 Nobuyuki Itoh
Nobuyuki Itoh Hiroya Ohta
Hiroya Ohta Morichika Konishi
Morichika Konishi- 1Medical Innovation Center, Kyoto University Graduate School of Medicine, Kyoto, Japan
- 2Department of Microbial Chemistry, Kobe Pharmaceutical University, Kobe, Japan
The human fibroblast growth factor (FGF) family comprises 22 structurally related polypeptides that play crucial roles in neuronal functions, development, and metabolism. FGFs are classified as intracrine, paracrine, and endocrine FGFs based on their action mechanisms. Paracrine and endocrine FGFs are secreted signaling molecules by acting via cell-surface FGF receptors (FGFRs). Paracrine FGFs require heparan sulfate as a cofactor for FGFRs. In contrast, endocrine FGFs, comprising FGF19, FGF21, and FGF23, require α-Klotho or β-Klotho as a cofactor for FGFRs. Endocrine FGFs, which are specific to vertebrates, lost heparan sulfate-binding affinity and acquired a systemic signaling system with α-Klotho or β-Klotho during early vertebrate evolution. The phenotypes of endocrine FGF knockout mice indicate that they play roles in metabolism including bile acid, energy, and phosphate/active vitamin D metabolism. Accumulated evidence for the involvement of endocrine FGFs in human genetic and metabolic diseases also indicates their pathophysiological roles in metabolic diseases, potential risk factors for metabolic diseases, and useful biomarkers for metabolic diseases. The therapeutic utility of endocrine FGFs is currently being developed. These findings provide new insights into the physiological and pathophysiological roles of endocrine FGFs and potential diagnostic and therapeutic strategies for metabolic diseases.
Introduction
Cell–cell communication with secreted signaling molecules ensures proper development and metabolism. Most fibroblast growth factors (FGFs) are well-established secreted signaling proteins that play critical roles in development and metabolism. The FGF family comprises 22 structurally and evolutionarily related proteins including FGF1–FGF23 in humans and mice (1). The human and mouse FGF families do not include FGF15 and FGF19, respectively, as they are orthologs. FGFs are classified as intracrine, paracrine, and endocrine FGFs by their action mechanisms (Figure 1A). Paracrine and endocrine FGFs, which comprise 15 and 3 FGFs, respectively, are secreted signaling proteins. In contrast, intracrine FGFs, which comprise four FGFs, are intracellular proteins that are not functionally related to paracrine or endocrine FGFs (1).
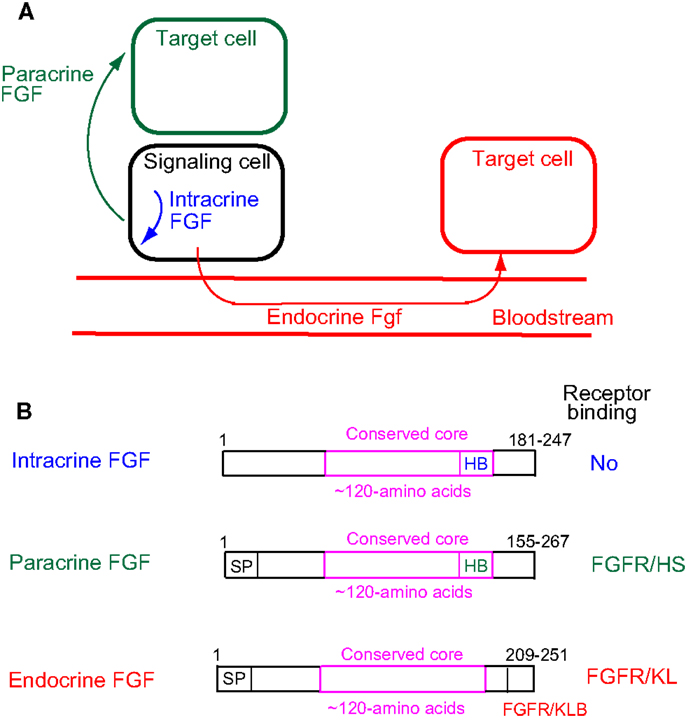
Figure 1. (A) Action mechanisms of FGFs. FGFs act on target cells in an intracrine, paracrine, or endocrine manner. (B) Schematic representations of FGF structures. SP, HB, and FGFR/KB indicate a secreted signal sequence, heparan sulfate-binding site, and FGFR/Klotho complex-binding site, respectively.
Endocrine FGFs comprise FGF19, FGF21, and FGF23. The mouse is a mammalian model that is useful for investigating gene functions through gene targeting. All endocrine FGF knockout mice have been generated and extensively analyzed, with their phenotypes indicating important roles in metabolism (1). Evidence to support the involvement of endocrine FGFs in human inherited and metabolic diseases has also been accumulated. We herein have reviewed the physiological/pathophysiological roles and diagnostic/therapeutic utility of endocrine FGFs.
Identification of Endocrine FGFS
FGF15 was originally identified as a downstream target of the chimeric homeodomain oncoprotein E2A-Pbx1 in mice (2). FGF19 was also originally identified by a homology-based DNA database search in humans (3, 4). However, FGF19 was later found to be the human ortholog of mouse FGF15 (5). Only rodent orthologs were named FGF15. Other vertebrate orthologs were named FGF19.
FGF21 was originally identified by a homology-based polymerase chain reaction in mice and humans (6). FGF21 was later found to be a stimulator of glucose uptake in cultured mouse adipocytes (7). FGF21 was also identified as a hepatic gene inducible by fasting or a ketogenic diet in mice (8, 9).
FGF23 was originally identified by a homology-based DNA database search in mice and humans (10). Human FGF23 was simultaneously identified as a gene responsible for autosomal dominant hypophosphatemic rickets (ADHR) characterized by low serum phosphate levels, rickets, osteomalacia, lower extremity deformities, a short stature, bone pain, and dental abscesses (11). Human FGF23 was subsequently found to be a causative humoral protein for human tumor-induced osteomalacia characterized by hypophosphatemia caused by renal phosphate wasting (12).
Unique Actions of Endocrine FGFS with FGF Receptors and Klotho Coreceptors
Paracrine and endocrine FGFs, which are extracellular signaling molecules, bind to and activate cell-surface FGF receptors (FGFRs). The human FGFR gene family comprises four members, FGFR1–FGFR4, that encode single-pass transmembrane proteins of ~800 amino acids with three extracellular immunoglobulin-like domains I, II, and III and a split intracellular tyrosine kinase domain. FGFR1, FGFR2, and FGFR3 genes encode two alternative splicing variants (b and c) of immunoglobulin-like domain III, which is a determinant of ligand-binding specificities (1). Thus, there are seven FGFR proteins, FGFRs 1b, 1c, 2b, 2c, 3b, 3c, and 4, with distinct ligand-binding specificity. The functional dimerization and transphosphorylation of FGFRs induced by FGF binding result in the activation of FGFRs, followed by the activation of intracellular FGF signaling pathways including RAS–RAF–MAPK, PI3K–AKT, STAT, and PLCγ pathways (1).
Paracrine FGFs bind to heparan sulfate proteoglycans, which extracellularly localize at neighboring cells as cofactors for FGFRs. Binding to heparan sulfate proteoglycans is essential for FGF signaling in a paracrine manner (Figure 1B) (1). In contrast, endocrine FGFs with reduced heparan sulfate-binding affinity are unable to function in a paracrine manner. Endocrine FGFs require α-Klotho or β-Klotho as a cofactor for FGFRs to function in an endocrine manner (1). α-Klotho and β-Klotho, which share structural similarities and characteristics with each other, are single-pass transmembrane proteins of ~1,000 amino acids with a short cytoplasmic domain (13). However, endocrine FGFs cannot efficiently bind to FGFR, α-Klotho, or β-Klotho alone; they efficiently bind to the FGFR–Klotho complex. The binding site for the FGFR–Klotho complex is localized in the C-terminal tail of endocrine FGF (Figure 1B) (14). FGF19 activates FGFR4 with β-Klotho and FGF21 activates FGFR1c with β-Klotho, whereas FGF23 activates FGFR1c with α-Klotho (1). FGF19 and FGF23 exhibit metabolic and proliferative activities. However, FGF21 is a unique FGF with metabolic activity, but no proliferative activity (15).
The Evolutionary History of Vertebrate-Specific Endocrine FGF Genes
The FGF signaling system has been conserved throughout metazoan evolution. The evolutionary history of the FGF gene family has already been proposed (5). The founder of the FGF family is an intracrine FGF. Intracrine FGFs, which are not secreted proteins, possess heparan sulfate-binding sites, but no secreted signal sequences. The prototype of the paracrine FGF subfamily was generated from the founder by gene duplication events early in metazoan evolution. During this evolution, paracrine FGFs acquired secreted signal sequences, thereby allowing them to function as extracellular FGFs in a paracrine manner. Intracrine and paracrine FGFs have been identified in invertebrates and vertebrates. In contrast, endocrine FGFs have been detected in vertebrates, but not invertebrates, indicating that they are specific to vertebrates (Figure 2). The prototypic endocrine FGF was generated from the prototype of paracrine FGF by local gene duplication early in vertebrate evolution. FGF19, FGF21, and FGF23 were subsequently generated from the prototype by two large-scale genome duplication events early in vertebrate evolution (5). Endocrine FGFs lost heparan sulfate-binding affinity and acquired FGFR/Klotho complex-binding affinity for a systemic signaling system in an endocrine manner during their evolution (5). FGF19 and FGF23 have been identified in all vertebrates examined, including teleosts, amphibians, reptiles, birds, and mammals. However, FGF21 was not detected in any birds examined, indicating that FGF21 was lost in the avian lineage (16, 17). The avian genome is principally characterized by its constrained size with the loss of many genes in the avian lineage, indicating that pan-avian genomic diversity covaries with adaptations to different lifestyles and the convergent evolution of traits (18).
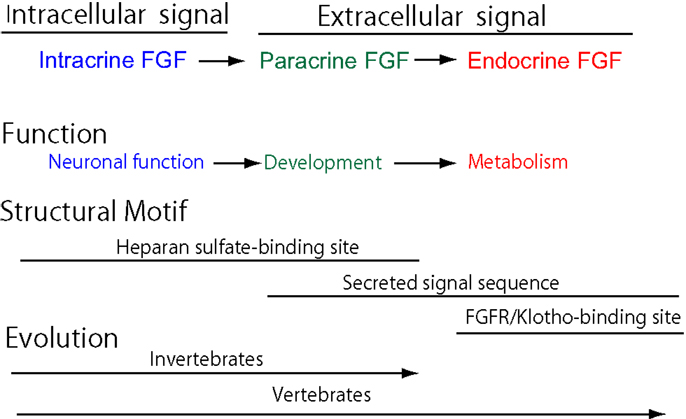
Figure 2. The functional evolutionary history of the FGF family.
Roles of Endocrine FGFs Indicated by Their Knockout Mouse Phenotypes
The mouse is a mammalian model that is useful for investigating gene functions. All endocrine FGF knockout mice have been extensively analyzed, indicating their important physiological roles in metabolism (Table 1).

Table 1. Roles of endocrine FGFs indicated by their knockout mouse phenotypes.
FGF15
FGF15 knockout mice survive until E10.5, but then gradually die due to defects in the cardiac outflow tract, indicating that FGF15 is required for proper morphogenesis of the cardiac outflow tract during embryonic development in the heart (2, 19). FGF15 is selectively expressed in the ileum at postnatal stages, and a small number of FGF15 knockout mice survive until postnatal stages, which suggests that FGF15 plays crucial roles in regulating hepatic bile acid synthesis and gallbladder filling in an endocrine manner (20, 21). Surviving FGF15 knockout mice also fail to properly maintain blood concentrations of glucose and normal postprandial amounts of liver glycogen, indicating that FGF15 is a postprandial, insulin-independent activator of hepatic protein and glycogen synthesis (22). Surviving hepatectomized FGF15 knockout mice exhibit severe defects in liver regeneration, indicating that FGF15 also plays a role in liver regeneration (23, 24). Ileal FGF15 expression is increased in mice undergoing carcinogenesis. Fibrosis-induced hepatocellular carcinogenesis results in fewer and smaller tumors in surviving FGF15 knockout mice than in wild-type mice, suggesting that ileal FGF15 enhances the development of carcinoma (25).
FGF21
FGF21 is expressed in the liver, pancreas, white adipose tissue, and muscle of mice (6, 26, 27). The phenotypes of FGF21 knockout mice, which are viable and fertile, indicate that FGF21 stimulates lipolysis in the white adipose tissue during feeding, but inhibits it during fasting (28). FGF21 knockout mice fed a ketogenic diet also indicate that FGF21 plays a role in the adaptation to a ketogenic diet and impairs adipocyte insulin sensitivity (29, 30). FGF21 knockout mice also indicate that FGF21 is an upstream effector of adiponectin in white adipocytes and that adiponectin also mediates many of the systemic effects of FGF21 on energy metabolism and insulin sensitivity in the liver and skeletal muscle (31, 32). Glucagon, an essential regulator of glucose homeostasis, modulates lipid metabolism and promotes weight loss. Glucagon receptor activation increases hepatic FGF21 expression in mice. FGF21 knockout mice indicate that FGF21 contributes, at least in part, to glucose, energy, and lipid metabolism controlled by glucagon (33). FGF21 knockout mice also exhibit insulin resistance, while being normoglycemic, and this is associated with increases in pancreatic beta-cell proliferation and insulin synthesis, which act as compensatory responses. This resistance results from enhanced growth hormone sensitivity in FGF21 knockout pancreatic islets, indicating that FGF21 is important in the regulation of pancreatic beta-cell proliferation and insulin synthesis, possibly via the modulation of growth hormone signaling (34). Endoplasmic reticulum (ER) stress leads to the development and progression of various diseases such as obesity and diabetes. ER stress induces hepatic FGF21 expression. ER stress and hepatic lipid accumulation are increased in FGF21 knockout mice, indicating that FGF21 plays a role in the adaptive responses to ER stress (35). FGF21 knockout mice also have an increased relative heart weight and develop enhanced signs of dilatation. FGF21 is markedly secreted by cardiac cells in response to cardiac stress. Thus, the heart appears to be a target of locally generated FGF21, indicating that FGF21 acts on cardiomyocytes, possibly in a paracrine manner (36). FGF21 prevents the production of reactive oxygen species in cardiac cells by acting as an antioxidant factor in the heart, thereby preventing pro-oxidative pathways under inflammatory or hypertrophic conditions (37). The expression of FGF21 in pancreatic acinar cells is markedly increased during cerulein-induced pancreatitis. FGF21 knockout mice have higher serum amylase levels and more tissue damage, suggesting a novel function for FGF21 as an immediate response gene protecting pancreatic acini from overt damage (38). FGF21 knockout mice with streptozotocin-induced diabetes exhibit severe cardiac dysfunction, increased cardiac lipid accumulation, and severe aortic remodeling, indicating that FGF21 plays roles in the development and progression of diabetic cardiomyopathy and aortic remodeling (39, 40).
FGF23
FGF23 is mainly expressed in osteocytes in mice. FGF23 knockout mice survive until birth, but then gradually die by 11 weeks after birth. The phenotypes of FGF23 knockout mice indicate that osteocytic FGF23 is a regulator for phosphate and active vitamin D metabolism in the kidney (41). FGF23 is also ubiquitously expressed throughout the cochlea. FGF23 heterozygous knockout mice are profoundly deaf with moderate hearing loss and a near-normal morphology. However, FGF23 homozygous knockout mice have dysplastic bulla and ossicles. These findings indicate that FGF23 is critical for normal development of the middle ear as well as the functions of the middle and inner ear (42).
Roles of Endocrine FGFs Indicated by Human Diseases
Evidence has been accumulated to support the involvement of FGF19 and FGF23 in human diseases and also indicates their crucial pathophysiological roles in diseases (Table 2).
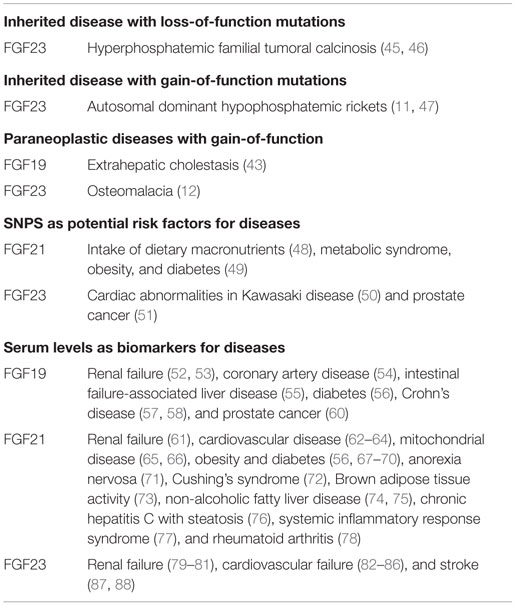
Table 2. Roles of endocrine FGFs indicated by human diseases.
FGF19
Paraneoplastic Diseases with Gain-of-Function
Extrahepatic cholestasis is a condition in which the release of bile acid is blocked in the bile ducts. Cholestasis is caused by a pancreatic tumor. Markedly increased serum FGF19 levels and the abundant expression of hepatic FGF19 are observed in cholestatic patients, indicating that FGF19 signaling is involved in some of the adaptations that protect the liver against bile acid toxicity (43).
FGF23
Inherited Disease with Loss-of-Function Mutations
Hyperphosphatemic familial tumoral calcinosis (HFTC) is a rare autosomal recessive metabolic disorder characterized by the deposition of calcium phosphate crystals. HFTC was initially attributed to loss-of function mutations in GLANT3 encoding a glycosyltransferase (44). Loss-of-function mutations in FGF23 are also associated with HFTC (45, 46).
Inherited Disease with Gain-of-Function Mutations
Autosomal dominant hypophosphatemic rickets is characterized by isolated renal phosphate wasting, hypophosphatemia, and inappropriately normal 1,25-dihydroxyvitamin D3 levels. Gain-of-function mutations in FGF23 result in AHDR. Cleaved FGF23 forms due to intracellular proteolysis lose their biological activities. FGF23 mutations in ADHR result in impaired proteolysis of FGF23, resulting in increased serum levels of active FGF23 (11, 47).
Paraneoplastic Diseases with Gain-of-Function
Osteomalacia is characterized by the softening of bones due to defective bone mineralization. Tumors that overexpress FGF23 induce tumor-induced osteomalacia characterized by hypophosphatemia, which is caused by renal phosphate wasting (12).
Single-Nucleotide Polymorphisms in Endocrine FGF Genes as Potential Risk Factors for Diseases
Single-nucleotide polymorphisms (SNPs) are DNA sequence variations that commonly occur within a population (e.g., 1%) with a single nucleotide in the genome. These SNP variations underlie differences in the susceptibility of humans to diseases. The severity of illnesses and the responses of the human body to treatments are also manifestations of SNPs. SNPs in endocrine FGFs also suggest their potential roles in risk factors for diseases (Table 2).
FGF21
The intake of dietary macronutrients such as carbohydrates, protein, and fat is associated with the risk of obesity and diabetes. A SNP located in the exon of FGF21 is significantly associated with the percentage of total caloric intake from protein and carbohydrates, indicating that FGF21 is a potential susceptibility gene for obesity and diabetes (48). SNPs in the 3′ untranslated region of FGF21 are also associated with metabolic syndrome, obesity, and diabetes (49). However, the mechanisms underlying these associations currently remain unclear.
FGF23
Kawasaki disease, which is a systemic panvasculitis that affects all medium/small-sized vessels, mainly occurs in early childhood and increases the risk of cardiac abnormalities. A SNP in the intron of FGF23 is significantly associated with higher serum FGF23 levels and cardiac abnormalities in children with Kawasaki disease (50). Three distinct SNPs in FGF23 are also associated with an increased risk of prostate cancer, indicating that FGF23 genetic variations increase prostate cancer susceptibility (51).
Serum Endocrine FGF Levels as Biomarkers for Diseases
Endocrine FGFs mainly play roles in an endocrine manner via the blood circulation system. Serum endocrine FGFs levels are sometimes altered in patients with metabolic and other diseases, indicating that endocrine FGFs are biomarkers for these diseases (Table 2).
FGF19
Renal Failure
Renal failure is a condition in which the kidneys fail to adequately filter waste products from the blood. Serum FGF19 levels are significantly increased in end-stage renal failure patients with chronic hemodialysis (52). The postprandial FGF19 response in serum is blunted in chronic kidney disease and is also associated with impaired insulin and C-peptide signaling (53).
Cardiovascular Disease
Coronary artery disease (CAD) is a group of diseases including stable angina, unstable angina, myocardial infarction, and sudden coronary death. Serum FGF19 levels are decreased in CAD in relation to its severity, suggesting that reductions in the expression of FGF19 are involved in the development and progression of CAD (54).
Intestinal Failure-Associated Liver Disease
In pediatric intestinal failure (IF), serum FGF19 levels are decreased corresponding to IF-associated liver disease (IFALD). This suggests that FGF19 contributes to the pathogenesis of IFALD (55).
Energy Metabolism Disorder
Serum FGF19 levels are decreased in patients with type 2 diabetes, indicating that the FGF19 pathway is significantly perturbed in severe diabetes (56).
Bile Acid Metabolism Disorder
Bile acids are reabsorbed by the terminal ileum. However, their reabsorption is frequently impaired in Crohn’s disease, resulting in secretory diarrhea. Serum FGF19 levels are decreased in Crohn’s disease associated with diarrhea and disease activity, indicating that FGF19 has potential as a biomarker for the functioning ileum in Crohn’s disease (57, 58). However, serum FGF19 levels are not affected in gallstone carriers (59).
Cancer
Serum FGF19 levels in the high Gleason grade of prostate cancer are higher than those in the low Gleason grade group, indicating that FGF19 promotes the progression of prostate cancer (60).
FGF21
Renal Failure
Serum FGF21 levels are increased in chronic kidney disease and acute kidney dysfunction, suggesting that renal excretion is a major route for the elimination of FGF21 (61).
Cardiovascular Disease
Coronary heart disease (CHD) is characterized by the narrowing of small blood vessels in the heart, which may lead to heart attacks. Serum FGF21 levels are significantly increased in CHD. Serum FGF21 levels in CHD patients with diabetes, hypertension, or both are higher than those in patients without these comorbidities. High serum FGF21 levels are associated with adverse lipid profiles in CHD patients, indicating that the paradoxical increase in serum FGF21 levels in CHD patients is a compensatory response or resistance to FGF21 (62). Serum FGF21 levels are positively correlated with carotid intima-media thickness (IMT) in women, but not in men, indicating that elevated serum FGF21 levels in women are an independent risk factor for increased carotid IMT (63). Preeclampsia, a serious cardiovascular complication in pregnancy, is associated with an increased future metabolic and cardiovascular risk. Serum FGF21 levels are significantly increased in patients with preeclampsia during pregnancy (64).
Mitochondrial Disease
Mitochondrial disease caused by dysfunctional mitochondria is a group of disorders with highly variable phenotypes including mitochondrial myopathy, diabetes mellitus, deafness, optic neuropathy, and multiple sclerosis-type disease. Serum FGF21 levels are significantly increased in human mitochondrial disease and the best predictor of mitochondrial disease among classical indicators including creatine kinase, lactate, and pyruvate (65, 66).
Energy Metabolism Disorder
Serum FGF21 levels are significantly increased in patients with obese and type 2 diabetes, suggesting the direct positive metabolic effects of FGF21 (56, 67–70). Anorexia nervosa (AN) is an eating disorder that is characterized by a low weight and food restriction. Serum FGF21 levels are significantly decreased in AN, suggesting that FGF21 is involved in the pathophysiology of or a complex adaptive response to AN (71). Cushing’s syndrome is caused by prolonged exposure to inappropriately high levels of glucocorticoids. Serum FGF21 levels are increased in patients with Cushing’s syndrome. Increased serum FGF21 levels are attributed to their excessive fat accumulation and related metabolic abnormalities, but not to the direct effects of cortisol on the production of FGF21 (72). Brown adipose tissue, which contains a markedly higher number of mitochondria, generates body heat in animals without shivering. Since brown adipose tissue is expected to markedly contribute to energy homeostasis in humans, it may represent a therapeutic target of obesity. Serum FGF21 levels are associated with brown adipose tissue activity, indicating a novel mechanism by which brown adipose tissue activity may be enhanced (73).
Lipid Metabolism Disorder
Serum FGF21 levels are increased in patients with non-alcoholic fatty liver disease (NAFLD), which supports the role of FGF21 as a key regulator of hepatic lipid metabolism (74). In contrast to adults with NAFLD, serum FGF19 and FGF21 levels are inversely associated with hepatic damage in children with NAFLD, providing insights for a better understanding of the progression of NAFLD (75).
Infection and Inflammation
Hepatitis C (HC) is an infectious disease that is caused by the HC virus. Serum FGF21 levels are increased in chronic HC (CHC) patients with steatosis and are associated with the steatosis grade, which may be a useful diagnostic marker for determining hepatic steatosis in CHC (76). Sepsis is an illness in which the body has a severe response to infection. Systemic inflammatory response syndrome (SIRS) is an inflammatory state in the whole body due to the response of the immune system to infection. SIRS is also closely related to sepsis. Serum FGF21 levels are significantly increased in patients with sepsis and SIRS, suggesting a role for FGF21 in inflammation (77). Rheumatoid arthritis (RA) is a chronic inflammatory disease in small hand and foot joints. Serum FGF21 levels are significantly increased in patients with RA, indicating the compensatory response of FGF21 to inflammation and immune response (78).
FGF23
Renal Failure
Serum FGF23 levels are significantly increased in chronic kidney disease, indicating the important role of FGF23 in maintaining a neutral phosphate balance as the glomerular filtration rate decreases (79–81).
Cardiovascular Disease
Serum FGF23 levels are positively correlated with the risk of heart failure (82). Serum FGF23 levels are increased in children with heart failure (83). These findings indicate that increased serum FGF23 levels are a risk factor for heart failure. Serum FGF23 levels are also increased in association with angiographic severity and the extent of CAD (84). Increased serum FGF23 levels are also a risk factor for myocardial infraction and hemorrhagic stroke (85). Serum FGF23 levels are strongly associated with left ventricular hypertrophy (86). These findings provide additional evidence for the role of FGF23 in cardiovascular disease.
Stroke
Stroke is a disease that is caused by cutting off the blood supply to part of the brain (ischemic) or by bleeding into the surrounding brain tissue from a weakened vessel (hemorrhagic). Serum FGF23 levels are increased in patients with stroke (87, 88).
Therapeutic Utility of Endocrine FGFs in Humans or Monkeys
FGF19
FGF19 is expected to prevent bile acid-induced liver damage by reducing hepatic bile acid levels through the modulation of bile acid synthesis. However, since FGF19 also stimulates the proliferation of hepatocytes, the potential risk associated with prolonged exposure to supraphysiological levels of FGF19 represents a major hurdle in the development of an FGF19-based therapy in hepatocellular carcinogenesis. A manufactured FGF19 variant, M70, which carries three amino acid substitutions and a five-amino acid deletion, regulates bile acid metabolism without tumorigenicity (89). The administration of M70 to healthy human volunteers potently reduces serum levels of 7α-hydroxy-4-cholesten-3-one, a surrogate marker for the hepatic activity of cholesterol 7α-hydroxylase (CYP7A1), which catalyzes the rate-limiting step in the bile acid synthetic pathway. These findings indicate that the development of non-tumorigenic FGF19 variants represents an effective approach for the prevention and treatment of cholestatic liver diseases and other disorders associated with bile acid dysregulation (89) (Table 3).
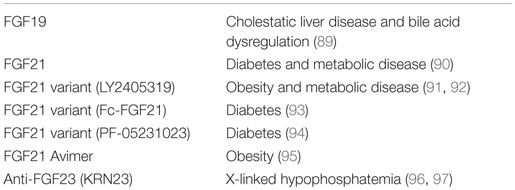
Table 3. Therapeutic utility of endocrine FGFs in humans or monkeys.
FGF21
The daily administration of FGF21 to diabetic rhesus monkeys for 6 weeks induces marked reductions in fasting serum glucose, triglyceride, insulin, and glucagon levels, significant improvements in lipoprotein profiles, and significant weight loss, thereby supporting the development of FGF21 for the treatment of diabetes and other metabolic diseases in humans (90). LY2405319 (LY) is a manufactured FGF21 variant with significantly improved biopharmaceutical properties (91). The administration of LY to patients with obesity and type 2 diabetes for 28 days produces significant improvements in dyslipidemia, a shift to a potentially less atherogenic apolipoprotein concentration profile, and favorable effects on body weight. These findings indicate that FGF21-based therapies may be effective for the treatment of selected metabolic disorders (92).
FGF21 has a short half-life (1–2 h). Fc-FGF21 is a long-acting FGF21 variant that is generated by fusing with an Fc fragment. In obese rhesus monkeys treated with FGF21 (once a day) or Fc-FGF21 (once a week), serum glucose, insulin, cholesterol, and triglyceride levels and body weight are markedly lower with Fc-FGF21 than with FGF21, indicating that the administration of Fc-FGF21 once a week leads to similar or greater efficacy than FGF21 administered daily (93). PF-05231023 is also a long-acting FGF21 variant generated by covalently conjugating FGF21 molecules to a non-targeting human IgG1 κ scaffold. Single intravenous injection of PF-05231023 to type 2 diabetes mellitus patients decreases serum triglyceride, total cholesterol, and low-density lipoprotein cholesterol levels and increases high-density lipoprotein cholesterol levels, but have no apparent effect on glucose levels (94).
As an alternative to FGF21, bispecific Avimer polypeptides that bind with high affinity and specificity to one of the receptor and coreceptor pairs used by FGF21, FGFR1c, and β-Klotho have been generated. These Avimers exhibit FGF21-like activity with greater potency than FGF21 in vitro. In obese male cynomolgus monkeys, a bispecific Avimer exhibits improved metabolic parameters and leads to greater reductions in body weight than those with FGF21, thereby supporting a novel therapeutic approach to target this potentially important pathway in the treatment of diabetes and obesity (95).
FGF23
X-linked hypophosphatemia (XLH) with elevated serum FGF23 levels is the most common heritable form of rickets and osteomalacia. KRN23 is a human anti-FGF23 antibody that has been developed as a potential treatment for XLH patients. KRN23 significantly increases the maximum renal tubular threshold for phosphate reabsorption, serum phosphate, and 1,25-dihydroxyvitamin D in XLH patients, suggesting utility for KRN23 in XLH patients (96, 97).
Conclusion
Endocrine FGFs comprising FGF19, FGF21, and FGF23 require FGFR and α-Klotho or β-Klotho as a cofactor for FGFR for their signaling in an endocrine manner. Endocrine FGFs lost heparan sulfate-binding affinity and acquired systemic signaling system with α-Klotho or β-Klotho during their evolution. The phenotypes of endocrine FGF knockout mice indicate that endocrine FGFs play important physiological roles in metabolism including bile acid metabolism, energy metabolism, and phosphate/active vitamin D metabolism. Evidence for the involvement of endocrine FGFs in human inherited and metabolic diseases has also been increasing, indicating their crucial pathophysiological roles in metabolic diseases, their SNPs as potential risk factors for diseases, and their serum levels as useful biomarkers for metabolic diseases. Furthermore, the therapeutic utility of endocrine FGFs is currently being developed. These findings provide new insights into the physiological and pathophysiological roles of endocrine FGFs and may provide potential diagnostic and therapeutic strategies for metabolic diseases.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1. Ornitz DM, Itoh N. The fibroblast growth factor signaling pathway. Wiley Interdiscip Rev Dev Biol (2005) 4:215–66. doi: 10.1002/wdev.176
2. McWhirter JR, Goulding M, Weiner JA, Chun J, Murre C. A novel fibroblast growth factor gene expressed in the developing nervous system is a downstream target of the chimeric homeodomain oncoprotein E2A-Pbx1. Development (1997) 124:3221–32.
3. Nishimura T, Utsunomiya Y, Hoshikawa M, Ohuchi H, Itoh N. Structure and expression of a novel human FGF, FGF-19, expressed in the fetal brain. Biochim Biophys Acta (1999) 1444:148–51. doi:10.1016/S0167-4781(98)00255-3
4. Xie MH, Holcomb I, Deuel B, Dowd P, Huang A, Vagts A, et al. FGF-19, a novel fibroblast growth factor with unique specificity for FGFR4. Cytokine (1999) 11:729–35. doi:10.1006/cyto.1999.0485
5. Itoh N, Ornitz DM. Functional evolutionary history of the mouse Fgf gene family. Dev Dyn (2008) 237:18–27. doi:10.1002/dvdy.21388
6. Nishimura T, Nakatake Y, Konishi M, Itoh N. Identification of a novel FGF, FGF-21, preferentially expressed in the liver. Biochim Biophys Acta (2000) 1492:203–6. doi:10.1016/S0167-4781(00)00067-1
7. Kharitonenkov A, Shiyanova TL, Koester A, Ford AM, Micanovic R, Galbreath EJ, et al. FGF-21 as a novel metabolic regulator. J Clin Invest (2005) 115:1627–35. doi:10.1172/JCI23606
8. Inagaki T, Dutchak P, Zhao G, Ding X, Gautron L, Parameswara V, et al. Endocrine regulation of the fasting response by PPARalpha-mediated induction of fibroblast growth factor 21. Cell Metab (2007) 25:415–25. doi:10.1016/j.cmet.2007.05.003
9. Badman MK, Pissios P, Kennedy AR, Koukos G, Flier JS, Maratos-Flier E. Hepatic fibroblast growth factor 21 is regulated by PPARalpha and is a key mediator of hepatic lipid metabolism in ketotic states. Cell Metab (2007) 5:426–37. doi:10.1016/j.cmet.2007.05.002
10. Yamashita T, Yoshioka M, Itoh N. Identification of a novel fibroblast growth factor, FGF-23, preferentially expressed in the ventrolateral thalamic nucleus of the brain. Biochem Biophys Res Commun (2000) 277:494–8. doi:10.1006/bbrc.2000.3696
11. Consortium ADHR. Autosomal dominant hypophosphataemic rickets is associated with mutations in FGF23. Nat Genet (2000) 26:345–8. doi:10.1038/81664
12. Shimada T, Mizutani S, Muto T, Yoneya T, Hino R, Takeda S, et al. Cloning and characterization of FGF23 as a causative factor of tumor-induced osteomalacia. Proc Natl Acad Sci U S A (2001) 98:6500–5. doi:10.1073/pnas.101545198
13. Kuro-o M. Klotho and βKlotho. Adv Exp Med Biol (2012) 728:25–40. doi:10.1007/978-1-4614-0887-1_2
14. Beenken A, Mohammadi M. The structural biology of the FGF19 subfamily. Adv Exp Med Biol (2012) 728:1–24. doi:10.1007/978-1-4614-0887-1_1
15. Wu X, Ge H, Lemon B, Vonderfecht S, Weiszmann J, Hecht R. FGF19-induced hepatocyte proliferation is mediated through FGFR4 activation. J Biol Chem (2010) 285:5165–70. doi:10.1074/jbc.M109.068783
16. Itoh N. Hormone-like (endocrine) Fgfs: their evolutionary history and roles in development, metabolism, and disease. Cell Tissue Res (2010) 342:1–11. doi:10.1007/s00441-010-1024-2
17. Oulion S, Bertrand S, Escriva H. Evolution of the FGF gene family. Int J Evol Biol (2012) 2012:298147. doi:10.1155/2012/298147
18. Zhang G, Li C, Li Q, Li B, Larkin DM, Lee C, et al. Comparative genomics reveals insights into avian genome evolution and adaptation. Science (2014) 346:1311–20. doi:10.1126/science.1251385
19. Vincentz JW, McWhirter JR, Murre C, Baldini A, Furuta Y. Fgf15 is required for proper morphogenesis of the mouse cardiac outflow tract. Genesis (2005) 41:192–201. doi:10.1002/gene.20114
20. Inagaki T, Choi M, Moschetta A, Peng L, Cummins CL, McDonald JG, et al. Fibroblast growth factor 15 functions as an enterohepatic signal to regulate bile acid homeostasis. Cell Metab (2005) 2:217–25. doi:10.1016/j.cmet.2005.09.001
21. Choi M, Moschetta A, Bookout AL, Peng L, Umetani M, Holmstrom SR, et al. Identification of a hormonal basis for gallbladder filling. Nat Med (2006) 12:1253–5. doi:10.1038/nm1501
22. Kir S, Beddow SA, Samuel VT, Miller P, Previs SF, Suino-Powell K, et al. FGF19 as a postprandial, insulin-independent activator of hepatic protein and glycogen synthesis. Science (2011) 331:1621–4. doi:10.1126/science.1198363
23. Uriarte I, Fernandez-Barrena MG, Monte MJ, Latasa MU, Chang HC, Carotti S, et al. Identification of fibroblast growth factor 15 as a novel mediator of liver regeneration and its application in the prevention of post-resection liver failure in mice. Gut (2013) 62:899–910. doi:10.1136/gutjnl-2012-302945
24. Kong B, Huang J, Zhu Y, Li G, Williams J, Shen S, et al. Fibroblast growth factor 15 deficiency impairs liver regeneration in mice. Am J Physiol Gastrointest Liver Physiol (2014) 306:G893–902. doi:10.1152/ajpgi.00337.2013
25. Uriarte I, Latasa MU, Carotti S, Fernandez-Barrena MG, Garcia-Irigoyen O, Elizalde M, et al. Ileal FGF15 contributes to fibrosis-associated hepatocellular carcinoma development. Int J Cancer (2015) 136:2469–75. doi:10.1002/ijc.29287
26. Kharitonenkov A, Shanafelt AB. FGF21: a novel prospect for the treatment of metabolic diseases. Curr Opin Investig Drugs (2009) 10:359–64.
27. Fon Tacer K, Bookout AL, Ding X, Kurosu H, John GB, Wang L, et al. Research resource: comprehensive expression atlas of the fibroblast growth factor system in adult mouse. Mol Endocrinol (2010) 24:2050–64. doi:10.1210/me.2010-0142
28. Hotta Y, Nakamura H, Konishi M, Murata Y, Takagi H, Matsumura S, et al. Fibroblast growth factor 21 regulates lipolysis in white adipose tissue but is not required for ketogenesis and triglyceride clearance in liver. Endocrinology (2009) 150:4625–33. doi:10.1210/en.2009-0119
29. Badman MK, Koester A, Flier JS, Kharitonenkov A, Maratos-Flier E. Fibroblast growth factor 21-deficient mice demonstrate impaired adaptation to ketosis. Endocrinology (2009) 150:4931–40. doi:10.1210/en.2009-0532
30. Murata Y, Nishio K, Mochiyama T, Konishi M, Shimada M, Ohta H, et al. Fgf21 impairs adipocyte insulin sensitivity in mice fed a low-carbohydrate, high-fat ketogenic diet. PLoS One (2013) 8:e69330. doi:10.1371/journal.pone.0069330
31. Lin Z, Tian H, Lam KS, Lin S, Hoo RC, Konishi M, et al. Adiponectin mediates the metabolic effects of FGF21 on glucose homeostasis and insulin sensitivity in mice. Cell Metab (2013) 17:779–89. doi:10.1016/j.cmet.2013.04.005
32. Holland WL, Adams AC, Brozinick JT, Bui HH, Miyauchi Y, Kusminski CM, et al. An FGF21-adiponectin-ceramide axis controls energy expenditure and insulin action in mice. Cell Metab (2013) 17:790–7. doi:10.1016/j.cmet.2013.03.019
33. Habegger KM, Stemmer K, Cheng C, Müller TD, Heppner KM, Ottaway N, et al. Fibroblast growth factor 21 mediates specific glucagon actions. Diabetes (2013) 62:1453–63. doi:10.2337/db12-1116
34. So WY, Cheng Q, Xu A, Lam KS, Leung PS. Loss of fibroblast growth factor 21 action induces insulin resistance, pancreatic islet hyperplasia and dysfunction in mice. Cell Death Dis (2015) 6:e1707. doi:10.1038/cddis.2015.80
35. Kim SH, Kim KH, Kim HK, Kim MJ, Back SH, Konishi M, et al. Fibroblast growth factor 21 participates in adaptation to endoplasmic reticulum stress and attenuates obesity-induced hepatic metabolic stress. Diabetologia (2015) 58:809–18. doi:10.1007/s00125-014-3475-6
36. Planavila A, Redondo I, Hondares E, Vinciguerra M, Munts C, Iglesias LA, et al. Fibroblast growth factor 21 protects against cardiac hypertrophy in mice. Nat Commun (2013) 4:2019. doi:10.1038/ncomms3019
37. Planavila A, Redondo-Angulo I, Ribas F, Garrabou G, Casademont J, Giralt M, et al. Fibroblast growth factor 21 protects the heart from oxidative stress. Cardiovasc Res (2015) 106:19–31. doi:10.1093/cvr/cvu263
38. Johnson CL, Weston JY, Chadi SA, Fazio EN, Huff MW, Kharitonenkov A, et al. Fibroblast growth factor 21 reduces the severity of cerulein-induced pancreatitis in mice. Gastroenterology (2009) 137:1795–804. doi:10.1053/j.gastro.2009.07.064
39. Yan X, Chen J, Zhang C, Zhou S, Zhang Z, Chen J, et al. FGF21 deletion exacerbates diabetic cardiomyopathy by aggravating cardiac lipid accumulation. J Cell Mol Med (2015) 19:1557–68. doi:10.1111/jcmm.12530
40. Yan X, Chen J, Zhang C, Zeng J, Zhou S, Zhang Z, et al. Fibroblast growth factor 21 deletion aggravates diabetes-induced pathogenic changes in the aorta in type 1 diabetic mice. Cardiovasc Diabetol (2015) 14:77. doi:10.1186/s12933-015-0241-0
41. Shimada T, Kakitani M, Yamazaki Y, Hasegawa H, Takeuchi Y, Fujita T, et al. Targeted ablation of Fgf23 demonstrates an essential physiological role of FGF23 in phosphate and vitamin D metabolism. J Clin Invest (2004) 113:561–8. doi:10.1172/JCI200419081
42. Lysaght AC, Yuan Q, Fan Y, Kalwani N, Caruso P, Cunnane M, et al. FGF23 deficiency leads to mixed hearing loss and middle ear malformation in mice. PLoS One (2014) 9:e107681. doi:10.1371/journal.pone.0107681
43. Schaap FG, van der Gaag NA, Gouma DJ, Jansen PL. High expression of the bile salt-homeostatic hormone fibroblast growth factor 19 in the liver of patients with extrahepatic cholestasis. Hepatology (2009) 49:1228–35. doi:10.1002/hep.22771
44. Topaz O, Shurman DL, Bergman R, Indelman M, Ratajczak P, Mizrachi M, et al. Mutations in GALNT3, encoding a protein involved in O-linked glycosylation, cause familial tumoral calcinosis. Nat Genet (2004) 36:579–81. doi:10.1038/ng1358
45. Chefetz I, Heller R, Galli-Tsinopoulou A, Richard G, Wollnik B, Indelman MA, et al. Novel homozygous missense mutation in FGF23 causes familial tumoral calcinosis associated with disseminated visceral calcification. Hum Genet (2005) 118:261–6. doi:10.1007/s00439-005-0026-8
46. Garringer HJ, Malekpour M, Esteghamat F, Mortazavi SM, Davis SI, Farrow EG, et al. Molecular genetic and biochemical analyses of FGF23 mutations in familial tumoral calcinosis. Am J Physiol Endocrinol Metab (2008) 295:E929–37. doi:10.1152/ajpendo.90456.2008
47. White KE, Carn G, Lorenz-Depiereux B, Benet-Pages A, Strom TM, Econs MJ. Autosomal-dominant hypophosphatemic rickets (ADHR) mutations stabilize FGF-23. Kidney Int (2001) 60:2079–86. doi:10.1046/j.1523-1755.2001.00064.x
48. Chu AY, Workalemahu T, Paynter NP, Rose LM, Giulianini F, Tanaka T, et al. Novel locus including FGF21 is associated with dietary macronutrient intake. Hum Mol Genet (2013) 22:1895–902. doi:10.1093/hmg/ddt032
49. Zhang M, Zeng L, Wang YJ, An ZM, Ying BW. Associations of fibroblast growth factor 21 gene 3′ untranslated region single-nucleotide polymorphisms with metabolic syndrome, obesity, and diabetes in a Han Chinese population. DNA Cell Biol (2012) 31:547–52. doi:10.1089/dna.2011.1302
50. Falcini F, Rigante D, Masi L, Covino M, Franceschelli F, Leoncini G, et al. Fibroblast growth factor 23 (FGF23) gene polymorphism in children with Kawasaki syndrome (KS) and susceptibility to cardiac abnormalities. Ital J Pediatr (2013) 39:69. doi:10.1186/1824-7288-39-69
51. Kim HJ, Kim KH, Lee J, Oh JJ, Cheong HS, Wong EL, et al. Single nucleotide polymorphisms in fibroblast growth factor 23 gene, FGF23, are associated with prostate cancer risk. BJU Int (2014) 114:303–10. doi:10.1111/bju.12396
52. Reiche M, Bachmann A, Lössner U, Blüher M, Stumvoll M, Fasshauer M. Fibroblast growth factor 19 serum levels: relation to renal function and metabolic parameters. Horm Metab Res (2010) 42:178–81. doi:10.1055/s-0029-1243249
53. Li M, Qureshi AR, Ellis E, Axelsson J. Impaired postprandial fibroblast growth factor (FGF)-19 response in patients with stage 5 chronic kidney diseases is ameliorated following antioxidative therapy. Nephrol Dial Transplant (2013) 28(Suppl 4):iv212–9. doi:10.1093/ndt/gft337
54. Hao Y, Zhou J, Zhou M, Ma X, Lu Z, Gao M, et al. Serum levels of fibroblast growth factor 19 are inversely associated with coronary artery disease in Chinese individuals. PLoS One (2013) 8:e72345. doi:10.1371/journal.pone.0072345
55. Mutanen A, Lohi J, Heikkilä P, Jalanko H, Pakarinen MP. Loss of ileum decreases serum fibroblast growth factor 19 in relation to liver inflammation and fibrosis in pediatric onset intestinal failure. J Hepatol (2015) 62:1391–7. doi:10.1016/j.jhep.2015.01.004
56. Roesch SL, Styer AM, Wood GC, Kosak Z, Seiler J, Benotti P, et al. Perturbations of fibroblast growth factors 19 and 21 in type 2 diabetes. PLoS One (2015) 10:e0116928. doi:10.1371/journal.pone.0116928
57. Pattni SS, Brydon WG, Dew T, Johnston IM, Nolan JD, Srinivas M. Fibroblast growth factor 19 in patients with bile acid diarrhoea: a prospective comparison of FGF19 serum assay and SeHCAT retention. Aliment Pharmacol Ther (2013) 38:967–76. doi:10.1111/apt.12466
58. Nolan JD, Johnston IM, Pattni SS, Dew T, Orchard TR, Walters JR. Diarrhea in Crohn’s disease: investigating the role of the ileal hormone fibroblast growth factor 19. J Crohns Colitis (2015) 9:125–31. doi:10.1093/ecco-jcc/jju022
59. Renner O, Harsch S, Matysik S, Lütjohann D, Schmitz G, Stange EF. Upregulation of hepatic bile acid synthesis via fibroblast growth factor 19 is defective in gallstone disease but functional in overweight individuals. United European Gastroenterol J (2014) 2:216–25. doi:10.1177/2050640614527938
60. Nagamatsu H, Teishima J, Goto K, Shikuma H, Kitano H, Shoji K, et al. FGF19 promotes progression of prostate cancer. Prostate (2015) 75:1092–101. doi:10.1002/pros.22994
61. Hindricks J, Ebert T, Bachmann A, Kralisch S, Lössner U, Kratzsch J, et al. Serum levels of fibroblast growth factor-21 are increased in chronic and acute renal dysfunction. Clin Endocrinol (Oxf) (2014) 80:918–24. doi:10.1111/cen.12380
62. Lin Z, Wu Z, Yin X, Liu Y, Yan X, Lin S, et al. Serum levels of FGF-21 are increased in coronary heart disease patients and are independently associated with adverse lipid profile. PLoS One (2010) 5:e15534. doi:10.1371/journal.pone.0015534
63. Chow WS, Xu A, Woo YC, Tso AW, Cheung SC, Fong CH, et al. Serum fibroblast growth factor-21 levels are associated with carotid atherosclerosis independent of established cardiovascular risk factors. Arterioscler Thromb Vasc Biol (2013) 33:2454–9. doi:10.1161/ATVBAHA.113.301599
64. Stepan H, Kley K, Hindricks J, Kralisch S, Jank A, Schaarschmidt W, et al. Serum levels of the adipokine fibroblast growth factor-21 are increased in preeclampsia. Cytokine (2013) 62:322–6. doi:10.1016/j.cyto.2013.02.019
65. Salehi MH, Kamalidehghan B, Houshmand M, Aryani O, Sadeghizadeh M, Mossalaeie MM. Association of fibroblast growth factor (FGF-21) as a biomarker with primary mitochondrial disorders, but not with secondary mitochondrial disorders (Friedreich ataxia). Mol Biol Rep (2013) 40:6495–9. doi:10.1007/s11033-013-2767-0
66. Davis RL, Liang C, Edema-Hildebrand F, Riley C, Needham M, Sue CM. Fibroblast growth factor 21 is a sensitive biomarker of mitochondrial disease. Neurology (2013) 81:1819–26. doi:10.1212/01.wnl.0000436068.43384.ef
67. Mraz M, Bartlova M, Lacinova Z, Michalsky D, Kasalicky M, Haluzikova D, et al. Serum concentrations and tissue expression of a novel endocrine regulator fibroblast growth factor-21 in patients with type 2 diabetes and obesity. Clin Endocrinol (Oxf) (2009) 71:369–75. doi:10.1111/j.1365-2265.2008.03502.x
68. Lin Y, Xiao YC, Zhu H, Xu QY, Qi L, Wang YB, et al. Serum fibroblast growth factor 21 levels are correlated with the severity of diabetic retinopathy. J Diabetes Res (2014) 2014:929756. doi:10.1155/2014/929756
69. Korwutthikulrangsri M, Mahachoklertwattana P, Chanprasertyothin S, Pongratanakul S, Poomthavorn P. Serum fibroblast growth factor 21 in overweight and obese Thai children and adolescents: its relation to glucose metabolism and its change after glucose loading. Clin Endocrinol (Oxf) (2015). doi:10.1111/cen.12808
70. Reinehr T, Karges B, Meissner T, Wiegand S, Fritsch M, Holl RW, et al. Fibroblast growth factor 21 and fetuin-A in obese adolescents with and without type 2 diabetes. J Clin Endocrinol Metab (2015) 100:3004–10. doi:10.1210/jc.2015-2192
71. Dostálová I, Kaválková P, Haluzíková D, Lacinová Z, Mráz M, Papezová H, et al. Plasma concentrations of fibroblast growth factors 19 and 21 in patients with anorexia nervosa. J Clin Endocrinol Metab (2008) 93:3627–32. doi:10.1210/jc.2008-0746
72. Durovcová V, Marek J, Hána V, Matoulek M, Zikán V, Haluzíková D, et al. Plasma concentrations of fibroblast growth factors 21 and 19 in patients with Cushing’s syndrome. Physiol Res (2010) 59:415–22.
73. Hanssen MJ, Broeders E, Samms RJ, Vosselman MJ, van der Lans AA, Cheng CC, et al. Serum FGF21 levels are associated with brown adipose tissue activity in humans. Sci Rep (2015) 5:10275. doi:10.1038/srep10275
74. Li H, Fang Q, Gao F, Fan J, Zhou J, Wang X, et al. Fibroblast growth factor 21 levels are increased in nonalcoholic fatty liver disease patients and are correlated with hepatic triglyceride. J Hepatol (2010) 53:934–40. doi:10.1016/j.jhep.2010.05.018
75. Alisi A, Ceccarelli S, Panera N, Prono F, Petrini S, De Stefanis C, et al. Association between serum atypical fibroblast growth factors 21 and 19 and pediatric nonalcoholic fatty liver disease. PLoS One (2013) 8:e67160. doi:10.1371/journal.pone.0067160
76. Kukla M, Berdowska A, Stygar D, Gabriel A, Mazur W, Łogiewa-Bazger B, et al. Serum FGF21 and RBP4 levels in patients with chronic hepatitis C. Scand J Gastroenterol (2012) 47:1037–47. doi:10.3109/00365521.2012.694901
77. Gariani K, Drifte G, Dunn-Siegrist I, Pugin J, Jornayvaz FR. Increased FGF21 plasma levels in humans with sepsis and SIRS. Endocr Connect (2013) 2:146–53. doi:10.1530/EC-13-0040
78. Hulejová H, Andrés Cerezo L, Kuklová M, Pecha O, Vondráček T, Pavelka K, et al. Novel adipokine fibroblast growth factor 21 is increased in rheumatoid arthritis. Physiol Res (2012) 61:489–94.
79. Ozeki M, Fujita S, Kizawa S, Morita H, Sohmiya K, Hoshiga M, et al. Association of serum levels of FGF23 and α-Klotho with glomerular filtration rate and proteinuria among cardiac patients. BMC Nephrol (2014) 15:147. doi:10.1186/1471-2369-15-147
80. Portale AA, Wolf M, Jüppner H, Messinger S, Kumar J, Wesseling-Perry K, et al. Disordered FGF23 and mineral metabolism in children with CKD. Clin J Am Soc Nephrol (2014) 9:344–53. doi:10.2215/CJN.05840513
81. Taal MW, Thurston V, McIntyre NJ, Fluck RJ, McIntyre CW. The impact of vitamin D status on the relative increase in fibroblast growth factor 23 and parathyroid hormone in chronic kidney disease. Kidney Int (2014) 86:407–13. doi:10.1038/ki.2013.537
82. di Giuseppe R, Buijsse B, Hirche F, Wirth J, Arregui M, Westphal S, et al. Plasma fibroblast growth factor 23, parathyroid hormone, 25-hydroxyvitamin D3, and risk of heart failure: a prospective, case-cohort study. J Clin Endocrinol Metab (2014) 99:947–55. doi:10.1210/jc.2013-2963
83. Isakova T, Houston J, Santacruz L, Schiavenato E, Somarriba G, Harmon WG, et al. Associations between fibroblast growth factor 23 and cardiac characteristics in pediatric heart failure. Pediatr Nephrol (2013) 28:2035–42. doi:10.1007/s00467-013-2515-7
84. Xiao Y, Peng C, Huang W, Zhang J, Xia M, Zhang Y, et al. Circulating fibroblast growth factor 23 is associated with angiographic severity and extent of coronary artery disease. PLoS One (2013) 8:e72545. doi:10.1371/journal.pone.0072545
85. di Giuseppe R, Kühn T, Hirche F, Buijsse B, Dierkes J, Fritsche A, et al. Plasma fibroblast growth factor 23 and risk of cardiovascular disease: results from the EPIC-Germany case-cohort study. Eur J Epidemiol (2015) 30:131–41. doi:10.1007/s10654-014-9982-4
86. Jovanovich A, Ix JH, Gottdiener J, McFann K, Katz R, Kestenbaum B, et al. Fibroblast growth factor 23, left ventricular mass, and left ventricular hypertrophy in community-dwelling older adults. Atherosclerosis (2013) 231:114–9. doi:10.1016/j.atherosclerosis.2013.09.002
87. Wright CB, Dong C, Stark M, Silverberg S, Rundek T, Elkind MS, et al. Plasma FGF23 and the risk of stroke: the Northern Manhattan study (NOMAS). Neurology (2014) 82:1700–6. doi:10.1212/WNL.0000000000000410
88. Panwar B, Jenny NS, Howard VJ, Wadley VG, Muntner P, Kissela BM, et al. Fibroblast growth factor 23 and risk of incident stroke in community-living adults. Stroke (2015) 46:322–8. doi:10.1161/STROKEAHA.114.007489
89. Luo J, Ko B, Elliott M, Zhou M, Lindhout DA, Phung V, et al. A nontumorigenic variant of FGF19 treats cholestatic liver diseases. Sci Transl Med (2014) 6:247ra100. doi:10.1126/scitranslmed.3009098
90. Kharitonenkov A, Wroblewski VJ, Koester A, Chen YF, Clutinger CK, Tigno XT, et al. The metabolic state of diabetic monkeys is regulated by fibroblast growth factor-21. Endocrinology (2007) 148:774–81. doi:10.1210/en.2006-1168
91. Adams AC, Halstead CA, Hansen BC, Irizarry AR, Martin JA, Myers SR, et al. LY2405319, an engineered FGF21 variant, improves the metabolic status of diabetic monkeys. PLoS One (2013) 8:e65763. doi:10.1371/journal.pone.0065763
92. Gaich G, Chien JY, Fu H, Glass LC, Deeg MA, Holland WL, et al. The effects of LY2405319, an FGF21 analog, in obese human subjects with type 2 diabetes. Cell Metab (2013) 18:333–40. doi:10.1016/j.cmet.2013.08.005
93. Véniant MM, Komorowski R, Chen P, Stanislaus S, Winters K, Hager T, et al. Long-acting FGF21 has enhanced efficacy in diet-induced obese mice and in obese rhesus monkeys. Endocrinology (2012) 153:4192–203. doi:10.1210/en.2012-1211
94. Dong JQ, Rossulek M, Somayaji VR, Baltrukonis D, Liang Y, Hudson K, et al. Pharmacokinetics and pharmacodynamics of PF-05231023, a novel long-acting FGF21 mimetic, in a first-in-human study. Br J Clin Pharmacol (2015). doi:10.1111/bcp.12676
95. Smith R, Duguay A, Bakker A, Li P, Weiszmann J, Thomas MR, et al. FGF21 can be mimicked in vitro and in vivo by a novel anti-FGFR1c/β-Klotho bispecific protein. PLoS One (2013) 8:e61432. doi:10.1371/journal.pone.0061432
96. Carpenter TO, Imel EA, Ruppe MD, Weber TJ, Klausner MA, Wooddell MM, et al. Randomized trial of the anti-FGF23 antibody KRN23 in X-linked hypophosphatemia. J Clin Invest (2014) 124:1587–97. doi:10.1172/JCI72829
Keywords: biomarker, disease, endocrine, FGF, Klotho, metabolism, mutation, polymorphism
Citation: Itoh N, Ohta H and Konishi M (2015) Endocrine FGFs: evolution, physiology, pathophysiology, and pharmacotherapy. Front. Endocrinol. 6:154. doi: 10.3389/fendo.2015.00154
Received: 27 July 2015; Accepted: 14 September 2015;
Published: 29 September 2015
Edited by:
Peter Blume-Jensen, Metamark Genetics, Inc., USAReviewed by:
Wendong Huang, Beckman Research Institute of City of Hope, USAMikhail Karganov, Russian Academy of Medical Sciences, Russia
Copyright: © 2015 Itoh, Ohta and Konishi. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Nobuyuki Itoh, Medical Innovation Center, Kyoto University Graduate School of Medicine, Shogoin-Kawara-cho, Sakyo, Kyoto 606-8507, Japan,bm9idXl1a2kuaXRvaC41NmNAc3Qua3lvdG8tdS5hYy5qcA==