 Helen Louise Robbins
Helen Louise Robbins Angela Hague
Angela Hague- 1Department of General Surgery, University Hospital Coventry and Warwickshire, Coventry, UK
- 2School of Oral and Dental Sciences, School of Cellular and Molecular Medicine, University of Bristol, Bristol, UK
The phosphatidylinositol 3-kinase (PI3K)/Akt pathway is a key driver in carcinogenesis. Defects in this pathway in human cancer syndromes such as Cowden’s disease and Multiple Endocrine Neoplasia result in tumors of endocrine tissues, highlighting its importance in these cancer types. This review explores the growing evidence from multiple animal and in vitro models and from analysis of human tumors for the involvement of this pathway in the following: thyroid carcinoma subtypes, parathyroid carcinoma, pituitary tumors, adrenocortical carcinoma, phaeochromocytoma, neuroblastoma, and gastroenteropancreatic neuroendocrine tumors. While data are not always consistent, immunohistochemistry performed on human tumor tissue has been used alongside other techniques to demonstrate Akt overactivation. We review active Akt as a potential prognostic marker and the PI3K pathway as a therapeutic target in endocrine neoplasia.
Introduction
The phosphatidylinositol 3-kinase (PI3K)/Akt pathway is recognized as a key pathway in carcinogenesis, with activating mutations in the PIK3CA gene (encoding the p110 catalytic subunit of PI3K) or inactivating mutation in the PTEN gene (encoding phosphatase and tensin homolog deleted on chromosome ten) occurring commonly in diverse human tumors (1). The PI3K/Akt pathway is explained in depth below; however, in brief: the actions of PI3K lead to phosphorylation (and hence activation) of Akt to p-Akt, an effect that is antagonized by PTEN. Akt represents a key signaling node: it phosphorylates a plethora of downstream cytoplasmic and nuclear targets, connecting it to a multitude of interrelated signaling pathways, and therefore it is responsible for modulating multiple processes – including cell survival, cell cycle progression, DNA repair, protein synthesis, glucose metabolism, differentiation, angiogenesis, and cellular migration (2–5). The central role of PI3K/Akt signaling in this complex network of cellular processes makes this pathway of great importance in cancer cells, and indeed p-Akt is known to be overexpressed in a multitude of human cancers, and overexpression appears to be related to poor overall survival in some cancer types (6). The PI3K/Akt pathway is perhaps less well studied in tumors of endocrine tissues than in other, more common, malignancies. Nonetheless, there is growing evidence from both human tumors and animal models that this pathway may play a significant role in tumors of endocrine tissues. In this review, we seek to explore the evidence relating to the role of the PI3K/Akt pathway in tumors of endocrine tissues, with particular focus on evidence from immunohistochemical studies.
PI3K/Akt Signaling Pathway
PI3K/Akt signaling in human cancer can be driven by tyrosine kinase receptors, G-coupled protein receptors, or mutant RAS. PI3K catalyzes the production of the lipid second messenger, phosphatidylinositol (3,4,5) trisphosphate (PIP3) from phosphatidylinositol (4,5) bisphosphate (PIP2). The action of PI3K is antagonized by PTEN, which dephosphorylates PIP3 at the 3′ position, returning it to its inactive form. Akt specifically binds the 3′-phosphorylated inositol lipids via its plekstrin homology domain, hence PIP3 recruits Akt to the cell membrane. Akt–PIP3 binding results in a conformational change that opens up the C-terminal kinase domain of Akt for activation by phosphorylation. The Akt family comprises three highly homologous serine-threonine kinases: Akt1, Akt2, and Akt3. For activation, each isoform requires phosphorylation at equivalent threonine and serine residues in the Akt1/2/3 molecule: first, close to the active site at Thr308/309/305 in a region termed the activation loop mediated by phosphoinositide-dependent kinase 1 (PDK1); and second, in a C-terminal hydrophobic motif at Ser473/474/472 (Figure 1). The kinase responsible for the serine phosphorylation has been debated, and kinases that can phosphorylate at this site include integrin-linked kinase (ILK), protein kinase Cβ2, DNA-dependent protein kinase (DNA-PK), and ataxia telangiectasia mutated (ATM) (7). Akt can also autophosphorylate at this residue if the plekstrin homology domain is exposed (allowing recruitment to the plasma membrane) and the threonine residue (Thr308 in Akt1) is phosphorylated (8). However, the mTORC2 complex is now believed to be the main kinase responsible for phosphorylation at the Ser473/474/472 position.
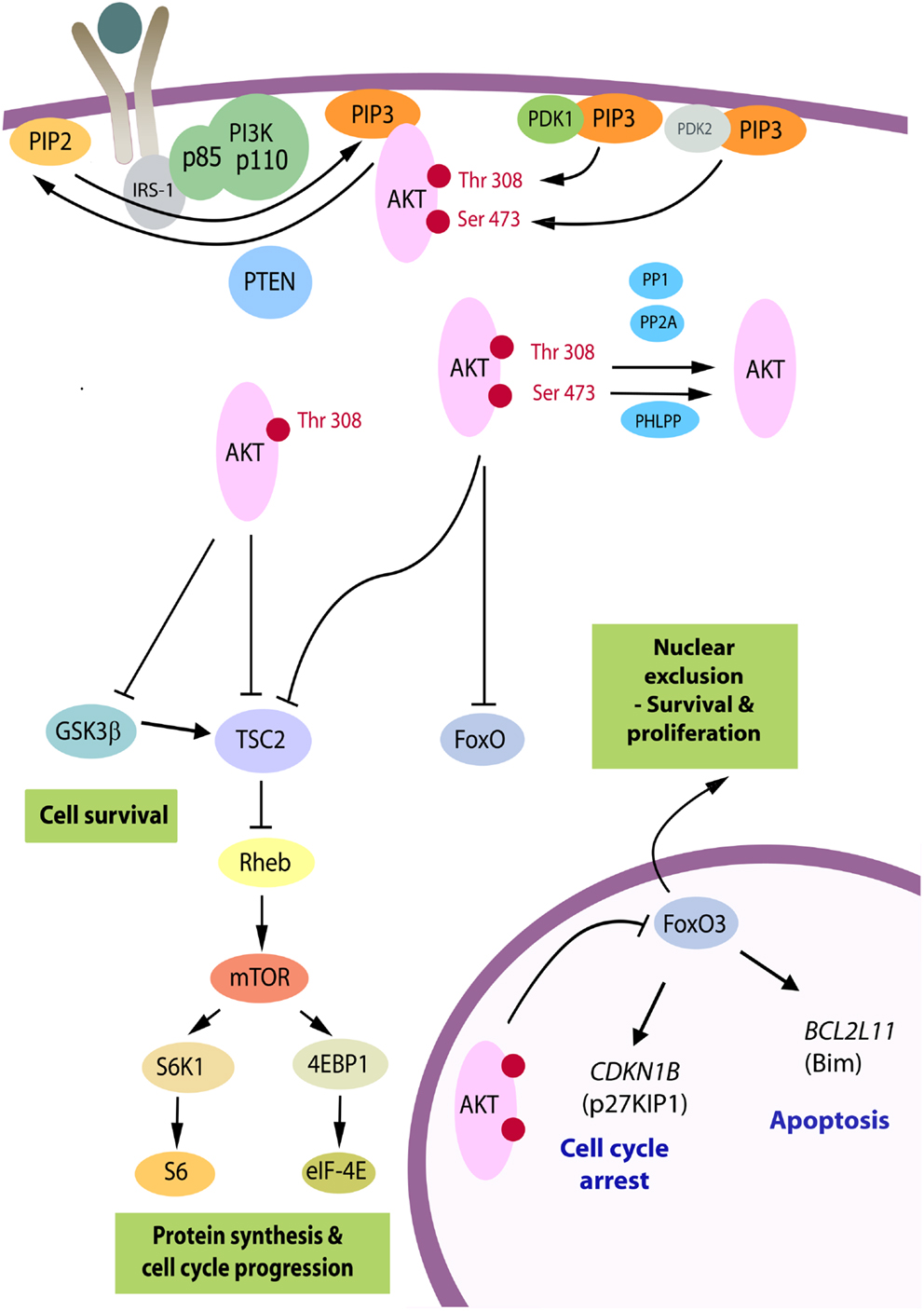
Figure 1. The PI3K/Akt pathway. Akt is recruited to the plasma membrane by phosphatidylinositol (3,4,5) trisphosphate (PIP3) produced from phosphatidylinositol (4, 5) bisphosphate (PIP2) by phosphatidylinositol 3-kinase (PI3K). PTEN is a phosphatase that promotes the reverse reaction. For activation, Akt is phosphorylated on Thr(308) by phosphoinositide-dependent kinase 1 (PDK1) and on Ser(473) primarily by the mTORC2 complex serving as PDK2. Upon full activation, Akt leaves the membrane and can adopt a nuclear or cytosolic localization. There are over 70 known molecular targets of the Akt kinase and the three targets believed to be the most important in carcinogenesis are shown. By phosphorylation of TSC2, Akt relieves its repressive effects on Rheb, resulting in downstream activation of the mTORC1 complex and enhanced RNA translation. GSK3β is a tumor suppressor, which targets a number of proliferation and survival regulators, including β-catenin and Mcl-1, and elevates activity of TSC2 [reviewed in Ref. (9)]. The family of forkhead transcription factors, FoxO, upregulates genes controlling cell cycle arrest and apoptosis and is inhibited by Akt. Whereas GSK3β and TSC2 can be phosphorylated by p-Akt(Thr308) in cells, phosphorylation at Ser(473) is critical to inactivate FoxO proteins [reviewed in Ref. (8)]. When these are phosphorylated they become transcriptionally inactive and subject to nuclear export. FoxO3A exhibits reduced nuclear expression in the majority of thyroid cancers in association with high levels of p-Akt(Ser473), and relevant FoxO3A downstream target genes have been identified in follicular rat thyroid cells (10) and subsequently in benign (FRTL-5) and malignant human thyrocytes (FTC-133) (11), as the CDKN1B gene encoding cyclin-dependent kinase inhibitor, p27KIP1, and BCL2L11, encoding Bim, a pro-apoptotic member of the Bcl-2 family of proteins.
Akt is phosphorylated at multiple sites by PI3K-independent mechanisms, for example tyrosine kinases such as Ack1/TNK2, Src, and protein tyrosine kinase 6 (PTK6) and serine/threonine kinases such as IKKϵ, TANK-binding kinase 1 (TBK1), Mre1/ATM, or DNA-PKcs. These modifications may alter Akt conformation and contribute to cancer development or tumor resistance to inhibition of PI3K [reviewed in Ref. (12)]. Recently, cyclin A/cyclin-dependent kinase-2 complexes have been reported to phosphorylate Akt1 at S477 and T479, stabilizing the C-terminal tail and promoting its kinase activity and ability to protect against apoptosis. Hence, Akt activity may be intimately linked with the cell cycle. S477/T479 phosphorylation may also prime Akt1 for mTORC2-mediated phosphorylation at S473 (13).
Akt is negatively regulated by the Pleckstrin homology domain leucine-rich repeat protein phosphatases 1 and 2 (PHLPP1/2), which selectively dephosphorylate Akt at the Akt1 Ser(473) site, while protein phosphatase PP2A dephosphorylates Akt at the Thr(308) site (8). PHLPP1 and PHLPP2 show relative specificity for different Akt isoforms. For example, in pancreatic adenocarcinoma cells, PHLPP1 selectively dephosphorylated Akt2, whereas PHLPP2 selectively dephosphorylated Akt1 (14).
In addition to its activation at the plasma membrane, Akt can be activated by nuclear pools of PI3K, and PDK1 and DNA-PK (15). In this regard, it is unsurprising that Akt is considered a signaling hub for tumor cells and the PI3K/Akt/mTOR pathway in particular has been the interest of targeted drug development (16).
Mammalian target of rapamycin (mTOR) is a serine/threonine kinase that forms two complexes, mTOR complex 1 (mTORC1) and mTOR complex 2 (mTORC2), implicated in carcinogenesis. Akt removes inhibition of mTORC1 (consisting of mTOR, mLST8, Raptor, PRAS40) by targeting the negative regulators tuberous sclerosis protein 2 (TSC2) and proline-rich Akt substrate of 40 kDa (PRAS40) by direct phosphorylation (4) and by indirect inhibition of TSC2 through GSK3β phosphorylation [reviewed in Ref. (17)]. mTORC1 phosphorylates ribosomal S6 kinase (70 kDa S6K1 or p70RSK), and eIF4E-binding proteins (4E-BPs) to enhance protein synthesis. mTORC2 (consisting of mTOR, mLST8, Rictor, Sin1) can act as the PDK2 for Akt and phosphorylate it at serine 473, but is not thought to function downstream of Akt (17).
There are a number of negative feedback mechanisms that control signaling through the PI3K/Akt/mTOR pathway that need to be considered when designing drug targeting strategies for human cancer. mTOR inhibition induces activation of Akt signaling pathways, through elimination of negative feedback loops. Ultimately, this may lead to growth escape and drug resistance, limiting the effectiveness of mTOR inhibition, and therefore combination treatments could be promising (18–20). The feedback loops that normally quench Akt signaling have been reviewed by Rozengurt et al. (21). mTOR inhibition prevents negative feedback mechanisms; in one of these S6K1 phosphorylates IRS-1 (insulin receptor substrate-1) causing it to dissociate from tyrosine kinase receptors; in another mTORC1 phosphorylates GRB10 which suppresses insulin and IGF receptor kinase activity and facilitates their degradation; and in a third mTORC2 assembly is prevented by phosphorylation of Sin1, mediated by S6K or Akt depending on the cell type. Whereas mTORC1 is sensitive to inhibition by rapamycin (sirolomus), mTORC2 is generally insensitive meaning Akt phosphorylation at Ser473 is enhanced with treatment because of inhibition of the latter feedback loop, although in some cell types long-term treatment with rapamycin can block mTORC2 assembly [reviewed in Ref. (22)]. Active site mTOR inhibitors function against mTORC1 and mTORC2, reducing phosphorylation of Akt at Ser473 but enhancing that at Thr308. Dual active site inhibitors of PI3K and mTOR are therefore useful to damp down the activation of the pathway; however, these elicit compensatory signaling through the MEK/ERK pathway. Hence researchers are currently investigating the potential for combining inhibition of tyrosine kinase receptors with mTOR targeting (21).
Immunohistochemical Analysis of Akt Activation
Immunohistochemistry for p-Akt has been used to assess the relevance and implications of the activation of the PI3K/Akt pathway in human cancers. Activation of Akt may be assessed through use of phospho-specific antibodies, raised against either phospho-Ser473-Akt or phospho-Thr308-Akt. These antibodies recognize all three phosphorylated Akt isoforms (23). Caution needs to be taken in assessing phospho-specific protein detection by immunohistochemistry, since delayed fixation can reduce the efficiency of staining and larger tumors can sometimes appear negative in the center of the fixed sample (24, 25). With this in mind, p-Akt has been used to determine a snapshot of Akt signaling in tumor specimens, with a view to its utility as a prognostic or predictive marker.
Ocana et al. (6) conducted a meta-analysis of reports in the literature of PI3K/Akt activation in solid tumors and overall survival. Eligibility criteria were the availability of survival data for at least 5 years in relation to three types of pathway aberrations (i) mutations in the PI3K gene, (ii) lack of PTEN expression by immunohistochemistry or western-blot, or (iii) evaluation of downstream components of the PI3K/Akt pathway such as Akt itself or downstream targets including phospho-S6, mTOR, phospho-mTOR, or phospho-4EBP1 by immunohistochemistry. Overall, they found an association between defects in the PI3K/Akt/mTOR pathway and poor 5-year survival. While the number of tumor types for which this information was available was small, the association was more marked in gastrointestinal tumors and gynecologic cancers than others. A recent meta-analysis of breast cancer patients showed significant association between p-Akt overexpression and worse overall survival (6,349 patients from 20 studies) and worse disease-free survival (8,683 patients from 24 studies) (26). There was no significant difference between patient groups according to stage of cancer, estrogen receptor status, progesterone receptor status, and HER2 status. Between-study heterogeneity was attributed by the authors at least in part to different scoring methods for p-Akt status and definitions of p-Akt overexpression. They called for a standardized assay methodology, which would help to determine whether Akt inhibition is likely to be an effective targeted cancer treatment.
The number and size of studies investigating the activation of the PI3K/Akt pathway in cancers of endocrine tissues are small and do not include 5-year overall survival data, but consideration of what is known on this subject will help to define the relevance of defects in this pathway to carcinogenesis and future approaches to therapy.
Thyroid Cancer
As the most common cancer of endocrine tissues, thyroid carcinomas are the most well studied. Immunohistochemical studies are corroborated by genetic analysis and animal models to provide strong evidence for an important function of the PI3K/Akt pathway in thyroid carcinogenesis. Thyroid carcinoma includes well-differentiated thyroid cancers of the papillary, follicular, and Hurthle cell types (of which papillary tumors are the most common), and undifferentiated or anaplastic thyroid cancer. Although uncommon, anaplastic thyroid cancer is aggressive with a high mortality rate and is believed to arise by anaplastic transformation from pre-existing follicular and papillary tumors through distinct genetic pathways (27). These cancers are derived from the follicular cells, whereas medullary carcinoma is derived from the C cells of the thyroid. A role for the PI3K/Akt pathway in thyroid carcinogenesis was first suggested by propensity of patients with Cowden’s syndrome, an autosomal dominant multi-organ hamartoma syndrome, to develop thyroid tumors. Germline mutations in PTEN underlie 80% of Cowden’s syndrome cases. Other cases result from germline promoter hypermethylation resulting in transcriptional down-regulation of KILLIN, a pro-apoptotic gene sharing PTEN promoter sequences, or germline mutations and variants in the succinate dehydrogenase genes (SDHx), which result in elevated Akt and MAPK signaling (28), or mutation in either PIK3CA or AKT1 (29).
The Cancer Genome Atlas Network reported that 61.7% of papillary thyroid cancer tumors had BRAF mutations, with a predominance of V600E substitutions (30) and 12.9% had mutations in RAS genes. “BRAFV600E-like tumors” had gene signatures distinctly different to “RAS-like tumors.” The former signal through MAPK to strongly activate ERK, whereas the latter signal through MAPK preferentially via c-RAF and additionally activate the PI3K pathway. The Catalogue of Somatic Mutations in Cancer (COSMIC, 2014, 31, 32) indicates that the incidence of BRAF mutation is strikingly higher in papillary than follicular or anaplastic cancers and that mutations in the RAS genes are more common in papillary thyroid cancers. Mutations in the PI3K/Akt pathway are a feature of follicular and anaplastic tumors and are less frequent in papillary tumors (Figure 2). Ricarte-Filho et al. (33) found BRAF mutation to be more common in radioactive iodine-resistant recurrent or metastatic thyroid carcinomas compared with poorly differentiated primary tumors, which more frequently harbored NRAS mutations. They reported that AKT1 mutations were detected only in metastases and that different metastases from the same individual could exhibit PIK3CA mutations or AKT1 mutations, leading them to suggest that defects in the PI3K/Akt pathway occur during tumor progression, and that use of primary cancer material to stratify patients for specific therapies may not be the most effective strategy. However, mutation tells only part of the story, and changes in gene copy number, expression and protein stability may contribute to over-activation of the PI3K/Akt pathway.
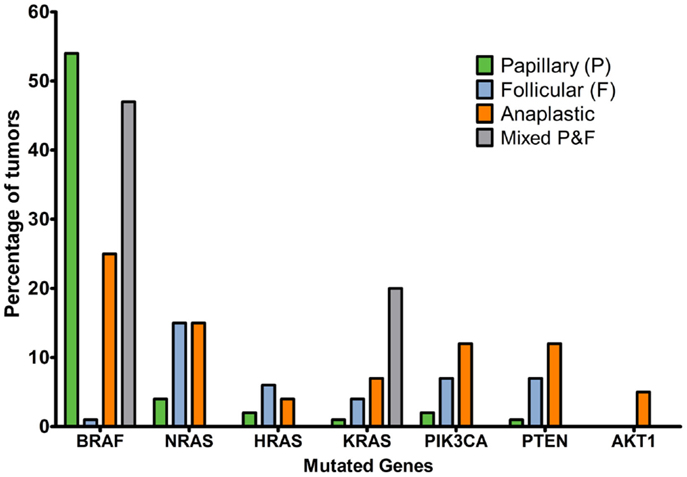
Figure 2. Percentage of tumors with mutations affecting MAPK or PI3K/Akt signaling pathways in thyroid cancer histotypes of follicular cell origin. Data obtained from the COSMIC, 2014 (32) database considering the top 20 mutations (accessed 16/12/2015). Numbers of samples: papillary, 19239; follicular, 253; anaplastic, 564; mixed papillary and follicular, 69.
Animal Models of Thyroid Cancer
Rodent models have provided evidence for elevated PI3K/Akt signaling in thyroid carcinogenesis. PTEN+/− mice serve as a model of Cowden’s disease and develop diverse types of tumor including prostate, endometrium, thyroid, adrenal medulla, and small intestine tumors. Chen et al. (34) crossed PTEN+/− mice with Akt1−/− mice to determine whether this would attenuate the propensity for tumor development. While the most marked reduction in tumor incidence was observed in the prostate, endometrium, and small intestine, Akt deficiency significantly reduced the incidence of thyroid tumors and high grade tumors of the adrenal medulla. Mice in which thyroid follicular cell specific PTEN deletion is achieved by thyroid peroxidase Cre-mediated recombination exhibit a marked increase in follicular cell p-Akt(Ser473) immunoreactivity and proliferation and develop follicular adenomas (35). The Akt target FoxO1, the mTOR target p70S6K1, and downstream S6 were highly phosphorylated in the mutant thyroids. Although PTEN+/−PHLPP1−/− mice are available (36), the role of PHLPP in thyroid and adrenal medulla carcinogenesis has yet to be explored. Neither have any studies addressed PHLPP expression in human thyroid cancer.
The TRPV mouse model makes use of the fact that the thyroid hormone receptor (TR) is a tumor suppressor for thyroid cancer (37). The PV mutation results in loss of thyroid hormone binding to the receptor and transcriptional activity and a knock-in mouse carrying two mutant TR alleles (TRβPV/PV) exhibited spontaneous follicular thyroid carcinomas, while TRβPV/+ heterozygous mice did not. Kim et al. (38) examined subcellular localization of Akt in the thyroid of such mice by both immunohistochemistry and confocal microscopy. All three Akt isoforms were overexpressed in the thyroid cancers of older TRβPV/PV mice compared to age-matched wild-type controls. In primary tumors, Akt1 was chiefly in the nucleus, Akt2 and Akt3 were primarily cytosolic and p-Akt(Ser473) was both nuclear and cytoplasmic. In metastases, all three Akt isoforms and p-Akt(Ser473) were primarily nuclear. Functional studies of the PI3K/Akt pathway were carried out by treating primary TRβPV/PV thyroid cells with the PI3K inhibitor LY294002 or transducing them with dominant negative Akt, both of which reduced cell motility in transwell assays.
Pringle et al. (39) generated mice with thyroid-specific knockout of both PRKAR1A and PTEN genes. PRKAR1A encodes the type 1A regulatory subunit of protein kinase A (PKA); when this gene is inactivated, PKA activity is enhanced. Single PRKAR1A knockout (R1a-TpoKO) mice develop thyroid cancer, but do not exhibit metastases. By contrast, the PRKAR1A-PTEN double knockout (DRP-TpoKO mice) developed aggressive follicular carcinomas, which frequently metastasized to the lungs. PTEN single knockout thyroids (pten-TpoKO) only developed benign follicular hyperplasia, but there was diffuse nuclear and cytoplasmic p-Akt staining and increases in phosphorylated mTOR and p70S6K. p-Akt staining in DRP-TpoKO tumors exhibited membranous localization. Pringle et al. (39) hypothesized that PKA may redirect Akt to a membranous localization and that, independently, PKA may contribute to the activation of mTOR. This is consistent with their immunohistochemical analysis suggesting that a higher proportion of follicular carcinomas overexpress p-p70S6K than p-Akt, although it would be useful to expand the number of follicular carcinomas analyzed.
While most studies of p-Akt have utilized genetic mouse models, a rat two-stage chemically induced thyroid cancer model has also been informative. The thyroid carcinogen N-bis(2-hydroxypropyl)nitrosamine followed by promotion using sulfadimethoxine for 10 and 15 weeks was sufficient to induce focal follicular cell hyperplasia, follicular cell adenomas, and carcinomas (40). This study compared staining using Akt1, Akt2, p-Akt(Ser473), and p-Akt(Thr308) antibodies as well as antibodies to PTEN, p-PTEN, GSK3β, and p-GSK3β(Ser9). Staining intensity and incidence for Akt1 and Akt2 were increased in carcinomas compared to focal follicular cell hyperplasia, as was p-Akt(Ser473) and p-Akt(Thr308). PTEN and GSK3β are both inactivated by phosphorylation. Although PTEN staining was higher, so was staining for inactive p-PTEN, suggesting that inactive p-PTEN accumulation may underlie Akt activation in these tumors. The elevation of p-GSK3β(Ser9) staining intensity observed in the tumors was further indication of increased Akt activation.
Evidence for Akt Activation in Human Thyroid Carcinoma
There is growing evidence from examination of Akt phosphorylation that Akt signaling has a role in sporadic thyroid cancers [reviewed in Ref. (40, 41)]. Ringel et al. (42) examined levels of p-Akt(Ser473) and Akt1, Akt2 and Akt3 in 8 follicular thyroid cancers, 9 papillary thyroid cancers, and 11 normal thyroid samples by western blotting and observed increased Akt1, Akt2 and p-Akt(Ser473) in follicular but not papillary cancers. By contrast, also using western blotting, Miyakawa et al. (43) compared seven matched pairs of papillary thyroid cancer and adjacent normal tissue and found total Akt levels unchanged, but there was significantly higher p-Akt in tumor tissue compared to surrounding tissues (five of seven pairs), with elevated levels of activated downstream targets p-Bad and p-p70S6K. Mandal et al. (44) also found elevated levels of p-Akt(Ser473) in six out of eight papillary thyroid cancers compared to adjacent normal tissue, whereas total Akt levels were similar. Krzeslak et al. (45) used an ELISA method, and found enhanced levels of cytoplasmic Akt1 in 23 thyroid carcinomas compared with 16 non-neoplastic lesions (nodular goiters). The majority of Akt1 was cytosolic in differentiated cancers whereas two of three anaplastic cancers showed lower cytosolic and higher nuclear Akt1 expression. However, when Akt1 immunoprecipitates were examined by immunoblotting, the ratio of p-Akt(Ser473) to total Akt1 was lower in cancers compared to normal tissues (although only a subset of the tumors were shown to be analyzed in this way and it is unclear if the anaplastic cancers with high nuclear Akt1 were included). On this basis, the authors proposed that either Akt2 or Akt3 might be activated in thyroid cancers rather than Akt1.
Levels of PTEN regulate the extent to which Akt is phosphorylated. Exogenous overexpression of PTEN in ARO or FB-1 cells caused decreased p-Akt(Ser473) and increased p27KIP1 expression leading to cell cycle arrest with no evidence of apoptosis. In a panel of 13 human thyroid carcinomas, there appeared to be an inverse correlation between PTEN and p-Akt(Ser437) levels as measured by western blotting, although with low numbers this was not quantified and analyzed statistically (46). Gimm et al. (47) stained 139 benign and malignant non-medullary thyroid tumors for PTEN and measured loss of heterozygosity in 92 tumors informative at the PTEN locus. This generated a picture of progressive loss of staining and increased frequency of PTEN allelic loss with progression to malignancy and an aggressive phenotype.
Results of p-Akt immunohistochemistry studies of human thyroid cancers are summarized in Table 1. Papillary, follicular, and anaplastic thyroid cancers exhibit increased p-Akt(Ser473) expression compared with normal tissue (48–50). One study reported clear p-Akt(Ser473) staining in only a minority or thyroid cancers, but this discrepant result appears to be due to cytoplasmic staining being judged as non-specific in this report (51). Detection of p-Akt(Ser473) in benign follicular adenomas is uncommon, but it is detectable in atypical cells of atypical adenomas, and frequent in follicular thyroid cancer – suggesting that Akt activation may play a role in the malignant phenotype (48, 52). This is further supported by the finding that p-Akt(Ser473) expression is greater at the invasive front of cancers, and also increased in cells invading the vasculature (48, 53).
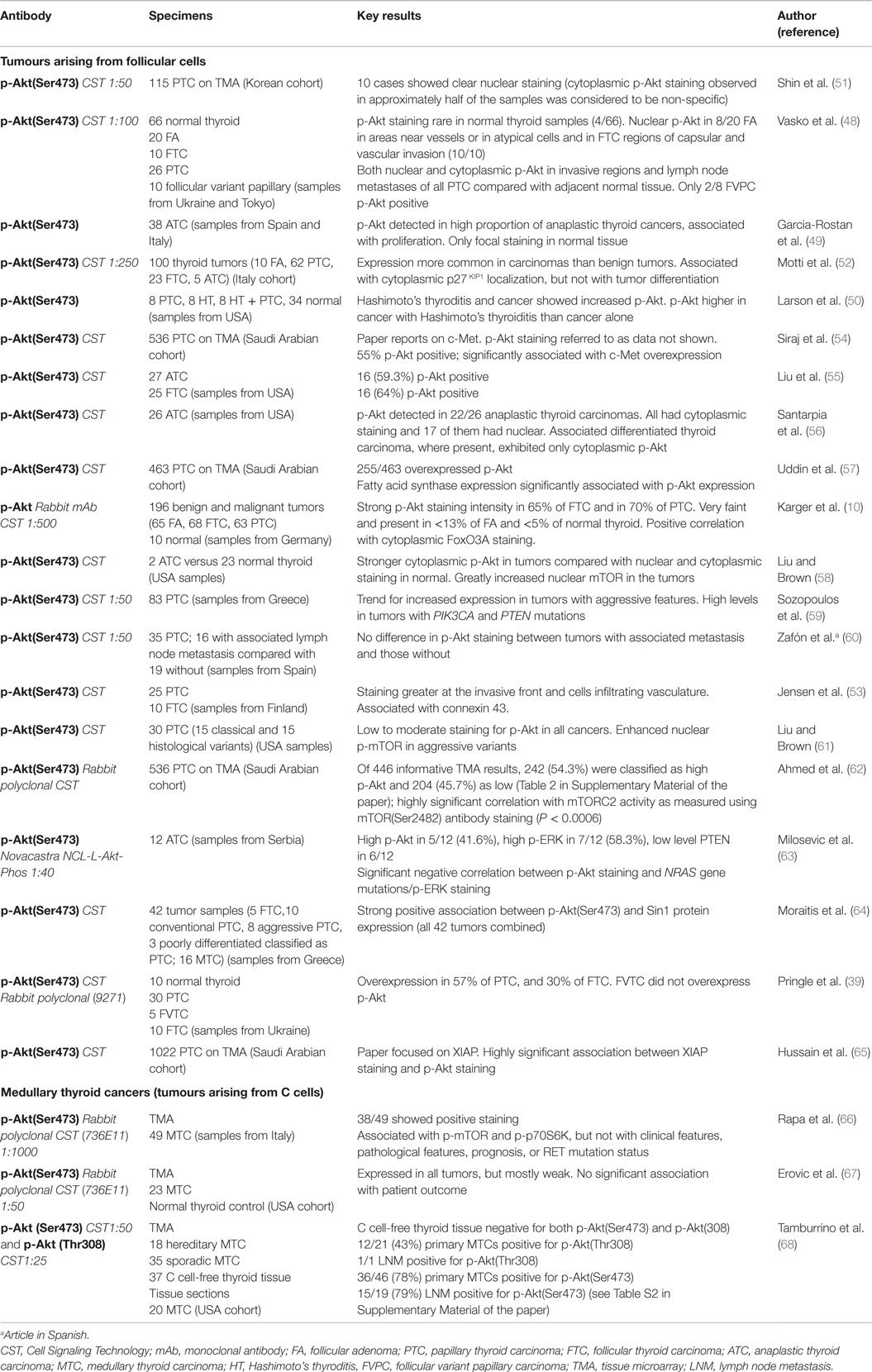
Table 1. Results of Immunohistochemical staining studies for p-Akt(Ser473) in thyroid tumors.
Liu et al. (55) observed p-Akt(Ser473) immunoreactivity in 16/25 follicular thyroid cancers, and found genetic alterations in the PI3K/Akt pathway to account for all of these, including copy gains of PIK3CA, PIK3CB, and AKT2. In anaplastic cancers 16/27 showed p-Akt(Ser473) immunoreactivity. The vast majority (95.8%) of anaplastic thyroid cancers had genetic alterations resulting in activation of both the PI3K and MAPK pathways, including frequent co-existence of receptor tyrosine kinase copy number gain and gene mutation, as well as PIK3CA, PIK3CB, and PDK1 copy number gains, PTEN mutation and BRAF mutation. Furthermore, RAS activation can result in activation of the PI3K/Akt pathway in thyroid carcinoma (69). However, a recent study of 12 anaplastic carcinomas reported that p-Akt(Ser473) staining inversely correlated with p-ERK staining and NRAS gene mutations, perhaps suggesting that only one of these two signaling pathways might drive progression to anaplastic tumors (63).
Overexpression of upstream receptors can elevate levels of p-Akt in thyroid cancers. Liu et al. (55) found that EGFR and VEGF1 copy number gain was particularly common in both follicular and anaplastic thyroid cancer. Rearranged during transfection (RET) is a tyrosine kinase receptor proto-oncogene, point mutation of which underlies Multiple Endocrine Neoplasia Types 2A and 2B (MEN2A/MEN2B) and familial medullary thyroid cancer. Some papillary thyroid carcinomas, including those arising after ionizing radiation exposure, develop through translocations resulting in fusion of RET with 1 of 13 other genes (RET/PTC rearrangements) (70) producing a protein with constitutively active tyrosine kinase signaling. There is evidence to show that a MEN2A-type mutant RET results in constitutive activation of the PI3K/Akt pathway (71), and papillary cancers with RET overexpression due to gene rearrangements demonstrated the greatest p-Akt levels and throughout the tumors (48). c-Met, the hepatocyte growth factor receptor, was associated with p-Akt expression in papillary thyroid carcinomas in a Middle Eastern population in which PIK3CA mutation was rare (54). The tyrosine kinase receptor EphA2 is overexpressed in benign and malignant thyroid tumors and has been shown to contribute to p-Akt activation (72).
Downstream targets have been associated with high levels of p-Akt(Ser473). The majority of thyroid cancers exhibit reduced nuclear FoxO3A (10), and FoxO3A downstream target genes CDKN1B encoding cyclin-dependent kinase inhibitor p27KIP1, and BCL2L11 encoding Bim, a pro-apoptotic member of the Bcl-2 family of proteins, have been identified as functionally relevant in follicular rat thyroid cells (10) (Figure 1). Inhibition of PI3K/Akt signaling by LY294002 resulted in reduced FoxO3A phosphorylation and its accumulation in the nucleus, and increased p27KIP1 and Bim mRNA in vitro. Vasco et al. (48) reported that invasive regions of papillary and thyroid cancer were characterized by both nuclear p-Akt and nuclear exclusion of p27KIP1 and used a human papillary thyroid cancer cell line (NPA) to link p-Akt and cytoplasmic exclusion of p27KIP1 with invasive capacity in a transwell assay. In addition, Bad and XIAP have been noted as relevant Akt targets in papillary thyroid carcinoma cells (62, 65), contributing to protection from apoptosis.
Enhanced nuclear p-mTOR suggests the activation of the PI3K/Akt/mTOR pathway in aggressive variants of papillary thyroid carcinomas (61). The Al-Kuraya group have performed tissue microarray immunohistochemistry for p-Akt(Ser473) and related proteins on a large series of papillary thyroid carcinomas of Middle Eastern origin, enabling correlative expression studies to be performed (albeit with some loss of informative spots on different staining runs). In a recent study, p-mTOR(Ser2448) was used as a marker of mTORC1 activity and was detected in 81% of tumors, while p-mTOR(Ser2481), a marker for mTORC2 activity was observed in 39%. There was a strong positive correlation with p-Akt(Ser473) and p-mTOR(Ser2481) staining, as well as activation of further Akt and mTORC1 target proteins. As evidence for the concept that both mTOR complexes are actively involved in regulating the survival of papillary thyroid carcinoma cells, Torin 2, a dual mTORC1/2 inhibitor, induced apoptosis in two papillary carcinoma cell lines, BCPAP and TPC, in association with reduced activity of Akt and downstream targets of p-Akt and mTOR complexes (62).
Strong positive association has been observed between staining for p-Akt(Ser473) and Sin1 protein, a component of the mTORC2 complex, in a series of 42 tumor samples, which included 16 medullary thyroid cancers (see below). This was a characteristic of aggressive papillary and medullary carcinomas and therefore further staining of larger cohorts for p-Akt in conjunction with Sin1 is worthwhile, since normal thyroid tissues showed no expression or <10% weakly Sin1 positive cells (64). In these cells, S6K appeared to be the relevant Sin1 kinase. In a series of 58 ovarian cancer specimens, p-Akt(Ser473) and p-Sin1(Thr86) staining were, if anything, negatively correlated, but this did not reach statistical significance. The generation of the phospho-specific Sin-1 antibodies, along with the discovery that mutation of the Sin1 phosphorylation sites occurs naturally in ovarian and skin cancers and abrogates the negative control on Akt activity (73), could be helpful in determining the mechanisms for the positive association between p-Akt(Ser473) and Sin1 in thyroid cancers.
Whether p-Akt(Ser473) expression is associated with actual patient prognosis has yet to be measured in thyroid cancer, and only inferences can be made from the literature. p-Akt(Ser473) was not associated with likely prognosis of papillary carcinoma (59). In a more recent study, the Al-Kuraya group stained for the anti-apoptotic protein XIAP on tissue microarray. Of 989 tumors producing informative staining for p-Akt(Ser473), 57.1% exhibited staining classed as high, and there was a highly significant positive association with staining for XIAP (65). XIAP staining was associated with poor disease free survival, but the study did not report on p-Akt directly in relation to prognosis.
Patterns of p-Akt staining have been reported to vary between follicular and papillary carcinomas, but more prominent nuclear p-Akt(Ser473) was reported in regions of invasion in both types (48). Similarly, while differentiated thyroid cancers demonstrate cytoplasmic staining, contiguous anaplastic thyroid cancers display both nuclear and cytoplasmic p-Akt(Ser473) (56). These observations may suggest a role of nuclear p-Akt in aggressive or invasive disease. This hypothesis is supported by findings that nuclear p-Akt is associated with migration in vitro (48), and that in the TRPV mouse model, p-Akt is both cytoplasmic and nuclear in primary tumors, but is predominantly nuclear in metastases (38). By contrast to these studies, Zafón et al. (60) did not find any difference in cytoplasmic or nuclear p-Akt(Ser473) staining between papillary thyroid cancers with or without associated lymph node metastases at the time of diagnosis, even though the group with associated metastases had a greater proportion overexpressing RET or EGFR.
Medullary Thyroid Cancer
p-Akt(Ser473) expression has also been investigated in medullary thyroid cancer, which unlike other subtypes of thyroid cancer is derived from parafollicular C-cells. Evidence from hereditary cancers and in vitro work suggests that the PI3K/Akt pathway may play a role in medullary thyroid carcinogenesis (74). Only three studies have so far reported p-Akt staining in human medullary thyroid cancers, and these used tissue microarrays (Table 1). p-Akt(Ser473) expression was strongly associated with p-mTOR and p-p70S6K in a series of 49 cancers, suggesting that the Akt/mTOR pathway is active in these tumors, and is therefore potentially amenable to therapy (66), but there was no association between p-Akt expression and prognosis or clinicopathological factors (66). Furthermore, in a study of 23 cancers, p-Akt(Ser473) and mTOR staining were predominantly weak, with no association between p-Akt(Ser473) and overall or disease-free survival (67). However, Tamburrino et al. (68) found significant correlations between p-Akt(Ser473) and p-ERK staining and Pennelli et al. (75) also remarked on the preferential activation of the PI3K/Akt pathway, detected as high levels of p-Akt(Ser473) on western blotting in medullary thyroid carcinoma cases which had mutant RAS.
In the Tamburrino study (68), a series of 53 human medullary thyroid carcinoma tissues (18 hereditary, 35 sporadic), ribosomal protein p-S6(Ser235/236) was frequently overexpressed compared to normal thyroid tissues. It should be noted that the normal thyroid used was largely C cell-free, and therefore the malignant cells were not being compared with their normal counterparts; however, other reports make no mention of positive C-cells in control normal thyroid tissue, which is classed as negative. Of the medullary tumors, 76% had detectable p-Akt(Ser473) and 96% expressed detectable levels of S6K. That S6K is more frequently detected than p-Akt is similar to findings of Pringle et al. (39) in their mouse model of follicular thyroid carcinoma. By contrast to p-Akt(Ser473), only 39% of tumors had detectable p-Akt(Thr308), perhaps due to difference in the sensitivity of the antibodies, as suggested by the authors, although far fewer tumors were stained for p-Akt(Thr308) than p-Akt(Ser473). A further relevant Akt target in medullary thyroid carcinoma may be the tumor suppressor, Programed Cell Death 4 (PDCD4), which Akt inactivates, which showed reduced expression in medullary thyroid carcinoma (75). Overall, immunohistochemistry results support previous observations of elevated Akt/mTOR signaling in medullary thyroid carcinoma. Furthermore, rapamycin reduced tumor volume of RET mutant MZ-CRC-1 xenografts in NOD/SCID mice (68).
Parathyroid Carcinoma
Parathyroid carcinomas are rare, accounting for <1% of primary hyperparathyroidism (76). These cancers are aggressive, with high risk of metastasis and recurrence. Parathyroid cancers remain poorly understood, and surgery remains the mainstay of treatment; little is known about the role of PI3K/Akt signaling in these tumors. p-Akt expression has been described in a cohort of 10 parathyroid carcinomas and 25 parathyroid adenomas (Table 2). In this study, p-Akt expression was reported to be variable; however, expression appeared to be lower in carcinomas: on average 28% of tumor cells were positive in carcinomas, with 87% positive in adenomas, although no statistical analysis was presented (77). p-mTOR and FoxO1 expression were also examined in this cohort, and the authors suggested that there was no marked alteration in their expression in parathyroid carcinoma compared to adenoma (77), but the exact picture remains unclear. The PI3K/Akt pathway has, to the best of our knowledge, remained otherwise unstudied in parathyroid neoplasia. However, mutation analysis from a single patient has identified an activating mutation in PIK3CA from the primary tumor, and RNA-seq datasets demonstrated overexpression of AKT2 mRNA in one of two areas of recurrence (78), perhaps suggesting that this pathway may be an important area for future research.
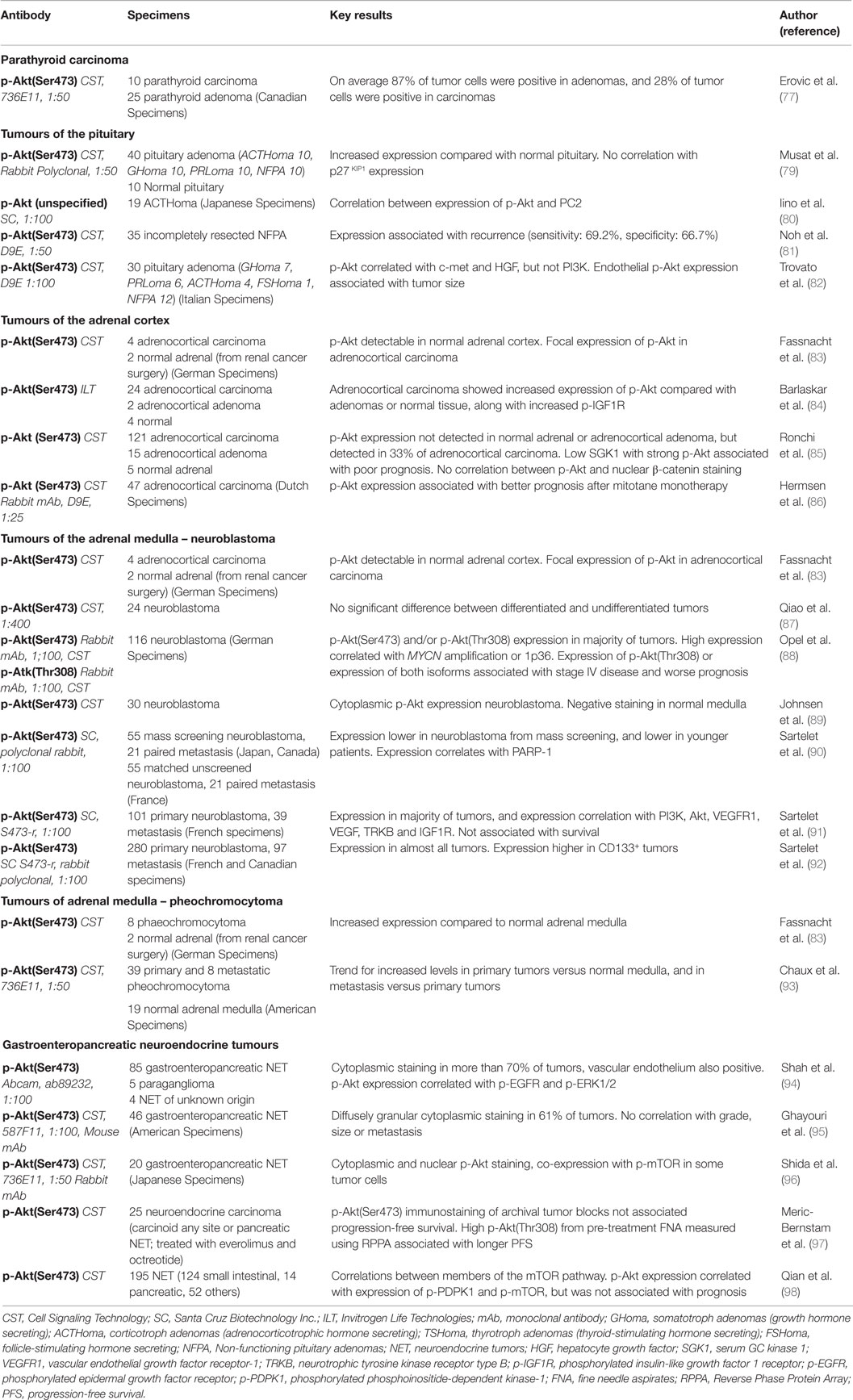
Table 2. Results of Immunohistochemical staining studies for p-Akt(Ser473) in non-thyroid tumors of endocrine tissue origin.
Pituitary Tumors
Pituitary adenomas are among the most common intracranial neoplasms. Approximately 60% of pituitary tumors are functionally active, of which prolactinomas [prolactin (PRL) secreting tumors] are the most common subtype. The remaining functional tumors are somatotroph adenomas [growth hormone (GH) secreting], corticotroph adenomas [adrenocorticotrophic hormone (ACTH) secreting], thyrotroph adenomas [thyroid-stimulating hormone (TSH) secreting], and gonadotroph adenomas [follicle-stimulating hormone (FSH) and luteinizing hormone (LH) secreting]. Non-functioning pituitary adenomas (NFPA) do not exhibit hypersecretion. Of note, some pituitary adenomas are endocrinologically “silent”: they produce hormones that are not secreted (99). Malignancy is rare, however, local invasion, recurrence, or treatment failure occur in a proportion of cases.
The PI3K/Akt pathway plays a role in maintaining viability of rat pituitary adenoma cell lines (GH3, LβT2) (100, 101), suggesting a potential role for Akt in pituitary tumorigenesis. This is further supported by work in TRβPV/PV mice and MENX (multiple endocrine neoplasia-like syndrome) rats. TRβPV/PV mice possess a knock-in mutation of the TRβ gene and spontaneously develop TSH-secreting pituitary adenomas (TSHoma) with age (102); MENX rats harbor biallelic frameshift mutations of the CDKN1B gene (encoding p27KIP1) and develop pituitary adenomas alongside multiple other endocrine tumors (103). Pituitary adenomas from TRβPV/PV mice show increased phosphorylation of Akt (Ser473), mTOR and S6 on western blotting, and PI3K/Akt inhibition decreases pituitary tumor mass and increases apoptosis (102). Likewise, immunohistochemistry demonstrated increased levels of p-Akt(Ser473) in pituitary adenomas from MENX rats, and increased p-Akt(Ser473), p-Akt(Thr308), and p-S6(Ser24/44) in primary cultures of tumor cells (104). The MENX rat model has been useful in demonstrating anti-proliferative and pro-apoptotic function of the dual PI3K/mTOR inhibitor NVP-BEZ235 in vivo and in organotypic culture (105).
Compared with normal tissue, human pituitary adenomas showed an increased percentage of cells immunostaining for p-Akt(Ser473) on immunohistochemistry, and increased p-Akt:total Akt ratios on western blotting suggest that Akt phosphorylation is increased in these tumors (79). Likewise, in a study of three gonadotropinomas, levels of p-Akt(Thr308) were elevated in protein lysates of tumors versus normal pituitaries (101), but to our knowledge there have been no larger studies examining Akt phosphorylation at Thr308 in human pituitary tumors. Few studies have investigated the prognostic role of p-Akt in pituitary tumors. However, in partially resected NFPA, p-Akt(Ser473) immunostaining was associated with tumor recurrence, suggesting that Akt phosphorylation may be of use in risk stratification in these cases (81). A recent study has also revealed that endothelial expression of p-Akt(Ser473) in pituitary tumors is positively associated with tumor size, leading the authors to hypothesize that Akt activation may play a role in angiogenesis in these tumors (82). A study of ACTH-producing pituitary adenomas has revealed significant associations between the immunostaining for p-Akt(Ser473) and prohormone convertase 2 (a serine protease involved in proteolytic cleavage of prohormones), but the relevance and implications of these findings remain unclear (80).
The mechanism for Akt over-activation in pituitary tumors is likely to be multifactorial. PIK3CA amplifications or mutations occur in around a third of pituitary tumors (106, 107), and PIK3CA mutations are associated with invasive disease (106). It is well established that there is altered or overexpression of growth factors and growth factor receptors in pituitary tumors, for example alterations in epidermal growth factor receptor (EGFR) and fibroblast growth factor receptor (FGFR) signaling, which could also drive Akt activation [reviewed in Ref. (108)]. In particular, Eps8 (Epidermal Growth Factor Receptor Pathway Substrate 8), which signals downstream of these receptors, promoted cell survival through modulation of Akt activity in Eps8-overexpressing LβT2 gonadotrope cells in vitro, and immunoblotting demonstrated that both p-Akt(Thr308) and Eps8 are overexpressed in human pituitary adenomas (101). Hepatocyte growth factor receptor (HGFR/c-met) signaling may act as a further driver for Akt activation: immunohistochemistry has demonstrated significant association between p-Akt(Ser473), c-met and HGF in human pituitary adenomas (82). The role for PTEN is less clear: while no PTEN mutations were identified in 33 pituitary tumors, and PTEN mRNA levels were comparable to normal tissue, nuclear PTEN expression was reduced in pituitary tumors (79). Investigations are needed to determine whether this results in activation of nuclear Akt pools. Interestingly, it has also been suggested that altered levels of microRNAs may play a role in pituitary tumorigenesis by modulating PI3K/Akt signaling via regulation of PTEN expression (109).
Downstream PI3K/Akt signaling in human pituitary tumors remains relatively poorly understood. Nuclear p27KIP1 is reduced in human pituitary adenomas, potentially consistent with cytoplasmic sequestration of p27KIP1 by Akt activity; however, the lack of correlation between p-Akt(Ser473) and p27KIP1 expression means conclusions must remain cautious (79). Pituitary adenomas have also been reported to have increased levels of mTOR activation, as measured by phosphorylation of downstream S6, with GHomas showing the most marked elevation of mTOR activity (110). However, mTOR activity appeared to be independent of PI3K/Akt signaling in pituitary adenoma cells on primary culture, and mTOR activity was not related to clinicopathological characteristics in the sample of 53 pituitary adenomas (110). Increased Cyclin D1 expression has been consistently reported in non-functioning pituitary tumors (111, 112). This is partially due to Cyclin D1 (CCND1) gene amplification, which occurs in around 25% of pituitary adenomas (111). Overexpression of Cyclin D1 in tumors without gene amplification may suggest upstream pathways such as PI3K/Akt signaling (112). However, although reduced levels of double phosphorylated c-Myc (Thr62/Ser58) are potentially consistent with activation of the PI3K/Akt pathway, phosphorylated and total TSC2 and mTOR levels were unchanged (112).
Targeting PI3K/Akt/mTOR signaling is of potential therapeutic interest in human pituitary tumors. mTOR inhibition reduces viability of human pituitary tumor cells in vitro (113). Interestingly, in human NFPA cells, a response to everolimus was more likely in cells originating from younger patients, of female gender, with invasive macroadenomas (113). As in other cell types, mTOR inhibition induces Akt activation in pituitary cells in vitro, and this may contribute to resistance to mTOR inhibitors, which may limit their clinical efficacy (114). Therefore, concomitantly targeting the upstream PI3K/Akt pathway is potentially promising: combining rapamycin with octreotide (a somatostatin analog) more effectively suppresses pituitary tumor cell proliferation than rapamycin alone, with combination treatment proving effective in NFPA cells resistant to single agent rapamycin (114). Likewise, in primary cultures of pituitary tumor cells from MENX rats, cell viability is more effectively reduced by the dual PI3K/mTOR inhibitor NVP-BEZ235 than by everolimus alone (104). Targeting the PI3K/Akt/mTOR pathway could also be combined with more traditional treatment modalities in pituitary tumors, potentially increasing efficacy and/or reducing toxicity by allowing dose reduction: mTOR inhibitors increase radiosensitivity of pituitary tumor cells in vitro (115), and temozolomide and XL765 (a dual PI3K/mTOR inhibitor) synergistically inhibit cellular proliferation in vitro and reduce tumor growth and hormone secretion in xenograft mice (116). Despite the promising preclinical work, experience in human pituitary tumors is limited to an isolated report of unsuccessful treatment with everolimus in a single patient with a pituitary carcinoma (a rare entity defined by presence of metastasis), and as such, no clear conclusions can be made about efficacy of PI3K/Akt/mTOR inhibition in human pituitary tumors (117).
Adrenal Cortex
Most adrenocortical tumors are sporadic, but adenomas or carcinomas may occur in association with familial cancer syndromes (118). Adrenocortical carcinomas (ACC) are generally highly aggressive tumors, with an incidence of approximately 1–2 cases per million per year (119). Complete surgical resection is potentially curative; however, recurrence is relatively common. Treatment of advanced disease remains challenging: mitotane plus etoposide, doxorubicin and cisplatin is the first-line cytotoxic therapy but response rate is relatively poor (119). The pathophysiology of these rare tumors remains poorly understood, but a greater understanding of key signaling pathways may provide much-needed treatment options.
Evidence suggests that IGF signaling may be of great clinical interest in ACC. Patients with Beckwith–Wiedemann Syndrome, in whom deregulated imprinting at 11p15.5 leads to biallelic expression of IGF2, have a 5% incidence of ACC (118). Furthermore, IGF2 is the most overexpressed gene in a large proportion of sporadic ACC and on transcriptome analysis forms part of a cluster of eight overexpressed genes characterizing malignant ACC rather than prevalent adrenocortical adenomas (ACA) (120). Early clinical trials of IGF-1R inhibitors showed potentially promising results in these rare cancers (121, 122), but unfortunately, the IGF-1R and insulin receptor inhibitor linsitinib showed limited therapeutic benefit in a recent phase III trial (123). Nonetheless, the majority of these tumors possess overactivation of the IGF pathway and/or enhanced EGFR signaling (124, 125), which could serve as further driver of Akt activation. Altered β-catenin expression may also regulate Akt activation in these tumors: β-catenin expression is known to be altered in 20% of ACC, and knockdown of β-catenin in human ACC cells in vitro decreases Akt phosphorylation (126), although no correlation was seen between p-Akt(Ser473) and β-catenin immunostaining in human tumors (85).
Akt activation appears to be enhanced in a subset of human ACC in vivo (Table 2). An early study by the Allolio group showed that total Akt and p-Akt(Ser473) were detectable in the adrenal cortex but not in the medulla, and ACC exhibited focal enhancement of p-Akt(Ser473), potentially suggesting local activation of Akt signaling in these cancers (83). The results from western blotting did not necessarily correlate with results obtained by immunohistochemistry, but this perhaps reflected the focal expression of p-Akt and also areas of stromal and necrotic tissue which are both negative for total Akt and p-Akt in ACC (83). A more recent study found elevation of both total Akt and p-Akt(Ser473) in six adrenocortical adenomas and four ACCs compared to normal adrenocortical controls on western blotting (127). Nakamura et al. (128) also detected p-Akt(Ser473) in the adrenal cortex in their immunohistochemical study, but comparing ACC (41 samples) with ACA (54 samples) there was a similar percentage of p-Akt(Ser473) positive cells. By contrast, another group demonstrated increased p-Akt(Ser473) expression in 24 ACC compared with 22 ACA or 4 normal tissue samples (84). Likewise, a more recent study of 121 ACC from the Allolio group describes enhanced p-Akt(Ser473) immunostaining in one-third of carcinomas, which was not seen in ACA or normal tissue (85).
The association between p-Akt(Ser473) expression and survival in ACC remains unclear. In one study, p-Akt(Ser473) expression alone was not associated with prognosis, but high p-Akt and low SGK1 (Serum/Glucocorticoid Regulated Kinase 1) expression identified a subgroup with extremely poor prognosis (85). In contrast, in tissue microarrays of 45 ACC patients who received mitotane monotherapy, in which p-Akt was detected in 31% of tumors, Hermsen et al. (86) unexpectedly found an association between p-Akt expression and longer overall survival, although there was no association with objective response to mitotane.
Early results suggest that targeting p-Akt signaling may be a promising therapeutic strategy in ACC. mTOR inhibitors sirolimus and temsirolimus inhibit ACC cell proliferation in vitro (129). Likewise, everolimus treatment results in antitumor effects in primary adrenocortical cultures, ACC cell lines, and in xenograft mice (130). Of note, significant synergism is seen when everolimus is combined with sorafenib (a tyrosine kinase inhibitor), suggesting that targeting signaling pathways at multiple levels may be of value in these tumors (130). Moreover, the dual PI3K/mTOR inhibitor, NVP-BEZ235 inhibits Akt phosphorylation, reduces ACC cell proliferation in vitro, and reduces tumor size in xenograft mice (131). The H295 cell line overproduces IGF2, which likely stimulates IGF-1R in an autocrine signaling loop, and this provided a useful model of the common IGF2 overproduction in ACC; IGF2-neutralizing antibody increased the anti-proliferative effects of sirolimus (129). Encouragingly, the combination of an IGF-1R inhibitor and mTOR inhibition with cixutumumab (monoclonal antibody targeting IGF-1R) and temsirolimus resulted in 42% of patients with ACC achieving stable disease of at least 6 months (132).
Adrenal Medulla: Phaeochromocytoma and Paraganglioma
Phaeochromocytomas and paragangliomas are catecholamine-secreting neuroendocrine tumors of chromaffin cells, either of the adrenal medulla (phaeochromocytoma) or extra-adrenal sympathetic or parasympathetic paraganglia (paraganglioma). Most phaeochromocytomas are apparently sporadic, although a significant proportion is associated with genetic syndromes, such as MEN2A, Neurofibromatosis (NF1), Von-Hippel Lindau (VHL), or germline mutations in SDHx (133). In the vast majority of cases, the disease is benign and surgical excision is curative. While estimates vary, approximately 10% are malignant, and in these cases current treatment options remain limited (134, 135).
In vitro evidence suggests that the PI3K/Akt pathway is of interest in phaeochromocytoma. It is well established that Akt signaling promotes survival of rat PC12 pheochromocytoma cells and neuronal cells in vitro, protecting against apoptosis induced by a range of stimuli, including cytotoxic chemotherapy (136–139). Furthermore, PI3K/Akt signaling may promote the neuroendocrine phenotype as inhibition of PI3K/Akt in PC12 cells reduces neuroendocrine marker expression and hormone secretion (139). PI3K/Akt signaling is also essential for anchorage-independent growth in rat fibroblasts expressing RET-MEN2A (71); RET-MEN2A germline mutations are responsible for MEN2A, which leads to pheochromocytoma in approximately 50% of patients – therefore these results suggest that this pathway may be critical in tumorigenesis in these patients.
Mice haplodeficient for PTEN develop bilateral tumors of the adrenal medulla with high penetrance; concomitant deletion of AKT1 (PTEN+/−AKT1−/−) in these rodents significantly reduces the size of the adrenal medulla and the tumors, and reduces the occurrence of high grade lesions, suggesting that Akt signaling plays a role at least in advanced tumors in these mice (34). However, PTEN mutation does not appear to be a major driver for Akt activation in human phaeochromocytomas, as PTEN mutations are reported to be rare in human tumors (83). A second rodent model, produced by ectopic overexpression of ErbB-2 in mice, leads to catecholamine-producing adrenal tumors with greatly reduced PTEN protein levels, and increased Akt phosphorylation and cyclin D1 (140). In this mouse model, while PTEN mRNA levels were somewhat reduced, this reduction did not appear to account for the marked reduction in protein levels, leading the authors to suggest that post-translational regulation of PTEN may play an important role (140). MENX rats represent a third rodent model of pheochromocytoma; these rats possess homozygous frameshift mutations in CDKN1B and develop tumors of the adrenal medulla alongside other endocrine tumors (141). Adrenal tumors of MENX rats overexpress the secretin receptor, and PI3K/Akt signaling acts downstream to promote cell proliferation (141). However, this pathway does not appear to be directly applicable to humans, as human phaeochromocytoma rarely overexpress the secretin receptor (141). MENX pheochromocytomas also overexpress secreted BMP7 (Bone Morphogenic Protein 7); this parallels elevated immunoreactivity for BMP7 in human pheochromocytoma samples, detected in 72% of 184 tumor samples on tissue microarray (150 pheochromocytomas and 34 paragangliomas) (142), and BMP7 knockdown was demonstrated to promote proliferation, migration, and invasion primary cultures of MENX-derived pheochromocytomas. From in vitro models, including PC12 cells transfected to overexpress BMP7, evidence was provided for non-canoncial signaling through the PI3K/Akt/mTOR pathway leading to overexpression of integrin β1, responsible for the BMP7-induced cancer phenotypes (142).
Two separate studies have shown elevated levels of p-Akt(Ser473) in human phaeochromocytoma using western blotting (83, 139). In one study, expression of p-Akt(Ser473) was further categorized in a smaller sample of human phaeochromocytoma using immunohistochemistry, demonstrating minimal or absent immunostaining in normal tissue, with increased expression in tumors (83). A larger sample of 39 primary and 8 unrelated metastatic pheochromocytoma has shown similar results, with higher levels of p-Akt(Ser473) in primary tumors than normal tissue, and higher levels in metastatic than primary tumors, but these differences were not statistically significant (93). Reports of PTEN immunostaining are less consistent, with reports of both increased (83) and decreased (93) levels in these tumors. Further evidence of altered PI3K/Akt/mTOR signaling is suggested by increased expression of S6K1 and reduced expression of p27KIP1 in human pheochromocytoma compared with normal tissue and elevated pS6 in metastatic tumors (93). Evidence also suggests that mTOR signaling may be driven by loss-of-function TMEM127 (transmembrane protein 127) mutations in some cases: germline TMEM127 mutations have been identified in a proportion of familial phaeochromocytomas, and in around 2% of apparently sporadic disease, with loss of heterozygosity in tumor specimens (143, 144). Functional TMEM127 appears to be act as a negative regulator of mTOR, with no apparent effects on Akt activation, therefore TMEM127 mutation may provide an alternative Akt-independent pathway for mTOR overactivation in these tumors (143).
Despite the apparent importance of PI3K/Akt/mTOR signaling in phaeochromocytoma, there has been limited experience and success in targeting this pathway clinically. Use of everolimus inhibitors in four patients with malignant pheochromocytoma showed disappointing results (135). However, dual inhibitors of mTORC1 and mTORC2 show promise in preclinical work. Dual mTORC1/2 inhibitors, unlike rapamycin, are able to significantly decrease Akt Ser473 phosphorylation. mTOR inhibitor AZD8055 significantly reduced the tumor and metastatic burden in athymic nude mice injected with cells of the metastatic mouse pheochromocytoma-derived cell line, MTT, and decreased survival of primary human pheochromocytoma cells in culture (145). Compared to rapamycin, dual mTORC1/2 inhibitor PP242 more effectively increased apoptosis of rat PC12 phaeochromocytoma xenografts, with a more marked increase in levels of pro-apoptotic Bax and reduction in levels of anti-apoptotic Bcl-2, leading to smaller tumors than control mice or those treated with rapamycin (124). Furthermore, dual mTORC1/2 inhibition more effectively suppressed VEGF (vascular endothelial growth factor) and angiogenesis (124). Knockdown of VEGFR-2 in PC12 cells reduced the ability of tyrosine kinase inhibitor sunitinib (which has strong affinity for VEGFRs) to inhibit Akt and mTOR phosphorylation and attenuated sunitinib-induced apoptosis (146). Together these observations suggest a positive feedback loop involving VEGF signaling through VEGF-R2 and PI3K/Akt/mTOR pathway activation. Inhibition of multiple hallmarks of cancer by dual mTORC1/2 inhibitors provides rationale for potential use in human pheochromocytoma.
Adrenal Medulla: Neuroblastoma
Neuroblastoma is derived from the neural crest and is the most common extracranial cancer of childhood, accounting for approximately 13% of pediatric cancer deaths (147). These tumors most commonly occur in the adrenal medulla, but may occur at any site of the sympathetic nervous system. Neuroblastomas show marked heterogeneity in their clinical course: while some spontaneously regress, others are widely metastatic, resistant to chemotherapy, and carry a high mortality. MYCN amplification occurs in 25% of primary tumors, and is an important marker of poor prognosis (148). Mass screening for neuroblastoma once appeared promising, however, such programs are now believed to result in overdiagnosis and overtreatment of biologically favorable tumors, at least a proportion of which would have spontaneously regressed or matured (148, 149).
In recent years, there has been increasing focus on the PI3K/Akt signaling pathway in neuroblastoma. In vitro work demonstrates that PI3K/Akt promotes neuroblastoma cell survival, protecting cells from various apoptotic stimuli including cytotoxic chemotherapy (88, 91, 150–152). Akt2 activity may also promote metastatic potential, as silencing Akt2 reduces anchorage-independent growth, migration, and invasion of neuroblastoma cells in vitro, and decreases their metastatic potential in mice (153). Furthermore, in vitro evidence suggests that Akt signaling interacts with key pathways in neuroblastoma. In neuroblastoma cells, Akt activation indirectly stabilizes N-Myc via negative regulation of GSK3β (154). Furthermore, N-Myc has been reported to regulate a microRNA, miR-184, that in turn targets the 3′UTR of AKT2 mRNA (155), suggesting that N-myc and PI3K/Akt signaling may form a positive feedback loop. Evidence also suggests that PI3K/Akt is activated downstream of Gastrin-Releasing Peptide (GRP)/Gastrin-Releasing Peptide Receptor (GRPR) and Brain-derived neurotrophic factor (BDNF)/TrkB signaling, which are reported to be overexpressed in human neuroblastoma (151, 153, 156).
A number of studies have used immunohistochemistry to assess Akt activation in human neuroblastoma (Table 2). p-Akt(Ser473) immunostaining is reported to be increased in neuroblastoma compared with normal adrenal medulla (89), and studies have consistently reported detection of p-Akt(Ser473) in a large proportion of tumors (88, 89, 91, 157). p-Akt(Thr308) is less well investigated; however, its expression has been reported in at least 60% of human neuroblastoma (88). Interestingly, p-Akt(Ser473) has been reported to be more strongly expressed in less differentiated cells (157). However, a separate study has reported that while PTEN was lower in undifferentiated neuroblastoma, p-Akt(Ser473) levels were comparable in differentiated and undifferentiated tumors (156).
Activation of the PI3K/Akt pathways is suggested by findings that p-Akt(Ser473) is correlated with downstream mediators, such as p-mTOR and cyclin-D1 (157) as well as potential upstream activators of the PI3K/Akt pathway, such as VEGFR and IGF-1R (91). Interestingly, p-Akt expression has also been reported to be higher in neuroblastoma expressing CD133 (91). CD133 has been identified as a cancer stem cell marker in other tumors and its expression is associated with worse prognosis in neuroblastoma (91, 158). The association between p-Akt and CD133 in human neuroblastoma is potentially important: CD133 appears to promote a stem-cell like phenotype and chemoresistance in vitro, and both these actions are at least partially mediated by the PI3K/Akt pathway (91, 158).
In a study of 116 neuroblastoma, p-Akt(Ser473) and p-Akt(Thr308) expression correlated with markers of poor prognosis, such as MYCN amplification, changes in 1p36, and stage IV disease. Furthermore, p-Akt(Ser473) and p-Akt(Thr308) expression were associated with worse event-free and overall survival in this sample (88). Notably, p-Akt(Ser473) immunostaining showed fewer associations with prognostic markers and showed a weaker association with survival than immunostaining for p-Akt(Thr308) or for both markers (88). While a second large study failed to show any association between p-Akt(Ser473) immunostaining and prognosis (91), expression of genes downstream of PI3K/Akt signaling has been associated with shorter overall survival (159). In addition, expression of p-Akt(Ser473) is significantly lower in tumors identified by mass screening than in other neuroblastomas (90). As discussed above, tumors identified by mass screening are rarely aggressive, have a substantially better prognosis, and may have otherwise have spontaneously regressed or matured (148, 149); it is possible that lower levels of Akt signaling could contribute to the favorable behavior of these tumors.
Gastroenteropancreatic Neuroendocrine Tumors
Gastroenteropancreatic neuroendocrine tumors (GEP-NETs) are a heterogeneous group of neoplasms. GEP-NETs are derived from neuroendocrine cells of the gastrointestinal tract and pancreatic islets, with intestinal NETs accounting for approximately 50% of cases, and pancreatic NETs accounting for approximately 30% (160). GEP-NETs are rare, but their incidence appears to be increasing (161). Most cases are sporadic; however, GEP-NET may occur in association with familial cancer syndromes such as MEN1, VHL, NF-1, and tuberous sclerosis. These tumors classically have an indolent course, but presentation often occurs at a late stage with metastases present at diagnosis (162). GEP-NETs can be divided into non-functional and functional tumors, with functional tumors resulting in clinical syndromes due to excessive hormone secretion. Small-intestinal NETs (carcinoids) present with carcinoid syndrome in 30% as a result of secretion of bioactive amines, resulting in an array of symptoms including vomiting and diarrhea (160). Approximately 50% of pancreatic neuroendocrine tumors (P-NET) are functional, producing gastrin (gastrinoma), insulin (insulinoma), glucagon (glucagonoma), or vasoactive intestinal peptide (VIPoma), or more rarely, other peptides/hormones (160). Surgery may cure localized GEP-NET disease, however, systemic therapies are currently limited due to the poor response to traditional cytotoxic chemotherapy. In recent years, there has been increasing interest in targeting PI3K/Akt in these cancers.
Work in GEP-NET cell lines demonstrates that the PI3K/Akt pathway is a promising target for therapy. Several studies have demonstrated that inhibiting this pathway in vitro (through PI3K inhibition, p-Akt inhibition, or siRNA) impairs cell proliferation and in some cases survival (163–166). Notably, the effects of Akt inhibition appear to be isoform specific: in human P-NET (BON1) cells, cell viability is impaired by knockdown of Akt1 or Akt3, but promoted by knockdown of Akt2 (165). Furthermore, inhibiting the PI3K/Akt pathway has been shown to reduce the expression of neuroendocrine tumor markers, such as ASCL1 and CgA in pancreatic BON1 cells (166) – this is potentially clinically relevant, as targeting the PI3K/Akt pathway may not only limit tumor cell proliferation or survival, but could conceivably control the troublesome symptoms resulting from hormone oversecretion (166). Evidence also suggests that the PI3K/Akt pathway may promote metastasis and invasion in these tumor cells: knockdown of Akt1, Akt2, or Akt3 impairs neuroendocrine cell invasion (165), and knockdown of PTEN promotes liver metastasis in mouse xenograft models (167).
Gene expression profiling of P-NET has demonstrated that PTEN and TSC2 are downregulated in 50 and 35% of patients, respectively, and downregulation is predictive of poor survival (168). Loss of heterozygosity at the PTEN locus is common in P-NET, occurring in 53% of malignant tumors (169). However, PTEN mutations are relatively uncommon in these tumors: in an analysis of 68 P-NETs, 7% were found to have mutations in PTEN (8% had mutations in TSC2, and 1.4% had mutations in PIK3CA) (170). The PI3K/Akt pathway is perhaps less well studied in intestinal NET than P-NET, however, in a cohort of 48 well-differentiated small intestinal NET (SI-NET), approximately one-third were found to have genetic alterations in the PI3K/Akt/mTOR pathway, with amplification of Akt1/2 being the most common anomaly in this pathway (171).
Immunohistochemistry is also suggestive of dysregulation of the PI3K/Akt pathway in these cancers. PTEN expression has been reported to be either reduced (168) or show altered subcellular localization (169) in P-NET. Lack of detection of PTEN and Akt2 positivity were both associated with failure to respond to systemic therapy in a mixed group of GEP-NETs, but despite this, Akt2 positivity was associated with longer overall survival (172). Low PTEN was associated with shorter time to progression and disease-free survival in a sample of 137 P-NET (168), and low PTEN expression was independently associated with shorter overall survival and metastasis-free survival in a sample of 160 low and intermediate grade P-NET (173).
Assessment of p-Akt expression in GEP-NET suggests that the PI3K/Akt pathway is active in these tumors (Table 2). In a sample of 46 GEP-NET, immunohistochemistry demonstrated that 61% of tumors were positive for p-Akt(Ser473) (95), and in a mixed sample of NET from various sites, 76% showed positive immunostaining (94). Furthermore, in mixed samples of NET, there was significant association between p-Akt(Ser473) and p-EGFR (94) and between mTOR, PIK3CA, or p-EIF4EBP1 and elevated Ki67 (98). Restriction of the analysis to intestinal NET revealed an association of these proteins with disease-free survival (98). Likewise, western blotting suggests that p-Akt(Ser473) correlates with cyclin D1 expression in P-NET (174), suggesting that these signaling pathways are actively promoting cell proliferation in these cancers, although cyclin D1 did not relate to tumor aggressiveness. Immunohistochemical studies have not demonstrated any significant association between p-Akt(Ser473) immunostaining and prognosis, tumor grade or other clinicopathological variables (94, 95, 97, 98), although this could perhaps reflect high levels of heterogeneity in samples examined. In a small sample of 14 gastrointestinal NET, high p-Akt levels measured by western blotting and a high p-Akt:PTEN ratio showed a non-significant association with shorter overall survival (163). The same study also reports a significant association between p-Akt expression and high levels of serum chromogranin A (CgA) expression (a serum marker of NET), and non-significant associations with the presence of carcinoid syndrome (163). While p-Akt(Ser473) has not convincingly been shown to be associated with prognosis, expression of its downstream target mTOR has been associated with tumor grade or worse prognosis (98, 175), but this has not been found in all studies (176).
There is increasing interest in targeting the PI3K/Akt/mTOR pathway, particularly in P-NET, which has been reviewed by Wolin (177). mTOR inhibitors have been investigated most extensively and have shown promise: a phase III trial of everolimus in advanced, low-, or intermediate-grade P-NET showed prolonged progression-free survival versus placebo (11 months versus 4.6 months) (178). Everolimus has now received Federal Drug Administration (FDA) approval for the treatment of advanced progressive P-NET. Markers of PI3K/Akt activation may be of use as biomarkers for sensitivity to mTOR inhibition in these tumors: cell lines with mutations in PIK3CA or PTEN are significantly more likely to be sensitive to rapamycin in vitro, and cells sensitive to rapamycin show higher baseline levels of p-Akt(Ser473) and p-Akt(Thr308), and greater increases in p-Akt(Ser473) and p-Akt(Thr308) following rapamycin treatment (97). Furthermore, in patients with NET treated on a phase II trial of everolimus and octreotide, pre-treatment and on-treatment p-Akt(Thr308) levels measured by RPPA from fine needle aspirates (FNA) were associated with longer progression-free survival, and increases in Akt phosphorylation from paired samples was associated with response to treatment (97). Interestingly, however there was no association between immunostaining for p-Akt(Ser473) from archival tumor blocks and prognosis (97). There are limited data from clinical research relating to novel inhibitors of the PI3K/Akt/mTOR pathway, although preclinical studies suggest that they may be of interest (177). A phase I trial of an allosteric oral Akt inhibitor, MK-2206, showed that treatment was safe with an acceptable side effect profile, furthermore, p-Akt(Ser473) immunostaining was reduced in post-treatment tumor biopsies compared with pre-treatment biopsies, and partial responses were noted in two patients with P-NET (179).
Discussion
Overall, a significant weight of evidence suggests that the PI3K/Akt pathway may have critical roles in multiple tumors of endocrine tissues. It is well established that Akt activation is a key mediator of cell proliferation, survival and motility, and, as discussed above, a significant body of in vitro work demonstrates that this is also applicable to endocrine cell lines. Furthermore, evidence from animal models, human cancer syndromes and immunohistochemical staining patterns in human endocrine tumors suggest that PI3K/Akt signaling may contribute to tumor progression, and, may even play a role in tumor initiation in some situations. The central role of the PI3K/Akt pathway in regulating many of the hallmarks of cancers makes it attractive as a potential indicator of prognosis, and a potential biomarker for response to therapies – especially in relation to novel agents targeting the PI3K/Akt pathway or downstream targets.
While immunohistochemical studies have undoubtedly provided invaluable insights into PI3K/Akt signaling in human cancers, concerns have been raised about the validity of p-Akt immunohistochemistry for assessment of Akt signaling because phospho-proteins are relatively unstable in tissue samples, with a reported half-life of 20 min (180). Since many studies rely on archival paraffin embedded material and the tumor collection process is not overseen by the researchers, the level of p-Akt activity in vivo could be underestimated. Sajjad et al. (110) caution against using cadaveric material for this reason, favoring histologically normal tissue adjacent to tumors as control samples. Furthermore, negative feedback regulation of the PI3K/Akt pathway may lead to strong mTOR staining accompanied by relatively modest p-Akt staining (61). While individual downstream targets are useful as a readout for p-Akt activity in vitro where the PI3K–Akt is modulated by specific treatments, use of downstream targets to validate the activation of the PI3K–Akt pathway in vivo requires a set of diverse targets, since each target is subject to additional regulation. For example, ERK, S6K, GSK3β, and AMP-activated protein kinase can all promote mTORC1 activity by phosphorylating TSC2 (1). It will also be valuable to identify which downstream targets are of relevance to specific cancers and tumor characteristics.
The vast majority of immunohistochemical studies of endocrine cancers have utilized p-Akt(Ser473) as a marker for Akt activation, with Cell Signaling Technology antibodies being favored. Unfortunately, relatively few papers report which specific p-Akt(Ser473) antibody is used. Currently two of four rabbit monoclonal antibodies (D9E and 736E11) are recommended by the company for immunohistochemistry; there is also a rabbit polyclonal, which has commonly been used for immunohistochemistry. In addition, while it is clear that Akt activation in tumors can be revealed by enhanced p-Akt(Ser473) immunohistochemical staining and outcomes have been supported by western blotting results, exploration of the relative usefulness of p-Akt(Ser473) versus p-Akt(Thr308) as an indicator of Akt activation may be informative. Interestingly, when protein lysates from individuals with PIK3CA or AKT1 mutations were compared with controls by western blotting using p-Akt(Ser473) and p-Akt(Thr308) antibodies, affected individuals showed significantly higher p-Akt(Thr308), but not p-Akt(Ser473) (29). In a study of non-small cell lung cancer, it was found that p-Akt(Thr308), but not p-Akt(Ser43) correlated with phosphorylation of three downstream substrates (PRAS40, TSC2, and TBC1D4), leading the authors to suggest that p-Akt(Thr308) represents a better measure of Akt activity in these tumors (181). However, an immunohistochemical study of colorectal carcinomas found that p-Akt(Ser473) staining correlated with phosphorylation of its downstream substrates GSK-3β and Bad, while p-Akt(Thr308) expression only correlated with phosphorylation of GSK-3β, and not that of Bad (182), perhaps suggesting substrate specific targeting. While in Figure 1, we depict activity of Akt in the absence of serine 473 phosphorylation toward TSC2 and GSK3β targets, Bhaskar and Hay (17) suggested that this may be conformation dependent, and put forward a suggestion that p-Akt(Thr308) may only be active in the membrane-bound form. No study has directly compared p-Akt(Ser473) and p-Akt(Thr308) in cancers of endocrine tissues. The Cancer Genome Atlas Research Network (30) compiled an RPPA data set including p-Akt(Thr308) and p-Akt(Ser473) to show that Akt is activated in RAS-like papillary thyroid tumors, however from these data, depicted as a heat map, it can be seen that Akt is not always highly phosphorylated on both sites.
Akt is subject to multiple additional post-translational modifications including phosphorylation at other residues, ubiquitination (lysine 63 linked), sumoylation, and glycosylation in the form of β-d-N-acetylglucosamine monosaccharide attachment; Akt2 can additionally be oxidated leading to intramolecular disulfide bonds between cysteine residues (183). These may regulate stability, trafficking, subcellular localization or substrate specificity of Akt. Sumoylation at Lys(276) is essential for Akt activation, which phosphorylates and promotes the activity of the sumoylation machinery, providing a positive feedback loop, which may be damped down by the sumoylation of PTEN (184). Glycosylation may occur at several sites, and Thr(305) and Thr(312) glycosylation may inhibit phosphorylation of Akt on Thr(308) (183). In murine pancreatic β-cells, glucosamine treatment led to decreased p-Akt(Ser473) in favor of O-linked N-acetylglucosamine at the same residue (185). Therefore, the complexities of regulation of Akt function by post-translational modification mean that we should be cautious in interpreting p-Akt(Ser473) as being fully active Akt.
Combination of PI3K inhibitors and mTOR inhibitors, dual mTORC1/2 inhibition, and mTOR and receptor tyrosine kinase inhibitors are strategies being explored for cancers of endocrine tissues in which the PI3K/Akt/mTOR pathway is hyperactivated (21). In thyroid carcinomas, inhibition of PI3K or mTOR enhances iodine uptake, raising the possibility of enhanced sensitivity to 131I therapy in cancers in which this pathway is activated (186). In addition, two types of Akt inhibitors are in clinical development, catalytic ATP-competitive inhibitors that target active Akt, preventing access of phosphatases, and allosteric inhibitors that prevent recruitment to the cell membrane and hence prevent activation. Using such inhibitors, the first type would lead to elevated levels of phosphorylated Akt, while the second type would prevent phosphorylation. Difficulties arise with activation of feedback pathways and compensatory pathways subsequent to Akt inhibition. Such issues, along with off target effects, need to be considered to ascertain dosing schedules that are sufficient to give long term inhibition. Further, it will be important to ascertain what level of active Akt would be required in a particular tumor before targeting of the PI3K/Akt pathway is likely to be of benefit (187).
Rather than selecting patients for treatment at random, better outcomes may result from a personalized approach to therapy by utilizing predictive biomarker screening strategies. To address this, Yan et al. (188) determined a set of biomarkers that can be used to predict patients likely to benefit from targeted Akt inhibition using GDC-0068, an inhibitor of the ATP competitive type. Of 100 protein and phospho-specific antibodies used in RPPA of lysates from tumor cell lines (PC3, prostate; U87, glioblastoma; MCF7.1, breast), p-Akt(Ser473) and p-Akt(Thr308) were elevated after treatment as expected, whereas substrates downstream of Akt, PRAS40, GSK-3β, mTOR, eIF4G, ATP citrate lyase, FoxO3A and S6, showed decreased phosphorylation. However, the array also revealed feedback activation of the MAPK pathway, with elevated p-ERK, and enhanced levels of HER3. Similar biomarker responses were obtained in mouse xenograft models and in human tumor material from a “first in human” trial of GDC-0068 in patients with advanced solid tumors. Interestingly, the tissue was removed by core needle biopsy and stroma and tumor cells were laser captured separately for RPPA analysis and validated by immunohistochemistry for p-Akt(Ser473), p-Akt(Thr308) and p-PRAS40(Thr246). While p-Akt(Ser473) and p-Akt(Thr308) were elevated in both tumor and stroma, p-PRAS40(Thr246) was only reduced in the tumor tissue, illustrating the importance of investigating the tumor cell population specifically. The authors propose that use of a biomarker set gives more robust readout for Akt inhibition than use of a single biomarker. Moreover, FNA or core needle biopsies allow comparison of pre-treatment and on-treatment functional proteomics by quantitative RPPA for components of the PI3K/Akt/mTOR pathway, particularly if tissue collection is optimized to maintain antigenicity of phosphorylated proteins such as p-Akt by fixation in cold formalin within minutes of removal from the patient, which is more successful for small core biopsies than larger tissue specimens (189, 190). Sampling replicates can be used to account for tumor heterogeneity, but microdissection may be needed for tumors with stromal or other tissue elements. With careful consideration of these limitations, these methods may be developed further to allow prediction of drug response.
A full understanding of how Akt and its targets are regulated is important for the development of effective targeted therapies, rational development of therapeutic combinations, and a better identification of biomarker panels in tumors of endocrine tissues. Future work will need to explore the efficacy of further treatment combinations, such as combinations with XIAP or ERK inhibition, and it will be important to explore how best to promote apoptotic responses over cytostatic effects of treatment without engendering intolerable toxicity. Alongside these developments, biomarkers of the PI3K/Akt pathway have the potential to predict and monitor tumor response. Overall, current knowledge suggests that this pathway plays a substantial role in tumors in endocrine tissues, and hence lends strong justification for further work in this area.
Author Contributions
HR and AH contributed equally to the conception and design, literature review, and final written review article.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We thank America Bennetts-Toledo for translation of Zafón et al. (60) from Spanish to English.
References
1. Fruman DA, Rommel C. PI3K and cancer: lessons, challenges and opportunities. Nat Rev Drug Discov (2014) 13:140–56. doi:10.1038/nrd4204
2. Saji M, Vasko V, Kada F, Allbritton EH, Burman KD, Ringel MD. Akt1 contains a functional leucine-rich nuclear export sequence. Biochem Biophys Res Commun (2005) 332:167–73. doi:10.1016/j.bbrc.2005.04.109
3. Manning BD, Cantley LC. AKT/PKB signaling: navigating downstream. Cell (2007) 129:1261–74. doi:10.1016/j.cell.2007.06.009
4. Hers I, Vincent EE, Tavare JM. Akt signalling in health and disease. Cell Signal (2011) 23:1515–27. doi:10.1016/j.cellsig.2011.05.004
5. Martelli AM, Tabellini G, Bressanin D, Ognibene A, Goto K, Cocco L, et al. The emerging multiple roles of nuclear Akt. Biochim Biophys Acta (2012) 1823:2168–78. doi:10.1016/j.bbamcr.2012.08.017
6. Ocana A, Vera-Badillo F, Al-Mubarak M, Templeton AJ, Corrales-Sanchez V, Diez-Gonzalez L, et al. Activation of the PI3K/mTOR/AKT pathway and survival in solid tumors: systematic review and meta-analysis. PLoS One (2014) 9:e95219. doi:10.1371/journal.pone.0095219
7. Liu P, Cheng H, Roberts TM, Zhao JJ. Targeting the phosphoinositide 3-kinase pathway in cancer. Nat Rev Drug Discov (2009) 8:627–44. doi:10.1038/nrd2926
8. Newton AC, Trotman LC. Turning off AKT: PHLPP as a drug target. Annu Rev Pharmacol Toxicol (2014) 54:537–58. doi:10.1146/annurev-pharmtox-011112-140338
9. McCubrey JA, Steelman LS, Bertrand FE, Davis NM, Sokolosky M, Abrams SL, et al. GSK-3 as potential target for therapeutic intervention in cancer. Oncotarget (2014) 5:2881–911. doi:10.18632/oncotarget.2037
10. Karger S, Weidinger C, Krause K, Sheu SY, Aigner T, Gimm O, et al. FOXO3a: a novel player in thyroid carcinogenesis? Endocr Relat Cancer (2009) 16:189–99. doi:10.1677/ERC-07-0283
11. Weidinger C, Karger S, Krause K, Schierle K, Steinert F, Gimm O, et al. Distinct regulation of intrinsic apoptosis in benign and malignant thyroid tumours. Horm Metab Res (2010) 42:553–6. doi:10.1055/s-0030-1253374
12. Mahajan K, Mahajan NP. PI3K-independent AKT activation in cancers: a treasure trove for novel therapeutics. J Cell Physiol (2012) 227:3178–84. doi:10.1002/jcp.24065
13. Liu P, Begley M, Michowski W, Inuzuka H, Ginzberg M, Gao D, et al. Cell-cycle-regulated activation of Akt kinase by phosphorylation at its carboxyl terminus. Nature (2014) 508:541–5. doi:10.1038/nature13079
14. Nitsche C, Edderkaoui M, Moore RM, Eibl G, Kasahara N, Treger J, et al. The phosphatase PHLPP1 regulates Akt2, promotes pancreatic cancer cell death, and inhibits tumor formation. Gastroenterology (2012) 142(377–387):e371–5. doi:10.1053/j.gastro.2011.10.026
15. Tzivion G, Dobson M, Ramakrishnan G. FoxO transcription factors; regulation by AKT and 14-3-3 proteins. Biochim Biophys Acta (2011) 1813:1938–45. doi:10.1016/j.bbamcr.2011.06.002
16. Owonikoko TK, Khuri FR. Targeting the PI3K/AKT/mTOR pathway: biomarkers of success and tribulation. Am Soc Clin Oncol Educ Book (2013). doi:10.1200/EdBook_AM.2013.33.e395
17. Bhaskar PT, Hay N. The two TORCs and Akt. Dev Cell (2007) 12:487–502. doi:10.1016/j.devcel.2007.03.020
18. O’Reilly KE, Rojo F, She QB, Solit D, Mills GB, Smith D, et al. mTOR inhibition induces upstream receptor tyrosine kinase signaling and activates Akt. Cancer Res (2006) 66:1500–8. doi:10.1158/0008-5472.CAN-05-2925
19. Wan X, Harkavy B, Shen N, Grohar P, Helman LJ. Rapamycin induces feedback activation of Akt signaling through an IGF-1R-dependent mechanism. Oncogene (2007) 26:1932–40. doi:10.1038/sj.onc.1209990
20. Svejda B, Kidd M, Kazberouk A, Lawrence B, Pfragner R, Modlin IM. Limitations in small intestinal neuroendocrine tumor therapy by mTor kinase inhibition reflect growth factor-mediated PI3K feedback loop activation via ERK1/2 and AKT. Cancer (2011) 117:4141–54. doi:10.1002/cncr.26011
21. Rozengurt E, Soares HP, Sinnet-Smith J. Suppression of feedback loops mediated by PI3K/mTOR induces multiple overactivation of compensatory pathways: an unintended consequence leading to drug resistance. Mol Cancer Ther (2014) 13:2477–88. doi:10.1158/1535-7163.MCT-14-0330
22. Guertin DA, Sabatini DM. Defining the role of mTOR in cancer. Cancer Cell (2007) 12:9–22. doi:10.1016/j.ccr.2007.05.008
23. Shtilbans V, Wu M, Burstein DE. Current overview of the role of Akt in cancer studies via applied immunohistochemistry. Ann Diagn Pathol (2008) 12:153–60. doi:10.1016/j.anndiagpath.2007.12.001
24. Mandell JW. Phosphorylation state-specific antibodies: applications in investigative and diagnostic pathology. Am J Pathol (2003) 163:1687–98. doi:10.1016/S0002-9440(10)63525-0
25. Pinhel IF, Macneill FA, Hills MJ, Salter J, Detre S, A’Hern R, et al. Extreme loss of immunoreactive p-Akt and p-Erk1/2 during routine fixation of primary breast cancer. Breast Cancer Res (2010) 12:R76. doi:10.1186/bcr2719
26. Yang ZY, Di MY, Yuan JQ, Shen WX, Zheng DY, Chen JZ, et al. The prognostic value of phosphorylated Akt in breast cancer: a systematic review. Sci Rep (2015) 5:7758. doi:10.1038/srep07758
27. Wiseman SM, Loree TR, Rigual NR, Hicks WL Jr, Douglas WG, Anderson GR, et al. Anaplastic transformation of thyroid cancer: review of clinical, pathologic, and molecular evidence provides new insights into disease biology and future therapy. Head Neck (2003) 25:662–70. doi:10.1002/hed.10277
28. Ngeow J, Mester J, Rybicki LA, Ni Y, Milas M, Eng C. Incidence and clinical characteristics of thyroid cancer in prospective series of individuals with Cowden and Cowden-like syndrome characterized by germline PTEN, SDH, or KLLN alterations. J Clin Endocrinol Metab (2011) 96:E2063–71. doi:10.1210/jc.2011-1616
29. Orloff MS, He X, Peterson C, Chen F, Chen JL, Mester JL, et al. Germline PIK3CA and AKT1 mutations in Cowden and Cowden-like syndromes. Am J Hum Genet (2013) 92:76–80. doi:10.1016/j.ajhg.2012.10.021
30. The Cancer Genome Atlas Research Network. Integrated genomic characterization of papillary thyroid carcinoma. Cell (2014) 159:676–90. doi:10.1016/j.cell.2014.09.050
31. Forbes SA, Beare D, Gunasekaren P, Leung K, Bindal N, Boutselakis H, et al. COSMIC: exploring the world’s knowledge of somatic mutations in human cancer. Nucl Acids Res (2015) 43(21):D805–11. doi:10.1093/nar/gku1075
32. COSMIC, 2014. Catalogue of Somatic Mutations in Cancer. Wellcome Trust Sanger Institute (2013). Accessed: December 2015. Available at: www.sanger.ac.uk/cosmic
33. Ricarte-Filho JC, Ryder M, Chitale DA, Rivera M, Heguy A, Ladanyi M, et al. Mutational profile of advanced primary and metastatic radioactive iodine-refractory thyroid cancers reveals distinct pathogenetic roles for BRAF, PIK3CA, and AKT1. Cancer Res (2009) 69:4885–93. doi:10.1158/0008-5472.CAN-09-0727
34. Chen ML, Xu PZ, Peng XD, Chen WS, Guzman G, Yang X, et al. The deficiency of Akt1 is sufficient to suppress tumor development in Pten+/− mice. Genes Dev (2006) 20:1569–74. doi:10.1101/gad.1395006
35. Yeager N, Klein-Szanto A, Kimura S, Di Cristofano A. Pten loss in the mouse thyroid causes goiter and follicular adenomas: insights into thyroid function and Cowden disease pathogenesis. Cancer Res (2007) 67:959–66. doi:10.1158/0008-5472.CAN-06-3524
36. Chen M, Pratt CP, Zeeman ME, Schultz N, Taylor BS, O’Neill A, et al. Identification of PHLPP1 as a tumor suppressor reveals the role of feedback activation in PTEN-mutant prostate cancer progression. Cancer Cell (2011) 20:173–86. doi:10.1016/j.ccr.2011.07.013
37. Kato Y, Ying H, Willingham MC, Cheng SY. A tumor suppressor role for thyroid hormone beta receptor in a mouse model of thyroid carcinogenesis. Endocrinology (2004) 145:4430–8. doi:10.1210/en.2004-0612
38. Kim CS, Vasko VV, Kato Y, Kruhlak M, Saji M, Cheng SY, et al. AKT activation promotes metastasis in a mouse model of follicular thyroid carcinoma. Endocrinology (2005) 146:4456–63. doi:10.1210/en.2005-0172
39. Pringle DR, Vasko VV, Yu L, Manchanda PK, Lee AA, Zhang X, et al. Follicular thyroid cancers demonstrate dual activation of PKA and mTOR as modeled by thyroid-specific deletion of Prkar1a and Pten in mice. J Clin Endocrinol Metab (2014) 99:E804–12. doi:10.1210/jc.2013-3101
40. Kemmochi S, Fujimoto H, Woo GH, Inoue K, Takahashi M, Mitsumori K, et al. Involvement of PTEN/Akt signaling in capsular invasive carcinomas developed in a rat two-stage thyroid carcinogenesis model after promotion with sulfadimethoxine. J Cancer Res Clin Oncol (2011) 137:723–32. doi:10.1007/s00432-010-0931-7
41. Saji M, Ringel MD. The PI3K-Akt-mTOR pathway in initiation and progression of thyroid tumors. Mol Cell Endocrinol (2010) 321:20–8. doi:10.1016/j.mce.2009.10.016
42. Ringel MD, Hayre N, Saito J, Saunier B, Schuppert F, Burch H, et al. Overexpression and overactivation of Akt in thyroid carcinoma. Cancer Res (2001) 61:6105–11.
43. Miyakawa M, Tsushima T, Murakami H, Wakai K, Isozaki O, Takano K. Increased expression of phosphorylated p70S6 kinase and Akt in papillary thyroid cancer tissues. Endocr J (2003) 50:77–83. doi:10.1507/endocrj.50.77
44. Mandal M, Kim S, Younes MN, Jasser SA, El-Naggar AK, Mills GB, et al. The Akt inhibitor KP372-1 suppresses Akt activity and cell proliferation and induces apoptosis in thyroid cancer cells. Br J Cancer (2005) 92:1899–905. doi:10.1038/sj.bjc.6602595
45. Krzeslak A, Pomorski L, Lipinska A. Expression, localization, and phosphorylation of Akt1 in benign and malignant thyroid lesions. Endocr Pathol (2011) 22:206–11. doi:10.1007/s12022-011-9177-4
46. Bruni P, Boccia A, Baldassarre G, Trapasso F, Santoro M, Chiappetta G, et al. PTEN expression is reduced in a subset of sporadic thyroid carcinomas: evidence that PTEN-growth suppressing activity in thyroid cancer cells mediated by p27kip1. Oncogene (2000) 19:3146–55. doi:10.1038/sj.onc.1203633
47. Gimm O, Perren A, Weng LP, Marsh DJ, Yeh JJ, Ziebold U, et al. Differential nuclear and cytoplasmic expression of PTEN in normal thyroid tissue, and benign and malignant epithelial thyroid tumors. Am J Pathol (2000) 156:1693–700. doi:10.1016/S0002-9440(10)65040-7
48. Vasko V, Saji M, Hardy E, Kruhlak M, Larin A, Savchenko V, et al. Akt activation and localisation correlate with tumour invasion and oncogene expression in thyroid cancer. J Med Genet (2004) 41:161–70. doi:10.1136/jmg.2003.015339
49. Garcia-Rostan G, Costa AM, Pereira-Castro I, Salvatore G, Hernandez R, Hermsem MJ, et al. Mutation of the PIK3CA gene in anaplastic thyroid cancer. Cancer Res (2005) 65:10199–207. doi:10.1158/0008-5472.CAN-04-4259
50. Larson SD, Jackson LN, Riall TS, Uchida T, Thomas RP, Qiu S, et al. Increased incidence of well-differentiated thyroid cancer associated with Hashimoto thyroiditis and the role of the PI3k/Akt pathway. J Am Coll Surg (2007) 204:764–73. doi:10.1016/j.jamcollsurg.2006.12.037
51. Shin E, Hong SW, Kim SH, Yang WI. Expression of down stream molecules of RET (p-ERK, p-p38 MAPK, p-JNK and p-AKT) in papillary thyroid carcinomas. Yonsei Med J (2004) 45:306–13. doi:10.3349/ymj.2004.45.2.306
52. Motti ML, Califano D, Troncone G, De Marco C, Migliaccio I, Palmieri E, et al. Complex regulation of the cyclin-dependent kinase inhibitor p27kip1 in thyroid cancer cells by the PI3K/AKT pathway: regulation of p27kip1 expression and localization. Am J Pathol (2005) 166:737–49. doi:10.1016/S0002-9440(10)62295-X
53. Jensen K, Patel A, Klubo-Gwiezdzinska J, Bauer A, Vasko V. Inhibition of gap junction transfer sensitizes thyroid cancer cells to anoikis. Endocr Relat Cancer (2011) 18:613–26. doi:10.1530/ERC-10-0289
54. Siraj AK, Bavi P, Abubaker J, Jehan Z, Sultana M, Al-Dayel F, et al. Genome-wide expression analysis of Middle Eastern papillary thyroid cancer reveals c-MET as a novel target for cancer therapy. J Pathol (2007) 213:190–9. doi:10.1002/path.2215
55. Liu Z, Hou P, Ji M, Guan H, Studeman K, Jensen K, et al. Highly prevalent genetic alterations in receptor tyrosine kinases and phosphatidylinositol 3-kinase/akt and mitogen-activated protein kinase pathways in anaplastic and follicular thyroid cancers. J Clin Endocrinol Metab (2008) 93:3106–16. doi:10.1210/jc.2008-0273
56. Santarpia L, El-Naggar AK, Cote GJ, Myers JN, Sherman SI. Phosphatidylinositol 3-kinase/akt and ras/raf-mitogen-activated protein kinase pathway mutations in anaplastic thyroid cancer. J Clin Endocrinol Metab (2008) 93:278–84. doi:10.1210/jc.2007-1076
57. Uddin S, Siraj AK, Al-Rasheed M, Ahmed M, Bu R, Myers JN, et al. Fatty acid synthase and AKT pathway signaling in a subset of papillary thyroid cancers. J Clin Endocrinol Metab (2008) 93:4088–97. doi:10.1210/jc.2008-0503
58. Liu J, Brown RE. Morphoproteomics demonstrates activation of mTOR pathway in anaplastic thyroid carcinoma: a preliminary observation. Ann Clin Lab Sci (2010) 40:211–7.
59. Sozopoulos E, Litsiou H, Voutsinas G, Mitsiades N, Anagnostakis N, Tseva T, et al. Mutational and immunohistochemical study of the PI3K/Akt pathway in papillary thyroid carcinoma in Greece. Endocr Pathol (2010) 21:90–100. doi:10.1007/s12022-010-9112-0
60. Zafón C, Castellvi J, Obiols G. Usefulness of the immunohistochemical analysis of several molecular markers in the characterization of papillary thyroid carcinoma with initial lymph node metastasis. Endocrinol Nutr (2010) 57:165–9. doi:10.1016/j.endonu.2010.02.005
61. Liu J, Brown RE. Morphoproteomics demonstrates activation of mammalian target of rapamycin pathway in papillary thyroid carcinomas with nuclear translocation of MTOR in aggressive histological variants. Mod Pathol (2011) 24:1553–9. doi:10.1038/modpathol.2011.121
62. Ahmed M, Hussain AR, Bavi P, Ahmed SO, Al Sobhi SS, Al-Dayel F, et al. High prevalence of mTOR complex activity can be targeted using Torin2 in papillary thyroid carcinoma. Carcinogenesis (2014) 35:1564–72. doi:10.1093/carcin/bgu051
63. Milosevic Z, Pesic M, Stankovic T, Dinic J, Milovanovic Z, Stojsic J, et al. Targeting RAS-MAPK-ERK and PI3K-AKT-mTOR signal transduction pathways to chemosensitize anaplastic thyroid carcinoma. Transl Res (2014) 164:411–23. doi:10.1016/j.trsl.2014.06.005
64. Moraitis D, Karanikou M, Liakou C, Dimas K, Tzimas G, Tseleni-Balafouta S, et al. SIN1, a critical component of the mTOR-Rictor complex, is overexpressed and associated with AKT activation in medullary and aggressive papillary thyroid carcinomas. Surgery (2014) 156:1542–8. doi:10.1016/j.surg.2014.08.095
65. Hussain AR, Bu R, Ahmed M, Jehan Z, Beg S, Al-Sobhi S, et al. Role of X-linked inhibitor of apoptosis as a prognostic marker and therapeutic target in papillary thyroid carcinoma. J Clin Endocrinol Metab (2015) 100:E974–84. doi:10.1210/jc.2014-4356
66. Rapa I, Saggiorato E, Giachino D, Palestini N, Orlandi F, Papotti M, et al. Mammalian target of rapamycin pathway activation is associated to RET mutation status in medullary thyroid carcinoma. J Clin Endocrinol Metab (2011) 96:2146–53. doi:10.1210/jc.2010-2655
67. Erovic BM, Kim D, Cassol C, Goldstein DP, Irish JC, Asa SL, et al. Prognostic and predictive markers in medullary thyroid carcinoma. Endocr Pathol (2012) 23:232–42. doi:10.1007/s12022-012-9225-8
68. Tamburrino A, Molinolo AA, Salerno P, Chernock RD, Raffeld M, Xi L, et al. Activation of the mTOR pathway in primary medullary thyroid carcinoma and lymph node metastases. Clin Cancer Res (2012) 18:3532–40. doi:10.1158/1078-0432.CCR-11-2700
69. Paes JE, Ringel MD. Dysregulation of the phosphatidylinositol 3-kinase pathway in thyroid neoplasia. Endocrinol Metab Clin North Am (2008) 37:375–87. doi:10.1016/j.ecl.2008.01.001
70. Romei C, Elisei R. RET/PTC translocations and clinico-pathological features in human papillary thyroid carcinoma. Front Endocrinol (2012) 3:54. doi:10.3389/fendo.2012.00054
71. Segouffin-Cariou C, Billaud M. Transforming ability of MEN2A-RET requires activation of the phosphatidylinositol 3-kinase/AKT signaling pathway. J Biol Chem (2000) 275:3568–76. doi:10.1074/jbc.275.5.3568
72. O’Malley Y, Lal G, Howe JR, Weigel RJ, Komorowski RA, Shilyansky J, et al. Invasion in follicular thyroid cancer cell lines is mediated by EphA2 and pAkt. Surgery (2012) 152:1218–24. doi:10.1016/j.surg.2012.08.041
73. Liu P, Gan W, Inuzuka H, Lazorchak AS, Gao D, Arojo O, et al. Sin1 phosphorylation impairs mTORC2 complex integrity and inhibits downstream Akt signalling to suppress tumorigenesis. Nat Cell Biol (2013) 15:1340–50. doi:10.1038/ncb2860
74. Pitt SC, Chen H. The phosphatidylinositol 3-kinase/akt signaling pathway in medullary thyroid cancer. Surgery (2008) 144:721–4. doi:10.1016/j.surg.2008.06.028
75. Pennelli G, Galuppini F, Barollo S, Cavedon E, Bertazza L, Fassan M, et al. The PDCD4/miR-21 pathway in medullary thyroid carcinoma. Hum Pathol (2015) 46:50–7. doi:10.1016/j.humpath.2014.09.006
76. Marcocci C, Cetani F, Rubin MR, Silverberg SJ, Pinchera A, Bilezikian JP. Parathyroid carcinoma. J Bone Miner Res (2008) 23:1869–80. doi:10.1359/jbmr.081018
77. Erovic BM, Harris L, Jamali M, Goldstein DP, Irish JC, Asa SL, et al. Biomarkers of parathyroid carcinoma. Endocr Pathol (2012) 23:221–31. doi:10.1007/s12022-012-9222-y
78. Kasaian K, Wiseman SM, Thiessen N, Mungall KL, Corbett RD, Qian JQ, et al. Complete genomic landscape of a recurring sporadic parathyroid carcinoma. J Pathol (2013) 230:249–60. doi:10.1002/path.4203
79. Musat M, Korbonits M, Kola B, Borboli N, Hanson MR, Nanzer AM, et al. Enhanced protein kinase B/Akt signalling in pituitary tumours. Endocr Relat Cancer (2005) 12:423–33. doi:10.1677/erc.1.00949
80. Iino K, Oki Y, Yamashita M, Matsushita F, Hayashi C, Yogo K, et al. Possible relevance between prohormone convertase 2 expression and tumor growth in human adrenocorticotropin-producing pituitary adenoma. J Clin Endocrinol Metab (2010) 95:4003–11. doi:10.1210/jc.2009-2716
81. Noh TW, Jeong HJ, Lee MK, Kim TS, Kim SH, Lee EJ. Predicting recurrence of nonfunctioning pituitary adenomas. J Clin Endocrinol Metab (2009) 94:4406–13. doi:10.1210/jc.2009-0471
82. Trovato M, Torre ML, Ragonese M, Simone A, Scarfi R, Barresi V, et al. /c-met system targeting PI3K/AKT and STAT3/phosphorylated-STAT3 pathways in pituitary adenomas: an immunohistochemical characterization in view of targeted therapies. Endocrine (2013) 44:735–43. doi:10.1007/s12020-013-9950-x
83. Fassnacht M, Weismann D, Ebert S, Adam P, Zink M, Beuschlein F, et al. AKT is highly phosphorylated in pheochromocytomas but not in benign adrenocortical tumors. J Clin Endocrinol Metab (2005) 90:4366–70. doi:10.1210/jc.2004-2198
84. Barlaskar FM, Spalding AC, Heaton JH, Kuick R, Kim AC, Thomas DG, et al. Preclinical targeting of the type I insulin-like growth factor receptor in adrenocortical carcinoma. J Clin Endocrinol Metab (2009) 94:204–12. doi:10.1210/jc.2008-1456
85. Ronchi CL, Sbiera S, Leich E, Tissier F, Steinhauer S, Deutschbein T, et al. Low SGK1 expression in human adrenocortical tumors is associated with ACTH-independent glucocorticoid secretion and poor prognosis. J Clin Endocrinol Metab (2012) 97:E2251–60. doi:10.1210/jc.2012-2669
86. Hermsen IG, Haak HR, de Krijger RR, Kerkhofs TM, Feelders RA, de Herder WW, et al. Mutational analyses of epidermal growth factor receptor and downstream pathways in adrenocortical carcinoma. Eur J Endocrinol (2013) 169:51–8. doi:10.1530/EJE-13-0093
87. Qiao J, Kang JH, Cree J, Evers BM, Chung DH. Ets1 transcription factor mediates gastrin-releasing peptide-induced IL-8 regulation in neuroblastoma cells. Neoplasia (2007) 9:184–91. doi:10.1593/neo.06841
88. Opel D, Poremba C, Simon T, Debatin KM, Fulda S. Activation of Akt predicts poor outcome in neuroblastoma. Cancer Res (2007) 67:735–45. doi:10.1158/0008-5472.CAN-06-2201
89. Johnsen JI, Segerstrom L, Orrego A, Elfman L, Henriksson M, Kagedal B, et al. Inhibitors of mammalian target of rapamycin downregulate MYCN protein expression and inhibit neuroblastoma growth in vitro and in vivo. Oncogene (2008) 27:2910–22. doi:10.1038/sj.onc.1210938
90. Sartelet H, Ohta S, Barrette S, Rougemont AL, Brevet M, Regairaz M, et al. High level of apoptosis and low AKT activation in mass screening as opposed to standard neuroblastoma. Histopathology (2010) 56:607–16. doi:10.1111/j.1365-2559.2010.03522.x
91. Sartelet H, Rougemont AL, Fabre M, Castaing M, Duval M, Fetni R, et al. Activation of the phosphatidylinositol 3′-kinase/AKT pathway in neuroblastoma and its regulation by thioredoxin 1. Hum Pathol (2011) 42:1727–39. doi:10.1016/j.humpath.2011.01.019
92. Sartelet H, Imbriglio T, Nyalendo C, Haddad E, Annabi B, Duval M, et al. CD133 expression is associated with poor outcome in neuroblastoma via chemoresistance mediated by the AKT pathway. Histopathology (2012) 60:1144–55. doi:10.1111/j.1365-2559.2012.04191.x
93. Chaux A, Brimo F, Gonzalez-Roibon N, Shah S, Schultz L, Rizk JM, et al. Immunohistochemical evidence of dysregulation of the mammalian target of rapamycin pathway in primary and metastatic pheochromocytomas. Urology (2012) 80:.e7–12. doi:10.1016/j.urology.2012.04.032
94. Shah T, Hochhauser D, Frow R, Quaglia A, Dhillon AP, Caplin ME. Epidermal growth factor receptor expression and activation in neuroendocrine tumours. J Neuroendocrinol (2006) 18:355–60. doi:10.1111/j.1365-2826.2006.01425.x
95. Ghayouri M, Boulware D, Nasir A, Strosberg J, Kvols L, Coppola D. Activation of the serine/theronine protein kinase Akt in enteropancreatic neuroendocrine tumors. Anticancer Res (2010) 30:5063–7.
96. Shida T, Kishimoto T, Furuya M, Nikaido T, Koda K, Takano S, et al. Expression of an activated mammalian target of rapamycin (mTOR) in gastroenteropancreatic neuroendocrine tumors. Cancer Chemother Pharmacol (2010) 65:889–93. doi:10.1007/s00280-009-1094-6
97. Meric-Bernstam F, Akcakanat A, Chen H, Do KA, Sangai T, Adkins F, et al. PIK3CA/PTEN mutations and Akt activation as markers of sensitivity to allosteric mTOR inhibitors. Clin Cancer Res (2012) 18:1777–89. doi:10.1158/1078-0432.CCR-11-2123
98. Qian ZR, Ter-Minassian M, Chan JA, Imamura Y, Hooshmand SM, Kuchiba A, et al. Prognostic significance of MTOR pathway component expression in neuroendocrine tumors. J Clin Oncol (2013) 31:3418–25. doi:10.1200/JCO.2012.46.6946
99. Arafah BM, Nasrallah MP. Pituitary tumors: pathophysiology, clinical manifestations and management. Endocr Relat Cancer (2001) 8:287–305. doi:10.1677/erc.0.0080287
100. Vender JR, Laird MD, Dhandapani KM. Inhibition of NFkappaB reduces cellular viability in GH3 pituitary adenoma cells. Neurosurgery (2008) 62:1122–7. doi:10.1227/01.neu.0000325874.82999.75
101. Xu M, Shorts-Cary L, Knox AJ, Kleinsmidt-DeMasters B, Lillehei K, Wierman ME. Epidermal growth factor receptor pathway substrate 8 is overexpressed in human pituitary tumors: role in proliferation and survival. Endocrinology (2009) 150:2064–71. doi:10.1210/en.2008-1265
102. Lu C, Willingham MC, Furuya F, Cheng SY. Activation of phosphatidylinositol 3-kinase signaling promotes aberrant pituitary growth in a mouse model of thyroid-stimulating hormone-secreting pituitary tumors. Endocrinology (2008) 149:3339–45. doi:10.1210/en.2007-1696
103. Pellegata NS, Quintanilla-Martinez L, Siggelkow H, Samson E, Bink K, Hofler H, et al. Germ-line mutations in p27Kip1 cause a multiple endocrine neoplasia syndrome in rats and humans. Proc Natl Acad Sci U S A (2006) 103:15558–63. doi:10.1073/pnas.0603877103
104. Lee M, Theodoropoulou M, Graw J, Roncaroli F, Zatelli MC, Pellegata NS. Levels of p27 sensitize to dual PI3K/mTOR inhibition. Mol Cancer Ther (2011) 10:1450–9. doi:10.1158/1535-7163.MCT-11-0188
105. Lee M, Wiedemann T, Gross C, Leinhauser I, Roncaroli F, Braren R, et al. Targeting PI3K/mTOR signaling displays potent antitumor efficacy against nonfunctioning pituitary adenomas. Clin Cancer Res (2015) 21:3204–15. doi:10.1158/1078-0432.CCR-15-0288
106. Lin Y, Jiang X, Shen Y, Li M, Ma H, Xing M, et al. Frequent mutations and amplifications of the PIK3CA gene in pituitary tumors. Endocr Relat Cancer (2009) 16:301–10. doi:10.1677/ERC-08-0167
107. Murat CB, Braga PB, Fortes MA, Bronstein MD, Correa-Giannella ML, Giorgi RR. Mutation and genomic amplification of the PIK3CA proto-oncogene in pituitary adenomas. Braz J Med Biol Res (2012) 45:851–5. doi:10.1590/S0100-879X2012007500115
108. Monsalves E, Juraschka K, Tateno T, Agnihotri S, Asa SL, Ezzat S, et al. The PI3K/AKT/mTOR pathway in the pathophysiology and treatment of pituitary adenomas. Endocr Relat Cancer (2014) 21:R331–44. doi:10.1530/ERC-14-0188
109. Palumbo T, Faucz FR, Azevedo M, Xekouki P, Iliopoulos D, Stratakis CA. Functional screen analysis reveals miR-26b and miR-128 as central regulators of pituitary somatomammotrophic tumor growth through activation of the PTEN-AKT pathway. Oncogene (2013) 32:1651–9. doi:10.1038/onc.2012.190
110. Sajjad EA, Zielinski G, Maksymowicz M, Hutnik L, Bednarczuk T, Wlodarski P. mTOR is frequently active in GH-secreting pituitary adenomas without influencing their morphopathological features. Endocr Pathol (2013) 24:11–9. doi:10.1007/s12022-012-9230-y
111. Hibberts NA, Simpson DJ, Bicknell JE, Broome JC, Hoban PR, Clayton RN, et al. Analysis of cyclin D1 (CCND1) allelic imbalance and overexpression in sporadic human pituitary tumors. Clin Cancer Res (1999) 5:2133–9.
112. Dworakowska D, Wlodek E, Leontiou CA, Igreja S, Cakir M, Teng M, et al. Activation of RAF/MEK/ERK and PI3K/AKT/mTOR pathways in pituitary adenomas and their effects on downstream effectors. Endocr Relat Cancer (2009) 16:1329–38. doi:10.1677/ERC-09-0101
113. Zatelli MC, Minoia M, Filieri C, Tagliati F, Buratto M, Ambrosio MR, et al. Effect of everolimus on cell viability in nonfunctioning pituitary adenomas. J Clin Endocrinol Metab (2010) 95:968–76. doi:10.1210/jc.2009-1641
114. Cerovac V, Monteserin-Garcia J, Rubinfeld H, Buchfelder M, Losa M, Florio T, et al. The somatostatin analogue octreotide confers sensitivity to rapamycin treatment on pituitary tumor cells. Cancer Res (2010) 70:666–74. doi:10.1158/0008-5472.CAN-09-2951
115. Sukumari-Ramesh S, Singh N, Dhandapani KM, Vender JR. mTOR inhibition reduces cellular proliferation and sensitizes pituitary adenoma cells to ionizing radiation. Surg Neurol Int (2011) 2:22. doi:10.4103/2152-7806.77029
116. Dai C, Zhang B, Liu X, Ma S, Yang Y, Yao Y, et al. Inhibition of PI3K/AKT/mTOR pathway enhances temozolomide-induced cytotoxicity in pituitary adenoma cell lines in vitro and xenografted pituitary adenoma in female nude mice. Endocrinology (2013) 154:1247–59. doi:10.1210/en.2012-1908
117. Jouanneau E, Wierinckx A, Ducray F, Favrel V, Borson-Chazot F, Honnorat J, et al. New targeted therapies in pituitary carcinoma resistant to temozolomide. Pituitary (2012) 15:37–43. doi:10.1007/s11102-011-0341-0
118. Mazzuco TL, Durand J, Chapman A, Crespigio J, Bourdeau I. Genetic aspects of adrenocortical tumours and hyperplasias. Clin Endocrinol (2012) 77:1–10. doi:10.1111/j.1365-2265.2012.04403.x
119. Mihai R. Diagnosis, treatment and outcome of adrenocortical cancer. Br J Surg (2015) 102:291–306. doi:10.1002/bjs.9743
120. Ragazzon B, Assie G, Bertherat J. Transcriptome analysis of adrenocortical cancers: from molecular classification to the identification of new treatments. Endocr Relat Cancer (2011) 18:R15–27. doi:10.1530/ERC-10-0220
121. Haluska P, Worden F, Olmos D, Yin D, Schteingart D, Batzel GN, et al. Safety, tolerability, and pharmacokinetics of the anti-IGF-1R monoclonal antibody figitumumab in patients with refractory adrenocortical carcinoma. Cancer Chemother Pharmacol (2010) 65:765–73. doi:10.1007/s00280-009-1083-9
122. Lerario AM, Worden FP, Ramm CA, Hesseltine EA, Stadler WM, Else T, et al. The combination of insulin-like growth factor receptor 1 (IGF1R) antibody cixutumumab and mitotane as a first-line therapy for patients with recurrent/metastatic adrenocortical carcinoma: a multi-institutional NCI-sponsored trial. Horm Cancer (2014) 5:232–9. doi:10.1007/s12672-014-0182-1
123. Fassnacht M, Berruti A, Baudin E, Demeure MJ, Gilbert J, Haak H, et al. Linsitinib (OSI-906) versus placebo for patients with locally advanced or metastatic adrenocortical carcinoma: a double-blind, randomised, phase 3 study. Lancet Oncol (2015) 16:426–35. doi:10.1016/S1470-2045(15)70081-1
124. Zhang X, Wang X, Qin L, Xu T, Zhu Z, Zhong S, et al. The dual mTORC1 and mTORC2 inhibitor PP242 shows strong antitumor activity in a pheochromocytoma PC12 cell tumor model. Urology (2015) 85(273):e271–7. doi:10.1016/j.urology.2014.09.020
125. Sasano H, Satoh F, Nakamura Y. Roles of the pathologist in evaluating surrogate markers for medical therapy in adrenocortical carcinoma. Endocr Pathol (2014) 25:366–70. doi:10.1007/s12022-014-9334-7
126. Salomon A, Keramidas M, Maisin C, Thomas M. Loss of beta-catenin in adrenocortical cancer cells causes growth inhibition and reversal of epithelial-to-mesenchymal transition. Oncotarget (2015). doi:10.18632/oncotarget.3222
127. Rubin B, Monticelli H, Redaelli M, Mucignat C, Barollo S, Bertazza L, et al. Mitogen-activated protein kinase pathway: genetic analysis of 95 adrenocortical tumors. Cancer Invest (2015):1–6. doi:10.3109/07357907.2015.1080832
128. Nakamura M, Miki Y, Akahira J, Morimoto R, Satoh F, Ishidoya S, et al. An analysis of potential surrogate markers of target-specific therapy in archival materials of adrenocortical carcinoma. Endocr Pathol (2009) 20:17–23. doi:10.1007/s12022-009-9058-2
129. De Martino MC, van Koetsveld PM, Feelders RA, Sprij-Mooij D, Waaijers M, Lamberts SW, et al. The role of mTOR inhibitors in the inhibition of growth and cortisol secretion in human adrenocortical carcinoma cells. Endocr Relat Cancer (2012) 19:351–64. doi:10.1530/ERC-11-0270
130. Mariniello B, Rosato A, Zuccolotto G, Rubin B, Cicala MV, Finco I, et al. Combination of sorafenib and everolimus impacts therapeutically on adrenocortical tumor models. Endocr Relat Cancer (2012) 19:527–39. doi:10.1530/ERC-11-0337
131. Doghman M, Lalli E. Efficacy of the novel dual PI3-kinase/mTOR inhibitor NVP-BEZ235 in a preclinical model of adrenocortical carcinoma. Mol Cell Endocrinol (2012) 364:101–4. doi:10.1016/j.mce.2012.08.014
132. Naing A, Lorusso P, Fu S, Hong D, Chen HX, Doyle LA, et al. Insulin growth factor receptor (IGF-1R) antibody cixutumumab combined with the mTOR inhibitor temsirolimus in patients with metastatic adrenocortical carcinoma. Br J Cancer (2013) 108:826–30. doi:10.1038/bjc.2013.46
133. Shuch B, Ricketts CJ, Metwalli AR, Pacak K, Linehan WM. The genetic basis of pheochromocytoma and paraganglioma: implications for management. Urology (2014) 83:1225–32. doi:10.1016/j.urology.2014.01.007
134. Eisenhofer G, Bornstein SR, Brouwers FM, Cheung NK, Dahia PL, de Krijger RR, et al. Malignant pheochromocytoma: current status and initiatives for future progress. Endocr Relat Cancer (2004) 11:423–36. doi:10.1677/erc.1.00829
135. Druce MR, Kaltsas GA, Fraenkel M, Gross DJ, Grossman AB. Novel and evolving therapies in the treatment of malignant phaeochromocytoma: experience with the mTOR inhibitor everolimus (RAD001). Horm Metab Res (2009) 41:697–702. doi:10.1055/s-0029-1220687
136. Yao R, Cooper GM. Requirement for phosphatidylinositol-3 kinase in the prevention of apoptosis by nerve growth factor. Science (1995) 267:2003–6. doi:10.1126/science.7701324
137. Dudek H, Datta SR, Franke TF, Birnbaum MJ, Yao R, Cooper GM, et al. Regulation of neuronal survival by the serine-threonine protein kinase Akt. Science (1997) 275:661–5. doi:10.1126/science.275.5300.661
138. Alvarez-Tejado M, Naranjo-Suarez S, Jimenez C, Carrera AC, Landazuri MO, del Peso L. Hypoxia induces the activation of the phosphatidylinositol 3-kinase/Akt cell survival pathway in PC12 cells: protective role in apoptosis. J Biol Chem (2001) 276:22368–74. doi:10.1074/jbc.M011688200
139. Adler JT, Hottinger DG, Kunnimalaiyaan M, Chen H. Inhibition of the PI3K pathway suppresses hormonal secretion and limits growth in pheochromocytoma cells. World J Surg (2009) 33:2452–7. doi:10.1007/s00268-009-0175-3
140. Lai EW, Rodriguez OC, Aventian M, Cromelin C, Fricke ST, Martiniova L, et al. ErbB-2 induces bilateral adrenal pheochromocytoma formation in mice. Cell Cycle (2007) 6:1946–50. doi:10.4161/cc.6.15.4521
141. Lee M, Waser B, Reubi JC, Pellegata NS. Secretin receptor promotes the proliferation of endocrine tumor cells via the PI3K/AKT pathway. Mol Endocrinol (2012) 26:1394–405. doi:10.1210/me.2012-1055
142. Leinhauser I, Richter A, Lee M, Hofig I, Anastasov N, Fend F, et al. Oncogenic features of the bone morphogenic protein 7 (BMP7) in pheochromocytoma. Oncotarget (2015) 17:39111–26. doi:10.18632/oncotarget.4912
143. Qin Y, Yao L, King EE, Buddavarapu K, Lenci RE, Chocron ES, et al. Germline mutations in TMEM127 confer susceptibility to pheochromocytoma. Nat Genet (2010) 42:229–33. doi:10.1038/ng.533
144. Yao L, Schiavi F, Cascon A, Qin Y, Inglada-Perez L, King EE, et al. Spectrum and prevalence of FP/TMEM127 gene mutations in pheochromocytomas and paragangliomas. JAMA (2010) 304:2611–9. doi:10.1001/jama.2010.1830
145. Giubellino A, Bullova P, Nolting S, Turkova H, Powers JF, Liu Q, et al. Combined inhibition of mTORC1 and mTORC2 signaling pathways is a promising therapeutic option in inhibiting pheochromocytoma tumor growth: in vitro and in vivo studies in female athymic nude mice. Endocrinology (2013) 154:646–55. doi:10.1210/en.2012-1854
146. Saito Y, Tanaka Y, Aita Y, Ishii KA, Ikeda T, Isobe K, et al. Sunitinib induces apoptosis in pheochromocytoma tumor cells by inhibiting VEGFR2/Akt/mTOR/S6K1 pathways through modulation of Bcl-2 and BAD. Am J Physiol Endocrinol Metab (2012) 302:E615–25. doi:10.1152/ajpendo.00035.2011
147. Louis CU, Shohet JM. Neuroblastoma: molecular pathogenesis and therapy. Annu Rev Med (2015) 66:49–63. doi:10.1146/annurev-med-011514-023121
148. Brodeur GM. Neuroblastoma: biological insights into a clinical enigma. Nat Rev Cancer (2003) 3:203–16. doi:10.1038/nrc1014
149. Maris JM. Recent advances in neuroblastoma. N Engl J Med (2010) 362:2202–11. doi:10.1056/NEJMra0804577
150. Liu L, Brown JC III, Webster WW, Morrisett RA, Monaghan DT. Insulin potentiates N-methyl-D-aspartate receptor activity in Xenopus oocytes and rat hippocampus. Neurosci Lett (1995) 192:5–8. doi:10.1016/0304-3940(95)11593-L
151. Li Z, Jaboin J, Dennis PA, Thiele CJ. Genetic and pharmacologic identification of Akt as a mediator of brain-derived neurotrophic factor/TrkB rescue of neuroblastoma cells from chemotherapy-induced cell death. Cancer Res (2005) 65:2070–5. doi:10.1158/0008-5472.CAN-04-3606
152. Bender A, Opel D, Naumann I, Kappler R, Friedman L, von Schweinitz D, et al. PI3K inhibitors prime neuroblastoma cells for chemotherapy by shifting the balance towards pro-apoptotic Bcl-2 proteins and enhanced mitochondrial apoptosis. Oncogene (2011) 30:494–503. doi:10.1038/onc.2010.429
153. Qiao J, Lee S, Paul P, Qiao L, Taylor CJ, Schlegel C, et al. Akt2 regulates metastatic potential in neuroblastoma. PLoS One (2013) 8:e56382. doi:10.1371/journal.pone.0056382
154. Chesler L, Schlieve C, Goldenberg DD, Kenney A, Kim G, McMillan A, et al. Inhibition of phosphatidylinositol 3-kinase destabilizes Mycn protein and blocks malignant progression in neuroblastoma. Cancer Res (2006) 66:8139–46. doi:10.1158/0008-5472.CAN-05-2769
155. Foley NH, Bray IM, Tivnan A, Bryan K, Murphy DM, Buckley PG, et al. MicroRNA-184 inhibits neuroblastoma cell survival through targeting the serine/threonine kinase AKT2. Mol Cancer (2010) 9:83. doi:10.1186/1476-4598-9-83
156. Qiao J, Kang J, Cree J, Evers BM, Chung DH. Gastrin-releasing peptide-induced down-regulation of tumor suppressor protein PTEN (phosphatase and tensin homolog deleted on chromosome ten) in neuroblastomas. Ann Surg (2005) 241:684–91. doi:10.1097/01.sla.0000161173.47717.71
157. Izycka-Swieszewska E, Drozynska E, Rzepko R, Kobierska-Gulida G, Grajkowska W, Perek D, et al. Analysis of PI3K/AKT/mTOR signalling pathway in high risk neuroblastic tumours. Pol J Pathol (2010) 61:192–8.
158. Takenobu H, Shimozato O, Nakamura T, Ochiai H, Yamaguchi Y, Ohira M, et al. CD133 suppresses neuroblastoma cell differentiation via signal pathway modification. Oncogene (2011) 30:97–105. doi:10.1038/onc.2010.383
159. Santo EE, Stroeken P, Sluis PV, Koster J, Versteeg R, Westerhout EM. FOXO3a is a major target of inactivation by PI3K/AKT signaling in aggressive neuroblastoma. Cancer Res (2013) 73:2189–98. doi:10.1158/0008-5472.CAN-12-3767
160. Oberg K, Knigge U, Kwekkeboom D, Perren A, Group EGW. Neuroendocrine gastro-entero-pancreatic tumors: ESMO clinical practice guidelines for diagnosis, treatment and follow-up. Ann Oncol (2012) 23(Suppl 7):vii124–30. doi:10.1093/annonc/mds267
161. Fraenkel M, Kim M, Faggiano A, de Herder WW, Valk GD, Knowledge N. Incidence of gastroenteropancreatic neuroendocrine tumours: a systematic review of the literature. Endocr Relat Cancer (2014) 21:R153–63. doi:10.1530/ERC-13-0125
162. Yao JC, Hassan M, Phan A, Dagohoy C, Leary C, Mares JE, et al. One hundred years after “carcinoid”: epidemiology of and prognostic factors for neuroendocrine tumors in 35,825 cases in the United States. J Clin Oncol (2008) 26:3063–72. doi:10.1200/JCO.2007.15.4377
163. Pitt SC, Chen H, Kunnimalaiyaan M. Inhibition of phosphatidylinositol 3-kinase/Akt signaling suppresses tumor cell proliferation and neuroendocrine marker expression in GI carcinoid tumors. Ann Surg Oncol (2009) 16:2936–42. doi:10.1245/s10434-009-0591-5
164. Gloesenkamp C, Nitzsche B, Lim AR, Normant E, Vosburgh E, Schrader M, et al. Heat shock protein 90 is a promising target for effective growth inhibition of gastrointestinal neuroendocrine tumors. Int J Oncol (2012) 40:1659–67. doi:10.3892/ijo.2012.1328
165. Zitzmann K, Vlotides G, Brand S, Lahm H, Spottl G, Goke B, et al. Perifosine-mediated Akt inhibition in neuroendocrine tumor cells: role of specific Akt isoforms. Endocr Relat Cancer (2012) 19:423–34. doi:10.1530/ERC-12-0074
166. Somnay Y, Simon K, Harrison AD, Kunnimalaiyaan S, Chen H, Kunnimalaiyaan M. Neuroendocrine phenotype alteration and growth suppression through apoptosis by MK-2206, an allosteric inhibitor of AKT, in carcinoid cell lines in vitro. Anticancer Drugs (2013) 24:66–72. doi:10.1097/CAD.0b013e3283584f75
167. Silva SR, Bowen KA, Rychahou PG, Jackson LN, Weiss HL, Lee EY, et al. VEGFR-2 expression in carcinoid cancer cells and its role in tumor growth and metastasis. Int J Cancer (2011) 128:1045–56. doi:10.1002/ijc.25441
168. Missiaglia E, Dalai I, Barbi S, Beghelli S, Falconi M, della Peruta M, et al. Pancreatic endocrine tumors: expression profiling evidences a role for AKT-mTOR pathway. J Clin Oncol (2010) 28:245–55. doi:10.1200/JCO.2008.21.5988
169. Perren A, Komminoth P, Saremaslani P, Matter C, Feurer S, Lees JA, et al. Mutation and expression analyses reveal differential subcellular compartmentalization of PTEN in endocrine pancreatic tumors compared to normal islet cells. Am J Pathol (2000) 157:1097–103. doi:10.1016/S0002-9440(10)64624-X
170. Jiao Y, Shi C, Edil BH, de Wilde RF, Klimstra DS, Maitra A, et al. DAXX/ATRX, MEN1, and mTOR pathway genes are frequently altered in pancreatic neuroendocrine tumors. Science (2011) 331:1199–203. doi:10.1126/science.1200609
171. Banck MS, Kanwar R, Kulkarni AA, Boora GK, Metge F, Kipp BR, et al. The genomic landscape of small intestine neuroendocrine tumors. J Clin Invest (2013) 123:2502–8. doi:10.1172/JCI67963
172. O’Toole D, Couvelard A, Rebours V, Zappa M, Hentic O, Hammel P, et al. Molecular markers associated with response to chemotherapy in gastro-entero-pancreatic neuroendocrine tumors. Endocr Relat Cancer (2010) 17:847–56. doi:10.1677/ERC-09-0204
173. Estrella JS, Broaddus RR, Mathews A, Milton DR, Yao JC, Wang H, et al. Progesterone receptor and PTEN expression predict survival in patients with low- and intermediate-grade pancreatic neuroendocrine tumors. Arch Pathol Lab Med (2014) 138:1027–36. doi:10.5858/arpa.2013-0195-OA
174. Guo SS, Wu X, Shimoide AT, Wong J, Moatamed F, Sawicki MP. Frequent overexpression of cyclin D1 in sporadic pancreatic endocrine tumours. J Endocrinol (2003) 179:73–9. doi:10.1677/joe.0.1790073
175. Kasajima A, Pavel M, Darb-Esfahani S, Noske A, Stenzinger A, Sasano H, et al. mTOR expression and activity patterns in gastroenteropancreatic neuroendocrine tumours. Endocr Relat Cancer (2011) 18:181–92. doi:10.1677/ERC-10-0126
176. Zhou CF, Ji J, Yuan F, Shi M, Zhang J, Liu BY, et al. mTOR activation in well differentiated pancreatic neuroendocrine tumors: a retrospective study on 34 cases. Hepatogastroenterology (2011) 58:2140–3. doi:10.5754/hge11212
177. Wolin EM. PI3K/Akt/mTOR pathway inhibitors in the therapy of pancreatic neuroendocrine tumors. Cancer Lett (2013) 335:1–8. doi:10.1016/j.canlet.2013.02.016
178. Yao JC, Shah MH, Ito T, Bohas CL, Wolin EM, Van Cutsem E, et al. Everolimus for advanced pancreatic neuroendocrine tumors. N Engl J Med (2011) 364:514–23. doi:10.1056/NEJMoa1009290
179. Yap TA, Yan L, Patnaik A, Fearen I, Olmos D, Papadopoulos K, et al. First-in-man clinical trial of the oral pan-AKT inhibitor MK-2206 in patients with advanced solid tumors. J Clin Oncol (2011) 29:4688–95. doi:10.1200/JCO.2011.35.5263
180. Baker AF, Dragovich T, Ihle NT, Williams R, Fenoglio-Preiser C, Powis G. Stability of phosphoprotein as a biological marker of tumor signaling. Clin Cancer Res (2005) 11:4338–40. doi:10.1158/1078-0432.CCR-05-0422
181. Vincent EE, Elder DJ, Thomas EC, Phillips L, Morgan C, Pawade J, et al. Akt phosphorylation on Thr308 but not on Ser473 correlates with Akt protein kinase activity in human non-small cell lung cancer. Br J Cancer (2011) 104:1755–61. doi:10.1038/bjc.2011.132
182. Khor TO, Gul YA, Ithnin H, Seow HF. Positive correlation between overexpression of phospho-BAD with phosphorylated Akt at serine 473 but not threonine 308 in colorectal carcinoma. Cancer Lett (2004) 210:139–50. doi:10.1016/j.canlet.2004.01.017
183. Risso G, Blaustein M, Pozzi B, Mammi P, Srebrow A. Akt/PKB: one kinase, many modifications. Biochem J (2015) 468:203–14. doi:10.1042/BJ20150041
184. Lin CH, Liu SY, Lee EH. SUMO modification of Akt regulates global SUMOylation and substrate SUMOylation specificity through Akt phosphorylation of Ubc9 and SUMO1. Oncogene (2015). doi:10.1038/onc.2015.115
185. Kang ES, Han D, Park J, Kwak TK, Oh MA, Lee SA, et al. GlcNAc modulation at Akt1 Ser473 correlates with apoptosis of murine pancreatic beta cells. Exp Cell Res (2008) 314:2238–48. doi:10.1016/j.yexcr.2008.04.014
186. Yi H, Long B, Ye X, Zhang L, Liu X, Zhang C. Autophagy: a potential target for thyroid cancer therapy (review). Mol Clin Oncol (2014) 2:661–5.
187. Sampath D, Malik A, Plunkett W, Nowak B, Williams B, Burton M, et al. Phase I clinical, pharmacokinetic, and pharmacodynamic study of the Akt-inhibitor triciribine phosphate monohydrate in patients with advanced hematologic malignancies. Leuk Res (2013) 37:1461–7. doi:10.1016/j.leukres.2013.07.034
188. Yan Y, Serra V, Prudkin L, Scaltriti M, Murli S, Rodriguez O, et al. Evaluation and clinical analyses of downstream targets of the Akt inhibitor GDC-0068. Clin Cancer Res (2013) 19:6976–86. doi:10.1158/1078-0432.CCR-13-0978
189. Meric-Bernstam F, Akcakanat A, Chen H, Sahin A, Tarco E, Carkaci S, et al. Influence of biospecimen variables on proteomic biomarkers in breast cancer. Clin Cancer Res (2014) 20:3870–83. doi:10.1158/1078-0432.CCR-13-1507
Keywords: thyroid tumors, parathyroid tumors, pituitary tumors, adrenocortical carcinoma, phaeochromocytoma, neuroblastoma, gastroenteropancreatic neuroendocrine tumors, Akt/PKB kinases
Citation: Robbins HL and Hague A (2016) The PI3K/Akt Pathway in Tumors of Endocrine Tissues. Front. Endocrinol. 6:188. doi: 10.3389/fendo.2015.00188
Received: 02 November 2015; Accepted: 07 December 2015;
Published: 11 January 2016
Edited by:
Rosamaria Lappano, University of Calabria, ItalyReviewed by:
Caterina Mian, University of Padova, ItalyFrancesca Peruzzi, Louisiana State University Health Sciences Center New Orleans, USA
Copyright: © 2016 Robbins and Hague. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Angela Hague, YS5oYWd1ZUBicmlzdG9sLmFjLnVr