 Seung-Hyun Ro
Seung-Hyun Ro Ian Semple
Ian Semple Allison Ho
Allison Ho Hwan-Woo Park
Hwan-Woo Park Jun Hee Lee
Jun Hee Lee- 1Department of Molecular and Integrative Physiology, University of Michigan, Ann Arbor, MI, USA
- 2Department of Cell Biology, College of Medicine, Konyang University, Daejeon, South Korea
Sestrin2 is a stress-inducible protein that functions as an antioxidant and inhibitor of mTOR complex 1. In a recent study, we found that Sestrin2 overexpression in brown adipocytes interfered with normal metabolism by reducing mitochondrial respiration through the suppression of uncoupling protein 1 (UCP1) expression. The metabolic effects of Sestrin2 in brown adipocytes were dependent on its antioxidant activity, and chemical antioxidants produced similar effects in inhibiting UCP1-dependent thermogenesis. These observations suggest that low levels of reactive oxygen species (ROS) in brown adipocytes can actually be beneficial and necessary for proper metabolic homeostasis. In addition, considering that Sestrins are ROS inducible and perform ROS detoxifying as well as other metabolism-controlling functions, they are potential regulators of mitohormesis. This is a concept in which overall beneficial effects result from low-level oxidative stress stimuli, such as the ones induced by caloric restriction or physical exercise. In this perspective, we incorporate our recent insight obtained from the Sestrin2 study toward a better understanding of the relationship between ROS, Sestrin2, and mitochondrial metabolism in the context of brown adipocyte physiology.
Introduction: Sestrins
Sestrins are a group of stress-inducible proteins, which are highly conserved across species and have two distinct biologically active functions (1). First, they function as an antioxidant that suppresses reactive oxygen species (ROS) accumulation (2, 3) through a poorly characterized biochemical mechanism (4), which may involve regulation of antioxidant transcription factors (5). Independently of this redox function, Sestrins also act as feedback inhibitors of mechanistic target of rapamycin complex 1 (mTORC1) through activation of AMP-activated protein kinase (AMPK) (6, 7) or through inhibition of Rag GTPases (8–11). Through these functions, Sestrins have been shown to attenuate multiple age- and obesity-associated metabolic pathologies, such as fat accumulation, glucose intolerance, insulin resistance, mitochondrial dysfunction, muscle degeneration, and cardiac malfunction (5–7, 12–15). Because the expression of Sestrins is induced upon a variety of environmental stresses, such as DNA damage, oxidative stress, and hypoxia (2), Sestrins are considered a mechanistic link between stress and aging (16).
ROS as Signaling Molecules
Reactive oxygen species are a group of free oxygen radicals and reactive non-radicals, such as superoxide , hydroxyl radical (OH⋅), nitric oxide (NO⋅), and hydrogen peroxide (H2O2) (17–19). In mammalian cells, ROS can be generated from various sources, such as mitochondria, peroxisomes, and ROS-producing cytosolic enzymes, such as NADPH oxidases (20–23). Under normal physiological conditions, the intracellular levels of ROS are homeostatically controlled. However, aberrantly increased ROS levels can damage intracellular organelles and critical macromolecules, such as proteins, DNA, and lipids. The ROS-induced oxidative damage can contribute to the development of numerous pathological disorders, such as cardiovascular disease, neurodegenerative diseases, mitochondrial disease, obesity, diabetes, cancer, and aging (24–30).
Although ROS were initially considered to be undesired byproducts of metabolism (31), a modernized view of ROS has emerged; ROS are now considered to be important signaling molecules (32–35). ROS can control diverse signaling pathways, including MAPK/ERK1/2 pathway (36, 37), PI3K/Akt pathway (38, 39), IKK/NF-kB pathway (40–42), and p38 MAPK pathway (43, 44), which are critically involved in cell growth, differentiation, metabolism, and inflammation. Through these pathways, ROS contribute to the maintenance of physiological homeostasis in cells and tissues (45–49). These new findings support the concept of mitohormesis, which explains how physiological ROS can be beneficial to the cells and organism; ROS serve as sub-lethal stressors that act as signaling molecules to induce endogenous defense mechanisms, which ultimately improve mitochondrial metabolism and promote stress resistance, metabolic health, and longevity (50–52).
Role of ROS in BAT Metabolism
BAT is the organ mainly responsible for non-shivering thermogenesis, which is mediated by uncoupling protein 1 (UCP1) (53–55). UCP1 is the key protein for thermogenesis, which is specifically induced in BAT upon exposure to cold temperature. Low temperatures activate p38 MAPK by stimulating sympathetic neurons and inducing cAMP accumulation in BAT (56–58). Activated p38 MAPK subsequently activates several transcription factors, such as ATF-2 and PGC-1α, which induces UCP1 expression in BAT (58, 59). By translocating into the mitochondrial inner membrane and dissipating the proton gradient, UCP1 uncouples mitochondrial respiration from ATP synthesis and generates heat (53, 60). UCP1-dependent thermogenesis in BAT increases energy expenditure reduces body fat and improves metabolic homeostasis (60, 61).
Chronic accumulation of ROS produced by dysfunctional mitochondria may deteriorate the BAT metabolism (62, 63), which can be one of the mechanisms of how aging and obesity interferes with BAT metabolism (64–66). In this respect, suppressing excessive ROS might be key to reinforcing BAT metabolism against aging. Chemical antioxidants, artificial or naturally occurring substances, serve to scavenge ROS, many of which are byproducts of cellular metabolism, which when in excess can cause oxidative damage and promote disease development and aging (26, 32). Most cells utilize various antioxidant enzymes, such as catalases, superoxide dismutases, glutathione peroxidases, peroxiredoxins, and Sestrins, which serve to reduce the negative consequences of ROS accumulation (3, 33, 67). We have recently investigated the role of ROS in BAT metabolism through the use of chemical antioxidants, such as butylated hydroxyanisole (BHA) and N-acetylcysteine (NAC), as well as forced overexpression of Sestrin2 (68).
Surprisingly, our recent results indicated that physiological levels of ROS could also play a critical role in thermogenic processes (68). BHA and NAC, as well as Sestrin2 overexpression, resulted in a strong reduction of UCP1 expression in BAT. Subsequent in vitro and in vivo studies showed that physiological ROS in BAT potentiate cAMP-induced p38 MAPK activation, which mediates cold-induced UCP1 expression. Correspondingly, Sestrin2-overexpressing mice were unable to upregulate UCP1 or generate heat in response to cold exposure. These results demonstrate that ROS are critical for proper BAT metabolism. Therefore, prolonged antioxidant treatment may interfere with thermogenesis and possibly other ROS-dependent physiological processes. This idea is consistent with recent reports where administered antioxidants had neutral or negative effects on health and life span in animals and humans (69, 70).
Caloric Restriction and Physical Exercise as Potential Inducers of ROS and Mitohormesis
Caloric restriction, being defined as a reduction in ad libitum calorie uptake, has been shown to extend life span in a variety of organisms (71). Notably, emerging evidence indicates that caloric restriction is capable of increasing mitochondrial metabolism, such as oxidative respiration, in yeast (72), Caenorhabditis elegans (73) and Drosophila (74, 75). Increased production of ROS as a consequence of increased mitochondrial respiration has been suggested to be a critical regulator of life span during caloric restriction (73, 76–79). Caloric restriction-induced ROS can stimulate antioxidant defense mechanisms, such as radical-scavenging enzymes, that mediate various mitohormetic responses (5, 73, 80–82). Carbohydrate-deficient diets also increase oxidative metabolism, subsequently resulting in enhanced ROS defense (83). Therefore, these recent studies suggest that caloric restriction increases longevity at least partially by inducing oxidative metabolism and a mitohormetic defense system.
Physical exercise undoubtedly provides positive effects on diverse diseases, such as obesity, type 2 diabetes, cardiovascular disease, cancer, and general aging (84–88). Similar to caloric restriction, exercise is capable of increasing mitochondrial biogenesis, oxidative metabolism, and mitochondrial ROS production (89–92). Many studies propose that exercise-induced ROS contributes to mitohormesis, which increases health span and mean life span (93–97). Because co-treatment with antioxidants prevented the beneficial mitohormetic response of physical exercise (85, 98), physiological ROS produced by exercise was considered critical for the benefits of exercise. Therefore, both caloric restriction and exercise seem to utilize physiological ROS induction as a means to induce antioxidant defense and promote the life and health span of an organism.
Sestrins as Potential Regulators of Mitohormesis
Sestrins have been identified as important regulators of age- and obesity-associated pathologies in diverse tissues including liver, adipose, and muscle (5–7, 12–15, 68). Sestrins are thought to attenuate tissue aging through their dual biological activities in reducing ROS and inhibiting mTORC1 (1, 2, 16). Excessive accumulation of ROS and chronic activation of mTORC1 signaling are well-known promoters of tissue aging. Considering that Sestrins are transcriptionally activated upon oxidative stress, it is highly likely that Sestrins may be regulators of mitohormesis. For example, upon a non-toxic level of ROS stimuli, Sestrins may be induced to perform antioxidant and mTORC1-suppressive functions to defend against oxidative damage and attenuate tissue aging.
It should also be noted that, although critical for attenuating tissue aging, the loss of Sestrins per se does not substantially reduce life span of Drosophila (1) or C. elegans (15). This could be because there are unknown compensatory mechanisms instigated by the loss of Sestrins, which are capable of maintaining life span, but are not sufficient to restore health span. However, Sestrin-deficient C. elegans is hypersensitive to oxidative stress (15), suggesting that Sestrin is indeed critical for the stress adaptation of an organism. It has been shown in the same organism that low levels of oxidative stress (conferred by a chemical that induces mitochondrial ROS) can increase the life span of C. elegans (99, 100). Caloric restriction in C. elegans extends life span partially by inducing mitochondrial production of ROS (73). Therefore, it would be very interesting to investigate if Sestrins are indeed regulators of the mitohormetic effect in these aging models.
Role of Sestrin2 in BAT Metabolism
Although the beneficial role of Sestrins in attenuating tissue aging and obesity-associated metabolic pathologies has been clear in several tissues, such as liver and skeletal/cardiac muscle (6, 7, 12, 13), the role of Sestrin2 in BAT metabolism seems to be more complicated. As discussed above, Sestrin2 overexpression in BAT interferes with proper UCP1 expression and mitochondrial uncoupling (68). Even though Sestrin2 overexpression activated AMPK and subsequently promoted mitochondrial biogenesis, these beneficial effects were nullified by the drastic effect of UCP1 loss. As a result, the Sestrin2-overexpressing mice were defective in BAT thermogenesis and exhibited increased fat accumulation (68).
Interestingly, loss of Sestrin2 also interfered with proper BAT metabolism. Although UCP1 expression was relatively increased in BAT of Sestrin2-deficient mice, the whitening of BAT due to fat accumulation is markedly increased upon Sestrin2 deficiency (68). Analysis of mRNA markers for mitochondrial biogenesis and quantification of mitochondrial DNA suggests that BAT from Sestrin2-deficient mice exhibited decreased mitochondrial contents (68). This could be because Sestrin2 plays a critical role in producing sufficient amounts of mitochondria in BAT, which would be necessary for proper energy dissipation and homeostatic BAT metabolism. Sestrin2 may promote mitochondrial biogenesis through activation of AMPK (7, 12, 101, 102) and subsequent upregulation of PGC-1α activity (103, 104). Therefore, although overexpression of Sestrin2 can interfere with the physiological level of ROS necessary for thermogenesis control, endogenous Sestrin2 still plays a critical role in maintaining mitochondrial homeostasis.
Conclusion
Upon diverse environmental stresses including oxidative stress, Sestrin-family proteins are transcriptionally upregulated to reduce pathogenic levels of ROS and suppress chronic activation of mTORC1 signaling. As chronic ROS accumulation and prolonged mTORC1 activation are both detrimental for metabolic homeostasis, Sestrins are potential regulators of mitohormesis, which is a beneficial metabolic effect of low-level ROS production. Sestrins may also play a critical metabolism-controlling role in BAT. However, because ROS are also critical for UCP1 expression and subsequent mitochondrial uncoupling, artificial overexpression of Sestrin2 and subsequent elimination of ROS interfered with non-shivering thermogenesis, which is one of the most critical physiological functions of BAT. Therefore, it is highly likely that ROS levels, Sestrin2 expression, and mitochondrial metabolism are connected to each other through a complicated and finely coordinated network, and a delicate balance between these components seems to be critical for proper BAT homeostasis (Figure 1).
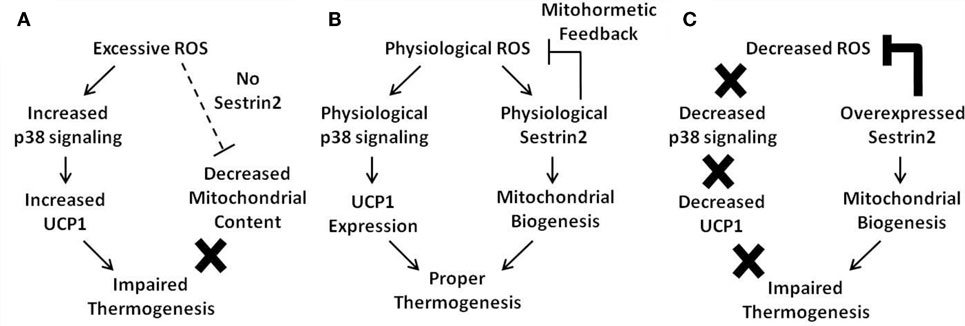
Figure 1. Current model of the mitohormetic relationship between ROS, Sestrin2, and mitochondrial thermogenesis. Under conditions of excessive or decreased ROS, BAT thermogenesis is impaired. However, a physiological level of ROS is able to maintain proper BAT metabolism. (A) In cases where antioxidant activity is low, such as in the absence of Sestrin2, ROS levels are upregulated. Excessive ROS may directly impair mitochondrial homeostasis by direct damage to the organelle. In addition, Sestrin2 is critical for mitochondrial biogenesis. Therefore, in the absence of Sestrin2, BAT mitochondrial content is decreased as well as its thermogenic capacity. (B) When Sestrin2 is physiologically expressed, an adequate level of ROS is produced to ensure proper p38 MAPK activation and UCP1 expression. Physiological Sestrin2 expression promotes mitochondrial biogenesis, and the mitohormetic mechanism conferred by Sestrin2 is necessary to maintain proper BAT metabolism such as thermogenesis. (C) When Sestrin2 is overexpressed, ROS are dramatically suppressed. Because ROS are critical for UCP1 expression in BAT, Sestrin2 overexpression decreases UCP1 expression. Although mitochondrial biogenesis is enhanced by Sestrin2 overexpression, decreased UCP1 expression prohibits proper thermogenesis in BAT.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
This work was supported by grants from the American Diabetes Association (Grant 1-13-BS-106) and Ellison Medical Foundation (Grant AG-NS-0932-12).
References
1. Lee JH, Budanov AV, Karin M. Sestrins orchestrate cellular metabolism to attenuate aging. Cell Metab (2013) 18(6):792–801. doi: 10.1016/j.cmet.2013.08.018
2. Budanov AV, Lee JH, Karin M. Stressin’ Sestrins take an aging fight. EMBO Mol Med (2010) 2(10):388–400. doi:10.1002/emmm.201000097
3. Budanov AV, Sablina AA, Feinstein E, Koonin EV, Chumakov PM. Regeneration of peroxiredoxins by p53-regulated sestrins, homologs of bacterial AhpD. Science (2004) 304(5670):596–600. doi:10.1126/science.1095569
4. Woo HA, Bae SH, Park S, Rhee SG. Sestrin 2 is not a reductase for cysteine sulfinic acid of peroxiredoxins. Antioxid Redox Signal (2009) 11(4):739–45. doi:10.1089/ARS.2008.2360
5. Bae SH, Sung SH, Oh SY, Lim JM, Lee SK, Park YN, et al. Sestrins activate Nrf2 by promoting p62-dependent autophagic degradation of Keap1 and prevent oxidative liver damage. Cell Metab (2013) 17(1):73–84. doi:10.1016/j.cmet.2012.12.002
6. Lee JH, Budanov AV, Park EJ, Birse R, Kim TE, Perkins GA, et al. Sestrin as a feedback inhibitor of TOR that prevents age-related pathologies. Science (2010) 327(5970):1223–8. doi:10.1126/science.1182228
7. Lee JH, Budanov AV, Talukdar S, Park EJ, Park HL, Park HW, et al. Maintenance of metabolic homeostasis by Sestrin2 and Sestrin3. Cell Metab (2012) 16(3):311–21. doi:10.1016/j.cmet.2012.08.004
8. Peng M, Yin N, Li MO. Sestrins function as guanine nucleotide dissociation inhibitors for Rag GTPases to control mTORC1 signaling. Cell (2014) 159(1):122–33. doi:10.1016/j.cell.2014.08.038
9. Chantranupong L, Wolfson RL, Orozco JM, Saxton RA, Scaria SM, Bar-Peled L, et al. The Sestrins interact with GATOR2 to negatively regulate the amino-acid-sensing pathway upstream of mTORC1. Cell Rep (2014) 9(1):1–8. doi:10.1016/j.celrep.2014.09.014
10. Parmigiani A, Nourbakhsh A, Ding B, Wang W, Kim YC, Akopiants K, et al. Sestrins inhibit mTORC1 kinase activation through the GATOR complex. Cell Rep (2014) 9(4):1281–91. doi:10.1016/j.celrep.2014.10.019
11. Kim JS, Ro SH, Kim M, Park HW, Semple IA, Park H, et al. Sestrin2 inhibits mTORC1 through modulation of GATOR complexes. Sci Rep (2015) 5:9502. doi:10.1038/srep09502
12. Morrison A, Chen L, Wang J, Zhang M, Yang H, Ma Y, et al. Sestrin2 promotes LKB1-mediated AMPK activation in the ischemic heart. FASEB J (2014) 29:408–17. doi:10.1096/fj.14-258814
13. Park HW, Park H, Ro SH, Jang I, Semple IA, Kim DN, et al. Hepatoprotective role of Sestrin2 against chronic ER stress. Nat Commun (2014) 5:4233. doi:10.1038/ncomms5233
14. Tao R, Xiong X, Liangpunsakul S, Dong XC. Sestrin 3 protein enhances hepatic insulin sensitivity by direct activation of the mTORC2-Akt signaling. Diabetes (2015) 64(4):1211–23. doi:10.2337/db14-0539
15. Yang YL, Loh KS, Liou BY, Chu IH, Kuo CJ, Chen HD, et al. SESN-1 is a positive regulator of lifespan in Caenorhabditis elegans. Exp Gerontol (2013) 48(3):371–9. doi:10.1016/j.exger.2012.12.011
16. Lee JH, Bodmer R, Bier E, Karin M. Sestrins at the crossroad between stress and aging. Aging (Albany NY) (2010) 2(6):369–74.
17. Murphy MP, Holmgren A, Larsson NG, Halliwell B, Chang CJ, Kalyanaraman B, et al. Unraveling the biological roles of reactive oxygen species. Cell Metab (2011) 13(4):361–6. doi:10.1016/j.cmet.2011.03.010
18. Dickinson BC, Chang CJ. Chemistry and biology of reactive oxygen species in signaling or stress responses. Nat Chem Biol (2011) 7(8):504–11. doi:10.1038/nchembio.607
19. Halliwell B, Whiteman M. Measuring reactive species and oxidative damage in vivo and in cell culture: how should you do it and what do the results mean? Br J Pharmacol (2004) 142(2):231–55. doi:10.1038/sj.bjp.0705776
21. Shabalina IG, Vrbacký M, Pecinová A, Kalinovich AV, Drahota Z, Houštek J, et al. ROS production in brown adipose tissue mitochondria: the question of UCP1-dependence. Biochim Biophys Acta (2014) 1837(12):2017–30. doi:10.1016/j.bbabio.2014.04.005
22. Sandalio LM, Romero-Puertas MC. Peroxisomes sense and respond to environmental cues by regulating ROS and RNS signalling networks. Ann Bot (2015). doi:10.1093/aob/mcv074
23. Wang B, Van Veldhoven PP, Brees C, Rubio N, Nordgren M, Apanasets O, et al. Mitochondria are targets for peroxisome-derived oxidative stress in cultured mammalian cells. Free Radic Biol Med (2013) 65:882–94. doi:10.1016/j.freeradbiomed.2013.08.173
24. Phaniendra A, Jestadi DB, Periyasamy L. Free radicals: properties, sources, targets, and their implication in various diseases. Indian J Clin Biochem (2015) 30(1):11–26. doi:10.1007/s12291-014-0446-0
25. Gupta RK, Patel AK, Shah N, Chaudhary AK, Jha UK, Yadav UC, et al. Oxidative stress and antioxidants in disease and cancer: a review. Asian Pac J Cancer Prev (2014) 15(11):4405–9. doi:10.7314/APJCP.2014.15.11.4405
26. Alfadda AA, Sallam RM. Reactive oxygen species in health and disease. J Biomed Biotechnol (2012) 2012:936486. doi:10.1155/2012/936486
27. El Assar M, Angulo J, Rodriguez-Manas L. Oxidative stress and vascular inflammation in aging. Free Radic Biol Med (2013) 65:380–401. doi:10.1016/j.freeradbiomed.2013.07.003
28. Maiese K. New insights for oxidative stress and diabetes mellitus. Oxid Med Cell Longev (2015) 2015:875961. doi:10.1155/2015/875961
29. Hayashi G, Cortopassi G. Oxidative stress in inherited mitochondrial diseases. Free Radic Biol Med (2015). doi:10.1016/j.freeradbiomed.2015.05.039
30. Cobb CA, Cole MP. Oxidative and nitrative stress in neurodegeneration. Neurobiol Dis (2015). doi:10.1016/j.nbd.2015.04.020
31. Harman D. Aging: a theory based on free radical and radiation chemistry. J Gerontol (1956) 11(3):298–300. doi:10.1093/geronj/11.3.298
32. Halliwell B, Gutteridge JMC. Free Radicals in Biology and Medicine. 4th ed. (Vol. xxxvi). Oxford: Oxford University Press (2007). 851 p.
33. Rhee SG, Woo HA. Multiple functions of peroxiredoxins: peroxidases, sensors and regulators of the intracellular messenger H(2)O(2), and protein chaperones. Antioxid Redox Signal (2011) 15(3):781–94. doi:10.1089/ars.2010.3393
34. Jiang F, Zhang Y, Dusting GJ. NADPH oxidase-mediated redox signaling: roles in cellular stress response, stress tolerance, and tissue repair. Pharmacol Rev (2011) 63(1):218–42. doi:10.1124/pr.110.002980
35. D’Autreaux B, Toledano MB. ROS as signalling molecules: mechanisms that generate specificity in ROS homeostasis. Nat Rev Mol Cell Biol (2007) 8(10):813–24. doi:10.1038/nrm2256
36. Chan DW, Liu VW, Tsao GS, Yao KM, Furukawa T, Chan KK, et al. Loss of MKP3 mediated by oxidative stress enhances tumorigenicity and chemoresistance of ovarian cancer cells. Carcinogenesis (2008) 29(9):1742–50. doi:10.1093/carcin/bgn167
37. McCubrey JA, Steelman LS, Chappell WH, Abrams SL, Wong EW, Chang F, et al. Roles of the Raf/MEK/ERK pathway in cell growth, malignant transformation and drug resistance. Biochim Biophys Acta (2007) 1773(8):1263–84. doi:10.1016/j.bbamcr.2006.10.001
38. Liu LZ, Hu XW, Xia C, He J, Zhou Q, Shi X, et al. Reactive oxygen species regulate epidermal growth factor-induced vascular endothelial growth factor and hypoxia-inducible factor-1alpha expression through activation of AKT and P70S6K1 in human ovarian cancer cells. Free Radic Biol Med (2006) 41(10):1521–33. doi:10.1016/j.freeradbiomed.2006.08.003
39. Higaki Y, Mikami T, Fujii N, Hirshman MF, Koyama K, Seino T, et al. Oxidative stress stimulates skeletal muscle glucose uptake through a phosphatidylinositol 3-kinase-dependent pathway. Am J Physiol Endocrinol Metab (2008) 294(5):E889–97. doi:10.1152/ajpendo.00150.2007
40. Li Q, Engelhardt JF. Interleukin-1beta induction of NFkappaB is partially regulated by H2O2-mediated activation of NFkappaB-inducing kinase. J Biol Chem (2006) 281(3):1495–505. doi:10.1074/jbc.M511153200
41. Wang Y, Huang X, Cang H, Gao F, Yamamoto T, Osaki T, et al. The endogenous reactive oxygen species promote NF-kappaB activation by targeting on activation of NF-kappaB-inducing kinase in oral squamous carcinoma cells. Free Radic Res (2007) 41(9):963–71. doi:10.1080/10715760701445045
42. Ruiz-Ramos R, Lopez-Carrillo L, Rios-Perez AD, De Vizcaya-Ruíz A, Cebrian ME. Sodium arsenite induces ROS generation, DNA oxidative damage, HO-1 and c-Myc proteins, NF-kappaB activation and cell proliferation in human breast cancer MCF-7 cells. Mutat Res (2009) 674(1–2):109–15. doi:10.1016/j.mrgentox.2008.09.021
43. Dolado I, Swat A, Ajenjo N, De Vita G, Cuadrado A, Nebreda AR. p38alpha MAP kinase as a sensor of reactive oxygen species in tumorigenesis. Cancer Cell (2007) 11(2):191–205. doi:10.1016/j.ccr.2006.12.013
44. Ushio-Fukai M, Alexander RW, Akers M, Griendling KK. p38 mitogen-activated protein kinase is a critical component of the redox-sensitive signaling pathways activated by angiotensin II. Role in vascular smooth muscle cell hypertrophy. J Biol Chem (1998) 273(24):15022–9. doi:10.1074/jbc.273.24.15022
45. Ray PD, Huang BW, Tsuji Y. Reactive oxygen species (ROS) homeostasis and redox regulation in cellular signaling. Cell Signal (2012) 24(5):981–90. doi:10.1016/j.cellsig.2012.01.008
46. Liou GY, Storz P. Reactive oxygen species in cancer. Free Radic Res (2010) 44(5):479–96. doi:10.3109/10715761003667554
47. Barazzoni R, Zanetti M, Gortan Cappellari G, Semolic A, Boschelle M, Codarin E, et al. Fatty acids acutely enhance insulin-induced oxidative stress and cause insulin resistance by increasing mitochondrial reactive oxygen species (ROS) generation and nuclear factor-kappaB inhibitor (IkappaB)-nuclear factor-kappaB (NFkappaB) activation in rat muscle, in the absence of mitochondrial dysfunction. Diabetologia (2012) 55(3):773–82. doi:10.1007/s00125-011-2396-x
48. Dozio E, Vianello E, Briganti S, Fink B, Malavazos AE, Scognamiglio ET, et al. Increased reactive oxygen species production in epicardial adipose tissues from coronary artery disease patients is associated with brown-to-white adipocyte trans-differentiation. Int J Cardiol (2014) 174(2):413–4. doi:10.1016/j.ijcard.2014.04.045
49. Gómez-Hernández A, Otero YF, de las Heras N, Escribano O, Cachofeiro V, Lahera V, et al. Brown fat lipoatrophy and increased visceral adiposity through a concerted adipocytokines overexpression induces vascular insulin resistance and dysfunction. Endocrinology (2012) 153(3):1242–55. doi:10.1210/en.2011-1765
50. Ristow M. Unraveling the truth about antioxidants: mitohormesis explains ROS-induced health benefits. Nat Med (2014) 20(7):709–11. doi:10.1038/nm.3624
51. Tapia PC. Sublethal mitochondrial stress with an attendant stoichiometric augmentation of reactive oxygen species may precipitate many of the beneficial alterations in cellular physiology produced by caloric restriction, intermittent fasting, exercise and dietary phytonutrients: “mitohormesis” for health and vitality. Med Hypotheses (2006) 66(4):832–43. doi:10.1016/j.mehy.2005.09.009
52. Sharma K. Mitochondrial hormesis and diabetic complications. Diabetes (2015) 64(3):663–72. doi:10.2337/db14-0874
53. Tews D, Wabitsch M. Renaissance of brown adipose tissue. Horm Res Paediatr (2011) 75(4):231–9. doi:10.1159/000324806
54. Poher AL, Altirriba J, Veyrat-Durebex C, Rohner-Jeanrenaud F. Brown adipose tissue activity as a target for the treatment of obesity/insulin resistance. Front Physiol (2015) 6:4. doi:10.3389/fphys.2015.00004
55. Enerback S. The origins of brown adipose tissue. N Engl J Med (2009) 360(19):2021–3. doi:10.1056/NEJMcibr0809610
56. Robidoux J, Cao W, Quan H, Daniel KW, Moukdar F, Bai X, et al. Selective activation of mitogen-activated protein (MAP) kinase kinase 3 and p38alpha MAP kinase is essential for cyclic AMP-dependent UCP1 expression in adipocytes. Mol Cell Biol (2005) 25(13):5466–79. doi:10.1128/MCB.25.13.5466-5479.2005
57. Cao W, Medvedev AV, Daniel KW, Collins S. beta-Adrenergic activation of p38 MAP kinase in adipocytes: cAMP induction of the uncoupling protein 1 (UCP1) gene requires p38 MAP kinase. J Biol Chem (2001) 276(29):27077–82. doi:10.1074/jbc.M101049200
58. Cao W, Daniel KW, Robidoux J, Puigserver P, Medvedev AV, Bai X, et al. p38 mitogen-activated protein kinase is the central regulator of cyclic AMP-dependent transcription of the brown fat uncoupling protein 1 gene. Mol Cell Biol (2004) 24(7):3057–67. doi:10.1128/MCB.24.7.3057-3067.2004
59. Silva JE, Rabelo R. Regulation of the uncoupling protein gene expression. Eur J Endocrinol (1997) 136(3):251–64. doi:10.1530/eje.0.1360251
60. Christian M, Parker MG. The engineering of brown fat. J Mol Cell Biol (2010) 2(1):23–5. doi:10.1093/jmcb/mjp035
61. Enerbäck S, Jacobsson A, Simpson EM, Guerra C, Yamashita H, Harper ME, et al. Mice lacking mitochondrial uncoupling protein are cold-sensitive but not obese. Nature (1997) 387(6628):90–4. doi:10.1038/387090a0
62. Marchi S, Giorgi C, Suski JM, Agnoletto C, Bononi A, Bonora M, et al. Mitochondria-ros crosstalk in the control of cell death and aging. J Signal Transduct (2012) 2012:329635. doi:10.1155/2012/329635
63. Wallace DC. A mitochondrial paradigm of metabolic and degenerative diseases, aging, and cancer: a dawn for evolutionary medicine. Annu Rev Genet (2005) 39:359–407. doi:10.1146/annurev.genet.39.110304.095751
64. Lecoultre V, Ravussin E. Brown adipose tissue and aging. Curr Opin Clin Nutr Metab Care (2011) 14(1):1–6. doi:10.1097/MCO.0b013e328341221e
65. Graja A, Schulz TJ. Mechanisms of aging-related impairment of brown adipocyte development and function. Gerontology (2015) 61(3):211–7. doi:10.1159/000366557
66. Shimizu I, Aprahamian T, Kikuchi R, Shimizu A, Papanicolaou KN, MacLauchlan S, et al. Vascular rarefaction mediates whitening of brown fat in obesity. J Clin Invest (2014) 124(5):2099–112. doi:10.1172/JCI71643
67. Mates JM, Sanchez-Jimenez F. Antioxidant enzymes and their implications in pathophysiologic processes. Front Biosci (1999) 4:D339–45. doi:10.2741/Mates
68. Ro SH, Nam M, Jang I, Park HW, Park H, Semple IA, et al. Sestrin2 inhibits uncoupling protein 1 expression through suppressing reactive oxygen species. Proc Natl Acad Sci U S A (2014) 111(21):7849–54. doi:10.1073/pnas.1401787111
69. Fusco D, Colloca G, Lo Monaco MR, Cesari M. Effects of antioxidant supplementation on the aging process. Clin Interv Aging (2007) 2(3):377–87.
70. Bjelakovic G, Nikolova D, Gluud C. Antioxidant supplements and mortality. Curr Opin Clin Nutr Metab Care (2014) 17(1):40–4. doi:10.1097/MCO.0000000000000009
71. Fontana L, Partridge L, Longo VD. Extending healthy life span – from yeast to humans. Science (2010) 328(5976):321–6. doi:10.1126/science.1172539
72. Lin SJ, Kaeberlein M, Andalis AA, Sturtz LA, Defossez PA, Culotta VC, et al. Calorie restriction extends Saccharomyces cerevisiae lifespan by increasing respiration. Nature (2002) 418(6895):344–8. doi:10.1038/nature00829
73. Schulz TJ, Zarse K, Voigt A, Urban N, Birringer M, Ristow M. Glucose restriction extends Caenorhabditis elegans life span by inducing mitochondrial respiration and increasing oxidative stress. Cell Metab (2007) 6(4):280–93. doi:10.1016/j.cmet.2007.08.011
74. Magwere T, Goodall S, Skepper J, Mair W, Brand MD, Partridge L. The effect of dietary restriction on mitochondrial protein density and flight muscle mitochondrial morphology in Drosophila. J Gerontol A Biol Sci Med Sci (2006) 61(1):36–47. doi:10.1093/gerona/61.1.36
75. Piper MD, Skorupa D, Partridge L. Diet, metabolism and lifespan in Drosophila. Exp Gerontol (2005) 40(11):857–62. doi:10.1016/j.exger.2005.06.013
76. Masoro EJ. Hormesis and the antiaging action of dietary restriction. Exp Gerontol (1998) 33(1–2):61–6. doi:10.1016/S0531-5565(97)00071-5
77. Agarwal S, Sharma S, Agrawal V, Roy N. Caloric restriction augments ROS defense in S. cerevisiae, by a Sir2p independent mechanism. Free Radic Res (2005) 39(1):55–62. doi:10.1080/10715760400022343
78. Sharma PK, Agrawal V, Roy N. Mitochondria-mediated hormetic response in life span extension of calorie-restricted Saccharomyces cerevisiae. Age (Dordr) (2011) 33(2):143–54. doi:10.1007/s11357-010-9169-1
79. Zuin A, Carmona M, Morales-Ivorra I, Gabrielli N, Vivancos AP, Ayté J, et al. Lifespan extension by calorie restriction relies on the Sty1 MAP kinase stress pathway. EMBO J (2010) 29(5):981–91. doi:10.1038/emboj.2009.407
80. Mahlke MA, Cortez LA, Ortiz MA, Rodriguez M, Uchida K, Shigenaga MK, et al. The anti-tumor effects of calorie restriction are correlated with reduced oxidative stress in ENU-induced gliomas. Pathobiol Aging Age Relat Dis (2011) 1:7189. doi:10.3402/pba.v1i0.7189
81. Schmeisser S, Priebe S, Groth M, Monajembashi S, Hemmerich P, Guthke R, et al. Neuronal ROS signaling rather than AMPK/sirtuin-mediated energy sensing links dietary restriction to lifespan extension. Mol Metab (2013) 2(2):92–102. doi:10.1016/j.molmet.2013.02.002
82. Zarse K, Schmeisser S, Groth M, Priebe S, Beuster G, Kuhlow D, et al. Impaired insulin/IGF1 signaling extends life span by promoting mitochondrial L-proline catabolism to induce a transient ROS signal. Cell Metab (2012) 15(4):451–65. doi:10.1016/j.cmet.2012.02.013
83. Nazarewicz RR, Ziolkowski W, Vaccaro PS, Ghafourifar P. Effect of short-term ketogenic diet on redox status of human blood. Rejuvenation Res (2007) 10(4):435–40. doi:10.1089/rej.2007.0540
84. Hu FB, Manson JE, Stampfer MJ, Colditz G, Liu S, Solomon CG, et al. Diet, lifestyle, and the risk of type 2 diabetes mellitus in women. N Engl J Med (2001) 345(11):790–7. doi:10.1056/NEJMoa010492
85. Lanza IR, Short DK, Short KR, Raghavakaimal S, Basu R, Joyner MJ, et al. Endurance exercise as a countermeasure for aging. Diabetes (2008) 57(11):2933–42. doi:10.2337/db08-0349
86. Manini TM, Everhart JE, Patel KV, Schoeller DA, Colbert LH, Visser M et al. Daily activity energy expenditure and mortality among older adults. JAMA (2006) 296(2):171–9. doi:10.1001/jama.296.2.171
87. Powers SK, Nelson WB, Hudson MB. Exercise-induced oxidative stress in humans: cause and consequences. Free Radic Biol Med (2011) 51(5):942–50. doi:10.1016/j.freeradbiomed.2010.12.009
88. Warburton DE, Nicol CW, Bredin SS. Health benefits of physical activity: the evidence. CMAJ (2006) 174(6):801–9. doi:10.1503/cmaj.051351
89. Alessio HM, Goldfarb AH. Lipid peroxidation and scavenger enzymes during exercise: adaptive response to training. J Appl Physiol (1985) (1988) 64(4):1333–6.
90. Chevion S, Moran DS, Heled Y, Shani Y, Regev G, Abbou B, et al. Plasma antioxidant status and cell injury after severe physical exercise. Proc Natl Acad Sci U S A (2003) 100(9):5119–23. doi:10.1073/pnas.0831097100
91. Davies KJ, Quintanilha AT, Brooks GA, Packer L. Free radicals and tissue damage produced by exercise. Biochem Biophys Res Commun (1982) 107(4):1198–205. doi:10.1016/S0006-291X(82)80124-1
92. Powers SK, Jackson MJ. Exercise-induced oxidative stress: cellular mechanisms and impact on muscle force production. Physiol Rev (2008) 88(4):1243–76. doi:10.1152/physrev.00031.2007
93. Ristow M, Zarse K. How increased oxidative stress promotes longevity and metabolic health: the concept of mitochondrial hormesis (mitohormesis). Exp Gerontol (2010) 45(6):410–8. doi:10.1016/j.exger.2010.03.014
94. Boveris A, Navarro A. Systemic and mitochondrial adaptive responses to moderate exercise in rodents. Free Radic Biol Med (2008) 44(2):224–9. doi:10.1016/j.freeradbiomed.2007.08.015
95. Crawford DR, Davies KJ. Adaptive response and oxidative stress. Environ Health Perspect (1994) 102(Suppl 10):25–8. doi:10.2307/3432208
96. Davies KJ. Intracellular proteolytic systems may function as secondary antioxidant defenses: an hypothesis. J Free Radic Biol Med (1986) 2(3):155–73. doi:10.1016/S0748-5514(86)80066-6
97. Gomez-Cabrera MC, Domenech E, Vina J. Moderate exercise is an antioxidant: upregulation of antioxidant genes by training. Free Radic Biol Med (2008) 44(2):126–31. doi:10.1016/j.freeradbiomed.2007.02.001
98. Ristow M, Zarse K, Oberbach A, Klöting N, Birringer M, Kiehntopf M, et al. Antioxidants prevent health-promoting effects of physical exercise in humans. Proc Natl Acad Sci U S A (2009) 106(21):8665–70. doi:10.1073/pnas.0903485106
99. Lee SJ, Hwang AB, Kenyon C. Inhibition of respiration extends C. elegans life span via reactive oxygen species that increase HIF-1 activity. Curr Biol (2010) 20(23):2131–6. doi:10.1016/j.cub.2010.10.057
100. Hwang AB, Ryu EA, Artan M, Chang HW, Kabir MH, Nam HJ, et al. Feedback regulation via AMPK and HIF-1 mediates ROS-dependent longevity in Caenorhabditis elegans. Proc Natl Acad Sci U S A (2014) 111(42):E4458–67. doi:10.1073/pnas.1411199111
101. Budanov AV, Karin M. p53 target genes sestrin1 and sestrin2 connect genotoxic stress and mTOR signaling. Cell (2008) 134(3):451–60. doi:10.1016/j.cell.2008.06.028
102. Sanli T, Linher-Melville K, Tsakiridis T, Singh G. Sestrin2 modulates AMPK subunit expression and its response to ionizing radiation in breast cancer cells. PLoS One (2012) 7(2):e32035. doi:10.1371/journal.pone.0032035
103. Atherton PJ, Babraj J, Smith K, Singh J, Rennie MJ, Wackerhage H. Selective activation of AMPK-PGC-1alpha or PKB-TSC2-mTOR signaling can explain specific adaptive responses to endurance or resistance training-like electrical muscle stimulation. FASEB J (2005) 19(7):786–8. doi:10.1096/fj.04-2179fje
Keywords: Sestrin2, brown adipose tissue, mitochondria metabolism, thermogenesis, reactive oxygen species, antioxidants, mitohormesis, aging
Citation: Ro S-H, Semple I, Ho A, Park H-W and Lee JH (2015) Sestrin2, a regulator of thermogenesis and mitohormesis in brown adipose tissue. Front. Endocrinol. 6:114. doi: 10.3389/fendo.2015.00114
Received: 27 May 2015; Accepted: 11 July 2015;
Published: 24 July 2015
Edited by:
Gary Sweeney, York University, CanadaReviewed by:
Jae B. Kim, Seoul National University, South KoreaWilliam T. Festuccia, University of São Paulo, Brazil
Copyright: © 2015 Ro, Semple, Ho, Park and Lee. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jun Hee Lee, Institute of Gerontology, Department of Molecular and Integrative Physiology, University of Michigan Medical School, 109 Zina Pitcher Place, 3019 BSRB, Ann Arbor, MI 48109, USA,bGVlanVAdW1pY2guZWR1