 Mariusz W. Szkudlinski
Mariusz W. Szkudlinski- Trophogen Inc., Rockville, MD, USA
Last two decades of structure–function studies performed in numerous laboratories provided substantial progress in understanding basic science, physiological, pathophysiological, pharmacological, and comparative aspects of glycoprotein hormones (GPHs) and their cognate receptors. Multiple concepts and models developed based on experimental data in the past stood the test of time and have been, at least in part, confirmed and/or remained compatible with the new structures resolved at the atomic level. Major advances in understanding of the ligand–receptor relationships are heralding the dawn of a new era for GPHs and their receptors, although many basic questions still remain unanswered. This article examines retrospectively several basic science aspects of GPH super-agonists and related “biosuperiors” in a broader context of the advances in the ligand–receptor structure–function relationships and new mechanistic models generated based on the structure elucidation. Due to selective focus of my comments and perspectives in certain parts, the reader is directed to the most relevant publications and reviews in the field for more comprehensive analyses.
Origins and Evolution of Function
The family of glycoprotein hormones (GPHs) consists of luteinizing hormone (LH), chorionic gonadotropin (CG), follicle-stimulating hormone (FSH), and thyroid-stimulating hormone (TSH), which are heterodimers formed by the non-covalent association of a common alpha (α) and a hormone-specific beta (β) subunit. Structurally, GPHs and their subunit ancestors belong to the cysteine-knot growth factor superfamily and due to relatively high glycosylation are recognized as the most complex protein hormone molecules (1–3). Their cognate GPH receptors (GPHRs) are type A leucine-rich repeat (LRR)-containing G-protein-coupled receptors (LGR) with a large glycoprotein extracellular domain (ECD). Early ancestors of GPHs and their receptors emerged at the origin of metazoan animals (multicellular mitochondrial eukaryotes) (4), although two domains of GPHRs, LRRs and 7-helix transmembrane domain (TMD), have much earlier evolutionary origin and are very well-diversified in extremely large number of functionally unrelated proteins in animals and plants (5). Parallel evolution of GPHs, their subunits, and cognate GPHRs was previously studied and discussed in detail (2, 6, 7). An evolution of the receptor–ligand interface likely progressed through the series of fine-tuning within the concave face of the LRRs and activating configuration within the hinge region located between the LRRs and TMD. Interestingly, as previously proposed (6), numerous early vertebrate GPHRs are functioning at least in part by utilizing their constitutive activity, which is determined in each individual cell by the number of receptors expressed in its cell membrane. Remarkably, nematode LGRs are constitutively activating only the Gs/cAMP, but not Gq/IP3 inositol phosphate pathway (8). In addition, comparative analysis of GPHRs signaling may suggest that Gs/cAMP pathway as the only mechanism of the agonist-dependent and -independent receptor activation has evolved into more diversified and complex signaling system (9, 10).
Significant level of inherent constitutive activity is present in various vertebrate GPHRs (11–13), including wild-type (WT) hTSHR. In sharp contrast, human LHRs and FSHRs are activated almost exclusively by their respective ligands and the number of natural or artificial receptor mutations in their respective ECDs causing constitutive activity is very low (14–16). With regard to the TSHR, there is an apparent correlation between a high level of basal constitutive activity and much more relaxed ligand specificity (promiscuity), which is exemplified by the prevalence of hCG-induced subclinical or overt hyperthyroidism in the first trimester of pregnancy (17).
Charge Cluster in the Common α-Subunit
Significant contribution of electrostatic interactions to high affinity receptor binding has been recognized for various ligand–receptor pairs, including different cysteine-knot growth factors and their respective receptors (18). Accordingly, a long-standing postulate held that charge–charge interactions are of major importance in the TSH–TSHR interactions (19). For the entire G-protein-coupled receptor, strong statistical evidence was provided that negatively charged amino acids are enriched in the ECDs, including extracellular loops (ECLs), but positively charged amino acids dominate within the intracellular domains (20). Design and sequential development of human TSH and gonadotropin super-agonists (Figure 1) were described previously in details (1, 21, 22). Our early mutagenesis studies, which has been recognized as “the advent of super hormone drugs” (21, 23) focused primarily on the 11–20 region of the human α-subunit. These studies have revealed that a basic charge cluster in this region, which has evolved in vertebrates and disappeared in apes and humans, is an important modulator of hormone–receptor binding and activation. Amino acid substitutions to positively charged lysine (K) or arginine (R) in the 11–20 region individually (T11K/R, Q13K/R, E14K/R, P16K/R, Q20K/R) and in various combinations increased the potency and efficacy of hTSH and hCG (21). Such human analogs remain highly specific for their respective receptors and inactive (up to 1000-fold higher concentration) at the other GPHRs (24). The effect of these substitutions on the in vitro bioactivity was highly correlated with their effects on the receptor binding activity. It was repeatedly demonstrated in media and buffers with various salt concentrations, and confirmed by studies in other laboratories (25, 26) as well as by using CHO-TSHR cells with largely depleted pool of the negatively charged cell surface proteoglycans. Notably, mutations to alanine did not alter hormone activity, indicating that only selective substitutions to K or R amino acid residues are causing an enhancement of cAMP and IP3 production, iodine uptake, proliferation of FRTL-5 cells, thyroxine and progesterone production, respectively (1, 21, 24). All our previous theoretical models of GPH–GPHR interaction derived from super-agonist studies were placing the mutagenized α-subunit αL1 and αL3 loops in a close proximity to the hinge region and the ECLs of the receptor TMD (1, 27), very similar to early structural predictions by Jiang et al. (28) and highly compatible with two epitope-mapping studies (29, 30).
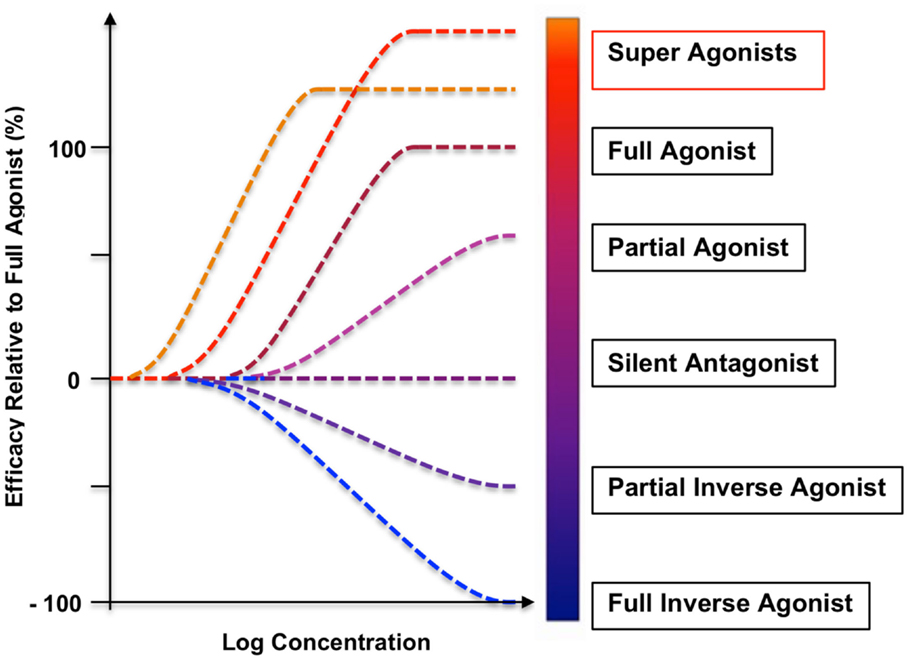
Figure 1. A range of dose–response curves and relative efficacy spectrum of GPHR ligands. The efficacies of the selected classes of ligands are illustrated by the in vitro stimulation of cAMP production and comparison with an endogenous, WT agonist (full agonist with 100% intrinsic efficacy). Although the term “super-agonist” has not been yet officially addressed in the NC-IUPHAR nomenclature, super-agonists show higher efficacy than full agonists, variable assay-dependent increases in receptor-binding affinity and potency, differences dependent on the receptor densities, differences related to the degree of signal amplification in the activation cascade, and significant enhancement of clinical efficacy in human and veterinary applications. High affinity super-agonists are especially desirable in various disorders with largely impaired receptor-binding and cell-surface expression (22, 24, 31). Multiple partial agonists of all GPHRs and TSHR-blocking anti-TSHR antibodies (silent or orthosteric antagonists) have been studied (32), but only one human monoclonal anti-TSHR antibody was recognized as an inverse agonist with a significant suppression of the basal constitutive activity of the WT TSHR (33).
Super-agonists of human GPHs, also named as “superactive analogs,” have been generated by introduction of positively charged amino acid residues in selected locations of the αL1, αL3, and βL3 beta-hairpin loops (1, 21, 27). Our “signal-enhancing αL1 loop mutations” were described as fully consistent with the model of receptor activation based on the structure of FSH bound to N-terminal cysteine cluster together with the LRRs proposed in 2005 (34). Highly unique position of the α-subunit αL1 13–20 mutations was more recently confirmed by Jiang et al. and the structure of hFSH bound to the entire ECD of hFSHR (35) (Figure 2). Moreover, Jiang et al. (36) discovered that such α-subunit αL1 loop amino acid substitutions to K (21) or R (37) are “concentrated near the top right side of the ‘activation pocket’ generating stronger electrostatic interaction to pull and lift the sulfated-tyrosine 335 (sTyr335) of the FSHR hairpin loop,” which is essential in the receptor activation.
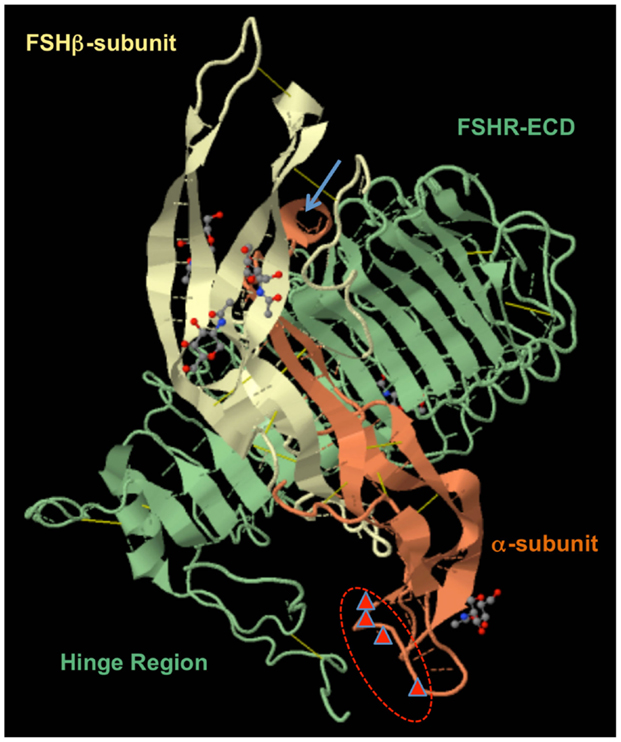
Figure 2. FSH–FSHR/ECD complex (PDB 4AY9) as reported by Jiang et al. (35). The locations of human α-subunit αL1 residues 13, 14, 16, and 20 are marked by red triangles and circled in red. The α-subunit α-helix is seen at the top as a light brown circle and is marked with a blue arrow. LRRs together with the hinge region are forming one large domain interconnected with three disulfide bridges. The recent “two-step model” subdivided this domain into the hormone-binding subdomain (HBSD) and signal-specificity subdomain (SSSD) (36).
GPH Super-Agonists – Tools in Structure–Function Studies
Two step activation mechanism proposed based on FSH–FSHR/ECD complexes (35, 36), and other structure-based models (38, 39) incorporated and explained, at least in part, several our previous findings as described below.
First, the new structure-based model by Jiang et al. (36) is placing much emphasis on the signal-specificity subdomain (SSSD). It explains, at least in part, why positive charge cluster in the αL1 loop, from our studies initiated in 1995, was rescuing (restoring) several major “loss-of-function” mutations including two mutations of K51 in the α-subunit (K51A and K51P) (40), which later have been found to be essential in the formation of salt bridge with highly conserved D153 within the receptor LRRs domain (34) and several others described elsewhere (1, 41) [see also Ref. (3, 38, 42, 43)]. Moreover, largely reduced binding activity and potency of single-chain hCG and its minimized variants were restored using αL1 loop substitutions (α4K and α4R) (25, 26). Remarkably, also the LH activity of the hTSH/hCG “seat-belt” “determinant loop” chimera was further increased by concomitant introduction of a cluster of K residues (α4K) into a highly distant from “seat-belt” αL1 13–20 domain (41). The results of several other published and unpublished studies further reinforced the role of αL1 loop basic charge cluster in the compensatory mechanism functioning within the SSSD.
Second, activation of the TSHR by free or fused homodimeric αL1 α-subunit analogs, but not the WT human α-subunits has been detected in a concentration range only 1–2 log orders higher than that of hTSH-WT (44, 45). Such agonistic activity of α-subunit analogs was observed only in free, non-tethered forms, but not in the yoked subunit-TSHR complexes constrained by the fusion of α-subunit and TSHR. These findings first challenged the most dominant at that time concept that the hormone-induced receptor activation is highly restricted to interactions primarily or exclusively within LRRs. In the light of new structures, the location and configuration of “signal-enhancing substitutions” in the activating complex is important and may not be altered, particularly considering much weaker binding of the free α-subunit analog than the hormone heterodimer. This explanation is also consistent with our data indicating that synthetic linear and cyclic peptides corresponding to the human α-subunit 11–20 residues with α4K substitutions and 10 μM concentrations are not inducing any significant cAMP responses (45).
Third, testing hTSH-WT in comparison to hTSH super-agonist TR1401 with the same α4K substitutions using mutated hTSHR in the hinge region, revealed that non-conservative substitutions E297, E303, and D382 are strongly reducing TR1401 binding and cAMP signaling based on TSHR cell surface expression normalization using FACS analysis (46). Two substitutions to the positively charged K residue (E297K and D382K) led to particularly strong decrease of TR1401 binding and cAMP signaling. Regardless of specific mechanism of electrostatic steering and repulsion involved, and considering well known limitations of simultaneous hormone and receptor mutagenesis (47–49), such studies indicated that the analogs with a limited number of “gain-of activity” substitutions can serve as an excellent tool for probing hormone–receptor interactions. Use of such minimally mutagenized human hTSH analogs (e.g., TR1401 with α4K) together with largely different bovine bTSH-WT (36 amino acid difference) combined with systematic receptor mutagenesis and TSHR expression normalization allowed to narrow interactions areas within the hinge region and SSSD (49, 50).
Fourth, heterothyrotropic activity of mammalian GPHs can be increased in goldfish by the same α4K substitutions and interactions within the SSSD and hinge region. Such interactions likely evolved long before diversification of LHR and TSHR receptors (51). Co-evolution of early GPHs and their receptors, driven by positive selection within the hormone-binding subdomain (HBSD), related to the need for adaptation of new functions, likely controlled “spillover” of hormone activity to other GPHRs.
Fifth, in addition to supergonists based on selective introduction of positively charged amino acid residues into the αL1 loop, additional substitutions to R or K in the αL3 and βL3 loops led to noticeable increases in the receptor binding both individually and in combination (21, 52–54). The oligomerization of GPHRs was observed in the past, but recent structures provided more convincing evidence for functional relevance of receptors oligomers, dimers, and trimers. An interaction of βL3 loop with three mutations to R (β3R) located in proximity to the FSHR trimer exosite, suggests potential role of such exosites for additional hTSH βL3 loop binding in trimeric receptors (36, 52, 55), which could be affected by the proportion of receptor trimers in different cells and in vitro bioassay conditions. Although, the trimeric FSH model does not precisely predicts the extend of cAMP increase due to signal amplification by the adenylate cyclase, it places again the location of both αL1 and αL3 loops near the TMD and predicts that upon dissociation of trimers into monomers both binding and signaling activities of glycosylated WT hFSH should increase threefold. Dual FSHR signaling by monomers and/or by trimers may serve as a part of evolutionarily based protection of reproductive functions, and constitute a possible compensatory mechanism preserving some minimum level of FSH induced signaling in various stress conditions affecting GnRH pulsatile secretion depressing pituitary hFSH production and secretion, observed during chronic malnutrition or starvation (56, 57). hFSHR and likely hLHR signaling by monomers or trimers may also modulate action of elevated endogenous gonadotropins during malignant cell transformation in the menopausal women and their ovarian epithelial and granulosa cells, later associated with the decreased FSHR expression, low receptor number in the cell membrane as well as altered receptor occupancy, trafficking, and biased signaling (58, 59).
Although, activation of GPHRs induces the coupling of different G proteins (60), most of physiological activities are mediated through a Gαs protein induced adenyl cyclase catalyzing the conversion of ATP into cAMP (1). However, as suggested by studies on TSHR deletions and others focusing on GPHRs signaling and trafficking, “biased agonism,” also referred to as “ligand directed signaling” is likely caused by a spectrum of different ligand–receptor complex conformations in combination with other cell-specific factors (1, 61–63). Recent studies indicated that different GPH glycoforms may have distinct effects on signaling and result in a biased agonism (64). Thus, it is expected that each GPH variant may have different and sometimes completely unique signaling pattern. However, there is also pharmacologically justified possibility that super-agonists are in general less capable of inducing multiple conformations and therefore much less biased (65). Selected super-agonists are known to have an extended receptor-residence time, which in turn may affect GPHR interactions with the cell adapter proteins, endosome signaling, and signal compartmentalization (61, 66–68).
In summary, new investigative strategies including “charge scanning and reversal mutagenesis,” “loss-of-function restoration by superagonist,” as well as “loss-of-superagonism” with mutated or truncated GPHRs constitute highly valuable tools in the structure-function studies both in the absence and presence of structural information (1, 46, 53, 69). We have first showed directly using large deletions that the ECD suppresses an inherent constitutive activity of the TMD of the human TSHR (14). Such an intrinsic property of the ECD acting on the TMD as a partial inverse agonist was recognized after introduction of the “driver hemagglutinin (HA) tag-sequence” at the N-terminus of the truncated constructs designed to improve and assess cell surface expression (14). We have demonstrated that the presence of such “driver sequence” in the TSHR and other GPHRs constructs with large ECD deletions in the ECD is absolutely necessary for an efficient cell surface expression (1, 14, 70). Similar “driver sequences” were subsequently used to express and normalize expression of various other GPHR constructs with several major ECD deletions, assess their interactions with analogs, and determine their constitutive activities in both cAMP and IP3 signaling pathways (1, 15, 62). Several newer studies analyzing the role of charged residues in the receptor hinge region attributed an inherent agonistic property to this domain and supported our early concept that the receptor charge clusters adjacent to the TMD are critical in a downstream signal transmission (49, 50, 71).
Development of GPH Biosuperiors and Other Related Molecules
“Biosuperiors” (biobetters or next-generation biological therapeutics) are defined as the second-generation products with substantial advantages over the originator molecules. Biosuperiors have the same receptor target and general mechanism of action as previously approved WT recombinant molecules but include structural changes and/or altered properties that result in an improvement in their clinical profile. GPH biosuperiors can be classified into three main categories: super-agonists, long-acting analogs, and WT molecules with optimized glycosylation, formulation, or delivery.
Long-acting bovine FSH super-agonists with much higher efficacy than all other products in veterinary markets have been developed at Trophogen Inc. and entered clinical trials for superovulation in cows and heifers (72). Human TSH and FSH super-agonists for diagnostic and therapeutic applications in the thyroid cancer of follicular origin and the treatment of infertility, including poor responders in the controlled ovarian stimulation (COS), respectively, have entered the late-phases of preclinical development (22, 24, 37, 73). Long-acting analog of WT hFSH (corifollitropin alfa; ElonvaR, Merck) has been approved in Europe for COS. It was constructed by fusion of the carboxy-terminal peptide of the β-subunit of hCG to the β-subunit of hFSH. Such additional 28 amino acid residues peptide containing 4-5 O-linked carbohydrate chains resulted in twofold increase in the FSH plasma half-life. Elonva® substituted for 7 days daily injection of the WT hormone in the standard COS procedure (74–76). Additional post-approval assessments are expected to determine its clinical convenience value and acceptance in the IVF market. Glyco-optimized highly sialylated WT FSH (FSH-GEX™) developed by Glycotope GmbH is based on its production in the human GT-5s cell line providing more optimal glycan structure pattern than standard CHO cell line (77). Similar efforts are now also directed to engineering CHO cells and generation of more homogenous FSH glycosylation, including “human-like” α2,6-terminal sialic acid linkages (78). Recent developments of various GPH biosimilars, improved formulations (79), injection frequency and convenience-focused preparations have not addressed several largely unmet needs for much higher efficacy-based biosuperiors (80, 81).
Clinical utility of thyrostimulin, GPH-related protein found in both vertebrates and invertebrates (82), still awaits full elucidation and rigorous assessment of specificity, selectivity and potential, if any, therapeutic benefits. Small molecules given orally may also lack sufficient specificity, but modified and/or minimized protein variants may provide sufficient balance between specificity, selectivity, half-life and convenience of enteral or topical administration (83). In contrast to large GPHs, there is largely incorrect perception that highly improved affinity of small molecules to GPHRs should result in parallel increase of their specificity. However, the specificity of small molecules is more relative to the degree of non-relevant binding than the strength of their interaction with specific receptor (84). These reservations aside, during the last 10 years, several new advances have been made in the development of GPHR small molecule ligands and allosteric modulators (85–87).
Closing Remarks and Future Perspectives
Glycoprotein hormone–glycoprotein hormone receptor structure–function research is evolving into new highly promising phase, which will likely culminate in elucidation of the entire active and inactive structure(s) of hormone–receptor complexes, including constitutively active receptors, entire receptors bound to super-agonists, antibodies and small molecule ligands. The advent of new optical techniques based on FRET and BRET sensors, as well as single-molecule microscopy, will allow more detailed analysis of real-time receptor activation and direct spatial assessment of signaling in the living cells. Such new optical techniques made already possible detection of TSHR signaling to cAMP after receptor internalization into endosomes (68, 88).
In analogy to human genome sequencing, full benefits of the structure–function achievements may not immediately translate into the new drugs and the third generation of GPH biosuperiors and theranostics. Further progress in the understanding of functional and therapeutic potential of signaling bias, receptor trimerization, trafficking, compartmentalization of signaling as well as detailed elucidation of the mechanism of super-agonists binding and signaling, should move this field to a new very exciting times of personalized drugs with predefined pharmacodynamics (PD), pharmacokinetics (PK), and signaling profiles (89).
Future third-generation recombinant protein biosuperiors will likely have even more advantages related to efficacy, potency and half-life, but also in relation to largely improved stability, formulation, bioavailability, and new methods of administration, eliminating the need for multiple injections (83, 90). New automated single-use sensor-based manufacturing technology platforms of biologics as well as largely improved purification and characterization methods should make a whole development process faster, safer, and more efficient, assuming necessary improvement and streamlining in the regulatory agencies, their flexibility, commitment to a case-by-case considerations and willingness to accept well justified unorthodox development strategies.
It is apparent to many biotechnology experts and market analysts that major biosuperiors, which are largely improved versions of the originator molecules, will be the next big opportunity in the entire field of biologics and GPCR protein ligands (80). It is predicted that biotech and pharma companies, well known for innovation and experience with the first-generation recombinant proteins and biosimilars, will be the best positioned to achieve early success with biosuperiors as well as with the biosuperior-based targeting conjugates and the nanoparticles with theranostic capabilities. New highly exciting frontier of precision medicine combining targeted and personalized interventions is already looming on the horizon. The sense of wonder, excitement, and anticipation of the future progress in the structure–function and novel drug design can be well expressed by Carl Sagan’s visionary quote: “Somewhere, something incredible is waiting to be known.”
Conflict of Interest Statement
The author is Executive Vice President, CSO, and Co-founder of Trophogen, Inc. discovering and developing biosuperior drugs for profit.
Acknowledgment
The author thanks Dr. Bruce D. Weintraub, President and CEO of Trophogen, Inc., for his visionary leadership, inspiration, and passion in the development of groundbreaking biosuperiors.
References
1. Szkudlinski MW, Fremont V, Ronin C, Weintraub BD. Thyroid-stimulating hormone and thyroid-stimulating hormone receptor structure-function relationships. Physiol Rev (2002) 82(2):473–502. doi:10.1152/physrev.00031.2001
2. Cahoreau C, Klett D, Combarnous Y. Structure-function relationships of glycoprotein hormones and their subunits’ ancestors. Front Endocrinol (2015) 6:26. doi:10.3389/fendo.2015.00026
3. Dias JA, Cohen BD, Lindau-Shepard B, Nechamen CA, Peterson AJ, Schmidt A. Molecular, structural, and cellular biology of follitropin and follitropin receptor. Vitam Horm (2002) 64:249–322. doi:10.1016/S0083-6729(02)64008-7
4. Roch GJ, Sherwood NM. Glycoprotein hormones and their receptors emerged at the origin of metazoans. Genome Biol Evol (2014) 6(6):1466–79. doi:10.1093/gbe/evu118
5. Taddese B, Upton GJ, Bailey GR, Jordan SR, Abdulla NY, Reeves PJ, et al. Do plants contain g protein-coupled receptors? Plant Physiol (2014) 164(1):287–307. doi:10.1104/pp.113.228874
6. Farid NR, Szkudlinski MW. Minireview: structural and functional evolution of the thyrotropin receptor. Endocrinology (2004) 145(9):4048–57. doi:10.1210/en.2004-0437
7. Moyle WR, Campbell RK, Myers RV, Bernard MP, Han Y, Wang X. Co-evolution of ligand-receptor pairs. Nature (1994) 368(6468):251–5. doi:10.1038/368251a0
8. Kudo M, Chen T, Nakabayashi K, Hsu SY, Hsueh AJ. The nematode leucine-rich repeat-containing, G protein-coupled receptor (LGR) protein homologous to vertebrate gonadotropin and thyrotropin receptors is constitutively active in mammalian cells. Mol Endocrinol (2000) 14(2):272–84. doi:10.1210/mend.14.2.0422
9. Ulloa-Aguirre A, Crépieux P, Poupon A, Maurel MC, Reiter E. Novel pathways in gonadotropin receptor signaling and biased agonism. Rev Endocr Metab Disord (2011) 12(4):259–74. doi:10.1007/s11154-011-9176-2
10. Landomiel F, Gallay N, Jégot G, Tranchant T, Durand G, Bourquard T, et al. Biased signalling in follicle stimulating hormone action. Mol Cell Endocrinol (2014) 382(1):452–9. doi:10.1016/j.mce.2013.09.035
11. Van Hiel MB, Vandersmissen HP, Proost P, Vanden Broeck J. Cloning, constitutive activity and expression profiling of two receptors related to relaxin receptors in Drosophila melanogaster. Peptides (2015) 68:83–90. doi:10.1016/j.peptides.2014.07.014
12. Van Hiel MB, Vandersmissen HP, Van Loy T, Vanden Broeck J. An evolutionary comparison of leucine-rich repeat containing G protein-coupled receptors reveals a novel LGR subtype. Peptides (2012) 34(1):193–200. doi:10.1016/j.peptides.2011.11.004
13. Van Loy T, Vandersmissen HP, Van Hiel MB, Poels J, Verlinden H, Badisco L, et al. Comparative genomics of leucine-rich repeats containing G protein-coupled receptors and their ligands. Gen Comp Endocrinol (2008) 155(1):14–21. doi:10.1016/j.ygcen.2007.06.022
14. Zhang M, Tong KP, Fremont V, Chen J, Narayan P, Puett D, et al. The extracellular domain suppresses constitutive activity of the transmembrane domain of the human TSH receptor: implications for hormone-receptor interaction and antagonist design. Endocrinology (2000) 141(9):3514–7. doi:10.1210/endo.141.9.7790
15. Vassart G, Pardo L, Costagliola S. A molecular dissection of the glycoprotein hormone receptors. Trends Biochem Sci (2004) 29(3):119–26. doi:10.1016/j.tibs.2004.01.006
16. Kreuchwig A, Kleinau G, Kreuchwig F, Worth CL, Krause G. Research resource: update and extension of a glycoprotein hormone receptors web application. Mol Endocrinol (2011) 25(4):707–12. doi:10.1210/me.2010-0510
17. Hershman JM. Physiological and pathological aspects of the effect of human chorionic gonadotropin on the thyroid. Best Pract Res Clin Endocrinol Metab (2004) 18(2):249–65. doi:10.1016/j.beem.2004.03.010
18. Szkudlinski MW, Grossmann M, Weintraub BD. Progress in understanding structure-function relationships of human thyroid-stimulating hormone. Curr Opin Endocrinol Diabetes (1997) 4(5):354–63. doi:10.1097/00060793-199710000-00007
19. Rees Smith B, McLachlan SM, Furmaniak J. Autoantibodies to the thyrotropin receptor. Endocr Rev (1988) 9(1):106–21. doi:10.1210/edrv-9-1-106
20. Wallin E, von Heijne G. Properties of N-terminal tails in G-protein coupled receptors: a statistical study. Protein Eng (1995) 8(7):693–8. doi:10.1093/protein/8.7.693
21. Szkudlinski MW, Teh NG, Grossmann M, Tropea JE, Weintraub BD. Engineering human glycoprotein hormone superactive analogues. Nat Biotechnol (1996) 14(10):1257–63. doi:10.1038/nbt1096-1257
22. Szkudlinski MW. Recombinant human thyrotropins of the twenty-first century. Expert Opin Pharmacother (2004) 5(12):2435–40. doi:10.1517/14656566.5.12.2435
24. Szkudlinski MW. Past, presence and future of thyroid-stimulating hormone (TSH) superactive analogs. Cancer Treat Res (2004) 122:345–56. doi:10.1007/1-4020-8107-3_19
25. Heikoop JC, Huisman-de Winkel B, Grootenhuis PD. Towards minimized gonadotropins with full bioactivity. Eur J Biochem (1999) 261(1):81–4. doi:10.1046/j.1432-1327.1999.00232.x
26. Setlur SR, Dighe RR. Single chain human chorionic gonadotropin, hCGalphabeta: effects of mutations in the alpha subunit on structure and bioactivity. Glycoconj J (2007) 24(1):97–106. doi:10.1007/s10719-006-9016-x
27. Grossmann M, Weintraub BD, Szkudlinski MW. Novel insights into the molecular mechanisms of human thyrotropin action: structural, physiological, and therapeutic implications for the glycoprotein hormone family. Endocr Rev (1997) 18(4):476–501. doi:10.1210/edrv.18.4.0305
28. Jiang X, Dreano M, Buckler DR, Cheng S, Ythier A, Wu H, et al. Structural predictions for the ligand-binding region of glycoprotein hormone receptors and the nature of hormone-receptor interactions. Structure (1995) 3(12):1341–53. doi:10.1016/S0969-2126(01)00272-6
29. Remy JJ, Couture L, Pantel J, Haertlé T, Rabesona H, Bozon V, et al. Mapping of HCG-receptor complexes. Mol Cell Endocrinol (1996) 125(1–2):79–91. doi:10.1016/S0303-7207(96)03955-X
30. Pantel J, Remy JJ, Salesse R, Jolivet A, Bidart JM. Unmasking of an immunoreactive site on the alpha subunit of human choriogonadotropin bound to the extracellular domain of its receptor. Biochem Biophys Res Commun (1993) 195(2):588–93. doi:10.1006/bbrc.1993.2086
31. Kenakin T. New concepts in drug discovery: collateral efficacy and permissive antagonism. Nat Rev Drug Discov (2005) 4(11):919–27. doi:10.1038/nrd1875
32. Sanders P, Young S, Sanders J, Kabelis K, Baker S, Sullivan A, et al. Crystal structure of the TSH receptor (TSHR) bound to a blocking-type TSHR autoantibody. J Mol Endocrinol (2011) 46(2):81–99. doi:10.1530/JME-10-0127
33. Sanders J, Miguel RN, Furmaniak J, Smith BR. TSH receptor monoclonal antibodies with agonist, antagonist, and inverse agonist activities. Methods Enzymol (2010) 485:393–420. doi:10.1016/B978-0-12-381296-4.00022-1
34. Fan QR, Hendrickson WA. Structure of human follicle-stimulating hormone in complex with its receptor. Nature (2005) 433(7023):269–77. doi:10.1038/nature03206
35. Jiang X, Liu H, Chen X, Chen PH, Fischer D, Sriraman V, et al. Structure of follicle-stimulating hormone in complex with the entire ectodomain of its receptor. Proc Natl Acad Sci U S A (2012) 109(31):12491–6. doi:10.1073/pnas.1206643109
36. Jiang X, Dias JA, He X. Structural biology of glycoprotein hormones and their receptors: insights to signaling. Mol Cell Endocrinol (2014) 382(1):424–51. doi:10.1016/j.mce.2013.08.021
37. Reinfelder J, Maschauer S, Foss CA, Nimmagadda S, Fremont V, Wolf V, et al. Effects of recombinant human thyroid-stimulating hormone superagonists on thyroidal uptake of 18F-fluorodeoxyglucose and radioiodide. Thyroid (2011) 21(7):783–92. doi:10.1089/thy.2010.0394
38. Núñez Miguel R, Sanders J, Chirgadze DY, Blundell TL, Furmaniak J, Rees Smith B. FSH and TSH binding to their respective receptors: similarities, differences and implication for glycoprotein hormone specificity. J Mol Endocrinol (2008) 41(3):145–64. doi:10.1677/JME-08-0040
39. Fan QR, Hendrickson WA. Assembly and structural characterization of an authentic complex between human follicle stimulating hormone and a hormone-binding ectodomain of its receptor. Mol Cell Endocrinol (2007) 26(0–262):73–82. doi:10.1016/j.mce.2005.12.055
40. Szkudlinski MW, Teh NG, Grossmann, M, Tropea JE, Witta, J, Weintraub BD, Role of the 40-51 region of the alpha-subunit in the bioactivity of human thyrotropin and gonadotropins: implications for the design of new hormone analogs based on simultaneous mutagenesis of multiple domains. Program & Abstracts, Abstract OR1-4. 78th Annual Endocrine Society Meeting. San Francisco, CA (1996).
41. Grossmann M, Szkudlinski MW, Wong R, Dias JA, Ji TH, Weintraub BD. Substitution of the seat-belt region of the thyroid-stimulating hormone (TSH) beta-subunit with the corresponding regions of choriogonadotropin or follitropin confers luteotropic but not follitropic activity to chimeric TSH. J Biol Chem (1997) 272(24):15532–40. doi:10.1074/jbc.272.24.15532
42. Bhowmick N, Huang J, Puett D, Isaacs NW, Lapthorn AJ. Determination of residues important in hormone binding to the extracellular domain of the luteinizing hormone/chorionic gonadotropin receptor by site-directed mutagenesis and modeling. Mol Endocrinol (1996) 10(9):1147–59. doi:10.1210/mend.10.9.8885249
43. Ulloa-Aguirre A, Zariñán T, Pasapera AM, Casas-González P, Dias JA. Multiple facets of follicle-stimulating hormone receptor function. Endocrine (2007) 32(3):251–63. doi:10.1007/s12020-008-9041-6
44. Angelova K, Fremont V, Jain R, Zhang M, Puett D, Narayan P, et al. Human alpha-subunit analogs act as partial agonists to the thyroid-stimulating hormone receptor: differential effects of free and yoked subunits. Endocrine (2004) 24(1):25–31. doi:10.1385/ENDO:24:1:025
45. Szkudlinski MW, Zhang M, Chen J, Tong KP, Leitolf H, Weintraub BD. Against the dogma: recombinant free alpha-subunit analogs with biological activity: implications for glycoprotein hormone minimization, evolution and hormone-receptor interaction. Program & Abstracts, Abstract – Hot Topic 9. 81th Annual Meeting of the Endocrine Society. San Diego, CA (1999).
46. Mueller S, Kleinau G, Szkudlinski MW, Jaeschke H, Krause G, Paschke R. The superagonistic activity of bovine thyroid-stimulating hormone (TSH) and the human TR1401 TSH analog is determined by specific amino acids in the hinge region of the human TSH receptor. J Biol Chem (2009) 284(24):16317–24. doi:10.1074/jbc.M109.005710
47. Ji I, Zeng H, Ji TH. Receptor activation of and signal generation by the lutropin/choriogonadotropin receptor. Cooperation of Asp397 of the receptor and alpha Lys91 of the hormone. J Biol Chem (1993) 268(31):22971–4.
48. Schaarschmidt J, Huth S, Meier R, Paschke R, Jaeschke H. Influence of the hinge region and its adjacent domains on binding and signaling patterns of the thyrotropin and follitropin receptor. PLoS One (2014) 9(10):e111570. doi:10.1371/journal.pone.0111570
49. Mueller S, Szkudlinski MW, Schaarschmidt J, Günther R, Paschke R, Jaeschke H. Identification of novel TSH interaction sites by systematic binding analysis of the TSHR hinge region. Endocrinology (2011) 152(8):3268–78. doi:10.1210/en.2011-0153
50. Kleinau G, Neumann S, Grüters A, Krude H, Biebermann H. Novel insights on thyroid-stimulating hormone receptor signal transduction. Endocr Rev (2013) 34(5):691–724. doi:10.1210/er.2012-1072
51. Miller TC, Jaques JT, Szkudlinski MW, Mackenzie DS. Thyrotropic activity of recombinant human glycoprotein hormone analogs and pituitary mammalian gonadotropins in goldfish (Carassius auratus): insights into the evolution of thyrotropin receptor specificity. Gen Comp Endocrinol (2012) 177(1):70–5. doi:10.1016/j.ygcen.2012.02.012
52. Grossmann M, Leitolf H, Weintraub BD, Szkudlinski MW. A rational design strategy for protein hormone superagonists. Nat Biotechnol (1998) 16(9):871–5. doi:10.1038/nbt0998-871
53. Leitolf H, Tong KP, Grossmann M, Weintraub BD, Szkudlinski MW. Bioengineering of human thyrotropin superactive analogs by site-directed “lysine-scanning” mutagenesis of the common βL3 loop of human glycoprotein hormones. Analysis of cooperative effects within and between peripheral β-hairpin loops. J Biol Chem (2000) 275(35):27457–65. doi:10.1074/jbc.M003707200
54. Szkudlinski MW, Leitolf H, Dong W, Grossmann M, Weintraub BD. Superagonists of human lutropin (hLH) and human chorionic gonadotropin (hCG) obtained by introduction of charged amino acids in the peripheral β hairpin loops. Program & Abstracts, Abstract OR28-2. 80th Annual Meeting of the Endocrine Society. New Orleans, LA (1998). 88 p.
55. Jiang X, Fischer D, Chen X, McKenna SD, Liu H, Sriraman V, et al. Evidence for follicle-stimulating hormone receptor as a functional trimer. J Biol Chem (2014) 289(20):14273–82. doi:10.1074/jbc.M114.549592
56. Campbell GA, Kurcz M, Marshall S, Meites J. Effects of starvation in rats on serum levels of follicle stimulating hormone, luteinizing hormone, thyrotropin, growth hormone and prolactin; response to LH-releasing hormone and thyrotropin-releasing hormone. Endocrinology (1977) 100(2):580–7. doi:10.1210/endo-100-2-580
57. Parrott JA, Doraiswamy V, Kim G, Mosher R, Skinner MK. Expression and actions of both the follicle stimulating hormone receptor and the luteinizing hormone receptor in normal ovarian surface epithelium and ovarian cancer. Mol Cell Endocrinol (2001) 172(1–2):213–22. doi:10.1016/S0303-7207(00)00340-3
58. Puett D, Angelova K, da Costa MR, Warrenfeltz SW, Fanelli F. The luteinizing hormone receptor: insights into structure-function relationships and hormone-receptor-mediated changes in gene expression in ovarian cancer cells. Mol Cell Endocrinol (2010) 329(1–2):47–55. doi:10.1016/j.mce.2010.04.025
59. Mertens-Walker I, Baxter RC, Marsh DJ. Gonadotropin signalling in epithelial ovarian cancer. Cancer Lett (2012) 324(2):152–9. doi:10.1016/j.canlet.2012.05.017
60. Laugwitz KL, Allgeier A, Offermanns S, Spicher K, Van Sande J, Dumont JE, et al. The human thyrotropin receptor: a heptahelical receptor capable of stimulating members of all four G protein families. Proc Natl Acad Sci U S A (1996) 93(1):116–20. doi:10.1073/pnas.93.1.116
61. Ulloa-Aguirre A, Dias JA, Bousfield G, Huhtaniemi I, Reiter E. Trafficking of the follitropin receptor. Methods Enzymol (2013) 521:17–45. doi:10.1016/B978-0-12-391862-8.00002-8
62. Fremont V, Zhang M, Weintraub BD, Szkudlinski MW. Novel insights into the molecular mechanism of glycoprotein hormone receptor activation. FASEB J (2001) 15(4):A175.
63. Nechamen CA, Thomas RM, Dias JA. APPL1, APPL2, Akt2 and FOXO1a interact with FSHR in a potential signaling complex. Mol Cell Endocrinol (2007) 260–262:93–9. doi:10.1016/j.mce.2006.08.014
64. Arey BJ, Lopez FJ. Are circulating gonadotropin isoforms naturally occurring biased agonists? Basic and therapeutic implications. Rev Endocr Metab Disord (2011) 12(4):275–88. doi:10.1007/s11154-011-9188-y
65. Langmead CJ, Christopoulos A. Supra-physiological efficacy at GPCRs: superstition or super agonists? Br J Pharmacol (2013) 169(2):353–6. doi:10.1111/bph.12142
66. Szkudlinski MW. Challenges and opportunities of trapping ligands. Mol Pharmacol (2007) 72(2):231–4. doi:10.1124/mol.107.038208
67. Dias JA, Mahale SD, Nechamen CA, Davydenko O, Thomas RM, Ulloa-Aguirre A. Emerging roles for the FSH receptor adapter protein APPL1 and overlap of a putative 14-3-3tau interaction domain with a canonical G-protein interaction site. Mol Cell Endocrinol (2010) 329(1–2):17–25. doi:10.1016/j.mce.2010.05.009
68. Calebiro D, Nikolaev VO, Lohse MJ. Imaging of persistent cAMP signaling by internalized G protein-coupled receptors. J Mol Endocrinol (2010) 45(1):1–8. doi:10.1677/JME-10-0014
69. Szkudlinski MW, Grossmann M, Leitolf H, Weintraub BD. Human thyroid-stimulating hormone: structure-function analysis. Methods (2000) 21(1):67–81. doi:10.1006/meth.2000.0976
70. Vlaeminck-Guillem V, Ho SC, Rodien P, Vassart G, Costagliola S. Activation of the cAMP pathway by the TSH receptor involves switching of the ectodomain from a tethered inverse agonist to an agonist. Mol Endocrinol (2002) 16(4):736–46. doi:10.1210/mend.16.4.0816
71. Chen CR, Salazar LM, McLachlan SM, Rapoport B. The thyrotropin receptor hinge region as a surrogate ligand: identification of loci contributing to the coupling of thyrotropin binding and receptor activation. Endocrinology (2012) 153(10):5058–67. doi:10.1210/en.2012-1376
72. Szkudlinski MW, Fremont V, Wolf V, Han Y, Wu D, Weintraub BD. Novel high affinity and long-acting recombinant bovine FSH analogs for veterinary superovulation. Program & Abstracts, Abstract 16. 18th Biennial AAVPT Symposium. Potomac, MD (2013).
73. Galli F, Manni I, Piaggio G, Balogh L, Weintraub BD, Szkudlinski MW, et al. (99m)Tc-labeled-rhTSH analogue (TR1401) for imaging poorly differentiated metastatic thyroid cancer. Thyroid (2014) 24(8):1297–308. doi:10.1089/thy.2013.0429
74. Boime I, Ben-Menahem D. Glycoprotein hormone structure-function and analog design. Recent Prog Horm Res (1999) 54:271–88.
75. Pouwer AW, Farquhar C, Kremer JA. Long-acting FSH versus daily FSH for women undergoing assisted reproduction. Cochrane Database Syst Rev (2012) 6:CD009577. doi:10.1002/14651858.CD009577.pub2
76. Croxtall JD, McKeage K. Corifollitropin alfa: a review of its use in controlled ovarian stimulation for assisted reproduction. BioDrugs (2011) 25(4):243–54. doi:10.2165/11206890-000000000-00000
77. Goletz S, Stockl L. Recombinant Human Follicle-Stimulating Hormone. USPTO US Patent Application 201301376636 (2013).
78. Yang Z, Wang S, Halim A, Schulz MA, Frodin M, Rahman SH, et al. Engineered CHO cells for production of diverse, homogeneous glycoproteins. Nat Biotechnol (2015) 33(8):842–4. doi:10.1038/nbt.3280
79. Clark E, Magner J, Skell J. Formulations for Therapeutic Administration of Thyroid Stimulating Hormone (TSH). USPTO, US Patent Application WO2008036271 (2008).
81. Ubaldi F, Vaiarelli A, D’Anna R, Rienzi L. Management of poor responders in IVF: is there anything new? Biomed Res Int (2014) 2014:352098. doi:10.1155/2014/352098
82. Bassett JH, van der Spek A, Logan JG, Gogakos A, Bagchi-Chakraborty J, Murphy E, et al. Thyrostimulin regulates osteoblastic bone formation during early skeletal development. Endocrinology (2015) 156(9):3098–113. doi:10.1210/en.2014-1943
83. Low SC, Nunes SL, Bitonti AJ, Dumont JA. Oral and pulmonary delivery of FSH-Fc fusion proteins via neonatal Fc receptor-mediated transcytosis. Hum Reprod (2005) 20(7):1805–13. doi:10.1093/humrep/deh896
84. Vance D, Martin J, Patke S, Kane RS. The design of polyvalent scaffolds for targeted delivery. Adv Drug Deliv Rev (2009) 61(11):931–9. doi:10.1016/j.addr.2009.06.002
85. Gershengorn MC, Neumann S. Update in TSH receptor agonists and antagonists. J Clin Endocrinol Metab (2012) 97(12):4287–92. doi:10.1210/jc.2012-3080
86. Palmer SS, McKenna S, Arkinstall S. Discovery of new molecules for future treatment of infertility. Reprod Biomed Online (2005) 10(Suppl 3):45–54. doi:10.1016/S1472-6483(11)60390-8
87. Kenakin TP. Biased signalling and allosteric machines: new vistas and challenges for drug discovery. Br J Pharmacol (2012) 165(6):1659–69. doi:10.1111/j.1476-5381.2011.01749.x
88. Calebiro D, Sungkaworn T, Maiellaro I. Real-time monitoring of GPCR/cAMP signalling by FRET and single-molecule microscopy. Horm Metab Res (2014) 46(12):827–32. doi:10.1055/s-0034-1384523
89. Kenakin T. A Pharmacology Primer: Theory, Applications, and Methods. London: Academic Press (2010).
Keywords: glycoprotein hormone, glycoprotein hormone receptor, structure–function, protein engineering, charge cluster, super–agonist, biosuperior, biobetter
Citation: Szkudlinski MW (2015) New frontier in glycoprotein hormones and their receptors structure–function. Front. Endocrinol. 6:155. doi: 10.3389/fendo.2015.00155
Received: 30 June 2015; Accepted: 18 September 2015;
Published: 19 October 2015
Edited by:
James A. Dias, University at Albany State University of New York, USAReviewed by:
Emma June Petrie, The Walter and Eliza Hall Institute of Medical Research, AustraliaLeigh Stoddart, University of Nottingham, UK
Copyright: © 2015 Szkudlinski. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Mariusz W. Szkudlinski, Trophogen Inc., 9714 Medical Center Drive, Rockville, MD, USA,bXN6a3VkbGluc2tpQHRyb3Bob2dlbi5jb20=