 Chandi C. Mandal
Chandi C. Mandal- Department of Biochemistry, School of Life Sciences, Central University of Rajasthan, Rajasthan, India
Many epidemiological studies show a positive connection between cardiovascular diseases and risk of osteoporosis, suggesting a role of hyperlipidemia and/or hypercholesterolemia in regulating osteoporosis. The majority of the studies indicated a correlation between high cholesterol and high LDL-cholesterol level with low bone mineral density, a strong predictor of osteoporosis. Similarly, bone metastasis is a serious complication of cancer for patients. Several epidemiological and basic studies have established that high cholesterol is associated with increased cancer risk. Moreover, osteoporotic bone environment predisposes the cancer cells for metastatic growth in the bone microenvironment. This review focuses on how cholesterol and cholesterol-lowering drugs (statins) regulate the functions of bone residential osteoblast and osteoclast cells to augment or to prevent bone deterioration. Moreover, this study provides an insight into molecular mechanisms of cholesterol-mediated bone deterioration. It also proposes a potential mechanism by which cellular cholesterol boosts cancer-induced bone metastasis.
Introduction
As the life expectancy increases, the risk of developing various diseases, such as cardiovascular diseases (CVD), osteoporosis, and cancer, increases with aging. Osteoporosis is a major health concern, especially for menopausal women, and it accounts for a significant economic burden worldwide. Epidemiological studies have linked an association of CVD with reduced bone mineral density (BMD) and with increased bone fracture (1–3). Low BMD is a strong predictor for osteoporosis-associated fracture risk.
Many, but not all, studies have suggested a positive link between dyslipidemia [such as high serum total cholesterol (TC) and high low-density lipoprotein cholesterol (LDL-C)] and low BMD, especially for postmenopausal women (4–8). Moreover, a reduction of BMD was reported, when rodents were kept on high fat diet (9). Various clinical and animal studies have evidenced that cholesterol-lowering statin drugs, reduce osteoporosis-associated fracture risk (10–12). Similarly, a relationship between cancer risk and hypercholesterolemia was found from many epidemiological and basic studies (13–17). In our studies, we have demonstrated the active role of statins in osteoblast differentiation, which supports bone formation, but inhibited osteolytic metastasis of breast cancer (18, 19).
In this review, we have attempted to address the effect of cholesterol and statins on the activities of osteoblast and osteoclast cells, and overall bone health. This report also highlights the underlying molecular mechanisms of high cholesterol-mediated bone deterioration.
Involvement of Various Cell Types in Bone Remodeling
Bone is an active and highly dynamic tissue, which is continuously remodeled by a balanced and coordinated action of bone-forming osteoblast and bone-resorbing osteoclast cells. In the osteoporotic condition, the rate of bone resorption exceeds that of bone formation either due to enhancement of osteoclast activity or impairment of osteoblast differentiation. Bone residential osteoblast, osteoclast, and osteocyte cells primarily regulate this physiological process. Mature osteoblast cells release several bone matrix proteins, such as collagen 1, osteocalcin (OCN), bone sialo protein (BSP), and osteopontin (OPN), to the extracellular environment. These components form osteoid (soft bone) (20). Osteoblasts also facilitate the formation of hydroxyapatite (HA; CaPO4) crystal inside the matrix vesicles (MVs). These MVs move to the extracellular environment, and unload the crystal molecules on the top of osteoid, which provides the cementing material, to make the matrix hard (21). In contrast to osteoblasts, multinucleated giant osteoclast cells release H+ ion to dissolve this cementing material, and secrete various proteolytic enzymes, such as tartrate resistant acid phosphatase (TRAP), collagenolytic cathepsin K, and matrix metalloproteinases (MMPs), to break down the bone matrix.
Osteoblasts/osteoblast lineage cells also produce osteoclastogenic factors, such as colony stimulating factor-1 (CSF-1), receptor activator of NFκB ligand (RANKL), and osteoprotegerin OPG (20). These CSF-1 and RANKL bind to their receptor CSF-1R and RANK present in osteoclast precursor monocyte/macrophage cells. These ligand–receptor interactions transduce different intracellular signaling cascades inside osteoclast cells, and subsequently promote survival, proliferation, and differentiation of osteoclast cells, and eventually activate osteoclast activity. Osteocytes are terminally differentiated osteoblast cells, and they are embedded into the bone matrix. Accumulating evidence documents that dendritic osteocytes interconnect with other bone cells through gap junction, and also sense the mechanical stress, minor crack, and changes in hormonal milieu of the bone (22, 23). Moreover, osteocyte cell-derived soluble factors, such as sclerostin, fibroblast growth factor (FGF) 23, OPG, and RANKL, regulate bone function, bone remodeling, and bone metabolism (24).
Recent studies have introduced novel molecular mechanisms by which osteoclast-derived factors, such as Wnt10, BMP-6, EphrinA2, and Semaphorin (SemaD4), regulate osteoblast activity (24, 25) (Figure 1). During the course of osteoclastic bone resorption, TGFβ and IGF-I are released in a large amount from the embedded bone matrix, and these growth factors may promote the recruitment and activation of osteoblasts in the bone multicellular units (26). Bone morphogenetic proteins (BMPs) are the most potent osteoinducers, which play a pivotal role in bone remodeling by regulating both osteoblast and osteoclast activity (27–29).
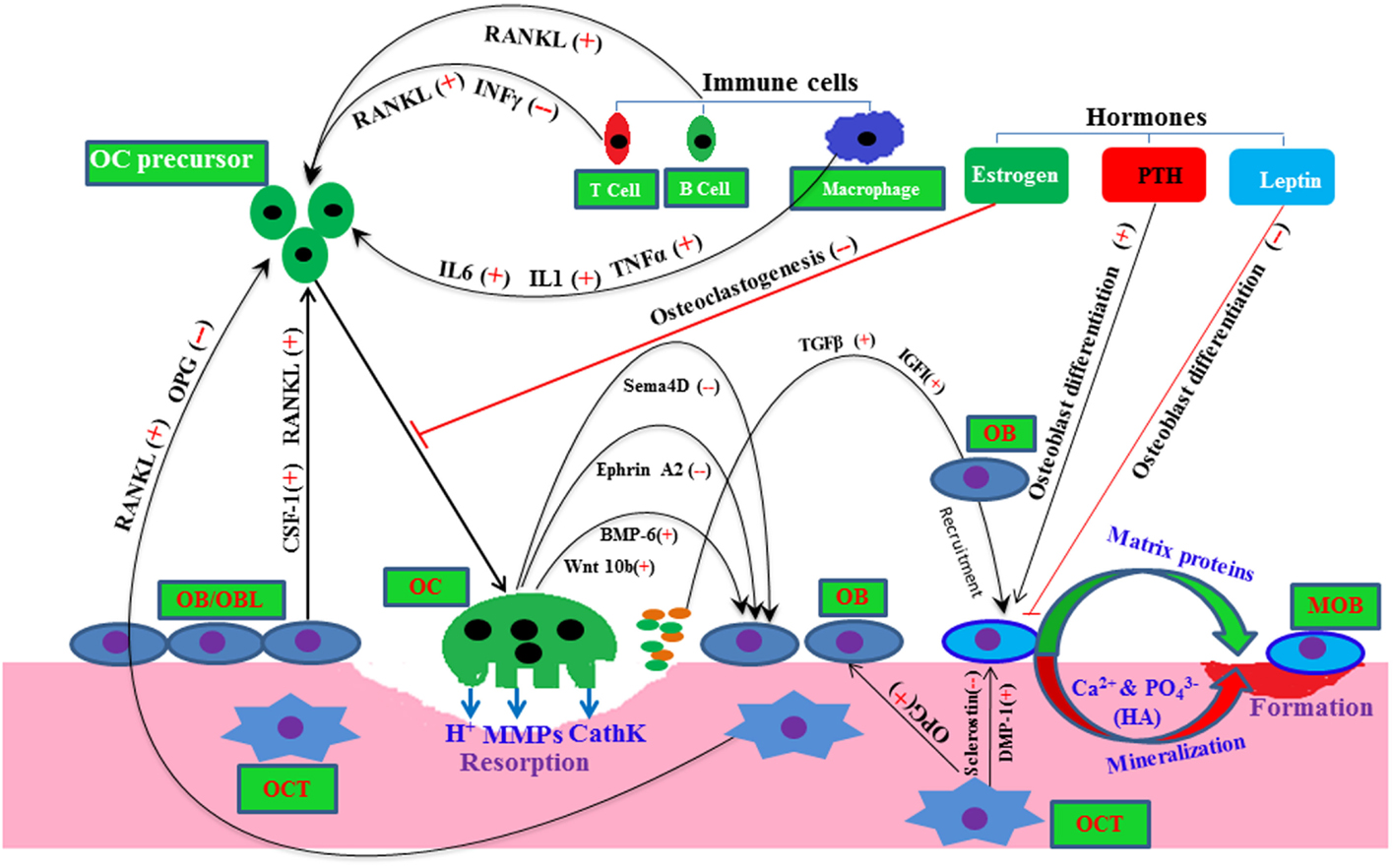
Figure 1. A molecular view of bone remodeling with a complex interaction among bone resident cells and other cells. Here, plus and minus signs depict positive and negative regulator, respectively, line arrow head and blunt ended line depict increased and decreased function, respectively. Abbreviations: OC, osteoclast; OB, osteoblast; OBL, osteoblast lineage; MOB, mature osteoblast; OCT, osteocyte; HA, hydroxyapatite; Sema4D, semasporin 4D; CathK, cathepsin K; PTH, parathyroid hormone.
Other cell types, such as immune T cells, B cells, and stromal cells, also influence this bone remodeling process by supplying osteoclastogenic RANKL and anti-osteoclastogenic interferon gamma (INFγ) to the osteoclast precursor cells (Figure 1) (30, 31). In pathophysiology condition-like arthritis, T and B cells both may increase osteoclastogenesis presumably by supplying RANKL to osteoclast cells (30–32). Various hormones, such as leptin, parathyroid hormone (PTH), and sex hormones, have been shown to regulate osteoblastic/osteoclastic function (23, 26, 33) (Figure 1). Thus, systemic and local environments manipulate the bone function, bone metabolism, and bone remodeling (34).
Cholesterol-Lowering Statin Drugs and Health Benefit
Statins are the most prescribed cholesterol-lowering drugs. Different members of statins, such as simvastatin, lovastatin, atorvastatin, pravastatin, and mevastatin, are therapeutically used for the treatment of CVD-like atherosclerosis. In all, statin treatment lowers 25–35% of serum LDL-cholesterol and reduces 25–30% frequency of heart attack (35). Statins competitively inhibit the activity of 3-hydroxy-3-methylglutaryl-coenzyme A (HMGCOA) reductase, a rate limiting enzyme of the cholesterol biosynthesis pathway in a reversible fashion (36, 37). In vitro, in vivo, and clinical studies show very promising results in reducing osteoporosis and cancers for statin users (10–12, 38). However, many studies have reported conflicting findings in cases of type 2 diabetes for statin users (39, 40).
Cholesterol and Osteoblasts
Research studies were carried out in cells, animal models, and clinical samples to evaluate the influence of serum cholesterol and/or cellular cholesterol on osteoblast cell proliferation, differentiation, and bone health. Findings of these studies are summarized in Table 1. In 1999, it was shown for the first time that the treatment of rodents with cholesterol-lowering drugs simvastatin and lovastatin both increased bone formation (10). They had found that intra-calvaria injection of these statins showed an increased calvarial bone formation, and similarly, oral administration increased cancellous bone formation (10). Later on, our research group had reported that treatment of osteoblast 2T3 cells with simvastatin and lovastatin increased alkaline phosphatase (ALP) activity (an early osteoblast differentiation marker), and expression of collagen and OPN (late osteoblast differentiation markers) (18). As a mechanism, it was found that simvastatin treatment increases the expression of the potent osteoinducer BMP-2, by upregulating Akt and Erk1/2 activity to enhance osteoblast differentiation. We and others have reported that statins upregulate BMP-2 expression to increase osteoblastic ALP activity and nodule formation (10, 18, 41, 42, 43). Similarly, simvastatin treatment was also found to increase cell proliferation and osteoblast differentiation of human periodontal ligament cells (PDLs) with concomitant increase of ALP, OPN, and mineralization (44). However, simvastatin was not found to influence proliferation/viability of PDLs (45). However, proliferation/viability of primary alveolar osteoblast cells (AOBs) was significantly decreased by the treatment of simvastatin at different concentrations (45). Simvastatin treatment (1 and 100 nM) was able to increase ALP activity of both AOBs and PDLs. Simvastatin and pravastatin treatments were found to increase the activity of ALP and nodule formation (46, 47). However, these treatments showed reduction of ALP and mineralization in aortic valve myofibroblast cells (46), whereas mevastatin failed to increase ALP activity and calcium incorporation into cells (48). Thus, the substantial data showed an increased osteoblastic differentiation, when osteoblast or osteoblast-like cells were treated with statins. But, the cell proliferative response upon statin treatment depends on cell types, type of statins, its dosage, and duration of exposure.
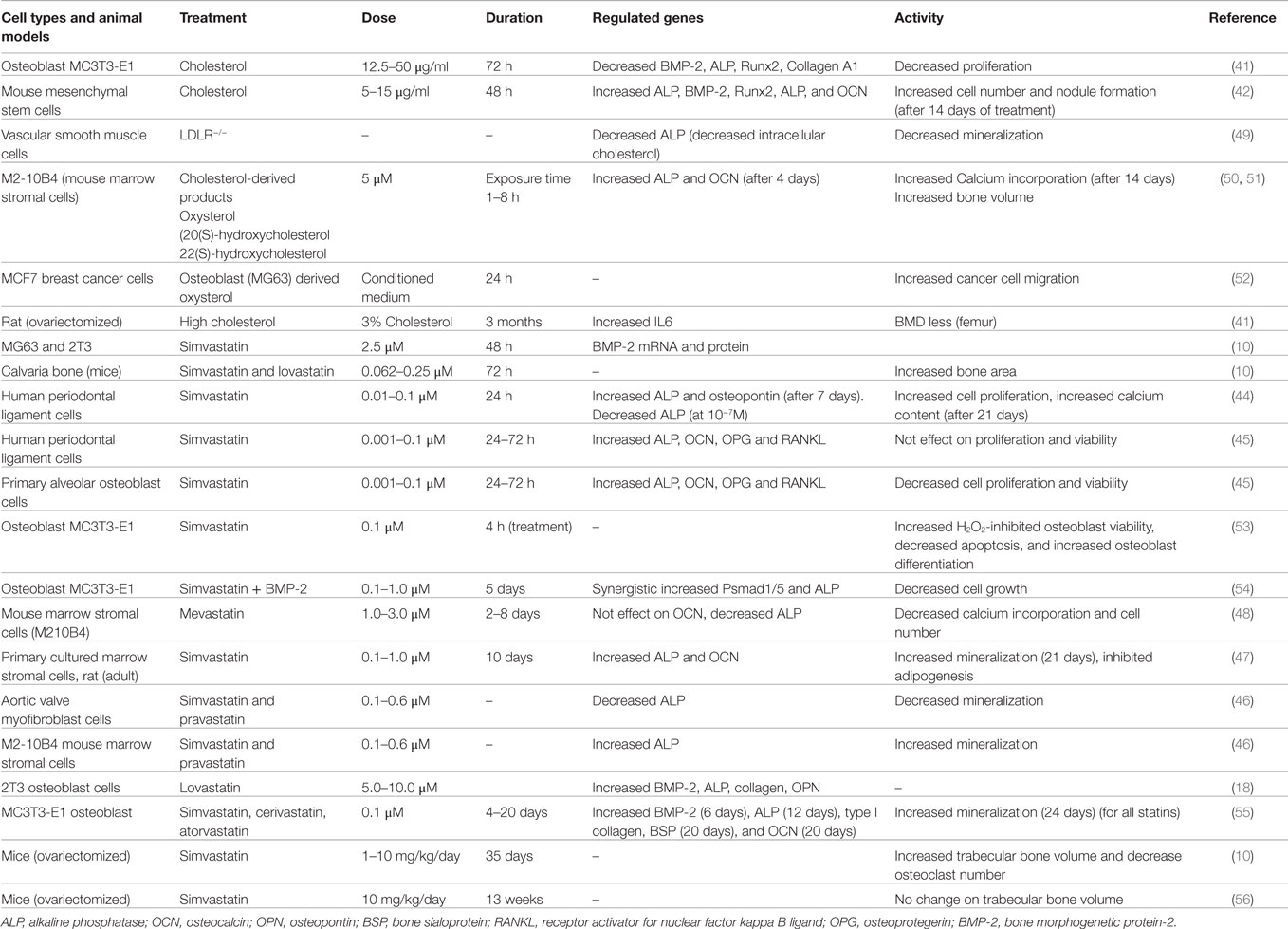
Table 1. Effect of cholesterol or cholesterol reducing drugs on osteoblast activity.
In animal studies, high cholesterol diet showed a reduction of BMD in femur with concomitant increase of serum OCN and carboxyterminal collagen crosslinks (CTX), which suggests that high cholesterol may increase bone turn over (41). Moreover, treatment of osteoblast MC3T3-E cells with different concentrations of cholesterol inhibited proliferation and differentiation (41). Cholesterol treatment reduced osteoblastic proteins Runx2, ALP, and collagen A1 with a concomitant decrease of BMP-2. These findings suggest that cholesterol may target BMP-2 to block the expression of Runx2, ALP, and collagen 1A in osteoblast cells, which in turn inhibits osteoblast differentiation. However, recent evidence showed that a high cholesterol diet was capable of masking the type 2 diabetes-induced bone loss (57). Other evidence documented that cholesterol-treated mesenchymal stem cells (MSCs) showed an increased expression of osteogenic lineage markers, increased ALP activity, and more mineralized nodule formation. It was noted that osteogenic potency of cholesterol was mostly due to elevation of cholesterol ester (42). These contrasting findings suggest that functional activity of cholesterol depends on disease condition, types of cholesterol (free cholesterol and cholesterol ester), and cell types, akin to statins.
Similarly, oxysterol, a cholesterol-derived product, showed induction of ALP, OCN, and mineralization in marrow stromal cells, and decreased adipocyte differentiation (50, 51). As a mechanism, it was found that oxysterol increased osteogenic markers by inhibiting DKK-1, a known Wnt inhibitor, but the blocking of PI3K pathway was shown to reverse the oxysterol-induced osteogenesis in these stromal cells (58). Moreover, oxysterol prevented oxidized LDL-inhibited osteoblast differentiation of marrow stromal cells (59). However, the primary cholesterol metabolite, 27 hydroxycholesterol, through its actions on both estrogen receptors and liver X receptors, decreased osteoblast differentiation and enhanced osteoclastogenesis, resulting in increased bone resorption (60).
Vascular smooth muscle cells isolated from LDL receptor (LDLR) knock-out mice showed reduction of ALP activity and matrix mineralization with a concomitant decrease of intracellular cholesterol when compared with wild type cells (49). Schilling et al. showed that apolipoprotein E (APOE) deficient mice showed an increased bone mass as compared to control mice (61).
These data indicate that different cholesterol products may have distinct roles in osteoblast- or osteoblast-like differentiation, but decreased of cellular cholesterol may prevent vascular calcification, whereas increased cholesterol may inhibit osteoblast differentiation to prevent bone formation.
Cholesterol and Osteoclasts
Almost two decades ago, the Mundy research group suggested, based on rodent model experiments that statins might prevent osteoporosis by increasing osteoblastic differentiation and decreasing bone resorption (10). At that time, some clinical data supported the concept that cholesterol-lowering statin drugs might decrease the fracture risk of bone (62). Von Stechow et al. observations did not correlate with these findings (56). It was reported that treatment of macrophages with cholesterol showed an enhancement of osteoclast activity by increasing the expression of the interleukin alpha gene (63). Similarly, the removal of cellular cholesterol from osteoclast precursor cells by treating with cyclodextrin or high-density lipoprotein (HDL) was found to increase apoptosis of osteoclast cells (64). Depletion of cellular cholesterol has prevented RANKL- or CSF-1-driven osteoclast activity, presumably by inhibiting the cell survival Akt–mTOR–S6 pathway (64). Moreover, LDLR-deficient osteoclast cells that might have low cellular cholesterol showed reduced size and life span when compared to wild type cells. Similarly, blocking the mevalonate pathway inhibited osteoclast activity, and also prevented apoptosis of osteoblast (65). More evidence showed that the depletion of lipid raft-cholesterol inhibits H-RAS-induced expression of RANKL to mitigate osteoclast activity (66). Similarly, the integrity of lipid rafts regulate V-ATPase activity in osteoclast, indicating that the removal of cholesterol from lipid rafts may impair osteoclast function (67). All these data together suggest that the cellular cholesterol of osteoclast cells might positively influence the bone deterioration by enhancing osteoclast activity and preventing osteoclast apoptosis. However, cellular cholesterol may have a contrasting function in regulating apoptosis of osteoblast and osteoclast cells (64, 65).
Lovastatin treatment inhibited osteoclast differentiation by reducing of geranylgeranylation of prenylated proteins that inhibition of geranylation and farnesylation has been shown to be the mechanism of action of nitrogen containing bisphosphonates, which are used to inhibit osteoclastic bone resorption (68, 69). Moreover, atorvastatin treatment to primary osteoblast cells showed an increased level of OPG, decoy receptor of RANKL, which prevents osteoblast-aided osteoclast activity by blocking the binding activity of RANKL to RANK receptor of osteoclast cells (70). It was also reported that RANKL expression of AOBs was increased with the treatment of simvastatin at different concentrations (low to high), but this expression of PDLs was increased only when higher concentrations of simvastatin (10–100 nM) were used (45). These findings indicate that statins may have a different cellular response in regulating RANKL expression. In these studies, they did not check the effect of simvastatin on osteoclast activity, but the expression of OPG level was increased in both cell types in response to simvastatin treatment (45). Since earlier studies documented that statin treatments might also increase the OPG level to block RANKL function (70). Thus, the RANKL/OPG ratio upon statin treatment can only predict the osteoclast activity.
Animal-based studies further indicate that the mice with hypercholesterolemia reduce the formation of cortical and trabecular bone in the femurs and vertebrae by promoting osteoclastogenesis (71). Moreover, under hypercholesterolemia condition, mice showed a higher risk of fracture. Similarly, high fat diet also induces cathepsin K positive osteoclast cells and RANKL expression, leading to enhanced osteoclastogenesis (72). Interestingly, ovariectomized (OVX) mice exhibit enhanced bone resorption and bone loss due to deficiency of estrogen, but simvastatin-treated OVX mice showed a reduction of bone loss by decreasing osteoclast activity (73). Similarly, in a rat model, a high cholesterol diet caused an enhancement of bone resorption with a concomitant increase of TRAP positive osteoclast cells (74).
Thus, the substantial data of cellular, animal, and clinical research studies suggest that the reduction of cholesterol might prevent bone loss by inhibiting osteoclast activity. Results of these studies are summarized in Table 2.
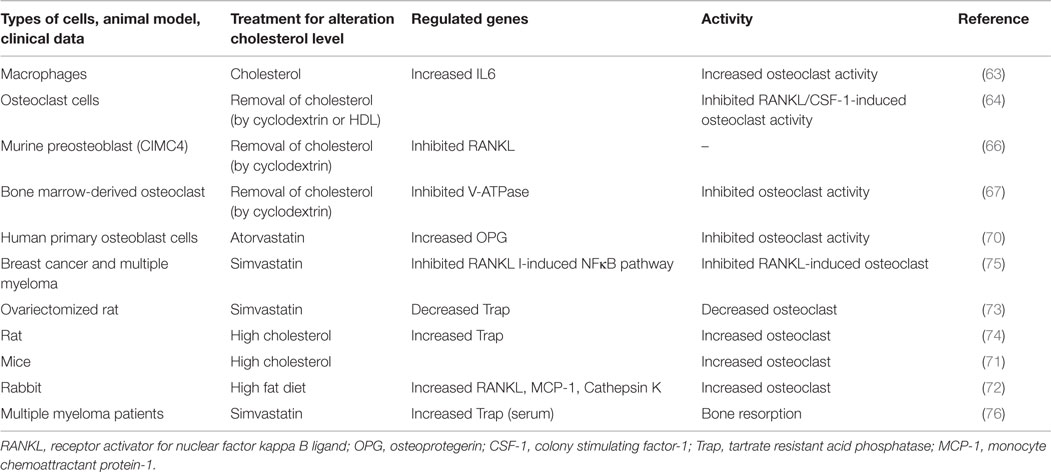
Table 2. Effect of cholesterol or cholesterol reducing drugs on osteoclast function.
Cholesterol and Bone Metastasis
Bone is one of the most common sites for the metastasis of many cancer types, such as breast, prostate, ovarian, and lungs, since bone provides a fertile soil to disseminated tumor cells (77). During bone metastasis, metastasized cancer cells interrupt the normal bone remodeling program either by increasing osteoclast activity or by altering the osteoblast activity, as depicted in Figure 2. For example, breast cancers mostly show osteolytic, and prostate cancers often undergo osteoblastic metastasis. But in both cases, either abnormal osteoclast or osteoblast activity leads to increase the risk of bone fracture. Many growth factors and cytokines have been shown to participate in regulating bone metastasis. For example, osteoclastogenic CSF-1 plays a pivotal role in bone remodeling, but the abnormal expression of this cytokine correlates with bone metastasis and osteoporosis (78–81).
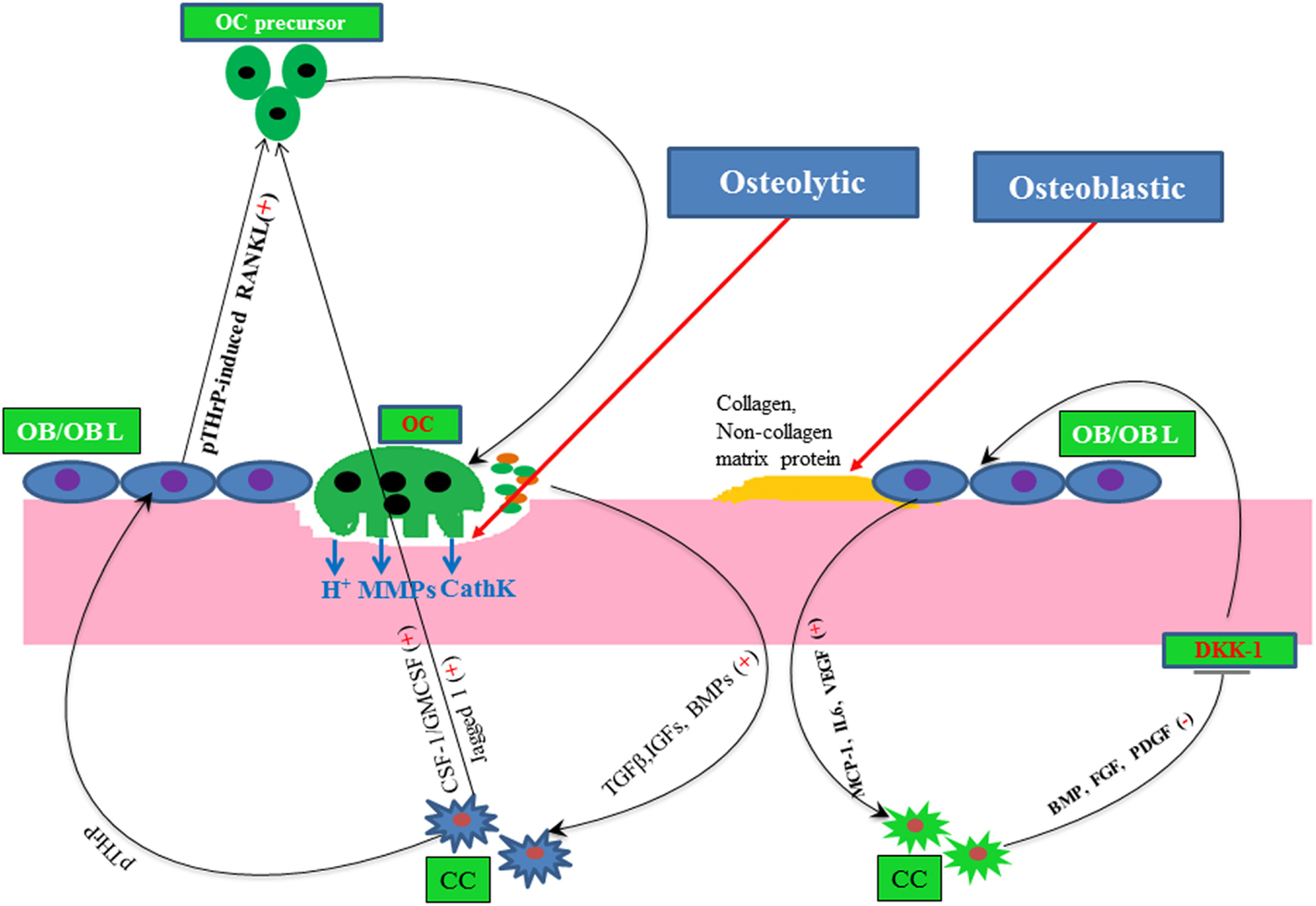
Figure 2. A simple molecular view of osteolytic and osteoblastic metastasis of cancer. Here, plus and minus signs depict positive and negative regulator, respectively, line arrow head and blunt ended line depict increased and decreased function, respectively. Abbreviations: OC, osteoclast; OB, osteoblast; OBL, osteoblast lineage; CC, cancer cells; CathK, cathepsin K.
It has been demonstrated that hyperlipidemia and high cholesterol promote cell migration and metastasis (13, 15). The detailed molecular mechanism is not yet known as to how high cholesterol increases tumorigenesis. Although, it was hypothesized that statins may prevent skeletal metastasis of cancers (82). Our study, for the first time, experimentally documented that simvastatin treatment inhibits osteolytic bone metastasis, which was developed after intracardiac inoculation of human metastatic breast cancer MDA-MB-231 cells in mice (19).
It was suggested that simvastatin treatment increased tumor suppressor protein p53 levels to transcriptionally repress breast cancer stem cell marker CD44 expression. This in turn inhibits the breast cancer cell migration and invasion, resulting in the inhibition of osteolytic metastasis (19). Later on, another research group had demonstrated that simvastatin treatment prevents osteolytic lesions of the spinal cord, which was produced due to injection of the lung cancer A549 into nude mice. Similar to our observation, they had also suggested that simvastatin treatment upregulated p53 protein with concomitant increase of CD44, MMP-2, and MMP-9 (83). Our research study also proved that metastatic breast cancer cells secrete more CSF-1 as compared to non-metastatic breast cancer cells (84). Other study reported that statins inhibit CSF-1 expression (85). These data suggest that breast cancer cell-derived CSF-1 might potentiate osteolytic bone loss, which could be inhibited by the treatment of statins. Recently, a study has shown that the combination of simvastatin and the anti-diabetic drug metformin synergistically reduced the metastatic property of cancer cells as well as metastasis to the spinal cord and femurs. In this study, prostate cancer C4-2B cells were injected into castrated aythmic mice to develop metastasis (86). The combination of these two drugs inhibited AKT and AMPKα activity (86). We had reported earlier that simvastatin treatment upregulated the tumor suppressor protein PTEN. PTEN inhibits AKT activity in preventing cancer growth both in in vitro and an animal model (87). These findings indicate that cholesterol may positively modulate bone metastasis of cancers. Clinical evidence supports this concept, since the mean cholesterol level was found to be increased in bone metastases when compared to healthy controls (88).
Altogether, these data support the idea that cellular cholesterol might potentiate bone metastasis of cancers. Based on these findings, a potential molecular mechanism by which statins inhibit cholesterol-mediated bone metastasis has been proposed (Figure 3).
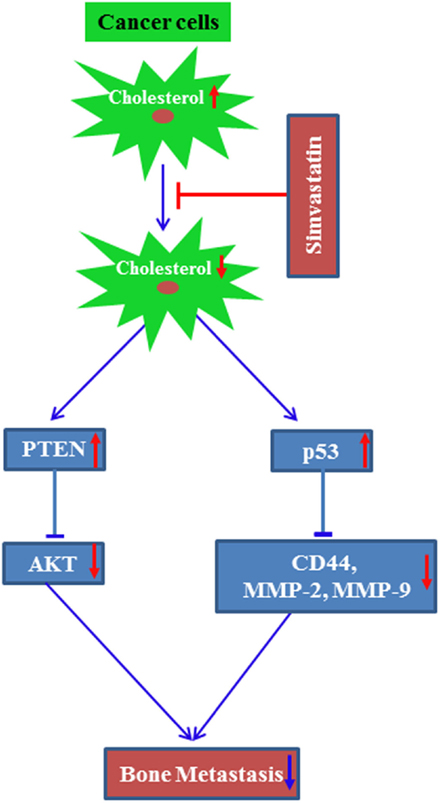
Figure 3. A proposed molecular mechanism by which simvastatin inhibits bone metastasis of cancer. Line arrow head and blunt ended line depict increased and decreased function, respectively, and up and bottom arrow show increased and decreased level/expression, respectively.
Cholesterol and Multiple Myeloma
Clinical findings suggest that serum cholesterol level of myeloma patients is low, since myeloma cancer cells consume more serum cholesterol for their growth (89). Exogenous treatment of cholesterol increases cell survival of myeloma cells (90). Myeloma cancer cells may increase cellular cholesterol either by inhibiting cholesterol efflux or increasing sterol regulatory element binding (SERB) transcription factors, which transcriptionally increase HMGCOA reductase and LDLR expression (91). More evidence showed that simvastatin treatment was quite capable to inhibit myeloma driven osteoclast activity, which was promoted by RANKL treatment (75). In contrast to the above studies, Sondergaard et al. reported that a high dose of simvastatin treatment to multiple myeloma patients showed elevated levels of TRAP and CTX in serum, indicating that simvastatin treatment might increase bone resorption, instead of inhibition (76).
These conflicting results indicate that cholesterol-lowering drugs may prevent myeloma-induced bone diseases, provided the drugs reduce the cellular cholesterol of the myeloma cells.
Conclusion
Low BMD and the occurrence of bone fractures are strong predictors for osteoporosis. Many research studies have strengthened the link between the serum lipid profile and BMD. Here, the relationship between cholesterol and BMD has been summarized in Table 3. Substantial data showed a negative association between BMD and serum TC, and LDL cholesterol (5, 7, 8, 92–97). A few studies found no association and/or a positive association with serum cholesterol and LDL cholesterol (98–101). The summarized data from all the studies indicate that there is an existence of an inconsistent relationship for the case of HDL cholesterol and BMD (6, 99, 100, 102). In several clinical trials, but not in all, the use of statins have been associated with a reduction of fracture risk, and the patients taking statins having a higher BMD than those who do not (103–105). In brief, a high cholesterol level is inversely associated with BMD, and statin treatment shows an enhancement of BMD, indicating high cholesterol as a negative regulator of bone health.
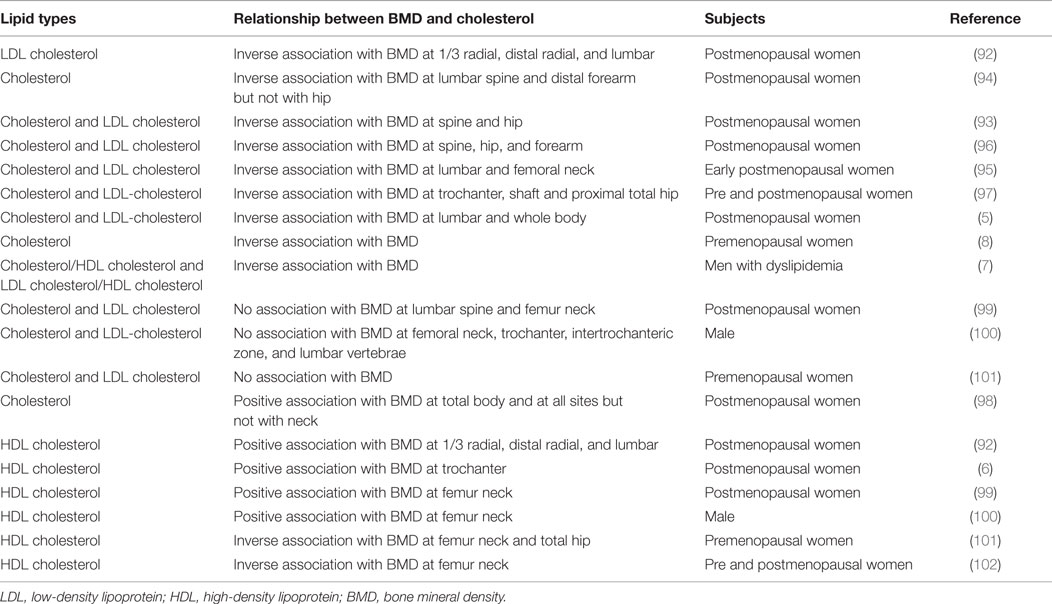
Table 3. Relationship between cholesterol and BMD.
However, the existence of this inverse relationship was not always found (Table 3). Thus, it could be that serum cholesterol or cholesterol-lowering drugs might have a variable influence in regulating BMD at different sites. The effect of serum cholesterol on BMD may vary among individuals, by gender, with age, and menopausal status of women. However, if we summarize all results of cell culture and animal experiments (Tables 1 and 2), it can be concluded that high cholesterol often increases osteoclast activity and decreases osteoblast function, especially in bone residential cells. It can also be concluded that targeting cellular cholesterol might decrease osteoclastogenesis with an enhancement of osteoblast function of bone cells (Tables 1 and 2). However, further research indicates that osteoblast activity in response to statin treatment markedly depends on cell types, dosage, and type of statins, but in most cases, statin treatment has been found to inhibit osteoclast or bone resorption activity (Tables 1 and 2). In cancers, most of the literature suggests that instead of serum cholesterol, cellular cholesterol might influence the cancer progression and metastasis (16, 38). Moreover, bone metastases contained an elevated level of cholesterol when compared to normal tissues (88). Similarly, depletion of cellular cholesterol prevents apoptosis of osteoblast cells, and increases apoptosis of osteoclast cells (64, 65).
Therefore, cellular cholesterol levels of bone occupant cells (such as osteoblast, osteoclast, osteocyte, myeloma, and bone metastasized cancer cells) might be a good predictor of the patient’s bone health, and hence, targeting the intracellular cholesterol might improve bone health.
Conflict of Interest Statement
The author declares that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
CM is supported by UGC Start-UP-Grant [30-49/2014 (BSR)], DBT [6242 P9/RGCB/PMD/DBT/CCML/2015], and Central University of Rajasthan, India. Author thanks to Prof. James A. Radosevich (Department of Oral Medicine and Diagnostic Sciences, College of Dentistry, University of Illinois at Chicago, Chicago, IL, USA-60612) for his valuable comments and suggestions.
References
1. Baldini V, Mastropasqua M, Francucci C, D’Erasmo E. Cardiovascular disease and osteoporosis. J Endocrinol Invest (2004) 28:69–72.
2. Tankó LB, Christiansen C, Cox DA, Geiger MJ, McNabb MA, Cummings SR. Relationship between osteoporosis and cardiovascular disease in postmenopausal women. J Bone Miner Res (2005) 20:1912–20. doi: 10.1359/JBMR.050711
3. Sennerby U, Melhus H, Gedeborg R, Byberg L, Garmo H, Ahlbom A, et al. Cardiovascular diseases and risk of hip fracture. JAMA (2009) 302:1666–73. doi:10.1001/jama.2009.1463
4. Poli A, Bruschi F, Cesana B, Rossi M, Paoletti R, Crosignani PG. Plasma low-density lipoprotein cholesterol and bone mass densitometry in postmenopausal women. Obstet Gynecol (2003) 102:922–6. doi:10.1016/j.obstetgynecol.2003.07.004
5. Makovey J, Chen JS, Hayward C, Williams FM, Sambrook PN. Association between serum cholesterol and bone mineral density. Bone (2009) 44:208–13. doi:10.1016/j.bone.2008.09.020
6. Kim K-C, Shin D-H, Lee S-Y, Im J-A, Lee D-C. Relation between obesity and bone mineral density and vertebral fractures in Korean postmenopausal women. Yonsei Med J (2010) 51:857–63. doi:10.3349/ymj.2010.51.6.857
7. Kim Y-H, Nam G-E, Cho K-H, Choi YS, Kim S-M, Han B-D, et al. Low bone mineral density is associated with dyslipidemia in South Korean men: The 2008–2010 Korean National Health and Nutrition Examination Survey. Endocr J (2013) 60:1179–89. doi:10.1507/endocrj.EJ13-0224
8. Jeong T-D, Lee W, Choi S-E, Kim JS, Kim H-K, Bae SJ, et al. Relationship between serum total cholesterol level and serum biochemical bone turnover markers in healthy pre-and postmenopausal women. Biomed Res Int (2014) 2014:398397. doi:10.1155/2014/398397
9. Parhami F, Tintut Y, Beamer WG, Gharavi N, Goodman W, Demer LL. Atherogenic high-fat diet reduces bone mineralization in mice. J Bone Miner Res (2001) 16:182–8. doi:10.1359/jbmr.2001.16.1.182
10. Mundy G, Garrett R, Harris S, Chan J, Chen D, Rossini G, et al. Stimulation of bone formation in vitro and in rodents by statins. Science (1999) 286:1946–9. doi:10.1126/science.286.5446.1946
11. Edwards C, Hart D, Spector T. Oral statins and increased bone-mineral density in postmenopausal women. Lancet (2000) 355:2218–9. doi:10.1016/S0140-6736(00)02408-9
12. Meier CR, Schlienger RG, Kraenzlin ME, Schlegel B, Jick H. HMG-CoA reductase inhibitors and the risk of fractures. JAMA (2000) 283:3205–10. doi:10.1001/jama.283.24.3205
13. Alikhani N, Ferguson RD, Novosyadlyy R, Gallagher EJ, Scheinman E, Yakar S, et al. Mammary tumor growth and pulmonary metastasis are enhanced in a hyperlipidemic mouse model. Oncogene (2013) 32:961–7. doi:10.1038/onc.2012.113
14. Kiberstis PA. Cholesterol and cancer. Sci Signal (2013) 6:ec292–292. doi:10.1126/scisignal.2004958
15. Nelson ER, Wardell SE, Jasper JS, Park S, Suchindran S, Howe MK, et al. 27-Hydroxycholesterol links hypercholesterolemia and breast cancer pathophysiology. Science (2013) 342:1094–8. doi:10.1126/science.1241908
16. Dos Santos CR, Fonseca I, Dias S, De Almeida JM. Plasma level of LDL-cholesterol at diagnosis is a predictor factor of breast tumor progression. BMC Cancer (2014) 14:132. doi:10.1186/1471-2407-14-132
17. McDonnell D, Chang C-Y, Nelson E. The estrogen receptor as a mediator of the pathological actions of cholesterol in breast cancer. Climacteric (2014) 17:60–5. doi:10.3109/13697137.2014.966949
18. Ghosh-Choudhury N, Mandal CC, Choudhury GG. Statin-induced Ras activation integrates the phosphatidylinositol 3-kinase signal to Akt and MAPK for bone morphogenetic protein-2 expression in osteoblast differentiation. J Biol Chem (2007) 282:4983–93. doi:10.1074/jbc.M606706200
19. Mandal CC, Ghosh-Choudhury N, Yoneda T, Choudhury GG, Ghosh-Choudhury N. Simvastatin prevents skeletal metastasis of breast cancer by an antagonistic interplay between p53 and CD44. J Biol Chem (2011) 286:11314–27. doi:10.1074/jbc.M110.193714
20. Raggatt LJ, Partridge NC. Cellular and molecular mechanisms of bone remodeling. J Biol Chem (2010) 285:25103–8. doi:10.1074/jbc.R109.041087
21. Anderson HC. Matrix vesicles and calcification. Curr Rheumatol Rep (2003) 5:222–6. doi:10.1007/s11926-003-0071-z
22. Civitelli R. Cell–cell communication in the osteoblast/osteocyte lineage. Arch Biochem Biophys (2008) 473:188–92. doi:10.1016/j.abb.2008.04.005
23. Zuo C, Huang Y, Bajis R, Sahih M, Li Y-P, Dai K, et al. Osteoblastogenesis regulation signals in bone remodeling. Osteoporos Int (2012) 23:1653–63. doi:10.1007/s00198-012-1909-x
24. Schaffler MB, Kennedy OD. Osteocyte signaling in bone. Curr Osteoporos Rep (2012) 10:118–25. doi:10.1007/s11914-012-0105-4
25. Henriksen K, Karsdal MA, Martin TJ. Osteoclast-derived coupling factors in bone remodeling. Calcif Tissue Int (2014) 94:88–97. doi:10.1007/s00223-013-9741-7
26. Lerner UH. Bone remodeling in post-menopausal osteoporosis. J Dent Res (2006) 85:584–95. doi:10.1177/154405910608500704
27. Mandal CC, Ghosh-Choudhury G, Ghosh-Choudhury N. Phosphatidylinositol 3 kinase/Akt signal relay cooperates with smad in bone morphogenetic protein-2-induced colony stimulating factor-1 (CSF-1) expression and osteoclast differentiation. Endocrinology (2009) 150:4989–98. doi:10.1210/en.2009-0026
28. Mandal CC, Drissi H, Choudhury GG, Ghosh-Choudhury N. Integration of phosphatidylinositol 3-kinase, Akt kinase, and Smad signaling pathway in BMP-2-induced osterix expression. Calcif Tissue Int (2010) 87:533–40. doi:10.1007/s00223-010-9419-3
29. Ghosh-Choudhury N, Mandal CC, Das F, Ganapathy S, Ahuja S, Choudhury GG. c-Abl-dependent molecular circuitry involving Smad5 and phosphatidylinositol 3-kinase regulates bone morphogenetic protein-2-induced osteogenesis. J Biol Chem (2013) 288:24503–17. doi:10.1074/jbc.M113.455733
30. Onal M, Xiong J, Chen X, Thostenson JD, Almeida M, Manolagas SC, et al. Receptor activator of nuclear factor κB ligand (RANKL) protein expression by B lymphocytes contributes to ovariectomy-induced bone loss. J Biol Chem (2012) 287:29851–60. doi:10.1074/jbc.M112.377945
31. Takayanagi H. New developments in osteoimmunology. Nat Rev Rheumatol (2012) 8:684–9. doi:10.1038/nrrheum.2012.167
32. Horowitz MC, Fretz JA, Lorenzo JA. How B cells influence bone biology in health and disease. Bone (2010) 47:472–9. doi:10.1016/j.bone.2010.06.011
33. Ducy P, Amling M, Takeda S, Priemel M, Schilling AF, Beil FT, et al. Leptin inhibits bone formation through a hypothalamic relay: a central control of bone mass. Cell (2000) 100:197–207. doi:10.1016/S0092-8674(00)81558-5
34. Teti A. Bone development: overview of bone cells and signaling. Curr Osteoporos Rep (2011) 9:264–73. doi:10.1007/s11914-011-0078-8
35. Akira E. A historical perspective on the discovery of statins. Proc Jpn Acad Ser B Phys Biol Sci (2010) 86:484. doi:10.2183/pjab.86.484
36. Brown MS, Dana SE, Goldstein JL. Regulation of 3-hydroxy-3-methylglutaryl coenzyme A reductase activity in human fibroblasts by lipoproteins. Proc Natl Acad Sci U S A (1973) 70:2162–6. doi:10.1073/pnas.70.7.2162
37. Endo A, Kuroda M, Tanzawa K. Competitive inhibition of 3-hydroxy-3-methylglutaryl coenzyme A reductase by ML-236A and ML-236B fungal metabolites, having hypocholesterolemic activity. FEBS Lett (1976) 72:323–6. doi:10.1016/0014-5793(76)80996-9
38. Mandal CC, Rahman MM. Targeting intracellular cholesterol is a novel therapeutic strategy for cancer treatment. J Cancer Sci Ther (2014) 6:510–3. doi:10.4172/1948-5956.1000316
39. Sattar N, Preiss D, Murray HM, Welsh P, Buckley BM, De Craen AJ, et al. Statins and risk of incident diabetes: a collaborative meta-analysis of randomised statin trials. Lancet (2010) 375:735–42. doi:10.1016/S0140-6736(09)61965-6
40. Swerdlow DI, Preiss D, Kuchenbaecker KB, Holmes MV, Engmann JE, Shah T, et al. HMG-coenzyme A reductase inhibition, type 2 diabetes, and bodyweight: evidence from genetic analysis and randomised trials. Lancet (2015) 385:351–61. doi:10.1016/S0140-6736(14)61183-1
41. You L, Sheng Z-Y, Tang C-L, Chen L, Pan L, Chen J-Y. High cholesterol diet increases osteoporosis risk via inhibiting bone formation in rats. Acta Pharmacol Sin (2011) 32:1498–504. doi:10.1038/aps.2011.135
42. Li H, Guo H, Li H. Cholesterol loading affects osteoblastic differentiation in mouse mesenchymal stem cells. Steroids (2013) 78:426–33. doi:10.1016/j.steroids.2013.01.007
43. Brittain SB, Gohil SV, Nair LS. Statins as bioactive molecules to support bone regeneration. Curr Med Chem (2014) 21:2980–8. doi:10.2174/0929867321666140601202933
44. Yazawa H, Zimmermann B, Asami Y, Bernimoulin J-P. Simvastatin promotes cell metabolism, proliferation, and osteoblastic differentiation in human periodontal ligament cells. J Periodontol (2005) 76:295–302. doi:10.1902/jop.2005.76.2.295
45. Liu S, Bertl K, Sun H, Liu Z-H, Andrukhov O, Rausch-Fan X. Effect of simvastatin on the osteogenetic behavior of alveolar osteoblasts and periodontal ligament cells. Hum cell (2012) 25:29–35. doi:10.1007/s13577-011-0028-x
46. Wu B, Elmariah S, Kaplan FS, Cheng G, Mohler ER. Paradoxical effects of statins on aortic valve myofibroblasts and osteoblasts implications for end-stage valvular heart disease. Arterioscler Thromb Vasc Biol (2005) 25:592–7. doi:10.1161/01.ATV.0000154278.01871.64
47. Liu M, Wang K, Tang T, Dai K, Zhu Z. The effect of simvastatin on the differentiation of marrow stromal cells from aging rats. Pharmazie (2009) 64:43–8.
48. Viccica G, Vignali E, Marcocci C. Role of the cholesterol biosynthetic pathway in osteoblastic differentiation. J Endocrinol Invest (2006) 30:8–12.
49. Geng Y, Hsu JJ, Lu J, Ting TC, Miyazaki M, Demer LL, et al. Role of cellular cholesterol metabolism in vascular cell calcification. J Biol Chem (2011) 286:33701–6. doi:10.1074/jbc.M111.269639
50. Aghaloo TL, Amantea CM, Cowan CM, Richardson JA, Wu BM, Parhami F, et al. Oxysterols enhance osteoblast differentiation in vitro and bone healing in vivo. J Orthop Res (2007) 25:1488–97. doi:10.1002/jor.20437
51. Kha HT, Basseri B, Shouhed D, Richardson J, Tetradis S, Hahn TJ, et al. Oxysterols regulate differentiation of mesenchymal stem cells: pro-bone and anti-fat. J Bone Miner Res (2004) 19:830–40. doi:10.1359/jbmr.040115
52. Silva J, Beckedorf A, Bieberich E. Osteoblast-derived oxysterol is a migration-inducing factor for human breast cancer cells. J Biol Chem (2003) 278:25376–85. doi:10.1074/jbc.M301233200
53. Huang W, Shang W-L, Li D-H, Wu W-W, Hou S-X. Simvastatin protects osteoblast against H2O2-induced oxidative damage via inhibiting the upregulation of Nox4. Mol Cell Biochem (2012) 360:71–7. doi:10.1007/s11010-011-1045-5
54. Park J-B. Combination of simvastatin and bone morphogenetic protein-2 enhances the differentiation of osteoblasts by regulating the expression of phospho-Smad1/5/8. Exp Ther Med (2012) 4:303–6. doi:10.3892/etm.2012.590
55. Maeda T, Matsunuma A, Kurahashi I, Yanagawa T, Yoshida H, Horiuchi N. Induction of osteoblast differentiation indices by statins in MC3T3-E1 cells. J Cell Biochem (2004) 92:458–71. doi:10.1002/jcb.20074
56. Von Stechow D, Fish S, Yahalom D, Bab I, Chorev M, Müller R, et al. Does simvastatin stimulate bone formation in vivo? BMC Musculoskelet Disord (2003) 4:8. doi:10.1186/1471-2474-4-8
57. Lapmanee S, Charoenphandhu N, Aeimlapa R, Suntornsaratoon P, Wongdee K, Tiyasatkulkovit W, et al. High dietary cholesterol masks type 2 diabetes-induced osteopenia and changes in bone microstructure in rats. Lipids (2014) 49:975–86. doi:10.1007/s11745-014-3950-3
58. Amantea CM, Kim WK, Meliton V, Tetradis S, Parhami F. Oxysterol-induced osteogenic differentiation of marrow stromal cells is regulated by Dkk-1 inhibitable and PI3-kinase mediated signaling. J Cell Biochem (2008) 105:424–36. doi:10.1002/jcb.21840
59. Shouhed D, Kha HT, Richardson JA, Amantea CM, Hahn TJ, Parhami F. Osteogenic oxysterols inhibit the adverse effects of oxidative stress on osteogenic differentiation of marrow stromal cells. J Cell Biochem (2005) 95:1276–83. doi:10.1002/jcb.20497
60. Nelson ER, Dusell CD, Wang X, Howe MK, Evans G, Michalek RD, et al. The oxysterol, 27-hydroxycholesterol, links cholesterol metabolism to bone homeostasis through its actions on the estrogen and liver X receptors. Endocrinology (2011) 152:4691–705. doi:10.1210/en.2011-1298
61. Schilling AF, Schinke T, Münch C, Gebauer M, Niemeier A, Priemel M, et al. Increased bone formation in mice lacking apolipoprotein E. J Bone Miner Res (2005) 20:274–82. doi:10.1359/JBMR.041101
62. Ferber D. Cholesterol drugs show promise as bone builders. Science (2000) 288:2297–8. doi:10.1126/science.288.5475.2297a
63. Sjögren U, Mukohyama H, Roth C, Sundqvist G, Lerner U. Bone-resorbing activity from cholesterol-exposed macrophages due to enhanced expression of interleukin-1α. J Dent Res (2002) 81:11–6. doi:10.1177/154405910208100104
64. Luegmayr E, Glantschnig H, Wesolowski G, Gentile M, Fisher J, Rodan G, et al. Osteoclast formation, survival and morphology are highly dependent on exogenous cholesterol/lipoproteins. Cell Death Differ (2004) 11:S108–18. doi:10.1038/sj.cdd.4401399
65. Edwards CJ, Spector TD. Statins as modulators of bone formation. Arthritis Res (2002) 4:151–3. doi:10.1186/ar399
66. Rubin J, Murphy TC, Rahnert J, Song H, Nanes MS, Greenfield EM, et al. Mechanical inhibition of RANKL expression is regulated by H-Ras-GTPase. J Biol Chem (2006) 281:1412–8. doi:10.1074/jbc.M508639200
67. Ryu J, Kim H, Chang E-J, Kim HJ, Lee Y, Kim H-H. Proteomic analysis of osteoclast lipid rafts: the role of the integrity of lipid rafts on V-ATPase activity in osteoclasts. J Bone Miner Metab (2010) 28:410–7. doi:10.1007/s00774-009-0150-y
68. Grasser W, Baumann A, Petras S, Harwood H, Devalaraja R, Renkiewicz R, et al. Regulation of osteoclast differentiation by statins. J Musculoskelet Neuronal Interact (2003) 3:53–62.
69. Kavanagh KL, Guo K, Dunford JE, Wu X, Knapp S, Ebetino FH, et al. The molecular mechanism of nitrogen-containing bisphosphonates as antiosteoporosis drugs. Proc Natl Acad Sci U S A (2006) 103:7829–34. doi:10.1073/pnas.0601643103
70. Viereck V, Gründker C, Blaschke S, Frosch KH, Schoppet M, Emons G, et al. Atorvastatin stimulates the production of osteoprotegerin by human osteoblasts. J Cell Biochem (2005) 96:1244–53. doi:10.1002/jcb.20598
71. Pelton K, Krieder J, Joiner D, Freeman MR, Goldstein SA, Solomon KR. Hypercholesterolemia promotes an osteoporotic phenotype. Am J Pathol (2012) 181:928–36. doi:10.1016/j.ajpath.2012.05.034
72. Prieto-Potín I, Roman-Blas JA, Martínez-Calatrava MJ, Gómez R, Largo R, Herrero-Beaumont G. Hypercholesterolemia boosts joint destruction in chronic arthritis. An experimental model aggravated by foam macrophage infiltration. Arthritis Res Ther (2013) 15:R81. doi:10.1186/ar4261
73. Oxlund H, Andreassen TT. Simvastatin treatment partially prevents ovariectomy-induced bone loss while increasing cortical bone formation. Bone (2004) 34:609–18. doi:10.1016/j.bone.2003.12.014
74. Sanbe T, Tomofuji T, Ekuni D, Azuma T, Tamaki N, Yamamoto T. Oral administration of vitamin C prevents alveolar bone resorption induced by high dietary cholesterol in rats. J Periodontol (2007) 78:2165–70. doi:10.1902/jop.2007.070181
75. Ahn KS, Sethi G, Chaturvedi MM, Aggarwal BB. Simvastatin, 3-hydroxy-3-methylglutaryl coenzyme A reductase inhibitor, suppresses osteoclastogenesis induced by receptor activator of nuclear factor-κB ligand through modulation of NF-κB pathway. Int J Cancer (2008) 123:1733–40. doi:10.1002/ijc.23745
76. Sondergaard T, Pedersen P, Andersen T, Søe K, Lund T, Østergaard B, et al. A phase II clinical trial does not show that high dose simvastatin has beneficial effect on markers of bone turnover in multiple myeloma. Hematol Oncol (2009) 27:17–22. doi:10.1002/hon.869
77. Chirgwin JM, Guise TA. Molecular mechanisms of tumor-bone interactions in osteolytic metastases. Crit Rev Eukaryot Gene Expr (2000) 10:159–78. doi:10.1615/CritRevEukarGeneExpr.v10.i2.50
78. Udagawa N, Takahashi N, Jimi E, Matsuzaki K, Tsurukai T, Itoh K, et al. Osteoblasts/stromal cells stimulate osteoclast activation through expression of osteoclast differentiation factor/RANKL but not macrophage colony-stimulating factor: receptor activator of NF-kappa B ligand. Bone (1999) 25:517–23. doi:10.1016/S8756-3282(99)00210-0
79. Lin EY, Gouon-Evans V, Nguyen AV, Pollard JW. The macrophage growth factor CSF-1 in mammary gland development and tumor progression. J Mammary Gland Biol Neoplasia (2002) 7:147–62. doi:10.1023/A:1020399802795
80. Sapi E. The role of CSF-1 in normal physiology of mammary gland and breast cancer: an update. Exp Biol Med (2004) 229:1–11.
81. Patsialou A, Wyckoff J, Wang Y, Goswami S, Stanley ER, Condeelis JS. Invasion of human breast cancer cells in vivo requires both paracrine and autocrine loops involving the colony-stimulating factor-1 receptor. Cancer Res (2009) 69:9498–506. doi:10.1158/0008-5472.CAN-09-1868
82. Dincer M, Bulut N, Harputluoglu H, Altundag K. Statins may decrease skeletal-related events in breast cancer patients with bone metastases. Med Hypotheses (2006) 67:1146–7. doi:10.1016/j.mehy.2006.05.009
83. Liu H, Wang Z, Li Y, Li W, Chen Y. Simvastatin prevents proliferation and bone metastases of lung adenocarcinoma in vitro and in vivo. Neoplasma (2012) 60:240–6. doi:10.4149/neo_2013_032
84. Mandal CC, Ghosh-Choudhury T, Dey N, Choudhury GG, Ghosh-Choudhury N. miR-21 is targeted by omega-3 polyunsaturated fatty acid to regulate breast tumor CSF-1 expression. Carcinogenesis (2012) 33(10):1897–908. doi:10.1093/carcin/bgs198
85. Tsubaki M, Satou T, Itoh T, Imano M, Yanae M, Kato C, et al. Bisphosphonate-and statin-induced enhancement of OPG expression and inhibition of CD9, M-CSF, and RANKL expressions via inhibition of the Ras/MEK/ERK pathway and activation of p38MAPK in mouse bone marrow stromal cell line ST2. Mol Cell Endocrinol (2012) 361:219–31. doi:10.1016/j.mce.2012.05.002
86. Babcook MA, Shukla S, Fu P, Vazquez EJ, Puchowicz MA, Molter JP, et al. Synergistic simvastatin and metformin combination chemotherapy for osseous metastatic castration-resistant prostate cancer. Mol Cancer Ther (2014) 13:2288–302. doi:10.1158/1535-7163.MCT-14-0451
87. Ghosh-Choudhury N, Mandal CC, Ghosh-Choudhury N, Choudhury GG. Simvastatin induces derepression of PTEN expression via NFκB to inhibit breast cancer cell growth. Cell Signal (2010) 22:749–58. doi:10.1016/j.cellsig.2009.12.010
88. Thysell E, Surowiec I, Hörnberg E, Crnalic S, Widmark A, Johansson AI, et al. Metabolomic characterization of human prostate cancer bone metastases reveals increased levels of cholesterol. PLoS One (2010) 5:e14175. doi:10.1371/journal.pone.0014175
89. Yavasoglu I, Tombuloglu M, Kadikoylu G, Donmez A, Cagırgan S, Bolaman Z. Cholesterol levels in patients with multiple myeloma. Ann Hematol (2008) 87:223–8. doi:10.1007/s00277-007-0375-6
90. Tirado-Vélez JM, Benítez-Rondán A, Cózar-Castellano I, Medina F, Perdomo G. Low-density lipoprotein cholesterol suppresses apoptosis in human multiple myeloma cells. Ann Hematol (2012) 91:83–8. doi:10.1007/s00277-011-1246-8
91. Jones RJ, Gu D, Bjorklund CC, Kuiatse I, Remaley AT, Bashir T, et al. The novel anticancer agent JNJ-26854165 induces cell death through inhibition of cholesterol transport and degradation of ABCA1. J Pharmacol Exp Ther (2013) 346:381–92. doi:10.1124/jpet.113.204958
92. Yamaguchi T, Sugimoto T, Yano S, Yamauchi M, Sowa H, Chen Q, et al. Plasma lipids and osteoporosis in postmenopausal women. Endocr J (2002) 49:211–7. doi:10.1507/endocrj.49.211
93. Bloch-Thomsen A, Silvestri S, Christiansen C, Bjarnason N. Associated response in bone mineral density and atherogenic lipid profile during treatment with two different selective estrogen receptor modulators: levormeloxifene and raloxifene. Climacteric (2003) 6:159–67. doi:10.1080/cmt.6.2.159.167
94. Tanko L, Bagger Y, Nielsen S, Christiansen C. Does serum cholesterol contribute to vertebral bone loss in postmenopausal women? Bone (2003) 32:8–14. doi:10.1016/S8756-3282(02)00918-3
95. Orozco P. Atherogenic lipid profile and elevated lipoprotein (a) are associated with lower bone mineral density in early postmenopausal overweight women. Eur J Epidemiol (2004) 19:1105–12. doi:10.1007/s10654-004-1706-8
96. Thomsen AB, Silvestri S, Haarbo J, Christiansen C, Bjarnason NH. Associated response in bone and lipids during hormone replacement therapy. Maturitas (2004) 47:39–45. doi:10.1016/S0378-5122(03)00225-1
97. Cui L-H, Shin M-H, Chung E-K, Lee Y-H, Kweon S-S, Park K-S, et al. Association between bone mineral densities and serum lipid profiles of pre-and post-menopausal rural women in South Korea. Osteoporos Int (2005) 16:1975–81. doi:10.1007/s00198-005-1977-2
98. Brownbill R, Ilich J. Lipid profile and bone paradox: higher serum lipids are associated with higher bone mineral density in postmenopausal women. J Womens Health (2006) 15:261–70. doi:10.1089/jwh.2006.15.261
99. Go J-H, Song Y-M, Park J-H, Park J-Y, Choi Y-H. Association between serum cholesterol level and bone mineral density at lumbar spine and femur neck in postmenopausal Korean women. Korean J Fam Med (2012) 33:166–73. doi:10.4082/kjfm.2012.33.3.166
100. Sabour H, Norouzi Javidan A, Latifi S, Hadian MR, Emami Razavi S-H, Shidfar F, et al. Is lipid profile associated with bone mineral density and bone formation in subjects with spinal cord injury? J Osteoporos (2014) 2014:695014. doi:10.1155/2014/695014
101. Li S, Guo H, Liu Y, Wu F, Zhang H, Zhang Z, et al. Relationships of serum lipid profiles and bone mineral density in postmenopausal Chinese women. Clin Endocrinol (2015) 82:53–8. doi:10.1111/cen.12616
102. Indhavivadhana S, Rattanasrithong P. The relationship between bone mineral density and metabolic syndrome in peri-and post-menopausal Thai women. Arch Gynecol Obstet (2015) 292(5):1127–33. doi:10.1007/s00404-015-3698-x
103. Edwards C, Russell R, Spector T. Statins and bone: myth or reality? Calcif Tissue Int (2001) 69:63–6. doi:10.1007/s00223-001-2017-7
104. Schoofs MW, Sturkenboom MC, Van Der Klift M, Hofman A, Pols HA, Stricker BH. HMG-CoA reductase inhibitors and the risk of vertebral fracture. J Bone Miner Res (2004) 19:1525–30. doi:10.1359/JBMR.040607
Keywords: osteoporosis, bone mineral density, bone remodeling, bone metastasis, osteoblast, osteoclast, cholesterol, statins
Citation: Mandal CC (2015) High cholesterol deteriorates bone health: new insights into molecular mechanisms. Front. Endocrinol. 6:165. doi: 10.3389/fendo.2015.00165
Received: 29 May 2015; Accepted: 08 October 2015;
Published: 23 October 2015
Edited by:
Jakob Starup-Linde, Aarhus Universitetshospital, DenmarkReviewed by:
Gudrun Stenbeck, Brunel University, UKArchana Sanjay, University of Connecticut Health Center, USA
Copyright: © 2015 Mandal. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Chandi C. Mandal, Y2hhbmRpY21hbmRhbEBnbWFpbC5jb20=