of estrogens in the cardiovascular system stays in the regulation of nitric oxide (NO) synthesis in endothelial cells (20–24).
Nitric oxide is a central controller of vascular function; it is a vasoactive molecule primarily produced by the vascular endothelium that exerts vascular-protective and anti-atherogenic effects on the vessel wall. NO decreases platelet aggregation and adhesion to the endothelium (25), limits vascular smooth muscle proliferation (26), inhibits neointima formation (27), prevents monocyte chemotaxis (28), and inhibits leukocyte adhesion to the endothelium by transcriptionally reducing adhesion molecule expression (29). Endothelial nitric oxide is produced by the endothelial nitric oxide synthase (eNOS) isoform upon conversion of the substrate l-arginine to l-citrulline (30). More recently, NO has been identified as a key agent in maintaining fetal oxygenation through the regulation of feto-placental blood flow. This is true in normal and even more in pregnancies where fetal oxygen delivery fails, such as during fetal growth restriction (31, 32).
We here characterized the effects of E4 on human umbilical vein endothelial cells (HUVEC), and specifically on eNOS activity and NO synthesis. Furthermore, we addressed the possible regulatory role of E4 on E2-related NO synthesis.
Materials and Methods
Cell Culture and Treatments
Human umbilical vein endothelial cells were obtained from umbilical veins from healthy women at the time of delivery. Written informed consent was obtained from each patient in accordance with the declaration of Helsinki. After collection, the umbilical cord was rapidly immersed in sterile saline solution and immediately processed for endothelial cell isolation. Under a laminar flow sterile hood, the umbilical vein was cannulated and thoroughly rinsed with sterile saline solution. After clamping the other extremity, the vein was filled for 30 min with 1 mg/mL type IA collagenase (Sigma-Aldrich, USA) pre-warmed at 37°C. The action of the collagenase solution was blocked with DMEM 10% FBS, the collected solutions were centrifuged at 4°C for 30 min at 1300 rpm, the supernatant was discarded, and the cell pellet was gently resuspended in phenol red-free DMEM (Gibco, Thermo Fisher Scientific) contained 10% FBS (Lonza Walkersville, Inc.), antibiotic–antimycotic (Gibco, Thermo Fisher Scientific), 2 mM l-glutamin (Gibco, Thermo Fisher Scientific), 25 mM HEPES (Gibco, Thermo Fisher Scientific), 0.2 μg/mL human epidermal growth factor (Sigma-Aldrich, USA), and 50 U/mL heparin. The cells were then plated on culture plates pre-coated with 1% sterile gelatin. All cell cultures were maintained in a humidified 5% CO2 atmosphere at 37°C and were grown to confluence and used between passage 3 and 4. Endothelial cell purity is extremely high after passage 2, due to elimination of contaminating macrophages and smooth muscle cells through replating. In confluent HUVEC, cell proliferation rate is negligible due to contact inhibition. Cell count and trypan blue exclusion were used to assess in preliminary experiments whether the experimental treatment may result in modified cell proliferation or death rates, and no significant changes were noted. Before experiments, medium was replaced for 24 h with steroid-deprived FBS (Lonza Walkersville, Inc.) and, when the inhibition treatment was required, the active treatments were done 30 min afterwards it. Control cells always received the same amount of ethanol (solvent for E2/E4, 0.01% final concentration). Estetrol was kindly provided by Herjian Coelingh Bennink (Pantarhei Biosciences), 17β-estradiol was from Sigma-Aldrich, USA-Aldrich, and ICI 182,780 from Tocris Cookson, UK.
Endothelial Nitric Oxide Synthase Activity Assay
Endothelial nitric oxide synthase activity was determined as conversion of 3H-arginine to 3H-citrulline in endothelial cell lysates with NOS assay kit (Calbiochem, Merck-Millipore, Germania). Briefly, HUVEC cells were harvested in ice-cold PBS and were pelleted in a microfuge at 13,000 rpm for 10 min at 4°C, then were homogenized in a buffer containing 25 mM Tris–HCl, pH 7.4, 1 mM EDTA and 1 mM EGTA, 1 mM DTT, 10 μg/mL leupeptin, 2 μg/mL aprotinin, and 100 μg/mL PMSF. About 0.001 mCi 3H-citrulline was separated with use of an acidic ion-exchange resin, as described in Ref. (33). Extracts incubated with the eNOS inhibitor, 1 mM N-nitro-l-arginine methyl ester, were used as blank. Results were expressed as picomoles of converted citrulline per milligram of assayed protein extracts.
Nitrite Assay
Nitric oxide production was determined as the overall amount of stable NO metabolites, nitrites, released in the cell culture medium during treatment. Briefly, the fluorescent product 1H-naphthotriazole was measured in the cell culture medium with excitation/emission wavelengths of 365 and 450 nm and was formed by the reaction of nitrosonium cation that forms spontaneously NO and 2,3-diaminonaphthalene (34). Standard curves were constructed with sodium nitrite. Non-specific fluorescence was determined in the presence of 3 mM NG-monomethyl-l-arginine.
Immunoblottings
After treatments, cells were collected on ice with lysis buffer containing 50 mM Tris–HCl, pH 7.4, 1 mM EDTA, 1% IGEPAL, protease inhibitor cocktail (Sigma-Aldrich, USA), and phosphatase inhibitor cocktail 3 (Sigma-Aldrich, USA). The concentration of total proteins was quantified by Pierce Micro BCA Assay (Thermo Fisher Scientific). Samples, containing 25 μg of protein, were separated on 10% SDS-page gels and transferred to a PVDF membrane (Immobilon-P, Millipore). Antibodies against the following proteins were used: eNOS (Transduction Laboratories, BD Biosciences), Ser1177-p-eNOS (Upstate Biotechnology, USA), Akt and Thr308-p-Akt (Upstate Biotechnology), ER alpha (sc-8005, Santa Cruz Biotechnology, USA), and ER beta (sc-390243, Santa Cruz Biotechnology, USA). Blots were blocked in 5% BSA and probed with primary antibody at 4°C overnight, then with an appropriate secondary antibody conjugated to horseradish peroxidase (Santa Cruz Biotechnology, USA) at room temperature for 2 h. Immunodetection of protein bands were visualized using chemiluminescence and were recorded with a quantitative digital imaging system (Quantity One, BioRad, USA). Meanwhile, the membranes were stripped and reproved with anti-actin antibody (Santa Cruz Biotechnology, USA) to confirm equal amounts of loaded samples. Densitometric analysis of the proteins bands was performed using the NIH ImageJ 1.40g software.
Statistical Analysis
Each experimental condition was reproduced in at least three independent experiments. All data are presented as mean ± SEM. Statistical analysis was performed using GraphPad Prism 5. Statistical differences between means were analyzed using one-way ANOVA followed by Bonferroni post-test. Differences at p < 0.05 were considered significant.
Results
E4 Increases eNOS Expression/Activity and NO Synthesis via ERs in HUVEC
To test the effects of E4 on endothelial NO level, we measured NO release in the cell culture medium as well as cellular expression and enzymatic activity of eNOS. HUVEC were treated for 48 h, with increasing concentration of E4 alone representing the physiological levels of E4 throughout pregnancy, or in combination with E2. E4 significantly induced the production of NO and eNOS enzymatic activity (Figure 1A). Protein levels of eNOS (Figure 1C) and of its phosphorylated active form (Figure 1D) were also increased during exposure to E4. However, E4 was significantly less effective compared to equimolar amounts of E2 (Figures 1A,C,D). Surprisingly, we did not find a linear relationship between the amount of E4 administered and the response in terms of NO synthesis, eNOS activity or eNOS expression, or phosphorylation. Indeed, the concentration–effect curve of E4 was bell-shaped, with increasing doses resulting in lower stimulation.
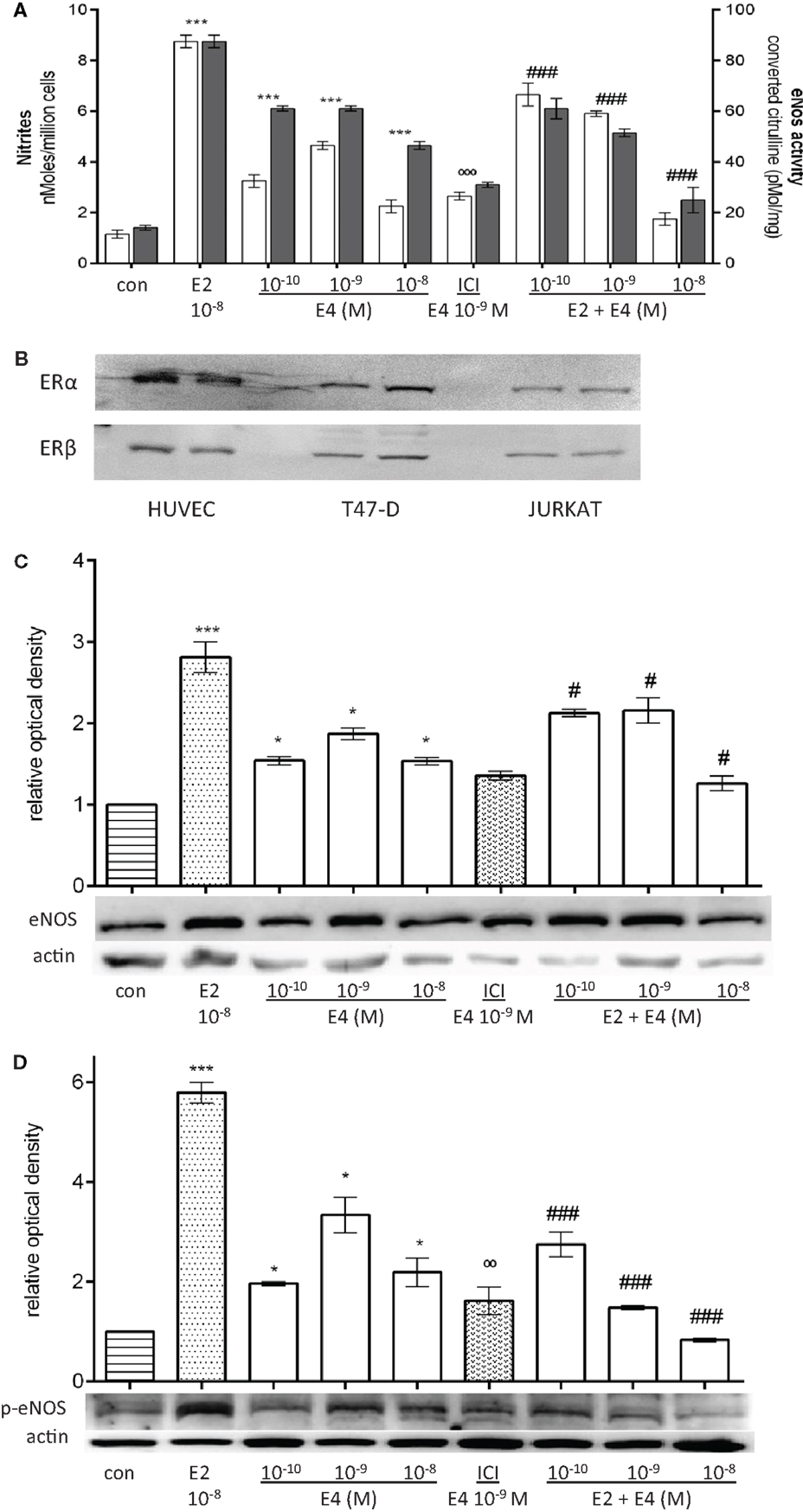
Figure 1. E4 modulates eNOS during prolonged administration via ER. Steroid-deprived HUVEC were treated for 48 h with 10−8M E2 or increasing concentrations of E4 (10−10, 10−9, and 10−8M) in the presence or absence of the pure estrogen antagonist ICI 182,780 (10−7M). (A) White bars represent nitrites levels and black bars represent enzymatic activity of eNOS. The experiments were repeated three times with comparable results. (B–D) Cell lysates were analyzed by western blotting for ER alpha, ER beta, eNOS, and Ser1177-p-eNOS. Quantification expressed as mean ± SEM for three independent experiments (upper panel) and representative blots are shown (lower panel). Values in the graph bar were obtained by the ratio of each protein band versus a loading reference. Significance of the observed effects was evaluated using one-way ANOVA followed by Bonferroni’s post hoc test (***p < 0.001, *p < 0.05 versus control, ˚˚˚p < 0.001, ˚˚p < 0.01 versus E4, ###p < 0.001, #p < 0.05 versus E2).
When HUVEC were treated with the pure estrogen receptor antagonist ICI 182,780, E4 effects were significantly reduced, suggesting that E4 acts at least in part via estrogen receptor recruitment (Figures 1A,C,D). ER alpha and ER beta are expressed in cultured HUVEC as shown by comparative western analysis with T47-D breast cancer cells and immortalized T lymphocyte Jurkat cells (Figure 1B).
Interestingly, the effects of an early-pregnancy-like concentration of E2 (10−8M) on NO synthesis as well as on cellular expression and enzymatic activity of eNOS and p-eNOS were significantly reduced when HUVEC were co-treated with E4 (Figures 1A,C,D). Inhibition of E2-dependent actions was related to the amount of E4 added (Figures 1A,C,D).
Differential Regulatory Actions of E4 on E2-Induced NO Production and eNOS Expression Based on the Relative Concentrations of the Two Steroids
Since in women concentration of E2 varies throughout reproductive life, three different concentrations of E2, mimicking early pregnancy (10−8M), follicular phase (10−9M), or post-menopause (10−10M) were used to study how NO production and eNOS expression are influenced in these three settings by E4. HUVEC were treated for 48 h, with E2 alone or in combination with increasing amounts of E4.
Addition of E4 to E2-treated cells resulted in a differential blockade of E2-dependent effects, but the extent of this action varied based on both E2 and E4 amounts. Indeed, when growing amounts of E4 were added to pregnancy-like E2 concentrations, a visible reduction of estradiol-mediated NO synthesis and eNOS expression was found (Figures 2A,B). On the opposite, when E4 was added to postmenopausal-like amounts of E2, a non-significant fall in NO synthesis and a lower reduction of eNOS expression was found (Figures 2A,B). This is particularly interesting, since it suggests that E4 is able to antagonize NO production in the presence of normal to high levels of E2, but not in the presence of low amounts, which deserves an explanation.
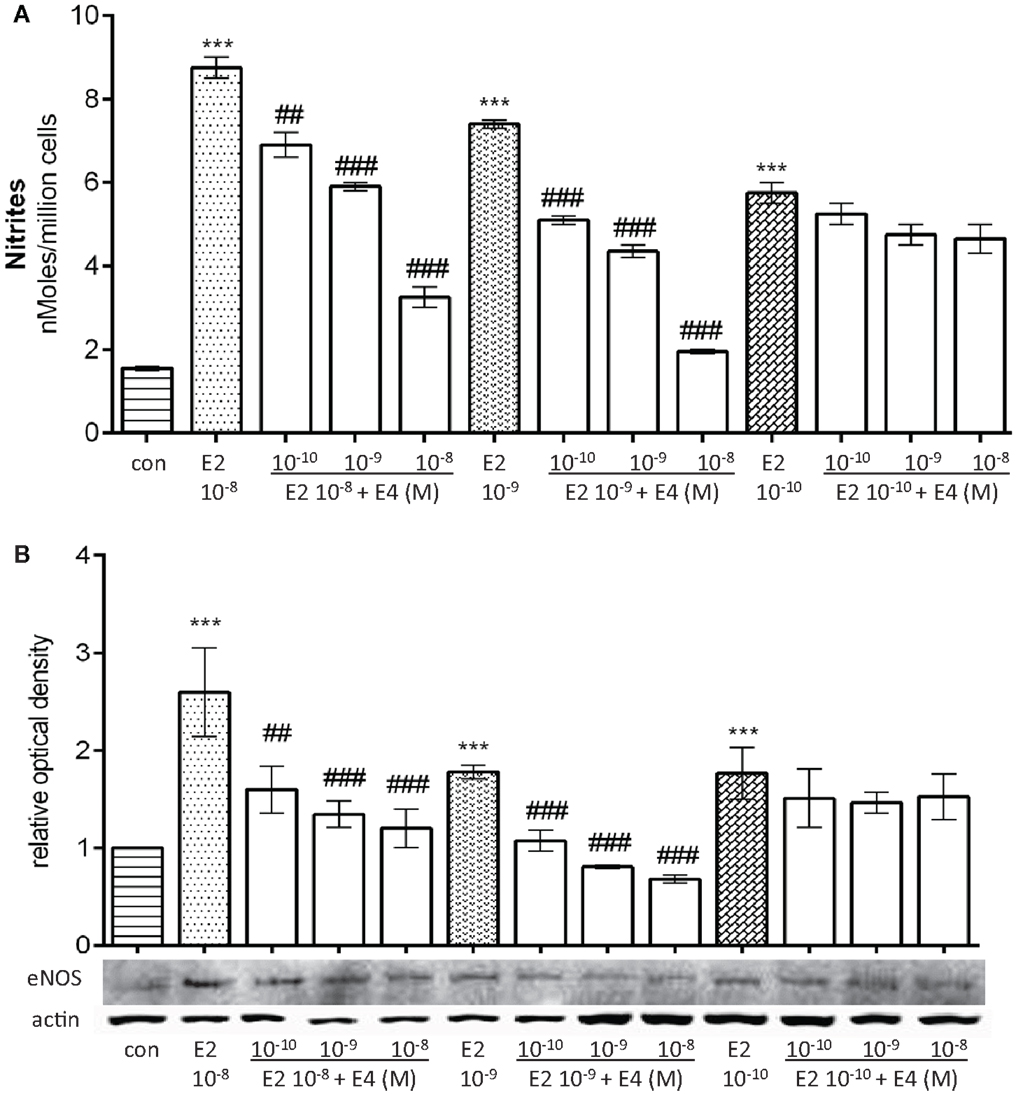
Figure 2. Differential regulatory actions of E4 on E2-induced NO production and eNOS expression based on the relative concentrations of the two steroids. Steroid-deprived HUVECs were treated for 48 h with increasing concentrations of E4 and E2. (A) Bars represent nitrites levels. The experiments were repeated three times with comparable results. (B) Cell lysates were analyzed by western blotting for eNOS. Values in the graph bar were obtained by the ratio of each protein band with a loading reference. Data are expressed as mean ± SEM for three independent experiments. Significance of the observed effects was evaluated using one-way ANOVA followed by Bonferroni’s post hoc test (***p < 0.001 versus control, ###p < 0.001, ##p < 0.01 versus E2).
E4 Modulates NO Synthesis via Rapid Extranuclear Signaling of ERs
Beyond being able to increase eNOS expression through conventional genomic mechanisms, estrogens also enhance NO synthesis through extranuclear-initiated signaling pathways in HUVEC (35–37). To assess whether E4 signaling to ERs also recruits this sort of mechanism, we exposed HUVEC for 30 min to E4 or E2.
Rapid E4 administration to HUVEC significantly increased NO release in the supernatant along with eNOS enzymatic activity (Figure 3A). During this type of E4 exposure, rapid phosphorylation of eNOS on Ser1177 was observed in the absence of any increase of eNOS expression (Figure 3B). When compared to E2, once more, E4 was less potent in this experimental setting (Figures 3A,B). Similar to the previous findings, increasing the concentrations of E4 over a 100-fold range did not turn into further increases in NO synthesis, eNOS activity nor of Ser1177-eNOS phosphorylation. On the opposite, a non-significant decrease in NO release and eNOS activity was noted with increasing E4 concentrations.
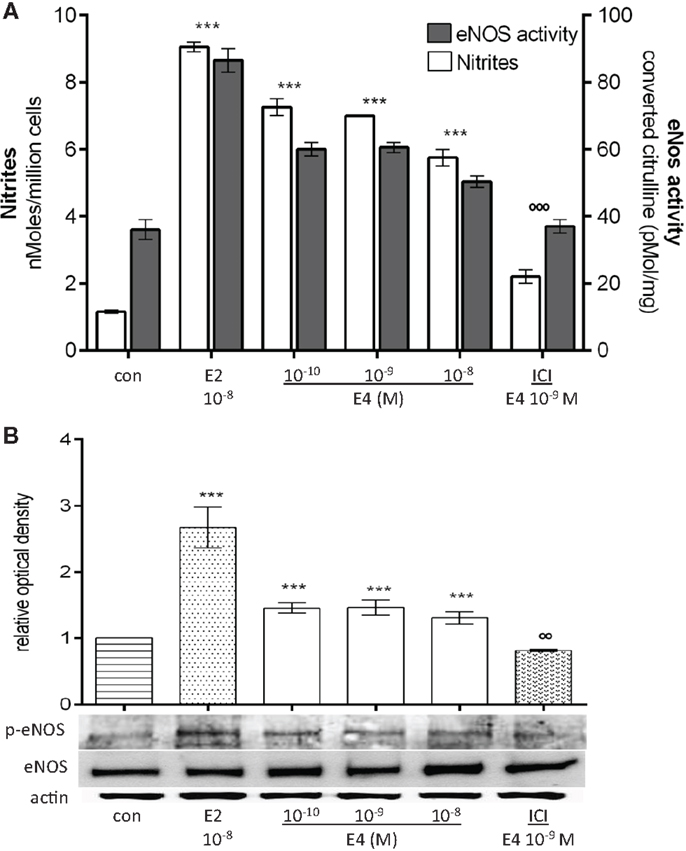
Figure 3. E4 rapidly modulates NO synthesis and eNOS enzymatic activity. Steroid-deprived HUVEC were treated for 30 min with 10−8M E2 or increasing concentrations of E4 in the presence or absence of the estrogen receptor antagonist ICI (10−7M). (A) White bars represent nitrites levels in HUVEC and the black bars represent the enzymatic activity of eNOS. The experiments were repeated three times with comparable results. (B) Cell lysates were analyzed by western blotting for eNOS and Ser1177-p-eNOS. Quantification expressed as mean ± SEM for three independent experiments (upper panel) and representative blots are shown (lower panel). Values in the graph bar were obtained by the ratio of eNOS/p-eNOS protein band intensity. Significance of the observed effects was evaluated using one-way ANOVA followed by Bonferroni’s post hoc test (***p < 0.001 versus control, ˚˚˚p < 0.001, ˚˚p < 0.01 versus E4).
Addition of ICI 182,780 dramatically reduced the action of E4, suggesting that also the rapid extranuclear actions of E4 occur at least in part via estrogen receptors (Figures 3A,B).
Extranuclear estrogen signaling to eNOS is primarily mediated by the lipid kinase phosphatidylinositol 3-OH kinase (PI3K) and by its downstream effector, the serine–threonine kinase Akt (23, 38–40). We therefore assessed whether E4 also uses this signaling pathway by treating HUVEC for 30 min with E4 or E2. While lower amounts of E4 (10−10M) resulted in increased phosphorylation of Akt, further increases in E4 concentrations resulted in a progressive decrease (Figure 4). Akt phosphorylation related to low doses of E4 was decreased by ICI 182,780 suggesting that E4 signaling to Akt relies in part on ER recruitment (Figure 4).
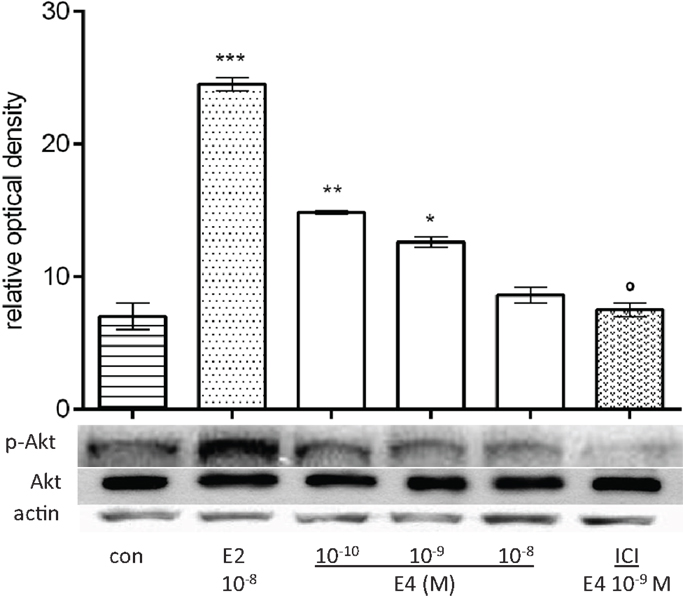
Figure 4. E4 regulates Akt. Steroid-deprived HUVECs were treated for 30 min with 10−8M E2 or increasing concentrations of E4 (10−10, 10−9, and 10−8M) in the presence or absence of ICI 182,780 (10−7M). Cell lysates were analyzed by western blotting for Akt and Thr308-p-Akt. Quantification expressed as mean ± SEM for three independent experiments (upper panel) and representative blots are shown (lower panel). Values in the graph bar were obtained by the ratio Akt/p-Akt protein band intensity. Significance of the observed effects was evaluated using one-way ANOVA followed by Bonferroni’s post hoc test (***p < 0.001, **p < 0.01, *p < 0.05 versus control, ˚p < 0.05 versus E4).
Differential Extranuclear Regulatory Actions of E4 on E2-Induced eNOS and Akt Phosphorylation Based on the Relative Concentrations of the Two Steroids
Similar to the previous set of experiments, we tested whether the modulation by differential combinations of E4 with pregnancy-like, follicular phase-like, or postmenopausal-like E2 amounts were also present when looking at extranuclear signaling to eNOS and Akt phosphorylation.
As shown in Figure 5, E4 decreased in a concentration-dependent manner eNOS and Akt phosphorylation induced by pregnancy-like amounts of E2, while it did not affect phosphorylation of these two targets when co-administered with postmenopausal amounts of E2 (Figures 5A,B).
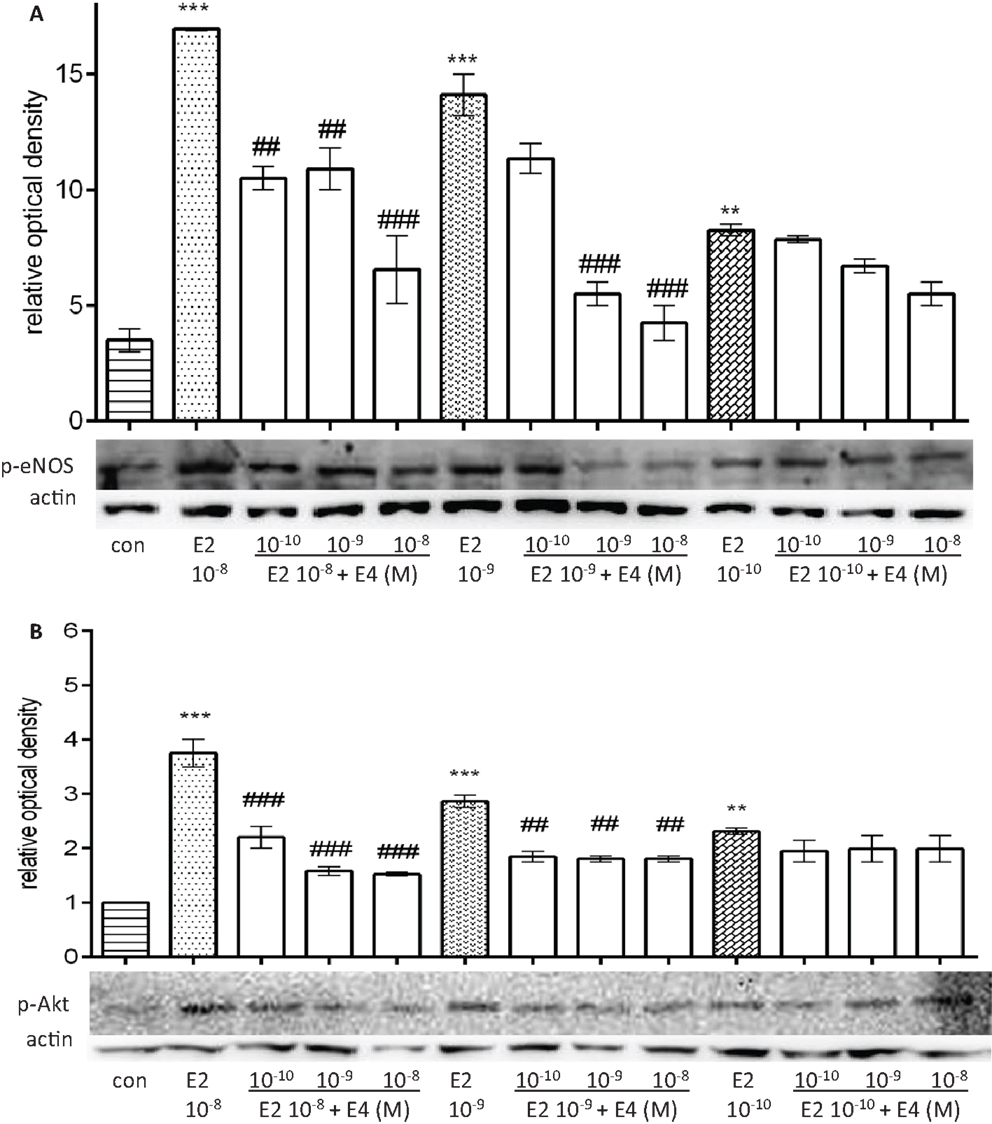
Figure 5. Differential regulatory actions of E4 on E2-induced eNOS and Akt phosphorylation based on the relative concentrations of the two steroids. Steroid-deprived HUVECs were treated for 30 min with increasing concentrations of E4 and E2. The total cell lysates were analyzed by western blotting for Ser1177-p-eNOS (A) and Thr308-p-Akt (B). Quantification expressed as mean ± SEM for three independent experiments (upper panel) and representative blots are shown (lower panel). Values in the graph bar were obtained by the ratio of each protein band versus a loading reference. Significance of the observed effects was evaluated using one-way ANOVA followed by Bonferroni’s post hoc test (***p < 0.001, **p < 0.01 versus control, ###p < 0.001, ##p < 0.01 versus E2).
Discussion
The main finding of this study is the identification of modulatory actions of estetrol on nitric oxide synthesis in human endothelial cells. E4 controls NO synthesis through seemingly complex signaling avenues. Indeed, the responses to increasing amounts of this steroid are not concentration-dependent. E4 administration to HUVEC turns into increased NO synthesis both because of enhanced eNOS expression and increased enzymatic activation through direct phosphorylation on Ser1177. Moreover, estetrol interferes with 17β-estradiol-dependent NO synthesis, but once again it does so with a non-conventional behavior. In the presence of pregnancy-like amounts of E2, E4 blunts NO activation. However, when lower E2 amounts are present, E4 does not interfere with E2 on NO synthesis. These observations may support the hypothesis that E4 could be a naturally occurring selective estrogen receptor modulator, but they also raise questions on its signaling mechanisms.
Estetrol is an interesting estrogen, produced only by the human fetal liver during pregnancy, and its physiological role or its mechanisms of action are still unknown. The present results suggest that regulation of vascular endothelial cells in the feto-placental unit may be a target for this steroid during pregnancy. Activation of endothelial NO synthesis in the placenta, umbilical vessels, and in the fetus is key to promote normal fetal development and to allow fetal adjustment to the stress of delivery (32, 41–43).
E4 concentrations are about 10-fold higher on the fetal side as compared to the maternal one. We here find that E4 results in differential regulation of NO synthesis in endothelial cells based (a) on its concentration and (b) on the amounts of estradiol available. This may suggest that the differential amounts of E4 in the mother and the fetus may result in a selective modulation of the powerful induction of NO synthesis by estradiol at these two sites. The high concentrations of E4 found throughout gestation (ranging between 400 and 1200 pg/mL) may thus represent a pregnancy-specific way to modulate NO production.
If this were true, it may be envisioned that on the maternal side, the lower amounts of E4 result in a lower inhibition of the E2-induced NO synthesis, therefore allowing for efficient vasodilatation toward the fetus. On the contrary, high amounts of E4 on the fetal side may be helpful to prevent excessive vascular dilatation, or possibly stimulation of other estrogen targets, such as the fetal breast. This is consistent with the recent data by Gerard et al. indicating that E4 antagonizes the stimulatory effects of E2 on breast glands in an animal model (44), where the antagonistic actions of E4 are similar in terms of concentration-dependency, to those we find in human endothelial cells.
While E4 is only found in humans during pregnancy, its peculiar chemical structure makes it interesting for exploitation as an endocrine tool in a variety of settings. For instance, its suggested estrogen-antagonistic action in breast tissue, along with its effectiveness in decreasing frequency and intensity of hot flashes (45), in preventing vulvo-vaginal atrophy (15) and osteoporosis (14), makes it attractive as a new form of menopausal hormone therapy. In this setting, the vascular actions of this steroid become particularly relevant. Estrogen replacement therapy has been shown in many studies to exert a vascular-protective action (19, 46), and most of these effects are attributed to the induction of NO synthesis in endothelial cells (35, 36, 47–50). Thus, should E4 result in decreased NO synthesis in endothelial cells, this would be undesirable. To this extent, our results indicate that in the presence of lower estradiol amounts, similar to those found after menopause, E4 does not significantly counteract E2-induced NO synthesis.
Why E4 does not increase eNOS expression and activity or NO synthesis in a linear concentration-dependent manner is unclear. Similarly, it is difficult to interpret why interference with NO synthesis induced by E2 does not happen in the presence of low estradiol concentrations. Given the high degree of similarity of cell responses through which E4 induces NO as compared to E2 (increased eNOS protein expression, increased eNOS activity, increased phosphorylation of eNOS), it would be tempting to attribute the interference of E4 with E2 signaling to competitive inhibition for ERs when the two estrogens are jointly present. To this extent, affinity of E4 for ER alpha is around 100-fold lower than that of E2 (11). Inhibition of ERs with a pure ER antagonist (ICI 182,780) blunts some of the effects of E4, thereby supporting the view that it is at least in part through ER binding that E4 affects NO synthesis in human endothelial cells.
However, this does neither seem to explain the peculiar E4 concentration-related responses of HUVEC in terms of NO nor the non-conventional inhibition of E2-related NO induction. Other signaling mechanisms may thus be involved, such as induction of a shift from the membrane of significant populations of ERs or alternatively an increased internalization and destruction of ERs. In a recent manuscript from Abot et al., E4 has been shown to be able to activate in vivo the ER alpha-dependent signaling that mediate protection from atherosclerosis development, while failing to activate the membrane-initiated signaling involved in ER alpha-dependent re-endothelialization and eNOS activation (51). The authors show that the three-dimensional structure of ER alpha ligand-binding domain in the presence of E2 or E4 is very similar, with only a minor shift in the orientation of helix 12 that is not expected to turn into significant modifications in the interaction with ER-interacting proteins (51). Interestingly, in the same manuscript, the authors show that both E2 and E4 are able to trigger the protein–protein interaction of ER alpha with one of its main signaling partners, c-Src. This small adapter protein is the upstream activator of ER alpha-dependent signaling to PI3K and MAPK, and is the key initiator of membrane-initiated estrogen signaling to eNOS (22). In the Abot paper, when E2 and E4 were provided together, ER alpha interaction with c-Src was significantly inhibited below baseline (51). Based on these observations, it is possible that part of the unexpected effects of higher concentrations of E4 or the selective inhibition of ER signaling associated with high but not low E2 concentrations may be ascribed to the ability of E4 to uncouple the recruitment of membrane-initiated signaling through interference with ER alpha/c-Src interaction.
Another potential explanation for the effects of E4 on NO synthesis in HUVEC may also be provided by parallel signaling through other receptors. Recently, Gerard et al. have generated preliminary data supporting the hypothesis that E4 may in part act via the G protein-coupled estrogen receptor 1 (GPER) in breast cancer cells (52). Thus, it is possible that interference with other receptors involved in signaling to eNOS may explain E4 actions in HUVEC. E4 signaling to eNOS in endothelial cells may thus represent a useful model to dissect the signaling mechanisms of this fetal steroid and its biological actions, and will be ground for future investigation.
In conclusion, our present findings that E4 modulates endothelial NO synthesis in HUVEC and that it also interferes with estradiol induction of NO. It is therefore tempting to speculate that E4 may represent a naturally occurring compound designed by nature to control the maternal and fetal vascular actions of estrogens during gestation.
Author Contributions
MM-G performed the molecular studies, bioinformatics statistical analysis, and drafted the manuscript. MG designed and performed the molecular studies and supervised the project. ER, AG, and PM performed the molecular studies. ARG and ADG supervised the project. TS designed and participated in the data interpretation, helped to draft the manuscript, and supervised the project. All authors read and approved the final manuscript.
Conflict of Interest Statement
The authors declare that there is no conflict of interest that could be perceived as prejudicing the impartiality of the research reported.
Acknowledgments
Estetrol was kindly provided by Herjian Coelingh Bennink, Pantarhei Biosciences, the Netherlands. This work has been supported by University of Pisa funds to TS, Pisa, Italy and by Italian Ministry of Education and Research Grant 20102CHST5 (to TS).
References
1. Hagen AA, Barr M, Diczfalusy E. Metabolism of 17b-oestradiol-4-14C in early infancy. Acta Endocrinol (Copenh) (1965) 49:207–20.
2. Zucconi G, Lisboa BP, Simonitsch E, Roth L, Hagen AA, Diczfalusy E. Isolation of 15a hydroxyl-oestriol from pregnancy urine and from the urine of newborn infants. Acta Endocrinol (1967) 56:413–23.
3. Schwers J, Eriksson G, Wiqvist N, Diczfalusy E. 15a-Hydroxylation: a new pathway of estrogen metabolism in the human fetus and newborn. Biochim Biophys Acta (1965) 100:313–6. doi: 10.1016/0304-4165(65)90464-2
4. Schwers J, Govaerts-Videtsky M, Wiqvist N, Diczfalusy E. Metabolism of oestrone sulphate by the previable human foetus. Acta Endocrinol (1965) 50:597–610.
5. Mancuso S, Benagiano G, Dell’Acqua S, Shapiro M, Wiqvist N, Diczfalusy E. Studies on the metabolism of C-19 steroids in the human foeto-placental unit. Acta Endocrinol (1968) 57:208–27.
6. Holinka CF, Diczfalusy E, Coelingh Bennink HJ. Estetrol: a unique steroid in human pregnancy. J Steroid Biochem Mol Biol (2008) 110(1–2):138–43. doi:10.1016/j.jsbmb.2008.03.027
7. Tulchinsky D, Frigoletto FD Jr, Ryan KJ, Fishman J. Plasma estetrol as an index of fetal well-being. J Clin Endocrinol Metab (1975) 40(4):560–7. doi:10.1210/jcem-40-4-560
8. Kundu N, Wachs M, Iverson GB, Petersen LP. Comparison of serum unconjugated estriol and estetrol in normal and complicated pregnancies. Obstet Gynecol (1981) 58(3):276–81.
9. Coelingh Bennink F, Holinka CF, Visser M, Coelingh Bennink HJ. Maternal and fetal estetrol levels during pregnancy. Climacteric (2008) 11(Suppl 1):69–72. doi:10.1080/13697130802056321
10. Hammond GL, Hogeveen KN, Visser M, Coelingh Bennink HJ. Estetrol does not bind sex hormone binding globulin or increase its production by human HepG2 cells. Climacteric (2008) 11(Suppl 1):41–6. doi:10.1080/13697130701851814
11. Visser M, Foidart JM, Coelingh Bennink HJ. In vitro effects of estetrol on receptor binding, drug targets and human liver cell metabolism. Climacteric (2008) 11(Suppl 1):64–8. doi:10.1080/13697130802050340
12. Visser M, Holinka CF, Coelingh Bennink HJ. First human exposure to exogenous single-dose oral estetrol in early postmenopausal women. Climacteric (2008) 11(Suppl 1):31–40. doi:10.1080/13697130802056511
13. Coelingh Bennink HJ, Holinka CF, Diczfalusy E. Estetrol review: profile and potential clinical applications. Climacteric (2008) 11(Suppl 1):47–58. doi:10.1080/13697130802073425
14. Coelingh Bennink HJ, Heegaard AM, Visser M, Holinka CF, Christiansen C. Oral bioavailability and bone-sparing effects of estetrol in an osteoporosis model. Climacteric (2008) 11(Suppl 1):2–14. doi:10.1080/13697130701798692
15. Heegaard AM, Holinka CF, Kenemans P, Coelingh Bennink HJ. Estrogenic uterovaginal effects of oral estetrol in the modified Allen-Doisy test. Climacteric (2008) 11(Suppl 1):22–8. doi:10.1080/13697130701842490
16. Coelingh BHJT, Singer C, Simoncini T, Genazzani AR, Holinka CF, Kubista E. Estetrol, a pregnancy-specific human steroid, prevents and suppresses mammary tumor growth in a rat model. Climacteric (2008) 11(1):29. doi:10.1515/hmbci-2012-0015
17. Visser M, Kloosterboer H, Coelingh Bennink HJ. Estetrol prevents and suppresses mammary tumors induced by DMBA in rat model. Horm Mol Biol Clin Investig (2012) 9(1):95–103. doi:10.1515/hmbci-2012-0015
18. Giretti MS, Montt Guevara MM, Cecchi E, Mannella P, Palla G, Spina S, et al. Effects of estetrol on migration and invasion in T47-D breast cancer cells through the actin cytoskeleton. Front Endocrinol (Lausanne) (2014) 5:80. doi:10.3389/fendo.2014.00080
19. Mendelsohn ME, Karas RH. The protective effects of estrogen on the cardiovascular system. N Engl J Med (1999) 340(23):1801–11. doi:10.1056/NEJM199906103402306
20. Hishikawa K, Nakaki T, Marumo T, Suzuki H, Kato R, Saruta T. Up-regulation of nitric oxide synthase by estradiol in human aortic endothelial cells. FEBS Lett (1995) 360(3):291–3. doi:10.1016/0014-5793(95)00124-R
21. Simoncini T, Genazzani AR. Direct vascular effects of estrogens and selective estrogen receptor modulators. Curr Opin Obstet Gynecol (2000) 12(3):181–7. doi:10.1097/00001703-200006000-00004
22. Simoncini T, Hafezi-Moghadam A, Brazil D, Ley K, Chin WW, Liao JK. Interaction of oestrogen receptor with the regulatory subunit of phosphatidylinositol-3-OH kinase. Nature (2000) 407:538–41. doi:10.1038/35035131
23. Haynes MP, Sinha D, Russell KS, Collinge M, Fulton D, Morales-Ruiz M, et al. Membrane estrogen receptor engagement activates endothelial nitric oxide synthase via the PI3-kinase-Akt pathway in human endothelial cells. Circ Res (2000) 87(8):677–82. doi:10.1161/01.RES.87.8.677
24. Simoncini T, Genazzani AR. Non-genomic actions of sex steroid hormones. Eur J Endocrinol (2003) 148(3):281–92. doi:10.1530/eje.0.1480281
25. Radomski MW, Palmer RM, Moncada S. An l-arginine/nitric oxide pathway present in human platelets regulates aggregation. Proc Natl Acad Sci USA (1990) 87:5193–7. doi:10.1073/pnas.87.13.5193
26. Radomski MW, Palmer RM, Moncada S. Endogenous nitric oxide inhibits human platelet adhesion to vascular endothelium. Lancet (1987) 2(8567):1057–8. doi:10.1016/S0140-6736(87)91481-4
27. Garg UC, Hassid A. Nitric oxide-generating vasodilators and 8-bromo-cyclic guanosine monophosphate inhibit mitogenesis and proliferation of cultured rat vascular smooth muscle cells. J Clin Invest (1989) 83(5):1774–7. doi:10.1172/JCI114081
28. Von der Leyen HE, Gibbons GH, Morishita R, Lewis NP, Zhang L, Nakajima M, et al. Gene therapy inhibiting neointimal vascular lesion: in vivo transfer of endothelial cell nitric oxide synthase gene. Proc Natl Acad Sci U S A (1995) 92(4):1137–41. doi:10.1073/pnas.92.4.1137
29. Bath PM, Hassall DG, Gladwin AM, Palmer RM, Martin JF. Nitric oxide and prostacyclin. Divergence of inhibitory effects on monocyte chemotaxis and adhesion to endothelium in vitro. Arterioscler Thromb (1991) 11(2):254–60. doi:10.1161/01.ATV.11.2.254
30. De Caterina R, Libby P, Peng HB, Thannickal VJ, Rajavashisth TB, Gimbrone MA Jr, et al. Nitric oxide decreases cytokine-induced endothelial activation. Nitric oxide selectively reduces endothelial expression of adhesion molecules and proinflammatory cytokines. J Clin Invest (1995) 96(1):60–8. doi:10.1172/JCI118074
31. Pisaneschi S, Strigini FA, Sanchez AM, Begliuomini S, Casarosa E, Ripoli A, et al. Compensatory feto-placental upregulation of the nitric oxide system during fetal growth restriction. PLoS One (2012) 7(9):e45294. doi:10.1371/journal.pone.0045294
32. Herrera EA, Krause B, Ebensperger G, Reyes RV, Casanello P, Parra-Cordero M, et al. The placental pursuit for an adequate oxidant balance between the mother and the fetus. Front Pharmacol (2014) 5:149. doi:10.3389/fphar.2014.00149
33. Knowles RG, Salter M. Measurement of NOS activity by conversion of radiolabeled arginine to citrulline using ion-exchange separation. In: Titheradge MA, editor. Nitric Oxide Protocols. Methods in Molecular Biology. (Vol. 100), Totowa, NJ: Humana Press (1998). p. 67–73.
34. Nussler AK, Glanemann M, Schirmeier A, Liu L, Nussler NC. Fluorometric measurement of nitrite/nitrate by 2,3-diaminonaphthalene. Nat Protoc (2006) 1(5):2223–6. doi:10.1038/nprot.2006.341
35. Chambliss KL, Shaul PW. Estrogen modulation of endothelial nitric oxide synthase. Endocr Rev (2002) 23(5):665–86. doi:10.1210/er.2001-0045
36. Simoncini T, Fornari L, Mannella P, Caruso A, Garibaldi S, Baldacci C, et al. Activation of nitric oxide synthesis in human endothelial cells by red clover extracts. Menopause (2005) 12(1):69–77. doi:10.1097/00042192-200512010-00013
37. Fu XD, Simoncini T. Non-genomic sex steroid actions in the vascular system. Semin Reprod Med (2007) 25(3):178–86. doi:10.1055/s-2007-973430
38. Dimmeler S, Fleming I, Fisslthaler B, Hermann C, Busse R, Zeiher AM. Activation of nitric oxide synthase in endothelial cells by Akt-dependent phosphorylation. Nature (1999) 399(6736):601–5. doi:10.1038/21224
39. Fulton D, Gratton JP, McCabe TJ, Fontana J, Fujio Y, Walsh K, et al. Regulation of endothelium-derived nitric oxide production by the protein kinase Akt. Nature (1999) 399(6736):597–601. doi:10.1038/21218
40. Hisamoto K, Ohmichi M, Kurachi H, Hayakawa J, Kanda Y, Nishio Y, et al. Estrogen induces the Akt-dependent activation of endothelial nitric-oxide synthase in vascular endothelial cells. J Biol Chem (2001) 276(5):3459–67. doi:10.1074/jbc.M005036200
41. Murphy VE, Smith R, Giles WB, Clifton VL. Endocrine regulation of human fetal growth: the role of the mother, placenta, and fetus. Endocr Rev (2006) 27(2):141–69. doi:10.1210/er.2005-0011
42. Poston L, Igosheva N, Mistry HD, Seed PT, Shennan AH, Rana S, et al. Role of oxidative stress and antioxidant supplementation in pregnancy disorders. Am J Clin Nutr (2011) 94(6 Suppl):1980S–5S. doi:10.3945/ajcn.110.001156
43. Hillman NH, Kallapur SG, Jobe AH. Physiology of transition from intrauterine to extrauterine life. Clin Perinatol (2012) 39(4):769–83. doi:10.1016/j.clp.2012.09.009
44. Gerard C, Blacher S, Communal L, Courtin A, Tskitishvili E, Mestdagt M, et al. Estetrol is a weak estrogen antagonizing estradiol-dependent mammary gland proliferation. J Endocrinol (2015) 224(1):85–95. doi:10.1530/JOE-14-0549
45. Holinka CF, Brincat M, Coelingh Bennink HJ. Preventive effect of oral estetrol in a menopausal hot flush model. Climacteric (2008) 11(Suppl 1):15–21. doi:10.1080/13697130701822807
46. Genazzani AR, Simoncini T. Pharmacotherapy: benefits of menopausal hormone therapy – timing is key. Nat Rev Endocrinol (2013) 9(1):5–6. doi:10.1038/nrendo.2012.228
47. Simoncini T, Genazzani AR, Liao JK. Nongenomic mechanisms of endothelial nitric oxide synthase activation by the selective estrogen receptor modulator raloxifene. Circulation (2002) 105(11):1368–73. doi:10.1161/hc1102.105267
48. Simoncini T, Varone G, Fornari L, Mannella P, Luisi M, Labrie F, et al. Genomic and nongenomic mechanisms of nitric oxide synthesis induction in human endothelial cells by a fourth-generation selective estrogen receptor modulator. Endocrinology (2002) 143(6):2052–61. doi:10.1210/endo.143.6.8749
49. Simoncini T, Mannella P, Fornari L, Caruso A, Varone G, Garibaldi S, et al. Tibolone activates nitric oxide synthesis in human endothelial cells. J Clin Endocrinol Metab (2004) 89(9):4594–600. doi:10.1210/jc.2003-032189
50. Simoncini T, Lenzi E, Zochling A, Gopal S, Goglia L, Russo E, et al. Estrogen-like effects of wine extracts on nitric oxide synthesis in human endothelial cells. Maturitas (2011) 70(2):169–75. doi:10.1016/j.maturitas.2011.07.004
51. Abot A, Fontaine C, Buscato M, Solinhac R, Flouriot G, Fabre A, et al. The uterine and vascular actions of estetrol delineate a distinctive profile of estrogen receptor alpha modulation, uncoupling nuclear and membrane activation. EMBO Mol Med (2014) 6(10):1328–46. doi:10.15252/emmm.201404112
52. Gérard C, Mestdagt M, Tskitishvili E, Communal L, Gompel A, Silva E, et al. Combined estrogenic and anti-estrogenic properties of estetrol on breast cancer may provide a safe therapeutic window for the treatment of menopausal symptoms. Oncotarget (2015). Available from: http://www.impactjournals.com/oncotarget/index.php?journal=oncotarget&page=article&op=view&path%5B%5D=4184&path%5B%5D=9310
Keywords: estetrol, estrogen, endothelial cells, nitric oxide, endothelial nitric oxide synthase
Citation: Montt-Guevara MM, Giretti MS, Russo E, Giannini A, Mannella P, Genazzani AR, Genazzani AD and Simoncini T (2015) Estetrol modulates endothelial nitric oxide synthesis in human endothelial cells. Front. Endocrinol. 6:111. doi: 10.3389/fendo.2015.00111
Received: 13 April 2015; Accepted: 06 July 2015;
Published: 22 July 2015
Edited by:
Michael Schumacher, INSERM, FranceReviewed by:
Subrata Chakrabarti, The University of Western Ontario, CanadaGuillermo Romero, University of Pittsburgh, USA
Copyright: © 2015 Montt-Guevara, Giretti, Russo, Giannini, Mannella, Genazzani, Genazzani and Simoncini. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Tommaso Simoncini, Molecular and Cellular Gynecological Endocrinology Laboratory (MCGEL), Department of Clinical and Experimental Medicine, Division of Obstetrics and Gynecology, University of Pisa, Via Roma 57, Pisa 56100, Italy,dG9tbWFzby5zaW1vbmNpbmlAbWVkLnVuaXBpLml0
†Maria Magdalena Montt-Guevara and Maria Silvia Giretti have contributed equally to this work.