 Craig Porter
Craig Porter Maria Chondronikola
Maria Chondronikola Labros S. Sidossis
Labros S. Sidossis- 1Metabolism Unit, Shriners Hospitals for Children-Galveston, Galveston, TX, USA
- 2Department of Surgery, University of Texas Medical Branch, Galveston, TX, USA
- 3Department of Preventive Medicine and Community Health, University of Texas Medical Branch, Galveston, TX, USA
- 4Department of Nutrition and Dietetics, Harokopio University of Athens, Athens, Greece
- 5Department of Internal Medicine, University of Texas Medical Branch, Galveston, TX, USA
Obesity and its metabolic consequences represent a significant clinical problem. From a thermodynamic standpoint, obesity results from a discord in energy intake and expenditure. To date, lifestyle interventions based on reducing energy intake and/or increasing energy expenditure have proved ineffective in the prevention and/or treatment of obesity, owing to poor long-term adherence to such interventions. Thus, an effective strategy to prevent or correct obesity is currently lacking. As the combustion engines of our cells, mitochondria play a critical role in energy expenditure. At a whole-body level, approximately 80% of mitochondrial membrane potential generated by fuel oxidation is used to produce ATP, and the remaining 20% is lost through heat-producing uncoupling reactions. The coupling of mitochondrial respiration to ATP production represents an important component in whole-body energy expenditure. Brown adipose tissue (BAT) is densely populated with mitochondria containing the inner mitochondrial proton carrier uncoupling protein 1 (UCP1). UCP1 uncouples oxidative phosphorylation, meaning that mitochondrial membrane potential is dissipated as heat. The recent rediscovery of BAT depots in adult humans has rekindled scientific interest in the manipulation of mitochondrial uncoupling reactions as a means to increase metabolic rate, thereby counteracting obesity and its associated metabolic phenotype. In this article, we discuss the evidence for the role BAT plays in metabolic rate and glucose and lipid metabolism in humans and the potential for UCP1 recruitment in the white adipose tissue of humans. While the future holds much promise for a therapeutic role of UCP1 expressing adipocytes in human energy metabolism, particularly in the context of obesity, tissue-specific strategies that activate or recruit UCP1 in human adipocytes represent an obligatory translational step for this early promise to be realized.
Introduction
Obesity and its associated metabolic complications (hyperlipidemia, insulin resistance, and glucose intolerance) have become significantly more prevalent in the United States and around the world in recent years (1, 2). One third of Americans are either obese (1) or exhibit symptoms of pre-diabetes (3). Worldwide, excessive adiposity is responsible for more than three million deaths and is a significant cause of disability (4). Increasing adiposity is associated with derangements in glucose and lipid metabolism and in the development of insulin resistance (5, 6). Therapeutic strategies aimed at preventing obesity or treating its metabolic complications typically target dietary intake and/or physical activity, but to date do not seem to exhibit a great deal of long-term efficacy. Mitochondrial proton leaks, i.e., respiration uncoupled from ATP production, accounts for around 20% of total mitochondrial respiration (7). The physiological role ascribed to this uncoupling of oxidative phosphorylation is heat production (thermogenesis). While thermogenesis represents a significant portion (~20%) of whole-body energy expenditure (7, 8), its therapeutic role in energy metabolism has only recently begun to receive attention.
Brown adipose tissue (BAT) is the major tissue responsible for non-shivering thermogenesis in mammals (9). The recent (re)discovery of BAT in human adults (10–12) has triggered scientific interest in the role of BAT in human energy metabolism. Unique features of BAT include an abundance of mitochondria that contain the transmembrane carrier protein thermogenin, most commonly referred to as uncoupling protein 1 (UCP1) (13). Upon activation by long-chain fatty acids, UCP1 acts as an inner mitochondrial proton carrier (14), allowing protons to reenter the mitochondrial matrix independently of ATP synthase, thus uncoupling mitochondrial respiration from ATP production (9). This process alters mitochondrial energy transduction, where mitochondrial membrane potential is lost as heat (Figure 1). The purpose of this review is to summarize the current evidence regarding the role of the human adipose tissue as a therapeutic target against obesity and its related metabolic complications.
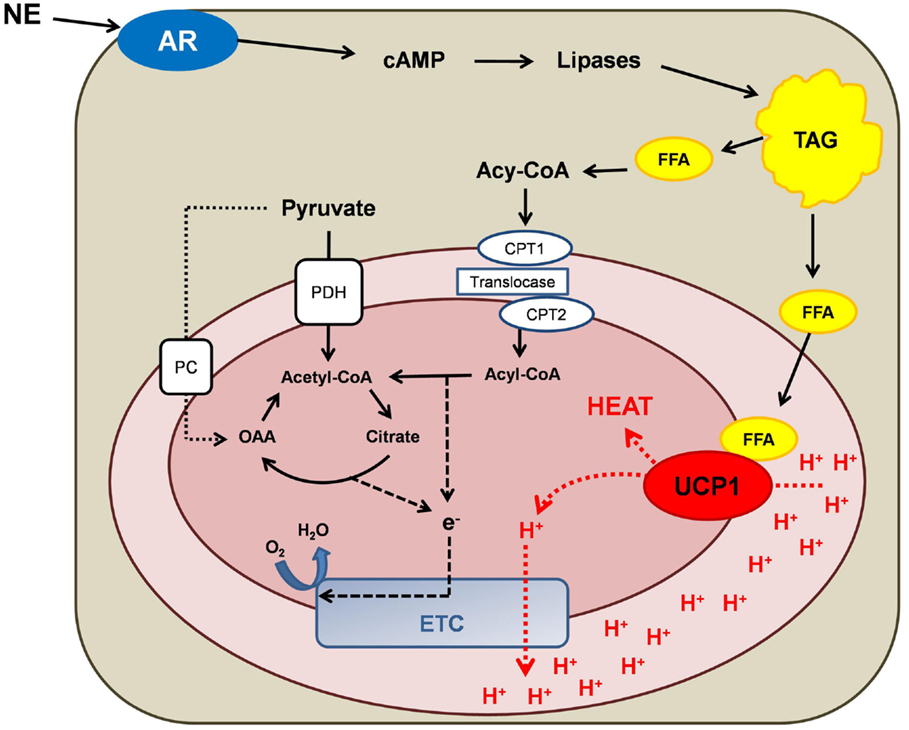
Figure 1. Schematic overview of mitochondrial energy transduction within a brown adipocyte mitochondrion. Norepinephrine (NE) activation of adrenergic receptors (AR) causes an increase in cytosolic cyclic adenosine monophosphate (cAMP) levels. The subsequent activation of lipases results in the lipolysis of triacylglycerol (TAG), which increases intracellular free fatty acid (FFA) concentrations. FFAs fulfill two principal metabolic fates: (i) FFAs bind to and activate uncoupling protein 1 (UCP1), thus switching on UCP1-mediated proton (H+) conductance. (ii) Activated FFAs (acyl-CoA) can be transported into the mitochondrion via the carnitine palmitoyl transferase (CPT) system and be oxidized to acetyl-CoA, thereby potentiating anaplerosis and providing reducing equivalents for the electron transport chain (ETC). Pyruvate also participates in mitochondrial anaplerosis and the production of reducing equivalent by being decarboxylated to acetyl-CoA by pyruvate dehydrogenase (PDH) or being carboxylated by pyruvate carboxylase (PC), forming oxaloacetate (OAA).
The Role of Brown Adipose Tissue in Energy Balance and Obesity
Although the role of BAT in energy metabolism and obesity has been studied in detail in rodents (15–17), our understanding of the role BAT plays in human energy metabolism remains in its infancy. For obvious reasons, it is more difficult to manipulate and study BAT in humans. Retrospective reviews of medical records and positron emission tomography–computed tomography (PET/CT) scans, originally performed for diagnostic purposes, demonstrate an association between BAT and body mass index (BMI) (10, 18, 19) and with non-alcoholic fatty liver disease (20). The retrospective nature of these studies along with the fact that the PET/CT scans were performed under non-standardized conditions limit the generalizability of these data. However, these findings are bolstered by prospective studies which report a correlation between BAT volume and activity with BMI (11, 21), total fat mass (11, 21), and both abdominal subcutaneous and visceral adiposity (21). Moreover, weight loss via bariatric surgery has been linked to increased BAT activity (22), further suggesting that BAT may contribute to energy balance in human beings.
Acute (non-shivering) cold exposure studies have been recently performed in an attempt to estimate the contribution of BAT to cold-induced energy expenditure. However, the reported results have been highly variable. These studies reported that acute cold exposure (2–4 h at 16–19°C) induces BAT activation, resulting in a 13–27% increase in resting energy expenditure (REE) (11, 23, 24). In contrast, Ouellet et al. reported an 80% increase in REE with acute cold exposure (for 2 h at ~18°C) (25), a result over threefold higher than those of other studies, possibly attributable to muscle shivering. Muzik et al. used oxygen 15 (15O)-PET/CT in an attempt to more directly quantify the contribution of BAT to energy expenditure (26, 27). In contrast to previous studies, these investigators found that BAT minimally contributed to the reported increase in energy expenditure (15–25 kcal/day). Although these two studies shed doubt on the role of BAT in energy expenditure in humans, the rather short cold stimulation protocol (30 min at 18°C) may not have been adequate to fully activate BAT. Indeed, BAT recruitment via daily mild cold exposure (17°C for 6 weeks) was associated with increased thermogenesis and decreased body fat, supporting a relationship between BAT activity and adiposity (28). Similarly, Lee et al. performed a crossover cold acclimation study in five healthy participants. These researchers showed that BAT recruitment was reversibly associated with increased post-prandial energy expenditure (diet-induced thermogenesis) after cold exposure, further supporting the role of BAT in energy expenditure (29).
When interpreting the results of these studies, one important point to consider is that chronic weight gain can result from even a small discordance in daily energy balance (e.g., an energy surplus of as little as 25 kcal/day in humans can result in a weight gain of 1 kg/year). Thus, even sporadic BAT activation for limited periods during the course of the day could conceivably have a significant cumulative impact on energy balance and adiposity over several months or years.
Can Human Brown Adipose Tissue Alter Glucose Homeostasis and Insulin Sensitivity?
Brown adipose tissue has been touted as therapeutic tissue which may protect against hyperglycemia in insulin-resistant individuals. In murine models of extreme cold exposure, BAT is responsible for the majority of whole-body glucose disposal (30), while transplantation of BAT into the abdominal visceral adipose tissue improves glucose tolerance and insulin sensitivity (31). In humans, the involvement of BAT in systemic glucose metabolism is evident from 18F-FDG uptake images from PET/CT scans (Figure 2) performed for diagnostic purposes (32). Moreover, evidence from retrospective medical record review studies indicates an inverse relationship between BAT activity, diabetes, and glycemia (10, 19, 33, 34). Further, prospective studies have shown that acute cold exposure increases glucose uptake in BAT, where BAT glucose uptake rate per unit of tissue was higher than that of muscle (25, 35, 36), underscoring the oxidative potential of BAT. Specifically, Orava et al. reported that cold exposure resulted in a 12-fold increase in glucose disposal in BAT only, but not in other tissues (35). Similar results have been reported by Ouellet et al. (25). These data suggest that upon activation, BAT clears plasma glucose. However, Ouellet et al. demonstrated that BAT minimally contributed to whole-body plasma glucose utilization, which might be due to the short duration of cold exposure (3 h at ~18°C) and the presence of mild shivering during cold exposure (25).
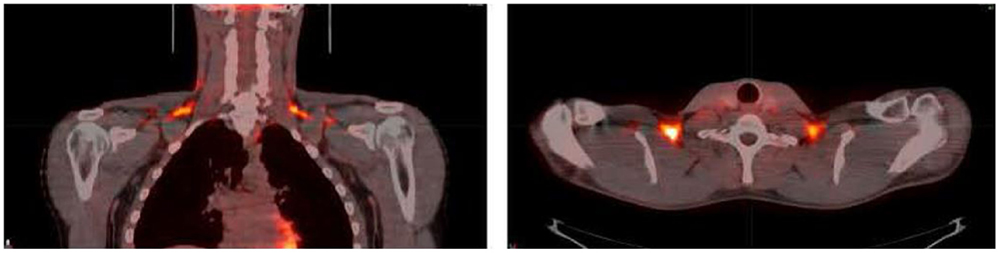
Figure 2. Coronal (left) and transverse (right) 2-deoxy-2-[18F]fluoro-d-glucose (18F-FDG) positron emission tomography (PET)-computed tomography (CT) images from a volunteer during cold exposure. The intense orange color in the supraclavicular area corresponds to brown adipose tissue. Image from Ref. (24). Reprinted with permission.
The opposing results of the aforementioned studies question the physiological significance of human BAT in systemic glucose metabolism. In an attempt to address this, we studied glucose metabolism in individuals with BAT (BAT+) or without BAT (BAT−) under cold exposure and thermoneutral conditions by infusing isotopically labeled glucose in the fasted state and during a hyperinsulinemic-euglycemic clamp. We found that cold exposure increased whole-body glucose disposal in the fasted and insulin-stimulated state only in BAT+ individuals (24), suggesting that BAT can indeed play a significant role in systemic glucose metabolism in humans. Theoretically, if BAT remained chronically active, it could dispose ~23 g of glucose in 24 h [equating to approximately 8.4 kg of glucose (33,600 kcal) in a year]. Similarly, Lee et al. recently reported that increased BAT activity via cold acclimation improved post-prandial glycemia (29). Collectively, these studies support the notion that upon activation, BAT can indeed play a significant role in glucose homeostasis. Therefore, BAT may represent a novel therapeutic target in the management of hyperglycemia.
Does Human Brown Adipose Tissue Play a Role in Systemic Lipid Metabolism?
Free fatty acids (FFAs) constitute the primary substrate for BAT (37). Moreover, FFAs bind to and activate UCP1, allowing proton transfer from the intermembrane space to the mitochondrial matrix resulting in heat production (9). Upon activation, BAT initially oxidizes intracellular lipid stores to produce heat (9). In contrast, prolonged BAT stimulation leads to increased uptake of FFA derived from circulating FFA and lipolysis of circulating lipoproteins, presumably when local lipid stores become depleted (9). BAT activation has been recently reported to increase the systemic clearance of TG-rich lipoproteins in rodents, lending credence to the notion that BAT may protect against hyperlipidemia (30, 38). Moreover, rodent BAT has been shown to release factors that increase vascular permeability in BAT, allowing TG-rich lipoproteins to enter the interstitial space (30).
The role of BAT in human lipid metabolism remains largely unexplored. Results from some (11, 35), but not all (23), cold exposure studies report a reduction in the respiratory gas exchange ratio during cold exposure, indicating increased fat oxidation in humans during BAT activation. Ouellet et al. (25) studied the role of BAT in lipid metabolism in more detail. Using PET/CT and a fatty acid tracer, 18F-fluoro-thiaheptadecanoic acid (18FTHA), they showed that 3 h of cold exposure increased BAT FFA uptake but that this accounted for <1% of the total FFA turnover (25). The conclusion of this study was that thermogenic BAT relies predominantly on the limited intracellular substrates (i.e., triglycerides stored inside BAT) to fuel mitochondrial thermogenesis (25), which questions the physiological significance of human BAT in whole-body lipid metabolism. However, the relatively short duration of the study might have been insufficient to deplete the intracellular lipid stores underestimating the contribution of BAT in the systemic lipid metabolism.
We recently studied BAT+ and BAT− individuals under mild cold exposure and thermoneutral conditions while infusing stable isotopes of glycerol and palmitate in order to directly trace lipid turnover in the fasted state and during a hyperinsulinemic-euglycemic clamp. We found that during cold exposure BAT+ individuals had greater whole-body lipolysis and FFA oxidation compared with BAT− participants, indicating that increased lipid mobilization from WAT provides FFA to sustain thermogenesis in BAT (39). Moreover, BAT+ individuals were protected from the cold-induced adipose tissue insulin resistance noted in the BAT− group (40). Our data suggest that BAT may indeed play an important role in the regulation of systemic lipid metabolism. The discord between our preliminary findings and those of others (25) may be related to the duration of cold exposure. For example, we studied individuals after ~5 h of cold exposure, while Ouellet et al. performed a 3 h cold exposure study. Whether there is a time-dependent depletion of BAT lipid stores, at which point BAT switches to using systemic FFAs as a fuel source, remains unknown.
Can Human WAT Really Brown?
More than 30 years ago, Young et al. demonstrated the appearance of brown adipocytes in the parametrial fat pad of mice following acclimation to severe cold exposure (41). This was confirmed when Cousin et al. demonstrated that severe cold exposure or treatment with a β3 adrenoreceptor agonist induced brown adipocytes within WAT (42). More recently, Shabalina et al. demonstrated that acclimation to cold (~5°) results in the browning of inguinal WAT. Importantly, these researchers showed the presence of UCP1 protein while demonstrating its function (via GDP-sensitive respiration) in inguinal WAT of cold-acclimated mice (43), thus demonstrating that these mitochondria had functional UCP1. Interestingly, per milligram of tissue, inguinal WAT from cold-acclimated mice developed a thermogenic capacity ~50% of that of intrascapular BAT (43). This striking plasticity of murine subcutaneous WAT to develop a thermogenic phenotype has potential implications for humans, given that most individuals have only 100 g or less of BAT, but many kilograms of subcutaneous WAT. If WAT in humans were able to exhibit the same plasticity as in mice, this would have huge implications for energy expenditure and substrate (glucose, lipids, and potentially amino acid) metabolism.
Unlike the inguinal subcutaneous WAT depot of mice, there is no evidence to suggest that human WAT has a comparable ability to alter its thermogenic capacity in response to an environmental stress. Indeed, chronic (10 days), albeit mild cold exposure, which activates BAT, does not alter the phenotype of WAT in humans (44). This suggests that a more profound and/or chronic adrenergic stress is likely needed to induce browning in humans. Indeed, this is perhaps reasonable considering that classic rodent models of browning typically acclimate rodents to extremely cold temperatures (~5°C) for several days, if not weeks (41–43). Considering that a thermoneutral temperature for a mouse is somewhere close to 30°C, it would be reasonable to theorize that chronic exposure to a temperature 25°C below thermoneutrality would be required to induce browning of WAT in humans.
Since cold exposure protocols used in rodents cannot be replicated in humans, researchers have looked to pathologies associated with chronic adrenergic stress. Over 30 years ago, it was reported that patients with pheochromocytoma, a tumor of the adrenal medulla which results in a chronic elevation in catecholamine secretion, had BAT surrounding their kidneys (45). More recently, Frontini et al. (46) reported the existence of multilocular adipocytes that stained positive for UCP1 within the omental adipose tissue of pheochromocytoma patients. This evidence suggests that human WAT may contain brown adipocytes, but unfortunately only histological and genomic measurements were made on these WAT samples. Whether these omental adipocytes, which showed morphological similarities to that of brown adipocyte and immune-reactivity to UCP1, had functionally thermogenic mitochondria was not addressed.
Burn trauma represents a unique injury model as it is accompanied by prolonged adrenergic stress and a hypermetabolic state. To determine if subcutaneous WAT can acquire a brown fat phenotype, we collected biopsy samples of WAT from severely burned individuals. We found that when prospectively following patients after burn injury, UCP1 mRNA and UCP1 protein were induced in subcutaneous WAT at ~2 weeks post-injury (47). Moreover, this was accompanied by a similar time-dependent increase in mitochondria respiratory capacity and whole-body metabolic rate (47). Thus, it appears that human subcutaneous WAT has the ability to brown. In the context of burn trauma, this makes physiological sense, since following the destruction of their skin barrier burn victims have difficulty in conserving heat and maintaining core temperature.
To the best of our knowledge, our recent data provide the first evidence of browning of WAT in humans (47). However, we should note that while we did indeed see induction of UCP1, morphological adaptations, and increased mitochondrial respiratory in permeabilized WAT samples in a time-dependent manner after burn, the increase in thermogenic capacity (~threefold) was rather modest when compared with those seen in rodent WAT after chronic cold exposure. For example, Shabalina et al. (43) showed that the mitochondrial respiratory capacity of mitochondria isolated from the BAT of mice kept at 30°C was ~10-fold greater than that of mitochondria isolated from inguinal WAT. However, when acclimated to 5°C, the respiratory capacity of BAT mitochondria was only ~1.7-fold greater than that of inguinal WAT mitochondria. Indeed, BAT mitochondrial respiratory capacity was only marginally greater in cold-acclimated mice compared with thermoneutral mice. In contrast, WAT mitochondrial respiratory capacity was ~500% greater in inguinal WAT of cold-acclimated mice compared with thermoneutral animals. This remarkable plasticity, where murine inguinal WAT can adopt a thermogenic capacity akin to that of BAT, does not appear to be conserved in other mammals such as humans. Indeed, while we saw a threefold increase in subcutaneous WAT respiratory capacity after burn injury, respiration per milligram of tissue was still ~20-fold lower than what we have measured in human intrascapular BAT (47).
The Therapeutic Potential of UCP1-Positive Mitochondria: Should we Activate BAT or Brown WAT?
Much emphasis has been placed on manipulating UCP1 in order to increase thermogenesis and thus energy expenditure, which over time would reduce adiposity and consequently correct metabolic abnormalities associated with obesity. Broadly speaking, manipulation of UCP1 can be classified into two distinct ways: (i) acute activation of existing UCP1 and (ii) induction of UCP1. Induction of UCP1 can take several forms, i.e., the expansion of existing BAT depots, producing more UCP1 per gram of BAT, or inducing UCP1 within WAT (browning). However, the significance of either activating UCP1 within classical BAT depots or browning WAT in humans in terms of energy expenditure remains unclear. Using available data on organ-specific metabolic rates, and our own data on (a) the metabolic rate of human BAT following acute cold exposure and (b) human WAT that has undergone browning, we present theoretical calculations to estimate the potential impact of UCP1 on energy expenditure in humans (Tables 1 and 2).
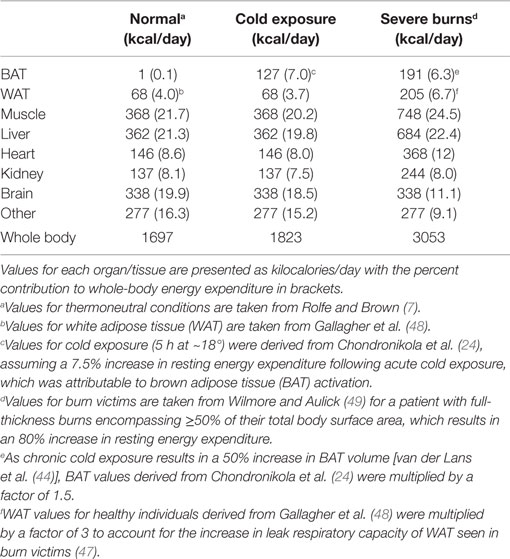
Table 1. Impact of acute non-shivering cold exposure and severe burns on whole-body energy expenditure.
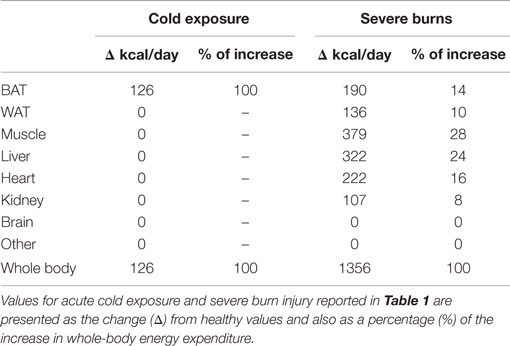
Table 2. The contribution of various tissues to the increase in whole-body metabolic rate accompanying acute non-shivering cold exposure and severe burns.
Since UCP1 is inactive in the presence of inhibitory purine nucleotides such as ATP (50), BAT makes a negligible contribution to whole-body REE given that ~100 g of BAT represents ~0.1% of total body mass of a 70 kg individual. Similarly, in an individual with ~20% total fat mass, WAT accounts for a small (~4%) portion of whole-body REE (48) (see Table 1). Following acute (5 h) acclimation to mild cold (5 h at ~18°), our data (24) suggest that on average there is a 7.5% increase in REE. This acute intervention transforms BAT from being a quiescent tissue to one that makes a comparable contribution to whole-body REE as the heart or the kidneys (Table 1). In a 70 kg individual, this activation of BAT increases REE by 127 kcal (Table 1), which if maintained for 30 days would combust 0.5 kg of adipose tissue.
Burn trauma results in a significant increase in REE (51), which persists for months if not years post injury (52). While many ATP-consuming processes increase after burn, increased ATP turnover only explains 50–60% of burn-induced hypermetabolism (53). This means that thermogenesis represents a significant portion of hypermetabolism in burn victims, which makes sense since these patients have a compromised skin barrier and inability to thermoregulate. In order to estimate the potential contribution of BAT used burn injury as a model of severe adrenergic stress which is accompanied by hypermetabolism. We made calculations of whole-body and tissue-specific energy expenditure based on a patient with a burn covering 50% of their total body surface area, which resulted in an 80% increase in REE (49).
While the impact of burn trauma on BAT is largely unknown, we theorized that like chronic intermittent cold exposure, prolonged adrenergic stress following severe burn trauma would expand BAT depots. Assuming an ~50% increase in BAT volume as seen with 10 days of mild cold exposure (44), BAT would account for 191 kcal of total REE (Table 1). Here, while BAT makes a greater contribution to REE in absolute terms, since burn injury results in a large increase in REE, the relative contribution of BAT to whole-body REE is slightly lower (6.3%) when compared to cold exposure (7%) (Table 1). Perhaps more strikingly, when considering WATs contribution to REE in burn victims, the threefold increase in thermogenic capacity we found in subcutaneous WAT of burn victims would increase WATs contribution to whole-body REE to ~7% (Table 1). Thus, WAT and BAT may make similar absolute and relative contributions to whole-body REE in burn survivors. Again though, we should point out that these calculations are based on an individual with around 100 g of BAT and 15 kg of WAT, underscoring the profound difference in thermogenic potential per gram of tissue between BAT and WAT.
While the chronic adrenergic stress accompanying severe burn trauma provides a unique model where human WAT undergoes browning, it also offers significant insight into the physiological impact of achieving UCP1 activation and recruitment. In Table 2, we present the values reported in Table 1 as the change from normal and also as a percentage in which each tissue contributes to the increase in whole-body REE. While the 127 kcal increase in REE following acute cold exposure is attributable to BAT, several organs contribute to the massive (1230 kcal) increase in REE following burn severe trauma. For example, while burn trauma may result in large absolute increases in BAT and WAT metabolic rates, their relative contributions to total REE are less impressive. Indeed, skeletal muscle (28%), the liver (24%), and the heart (16%) contribute most to burn-induced hypermetabolism, followed by BAT (14%) and WAT (10%) (Table 2).
We make the point above to underscore the fact that while the chronic stress response to burns may augment BAT and WAT thermogenesis, the systemic milieu which brings these adaptations in adipose tissue energy expenditure likely impacts many other organs. While our data on burn victims (47) provide novel evidence that human WAT can indeed undergo browning under very severe and extended adrenergic stress, this extreme pathophysiological state has an impact on nearly every organ system in the body. Thus, with all the available evidence to date, we suggest that the therapeutic potential of UCP1-positive mitochondria in combating obesity and its metabolic complications will likely be realized in humans by the acute activation of existing BAT and/or the expansion of BAT depots. If WAT browning holds any therapeutic potential in the context of obesity, the development of safe tissue-specific agents to induce browning is needed before this potential can be realized.
Summary
Adipocytes with an abundance of UCP1-positive mitochondria are unique in that gram for gram they have a respiratory capacity akin to that of muscle or liver, yet only minimal ability to produce ATP. The physiological role ascribed to this adaptation in mitochondrial energy transduction is heat production, facilitating thermoregulation in mammals. In the context of the global obesity epidemic, there is now much interest in the environmental and/or pharmacological exploitation of this thermoregulatory mechanism, since increased metabolic rate will combat obesity and the deleterious metabolic phenotype it incurs. In humans, a growing body of evidence suggests that acute activation of BAT can indeed have a meaningful impact on REE and intermediary glucose and lipid metabolism. However, whether brown adipocytes can be recruited in WAT and whether this alters REE or macronutrient metabolism remains unclear. Few human models mimic the extremity of confining rodents to near-freezing temperatures for several weeks, and in our study of severely burned patients, while prolonged stress does indeed brown WAT, several other organs are also significantly affected. Thus, to avoid the fate of the mitochondrial uncoupler dinitrophenol, future strategies aimed at augmenting UCP1 content and function in humans must strive to be tissue specific.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
This work was supported by grants from the National Institutes of Health (P50-GM60338, R01-GM05668, T32 GM008256, H133P110012, and a Clinical and Translational Science Award UL1TR000071) as well as grants from the Shriners Hospitals for Children (85310 and 84090), the John Sealy Memorial Endowment Fund for Biomedical Research (66992), ADA (1-14-TS-35), and the Sealy Center on Aging at UTMB. CP was supported, in part, by a National Institute of Disability and Rehabilitation Research Training Grant (H133P110012). MC is supported by the Onassis Foundation.
References
1. Centers for Disease Control and Prevention/National Center for Health Statistics/Office of Analysis and Epidemiology. Health Unites States, 2013 (2014). Available from: http://www.cdc.gov/nchs/data/hus/hus13.pdf[[x00023]]064
2. Ng M, Fleming T, Robinson M, Thomson B, Graetz N, Margono C, et al. Global, regional, and national prevalence of overweight and obesity in children and adults during 1980-2013: a systematic analysis for the Global Burden of Disease Study 2013. Lancet (2014) 384(9945):766–81. doi: 10.1016/S0140-6736(14)60460-8
3. Centers for Disease Control and Prevention. National Diabetes Statistics Report (2014). Available from: http://www.cdc.gov/diabetes/pubs/statsreport14/national-diabetes-report-web.pdf
4. Lim SS, Vos T, Flaxman AD, Danaei G, Shibuya K, Adair-Rohani H, et al. A comparative risk assessment of burden of disease and injury attributable to 67 risk factors and risk factor clusters in 21 regions, 1990–2010: a systematic analysis for the Global Burden of Disease Study 2010. Lancet (2012) 380(9859):2224–60. doi:10.1016/S0140-6736(12)61766-8
5. Pou KM, Massaro JM, Hoffmann U, Vasan RS, Maurovich-Horvat P, Larson MG, et al. Visceral and subcutaneous adipose tissue volumes are cross-sectionally related to markers of inflammation and oxidative stress: the Framingham Heart Study. Circulation (2007) 116(11):1234–41. doi:10.1161/CIRCULATIONAHA.107.710509
6. Gastaldelli A, Cusi K, Pettiti M, Hardies J, Miyazaki Y, Berria R, et al. Relationship between hepatic/visceral fat and hepatic insulin resistance in nondiabetic and type 2 diabetic subjects. Gastroenterology (2007) 133(2):496–506. doi:10.1053/j.gastro.2007.04.068
7. Rolfe D, Brown G. Cellular energy utilization and molecular origin of standard metabolic rate in mammals. Physiol Rev (1997) 77:731–58.
8. van Marken Lichtenbelt WD, Schrauwen P. Implications of nonshivering thermogenesis for energy balance regulation in humans. Am J Physiol Regul Integr Comp Physiol (2011) 301(2):R285–96. doi:10.1152/ajpregu.00652.2010
9. Cannon B, Nedergaard J. Brown adipose tissue: function and physiological significance. Physiol Rev (2004) 84(1):277–359. doi:10.1152/physrev.00015.2003
10. Cypess AM, Lehman S, Williams G, Tal I, Rodman D, Goldfine AB, et al. Identification and importance of brown adipose tissue in adult humans. N Engl J Med (2009) 360(15):1509–17. doi:10.1056/NEJMoa0810780
11. van Marken Lichtenbelt WD, Vanhommerig JW, Smulders NM, Drossaerts JM, Kemerink GJ, Bouvy ND, et al. Cold-activated brown adipose tissue in healthy men. N Engl J Med (2009) 360(15):1500–8. doi:10.1056/NEJMoa0808718
12. Virtanen KA, Lidell ME, Orava J, Heglind M, Westergren R, Niemi T, et al. Functional brown adipose tissue in healthy adults. N Engl J Med (2009) 360(15):1518–25. doi:10.1056/NEJMoa0808949
13. Nicholls D, Bernson V, Heaton G. The identification of the component in the inner membrane of brown adipose tissue mitochondria responsible for regulating energy dissipation. Experientia Suppl (1978) 32:89–93. doi:10.1007/978-3-0348-5559-4_9
14. Fedorenko A, Lishko P, Kirichok Y. Mechanism of fatty-acid-dependent UCP1 uncoupling in brown fat mitochondria. Cell (2012) 151:400–13. doi:10.1016/j.cell.2012.09.010
15. Feldmann HM, Golozoubova V, Cannon B, Nedergaard J. UCP1 ablation induces obesity and abolishes diet-induced thermogenesis in mice exempt from thermal stress by living at thermoneutrality. Cell Metab (2009) 9(2):203–9. doi:10.1016/j.cmet.2008.12.014
16. Liu X, Rossmeisl M, McClaine J, Riachi M, Harper ME, Kozak LP. Paradoxical resistance to diet-induced obesity in UCP1-deficient mice. J Clin Invest (2003) 111(3):399–407. doi:10.1172/JCI200315737
17. Inokuma K, Okamatsu-Ogura Y, Omachi A, Matsushita Y, Kimura K, Yamashita H, et al. Indispensable role of mitochondrial UCP1 for antiobesity effect of beta3-adrenergic stimulation. Am J Physiol Endocrinol Metab (2006) 290(5):E1014–21. doi:10.1152/ajpendo.00105.2005
18. Lee P, Greenfield JR, Ho KK, Fulham MJ. A critical appraisal of the prevalence and metabolic significance of brown adipose tissue in adult humans. Am J Physiol Endocrinol Metab (2010) 299(4):E601–6. doi:10.1152/ajpendo.00298.2010
19. Ouellet V, Routhier-Labadie A, Bellemare W, Lakhal-Chaieb L, Turcotte E, Carpentier AC, et al. Outdoor temperature, age, sex, body mass index, and diabetic status determine the prevalence, mass, and glucose-uptake activity of 18F-FDG-detected BAT in humans. J Clin Endocrinol Metab (2011) 96(1):192–9. doi:10.1210/jc.2010-0989
20. Yilmaz Y, Ones T, Purnak T, Ozguven S, Kurt R, Atug O, et al. Association between the presence of brown adipose tissue and non-alcoholic fatty liver disease in adult humans. Aliment Pharmacol Ther (2011) 34(3):318–23. doi:10.1111/j.1365-2036.2011.04723.x
21. Saito M, Okamatsu-Ogura Y, Matsushita M, Watanabe K, Yoneshiro T, Nio-Kobayashi J, et al. High incidence of metabolically active brown adipose tissue in healthy adult humans: effects of cold exposure and adiposity. Diabetes (2009) 58(7):1526–31. doi:10.2337/db09-0530
22. Vijgen GH, Bouvy ND, Teule GJ, Brans B, Hoeks J, Schrauwen P, et al. Increase in brown adipose tissue activity after weight loss in morbidly obese subjects. J Clin Endocrinol Metab (2012) 97(7):E1229–33. doi:10.1210/jc.2012-1289
23. Yoneshiro TAS, Matsushita M, Kameya T, Nakada K, Kawai Y, Saito M. Brown adipose tissue, whole-body energy expenditure, and thermogenesis in healthy adult men. Obesity (2011) 19(1):13–6. doi:10.1038/oby.2010.105
24. Chondronikola M, Volpi E, Borsheim E, Porter C, Annamalai P, Enerback S, et al. Brown adipose tissue improves whole-body glucose homeostasis and insulin sensitivity in humans. Diabetes (2014) 63(12):4089–99. doi:10.2337/db14-0746
25. Ouellet V, Labbe SM, Blondin DP, Phoenix S, Guerin B, Haman F, et al. Brown adipose tissue oxidative metabolism contributes to energy expenditure during acute cold exposure in humans. J Clin Invest (2012) 122(2):545–52. doi:10.1172/JCI60433
26. Muzik O, Mangner TJ, Leonard WR, Kumar A, Janisse J, Granneman JG. 15O PET measurement of blood flow and oxygen consumption in cold-activated human brown fat. J Nucl Med (2013) 54(4):523–31. doi:10.2967/jnumed.112.111336
27. Muzik O, Mangner TJ, Granneman JG. Assessment of oxidative metabolism in brown fat using PET imaging. Front Endocrinol (2012) 3:15. doi:10.3389/fendo.2012.00015
28. Yoneshiro T, Aita S, Matsushita M, Kayahara T, Kameya T, Kawai Y, et al. Recruited brown adipose tissue as an antiobesity agent in humans. J Clin Invest (2013) 123(8):3404–8. doi:10.1172/JCI67803
29. Lee P, Smith S, Linderman J, Courville AB, Brychta RJ, Dieckmann W, et al. Temperature-acclimated brown adipose tissue modulates insulin sensitivity in humans. Diabetes (2014) 63(11):3686–98. doi:10.2337/db14-0513
30. Bartelt A, Bruns OT, Reimer R, Hohenberg H, Ittrich H, Peldschus K, et al. Brown adipose tissue activity controls triglyceride clearance. Nat Med (2011) 17(2):200–5. doi:10.1038/nm.2297
31. Stanford KI, Middelbeek RJ, Townsend KL, An D, Nygaard EB, Hitchcox KM, et al. Brown adipose tissue regulates glucose homeostasis and insulin sensitivity. J Clin Invest (2013) 123(1):215–23. doi:10.1172/JCI62308
32. Hany TF, Gharehpapagh E, Kamel EM, Buck A, Himms-Hagen J, von Schulthess GK. Brown adipose tissue: a factor to consider in symmetrical tracer uptake in the neck and upper chest region. Eur J Nucl Med Mol Imaging (2002) 29(10):1393–8. doi:10.1007/s00259-002-0902-6
33. Jacene HA, Cohade CC, Zhang Z, Wahl RL. The relationship between patients’ serum glucose levels and metabolically active brown adipose tissue detected by PET/CT. Mol Imaging Biol (2011) 13(6):1278–83. doi:10.1007/s11307-010-0379-9
34. Matsushita M, Yoneshiro T, Aita S, Kameya T, Sugie H, Saito M. Impact of brown adipose tissue on body fatness and glucose metabolism in healthy humans. Int J Obes (Lond) (2013) 38(6): 812–7. doi:10.1038/ijo.2013.206
35. Orava J, Nuutila P, Lidell ME, Oikonen V, Noponen T, Viljanen T, et al. Different metabolic responses of human brown adipose tissue to activation by cold and insulin. Cell Metab (2011) 14(2):272–9. doi:10.1016/j.cmet.2011.06.012
36. Orava J, Nuutila P, Noponen T, Parkkola R, Viljanen T, Enerback S, et al. Blunted metabolic responses to cold and insulin stimulation in brown adipose tissue of obese humans. Obesity (Silver Spring) (2013) 21(11): 2279–87. doi:10.1002/oby.20456
37. Ma SW, Foster DO. Uptake of glucose and release of fatty acids and glycerol by rat brown adipose tissue in vivo. Can J Physiol Pharmacol (1986) 64(5):609–14. doi:10.1139/y86-101
38. Berbee JF, Boon MR, Khedoe PP, Bartelt A, Schlein C, Worthmann A, et al. Brown fat activation reduces hypercholesterolaemia and protects from atherosclerosis development. Nat Commun (2015) 6:6356. doi:10.1038/ncomms7356
39. Chondronikola M, Porter C, Hurren N, Chao T, Yfanti C, Labbé S, et al. The functional role of brown adipose tissue in whole-body lipid metabolism in humans (1160.3). FASEB J (2014) 28(1 Suppl).
40. Chondronikola M, Chao T, Porter C, Labbé S, Hurren N, Yfanti C, et al. Brown adipose tissue activation affects white adipose tissue insulin sensitivity in humans. 75th Scientific Sessions American Diabetes Association. Boston, MA (2015).
41. Young P, Arch J, Ashwell M. Brown adipose tissue in the parametrial fat pad of the mouse. FEBS Lett (1984) 167:10–4. doi:10.1016/0014-5793(84)80822-4
42. Cousin B, Cinti S, Morroni M, Raimbault S, Ricquier D, Pénicaud L, et al. Occurrence of brown adipocytes in rat white adipose tissue: molecular and morphological characterization. J Cell Sci (1992) 103:931–42.
43. Shabalina I, Petrovic N, de Jong J, Kalinovich A, Cannon B, Nedergaard J. UCP1 in brite/beige adipose tissue mitochondria is functionally thermogenic. Cell Rep (2013) 5:1196–203. doi:10.1016/j.celrep.2013.10.044
44. van der Lans A, Hoeks J, Brans B, Vijgen G, Visser M, Vosselman M, et al. Cold acclimation recruits human brown fat and increases nonshivering thermogenesis. J Clin Invest (2013) 123:3395–403. doi:10.1172/JCI68993
45. Ricquier D, Nechad M, Mory G. Ultrastructural and biochemical characterization of human brown adipose tissue in pheochromocytoma. J Clin Endocrinol Metab (1982) 54:803–7.
46. Frontini A, Vitali A, Perugini J, Murano I, Romiti C, Ricquier D, et al. White-to-brown transdifferentiation of omental adipocytes in patients affected by pheochromocytoma. Biochim Biophys Acta (2013) 1831:950–9. doi:10.1016/j.bbalip.2013.02.005
47. Sidossis L, Porter C, Saraf M, Borsheim E, Radhakrishnan R, Chao T, et al. Browning of subcutaneous white adipose tissue in humans after severe adrenergic stress. Cell Metab (2015) 22:219–27. doi:10.1016/j.cmet.2015.06.022
48. Gallagher D, Belmonte D, Deurenberg P, Wang Z, Krasnow N, Pi-Sunyer F, et al. Organ-tissue mass measurement allows modeling of REE and metabolically active tissue mass. Am J Physiol (1998) 275:249–58.
49. Wilmore D, Aulick L. Systemic responses to injury and the healing wound. JPEN J Parenter Enteral Nutr (1980) 4:147–51. doi:10.1177/0148607180004002147
50. Shabalina I, Ost M, Petrovic N, Vrbacky M, Nedergaard J, Cannon B. Uncoupling protein-1 is not leaky. Biochim Biophys Acta (2010) 1797:773–84. doi:10.1016/j.bbabio.2010.04.007
51. Wilmore D, Long J, Mason AJ, Skreen R, Pruitt BJ. Catecholamines: mediator of the hypermetabolic response to thermal injury. Ann Surg (1974) 180:653–69. doi:10.1097/00000658-197410000-00031
52. Jeschke MG, Gauglitz GG, Kulp GA, Finnerty CC, Williams FN, Kraft R, et al. Long-term persistance of the pathophysiologic response to severe burn injury. PLoS One (2011) 6:e21245. doi:10.1371/journal.pone.0021245
Keywords: adipose tissue, mitochondria, uncoupling protein 1, thermogenesis, obesity
Citation: Porter C, Chondronikola M and Sidossis LS (2015) The therapeutic potential of brown adipocytes in humans. Front. Endocrinol. 6:156. doi: 10.3389/fendo.2015.00156
Received: 30 June 2015; Accepted: 18 September 2015;
Published: 13 October 2015
Edited by:
Jun Wu, University of Michigan, USAReviewed by:
Elahu Gosney Sustarsic, University of Copenhagen, DenmarkSona Kang, Beth Israel Deaconess Medical Center, USA
Copyright: © 2015 Porter, Chondronikola and Sidossis. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Craig Porter and Labros S. Sidossis, University of Texas Medical Branch, 301 University Boulevard, Galveston, TX 77550, USA,Y3IycG9ydGVAdXRtYi5lZHU=;bGFzaWRvc3NAdXRtYi5lZHU=