 Parth J. Parekh
Parth J. Parekh Vipul R. Nayi2
Vipul R. Nayi2 David A. Johnson
David A. Johnson Aaron I. Vinik
Aaron I. Vinik- 1Division of Gastroenterology and Hepatology, Department of Internal Medicine, Tulane University, New Orleans, LA, USA
- 2Department of Internal Medicine, Montefiore Medical Center, Albert Einstein College of Medicine, Bronx, NY, USA
- 3Division of Gastroenterology, Department of Internal Medicine, Eastern Virginia Medical School, Norfolk, VA, USA
- 4Division of Endocrinology, Department of Internal Medicine, Eastern Virginia Medical School, Norfolk, VA, USA
The obesity epidemic has drastically impacted the state of health care in the United States. Paralleling this epidemic is the incidence of diabetes mellitus, with a notable shift toward a much younger age of onset. While central to the pathogenesis of diabetes associated with obesity is the role of inflammation attributed to “adiposopathy.” Emerging data suggest that changes in sympathetic/parasympathetic balance regulated by the brain precede changes in the inflammatory cascade. It has now been established that the gut microflora contributes significantly to the activation and inhibition of autonomic control and impact the set of the neuroinflammatory inhibitory reflex mediated by the cholinergic nervous system. There has been a paradigm shift toward further investigating commensal bacteria in the pathogenesis of obesity and diabetes mellitus and its complications, as dysbiosis is thought to play a pivotal role in diabetic-associated disorders. This paper is intended to evaluate the role of intestinal dysbiosis in the pathogenesis of diabetes mellitus and examine the potential for restoration of balance via use of probiotics.
Introduction
The incidence of diabetes mellitus continues to rise at historic rates, paralleling that of the obesity epidemic (1). Superimposed on this worldwide epidemic, clinicians are seeing a demographic trend to an even younger age of diagnosis of diabetes mellitus type 2 (T2DM) (2). Currently, therapeutic approaches mainly target the sequelae of disease, whereas research efforts are directed toward addressing the underlying cause of impaired metabolism (3, 4). There has been considerable interest in the role of autonomic balance (5, 6) and the intestinal microbiota may play in this disease, with emerging data implicating a brain–gut dysbiosis in the pathogenesis of obesity, diabetes, and metabolic syndrome (7). In this review, we examine the most current literature focusing on the emerging links between the gut microbiome brain axis and its role in the pathogenesis of diabetes mellitus and its complications.
“Metabolic Infection” – Microbiota and a Low-Grade Inflammatory State
The metabolic alterations associated with obesity also cause a chronic low-grade inflammatory state, which affects energy homeostasis and glucose metabolism (7). Diabetes mellitus has long been thought of as low-grade inflammatory state, secondary to adipocyte necrosis (8–10). The imbalance between caloric intake and energy expenditure results in adipocyte hypertrophy, culminating in local hypoxia and apoptosis (10). As a result, these adipocytes begin to secrete TNF-α in low quantities, thereby stimulating a chemotactic response (7, 10).
Cani et al. suggested that bacterial lipopolysaccharide (LPS) was responsible for this low-grade inflammatory state (11). They demonstrated that levels of endotoxemia fluctuated depending on whether or not the host was in a fasting or fed state. After a 4-week high-fat diet was introduced, levels of LPS-producing microbiota had significantly increased. In addition, continuous subcutaneous infusion of LPS resulted in fasting hyperglycemia and hyperinsulinemia, associated with whole-body, liver, and adipose tissue weight gain that was comparable to those mice fed a high-fat diet for 4 weeks. As the hepatic, but not whole-body, insulin resistance was seen in LPS-infused mice, the authors concluded that metabolic endotoxemia results in a low-grade inflammatory state and acts as a trigger for insulin resistance and the onset of diabetes and obesity. The findings by Cani et al. indicate that LPS is in fact a trigger for the early development of metabolic disease and is involved early in the inflammatory cascade as it stimulates a number of key cytokines (evidenced by increased expression of genes encoding IL-6, TNF-α, IL-1, and PAI-1 in adipose, liver, and muscle). This upregulation is pivotal in the development of insulin resistance via its effect on mCD14 and toll-like receptor (TLR)-4.
Remely et al. evaluated the impact of intestinal dysbiosis on the upregulation of pro-inflammatory cytokines, namely, TLRs 2 and 4 in three groups of subjects: subjects with T2DM receiving glucagon-like peptide-1 (GLP-1) agonist therapy, obese subjects without established insulin resistance, and a lean normative control group (12). They found there to be a significantly higher ratio of Firmicutes:Bacteroides in the diabetes cohort compared to their lean and obese counterparts. In addition, Faecalibacterium prausnitzii were least abundant in the diabetes cohort and most prevalent in lean controls. Methylation analysis demonstrated significantly lower methylation of TLRs 2 and 4 in the diabetic group compared to obese and lean controls, with levels significantly correlating with body mass index. The authors suggested that the changes in commensal microbiota-induced changes and subsequent alterations in cell components are involved in the epigenetic regulation of the inflammatory cascade. Endotoxin LPS is invariably associated with Gram-negative bacteria whether the organisms pathogenic or not. While normally LPS endotoxemia triggers the inflammatory process by binding to the TLR-4 complex at the surface of innate immune cells triggering the host inflammatory cascade, ultimately resulting in insulin resistance; counterintuitively, it was lower levels of Gram-negative Bacteroides that was associated with diabetes. One possible explanation is the impaired innate immune system. The commensal relationship between host and the gut microbiome is pivotal in maintaining the integrity of the immune system. Bacteroides has been demonstrated to influence the activity of defensins (e.g., angiogenin), which regulated the microbial system and innate immunity when secreted; thus, a populous deficient would be predisposed to intestinal dysbiosis and inflammation (13).
Apelin is hormone produced by adipose tissue that is thought to play a key role in the regulation of homeostasis and low-grade inflammation (14, 15). Geurts et al. investigated the microbial composition in obese and diabetic leptin-resistant mice (db/db) in order to establish the role of specific microbial strains and their gut-derived compounds, i.e., LPS, in adipose tissue metabolism via the endocannabinoid system (14). They found db/db mice to have a significantly higher populous of Firmicutes, Proteobacteria, and Fibrobacteres phyla compared to the lean cohort. In addition, they were able to demonstrate the roles of the endocannabinoid system and LPS in regulating apelinergic tone. Using in vitro and in vivo models, they found both the endocannabinoid system and low-grade inflammation to regulate apelin expression in adipose tissue. Lastly, microbiota profiling revealed the commensal microbiota of T2DM mice to significantly differ from that of their lean counterparts, indicating a specific relationship between the microbiome and regulation of the apelinergic system.
The next step was to demonstrate the role of intestinal dysbiosis in the occurrence of diabetes mellitus. Cani et al. administered antibiotics in order to alter commensal microbiota to evaluate whether intestinal dysbiosis is responsible for metabolic endotoxemia, low-grade inflammation, obesity, and diabetes and also to determine what underlying mechanisms are at play (16). They found that antibiotic-induced microbial changes had reduced metabolic endotoxemia and cecal content of LPS compared to high-fat fed mice and genetically obese mice (ob/ob). This correlated with reduced glucose tolerance, inflammation, oxidative stress, body weight gain, development of fat mass, and macrophage infiltration marker mRNA expression in visceral adipose tissue. Mice subjected to high-fat feeding strongly increased intestinal permeability and reduced expression of proteins responsible for tight junctions. Lastly, genetically altered mice that lack functional LPS receptors (CD14 knockout mice) exhibited similar characteristics as mice subjected to antibiotics (i.e., resistant to diet-induced obesity and hepatic insulin resistance). Subsequent metagenome-wide studies have demonstrated the presence of intestinal dysbiosis in patients with T2DM (17, 18), of interest a decrease in the abundance of some universal butyrate-producing bacteria (17), which can alter energy homeostasis, further supporting the concept of intestinal dysbiosis and metabolic endotoxemia and its sequelae set forth by Cani et al. (16).
More recent studies have aimed to investigate the impact of diet on alteration of commensal microbiota and potentially identify specific bacterial strains that predominate diabetes-induced low-grade inflammation. Fallucca et al. investigated the use of the macrobiotic Ma-Pi 2 diet in patients with T2DM for 21 days (19). The Ma-Pi2 diet is one that is low in fat, protein, and fructose containing no animal protein or sugar. It is rich in complex carbohydrates, natural fibers, and most significantly prebiotic and probiotic products (20). The investigators found there to be significant reduction of fasting blood glucose, plasma lipid fractions, plasma insulin, and energy homeostasis by implementing the Ma-Pi 2 diet supporting the role of intestinal dysbiosis in the development of diabetes mellitus and that manipulation of the gut microbiome may be a promising therapeutic option. Subsequent studies have also demonstrated the Ma-Pi 2 diet to reduce markers of insulin resistance, inflammation, and insulin growth factor-1, supporting its use in patients with T2DM (21–23). None of these studies aimed to understand the link between the intestinal dysbiosis and the metabolic derangements found.
Role of Specific Bacterial Strains
Several studies have suggested the protective benefit of Lactobacillus casei against diabetes mellitus (24–29). A recent study by Chen et al. evaluated the antidiabetic effects of L. casei on mice using a model of T2DM induced by a high-fat diet and low-dose streptozotocin (24). Administration of L. casei significantly reduced fasting and postprandial 2-h serum glucose levels, hemoglobin A1c levels, other markers of metabolic syndrome [e.g., triglycerides, total cholesterol, and low-density lipoprotein cholesterol (LDL-C)], and markers of inflammation (e.g., TNF-α, endotoxin) compared to control. In addition, islets of Langerhans were substantially protected against destruction when exposed to L. casei compared to controls, which led to the conclusion that oral administration of L. casei may have a role in the primary prevention of diabetes mellitus. A subsequent study by Zhang et al. postulated that L. casei might induce its antidiabetic effects by acting on the bile acid-chloride transport (25), while we will suggest an alternative mechanism as detailed in the enhancement of the neuroinflammatory reflex (see Figure 1).
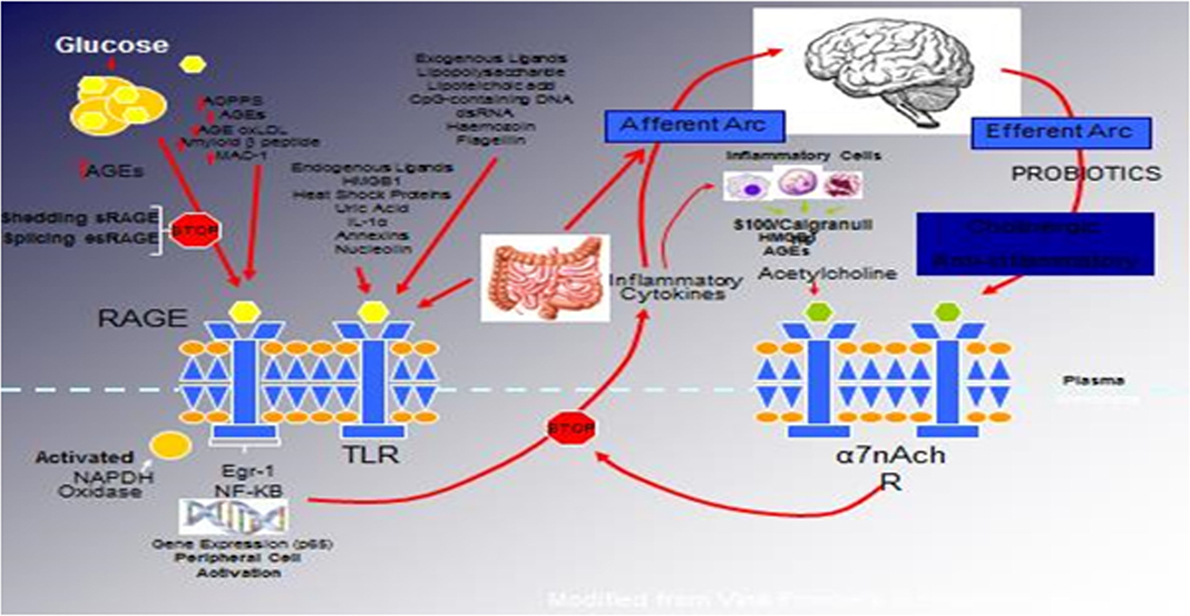
Figure 1. The relationship between binding of ligands to the pattern recognition AGE receptor (RAGE) and inflammation, gene expression, oxidative and nitrosative stress, and activation of an inflammatory cascade involving a cadre of inflammatory cytokines as an afferent arc impacting the brainstem nuclei initiating an efferent arc, in which acetylcholine binds to its receptor inhibiting the activation of the inflammatory cascade.
Unequivocal evidence demonstrates the impact of the gut microbiota on whole-body metabolism and energy homeostasis (7); however, the microbial composition and the underlying mechanisms of interaction between host and commensal microbiota that impact host–gut barrier function and metabolism remain unclear. Recent efforts have shifted focus onto the potential role Akkermansia muciniphila (30–37) and F. prausnitzii (12, 38–42) in mediating diet-induced obesity and T2DM. A study by Everard et al. isolated A. muciniphilia (a mucin-degrading Gram-negative bacterium) within the mucous layer (30). Studies have demonstrated there to be a relative decrease in the population of A. muciniphilia in obese mice and mice with T2DM compared to lean cohorts (30). In addition, the use of prebiotics to reconstitute the population of A. muciniphilia resulted in improved metabolic profiles, reversal of high-fat diet-induced metabolic disorders (e.g., obesity, metabolic endotoxemia, adipocyte hypertrophy, and insulin resistance), increased intestinal levels of endocannabinoids that regulate inflammation, gut mucosal integrity, and intestinal peptide secretion, which ultimately results in restoration of the intestinal barrier (29). At present, the role of A. muciniphilia in the pathogenesis of obesity and T2DM are not known; however, studies suggest that the proximity of A. muciniphilia to the intestinal epithelium allows it to control gut barrier function, fat mass storage, and regulate glucose homeostasis (30).
Faecalibacterium prausnitzii is considered the prototypical anti-inflammatory bacterium and, thus, is among the most studied in a variety of disease states characterized and driven by inflammation (43, 44). Remely et al. recently studied the potential interaction between commensal microbiota in obese patients and patients with T2DM (38). These patients were compared to a lean cohort and evaluated over a 4-month intervention period (interventions comprised a GLP-1 agonist for those with T2DM and nutritional counseling for both groups). Through high-throughput sequencing and pyrosequencing, they found microbial diversity and the populous of F. prausnitzii to be significantly decreased in obese patients and patients with T2DM when compared to their lean counterparts. In addition, analysis of CpGs in the promoter region of FFAR3 demonstrated markedly lower methylation in obese patients and patients with T2DM, which increased in obese patients over the intervention period, thus showing a significant correlation between a higher body mass index and lower methylation of FFAR3. These results suggest that differing microbial makeups affect epigenetic regulation of FFAR-3 and possibly LINE-1 in obese patients and patients with T2DM.
Targeted Therapy: Diet, Probiotics, Prebiotics, and Fecal Transplant
The composition and behavior of gut microbiota can be largely influenced via diet (45); therefore, the next logical question would be to see if diet has any contribution to the development or progression of diabetes. Since diabetes is known to be a multifactorial disorder, adjunctive therapeutic approaches beyond focused glycemic control may be helpful (46). Much of the research into this topic been focused on the use of prebiotics, probiotics, and fecal transplant.
Prebiotics
Prebiotics are defined as non-digestible, fermentable carbohydrates (45, 47) that have the ability to influence the gut microbiota in order to provide a health benefit to the host (48); some examples of such prebiotics are inulin, oligofructose, and resistant starch (47). There are many natural foods that contain prebiotics in their raw form, such as chicory root, Jerusalem artichoke, barley, garlic, onion, globe artichoke, rye bran or grain, wheat bran, and asparagus; chocolate and white bread are examples of cooked or processed foods that contain prebiotics (45). A recent review of several randomized control trials of the effect of prebiotics on patients with prediabetes and T2DM by Barengolts et al. found that dietary changes (which include prebiotic supplementation) may ameliorate the effects of impaired insulin signaling and secretion (45).
In obese humans and genetically obese mice, a decrease in Bacteroidetes and an increase in Firmicutes phyla have been observed when compared to lean humans/mice (31, 45, 49, 50). Using genetically obese and diabetic mice, Everard et al. reported that a prebiotic-enriched diet resulted in changes of 102 distinct taxa, 16 of which displayed a >10-fold change in abundance. Of these results, two of particular importance was an increase in Bacteroidetes and a decrease in Firmicutes phyla; the obese mice fed a prebiotic-enriched diet showed a shift in two dominant phyla toward a profile more similar to lean mice (51). Prebiotics were also shown to improve many metabolic parameters, including lower fasting glycemic levels, improved glucose tolerance (52), reduced plasma triglyceride levels (47, 51), muscle lipid infiltration, adipose tissue mass, and oxidative stress, and increased leptin sensitivity (51).
Outside of affecting the composition of bacterial phyla, these fermentable carbohydrates play a role in the pathogenesis of T2DM and low-grade inflammation. One example is Bifidobacteria, whose growth can be nurtured via the supplementation of prebiotics, specifically dietary fructans. Bifidobacteria express the enzyme β-fructofuranosidase that allows them to break down fructans for energy (53). This is important because the number of Bifidobacteria is decreased in patients with diabetes mellitus, when compared to non-diabetics (53). In mice fed a high-fat diet supplemented with the prebiotic oligofructose, Bifidobacterium was shown to significantly and positively correlate with improved glucose tolerance, insulin secretion, and decreased inflammatory markers (54).
Prebiotics have also been shown to modulate the enteroendocrine and neuroendocrine brain systems via gastrointestinal peptides and neural signaling. Studies have shown that prebiotic supplementation can increase plasma glucagon-like peptide 1 (GLP-1) and plasma glucagon-like peptide 2 (GLP-2) (52, 54–56). In the setting of gut microbiota and diabetes, GLP-1 is thought to play a role in reducing appetite, fat mass, and hepatic insulin resistance (52), while GLP-2 is believed to reduce metabolic endotoxemia by decreasing intestinal wall permeability (57). Prebiotic treatment has also been shown decrease satiety and postprandial plasma glucose response after a standardized meal (54). It is possible that one of the mechanisms in which prebiotics contribute to changes in satiety and postprandial glucose excursion responses is through the modifications of these gastrointestinal peptides.
Probiotics
Probiotics are living microorganisms that can improve the health of its host by altering the intestinal microflora when ingested (58). Research using in vivo and in vitro animal models has shown that probiotics may have a beneficial role in preventing and treating diabetes (46). L. casei is one example that has been proven to have anti-hyperglycemic capabilities in a diabetic mouse model. One group of researchers showed that supplementation of heat-killed L. casei cells exhibited decreased plasma glucose levels in a type 1 diabetic mouse model and prevented the onset of diabetes in a type 2 diabetic mouse model (26, 28). Another example included feeding Lactobacillus rhamnosus subtype GG to diabetic rats, which in 9 weeks had lowered the blood hemoglobin A1c level and improved glucose tolerance when compared to rats fed with normal diet (59). Along with improving diabetic parameters, probiotic supplementation in animals has also shown to decrease beta cell destruction, reduce oxidative damage to the pancreas, exhibit anti-inflammatory properties by increasing liver natural killer cells, reduce bacterial translocation from the intestine into the host, and decrease expression of pro-inflammatory cytokines (27, 60–62).
Unfortunately, there are few studies using probiotic therapy in diabetic human populations. Of the research available, it has been shown that probiotic supplementation can significantly reduce both the oxidative stress and associated inflammatory response, along with decreasing intestinal permeability (63). These favorable effects are thought to lead to greater insulin sensitivity, a decrease in autoimmune response (63), and an improvement in autonomic balance (see Figure 1).
A study in 2012 found that probiotic yogurt (already containing Lactobacillus bulgaricus and Streptococcus thermophiles) enriched with Lactobacillus acidophilus (La5) and Bifidobacterium lactis (Bb12) significantly decreased fasting blood glucose and hemoglobin A1c (58). Additionally, there was associated improved antioxidant status (increased erythrocyte superoxide dismutase, glutathione peroxidase activity) in patients with T2DM when compared to patients supplemented with regular, unenriched yogurt (containing only L. bulgaricus and S. thermophiles). The authors concluded that the decrease in oxidative stress likely resulted from anti-inflammatory and immunomodulatory properties of enriched probiotics. In that, oxidative/nitrosative stress plays a role in the initiation and progression of diabetes (64) and autonomic neuropathy (65); the addition of probiotics may be a potential adjunctive therapy in diabetics or perhaps a primary approach in pre-diabetics and in particular those with newly diagnosed neuropathy (66) and early neuropathy (65).
Cardiovascular disease is responsible for up to 65% of all deaths in diabetic patients (67). Probiotic supplementation can help improve the lipid profile of patients with T2DM. The same group from the study mentioned above showed that consuming probiotic yogurt enriched with L. acidophilus (La5) and B. lactis (Bb12) resulted in a 4.54% decrease in total cholesterol and a 7.45% decrease in LDL-C compared with patients who consumed regular, unenriched yogurt (58). There was no significant change in triglyceride or high-density lipoprotein cholesterol levels. Using the same probiotics, a different group of researchers found that supplementation with L. acidophilus (La5) and B. lactis (Bb12) caused a significant increase in HDL-C and a decrease in the ratio of LDL-C:HDL-C (68). Although these effects are mild compared with statins, the greatest predictor of cardiovascular events in diabetes is the loss of HRV combined with numb feet (6, 69). The effects of these probiotics on the ANS remain to be shown.
Fecal Microbiota Transplant
Changes in the gut microbiota contribute to energy homeostasis and, therefore, the pathophysiological progression to diabetes and obesity. Studies have shown that alterations that occur within the gut flora allow obese animals to harvest a greater portion of energy from their diet (7). It has also been demonstrated that mice with sterile intestines that were fed a high-fat “western diet” did not develop obesity, insulin resistance, or dyslipidemia (70). Animal studies have shown that fecal microbiota transplant from a genetically bred obese mouse to a genetically bred lean mouse can transform the recipient to be the habitus of the donor (71).
Conceptually, a potential rebalancing of the gut flora in obese, diabetic patients could positively influence their energy homeostasis. A recent double-blinded, RCT studied the therapeutic effects of allogeneic lean donor feces infusion on insulin resistance in male patients with the metabolic syndrome (or, in the case of the control group, an autologous transplant) (72). Six weeks later, they found that the patients who had received an allogeneic transplant from lean donors had improved insulin sensitivity (median rate of glucose disappearance increased from 26.2 to 45.3 μmol/kg/min). They also found an increased abundance of butyrate-producing gut microbiota, which they concluded could be the reason for the improved insulin sensitivity, given the role of butyrate in increasing energy expenditure and mitochondrial function. Of note, butyrate stimulates the release of GLP-1 and enhances the beneficial effects of vagal activation. These favorable alterations in glucose metabolism support the notion that reconstituting commensal microflora with lean microflora might be a novel therapeutic intervention by reestablishing a “healthier” host environment.
The Cholinergic Anti-Inflammatory Pathway in Diabetes and Obesity
The inflammatory response is controlled by neural circuitry of the autonomic nervous system. The afferent arc consists of nerves that sense the injury and infection, and this in turn activates a cholinergic anti-inflammatory pathway that modulates the response and is the potential target for future therapies of diabetes (73, 74). The lymphoid organs of the immune system are innervated by cholinergic, catecholaminergic, dopaminergic, and peptidergic neurons, and the neurotransmitters can interact with immune cells and alter their level of function. For instance, in the non-obese diabetic (NOD) mouse (an animal model for type 1 diabetes), neurons surrounding the pancreatic beta cells are lost before there is damage to the islet, and the loss of tonic inhibitory signals contributes to the subsequent beta cell destruction (75). In addition, destruction of the capsaicin-sensitive nerve fibers of the pancreas protect the beta cell from streptozotocin-induced beta cell injury (capsaicin and STZ) (76). Thus, it seems that to begin to understand the role of the autonomic nervous system in its more complex role in inflammation and autoimmunity, one needs to delve further into this complexity. Like St. Thomas, we need to probe deeper!
Watkins and colleagues discovered that sensory neurons detect the presence of inflammation in tissues (77). These responses to IL-1-induced inflammation were mediated by the vagus nerve and could be abolished by vagotomy or a selective competitive antagonist of the IL-1 receptor (78, 79).
There are a number of ligands other than IL-1 derived from macrophages, monocytes, and dendritic cells referred to as pathogen-associated molecular patterns (PAMPs), which activate TLRs, leading to increased expression of NF-κB and increased release of inflammatory cytokines such as TNFα and IL-6. Endogenous molecular products are also released from damaged cells and are referred to as damage-associated molecular patterns (DAMPs). Thus, the nervous system is capable of initiating a response to tissue injury and inflammation and can per se initiate a pro-inflammatory response.
The neurotransmitter of the efferent arc acetylcholine interacts with the innate immune cells that express the nicotinic acetylcholine receptor subunit α7 (α7nAChR). The receptor is widely expressed in neurons that function as ligand-gated ion channels encoded by CHRNA 7 on chromosome 15q14 and is a product of 10 exons yielding a mature protein of 50 kDa. α7nAChR has a tonic inhibitory role in the immune cells similar to the effects of the parasympathetic system on inhibition of heart rate. Exaggerated responses to inflammatory molecules are caused by vagotomy, whereas stimulation of the vagus downregulates the production of TNFα, IL-1, IL-6, and IL-8 but does not alter the anti-inflammatory cytokine IL-10 and TGFβ. This ACh activates the JAK/STAT pathway, affecting the inflammatory responses mediated by NF-κB and initiating the release of a variety of inflammatory cytokines. Thus, this is a defensive reflex protecting the organism from organ damage and death when exposed to syndromes of excess cytokine release such as infection, trauma, and stress.
We have hypothesized that in the metabolic syndrome and diabetes, there is a constant increase in low-grade inflammation mediated by a large cadre of exogenous and endogenous ligands (Figure 1). These, in turn, are capable of binding to the advanced glycation end product receptor (RAGE), thereby activating NF-κB pathway and increasing the production of inflammatory cytokines. In addition, there are a number of other ligands capable of activating the inflammatory cascade. DAMPs, for example, can stimulate innate immune responses culminating in NF-κB activation as with the ligands for RAGE. This is further compounded by the ligands binding to the TLRs, which potentiate the activation of NF-κB. The balance occurs by binding of acetylcholine to the α7nACHR receptor restraining the activation of NF-κB-mediated inflammation. Thus, the loss of autonomic control with reduction of parasympathetic activity, hallmark autonomic dysfunction in diabetes, initiates a cascade of inflammatory responses that if continued unabated will culminate in considerable morbidity and mortality. The possible approaches to enhancing parasympathetic function will be discussed below.
We (60) examined patients with newly diagnosed diabetes, established diabetes and healthy controls and analyzed a cadre of inflammatory markers in addition to time and frequency domain measures of autonomic function. Of great interest to us is the appearance before the advent of inflammation of loss of sympathetic/parasympathetic (S/P) balance. Early in the development of autonomic dysfunction, there is loss of S/P balance, and this correlates with an increase in circulating markers of inflammation, such as CRP and IL-6, and a reduction of the high molecular weight adiponectin/leptin ratios, which correlate with loss of parasympathetic function reflected by changes in the LF/a/HFa frequency ratios and a reduction in the RMSSD and SDNN in time and frequency evaluation of cardiac autonomic function (66, 80, 81). Activation of the efferent arm of the reflex arc (through the administration of an acetylcholine receptor agonist) causes a decrease in pro-inflammatory cytokine production and a reduction in disease severity (82, 83). In his review, Tracey points out a number of important clinical studies that show correlations between vagal nerve activity and inflammatory human diseases, such as rheumatoid arthritis and lupus (74). They considered that inhibition of macrophage function is mediated by Ach released by the vagus acting on specific alpha7nicotinic receptors expressed by the immune cell. Our results are in keeping with this research and suggest that such a reflex arc may be involved in the inflammation seen early in T2D. However, our studies implicate the hypothalamus as the conductor of the endocrine orchestra and show that the earliest changes that are detectable in the evolution of diabetes are abnormalities in autonomic balance. It is not beyond the realms of reason that we could reverse the unfortunate evolutionary profile by targeting the hypothalamic set point of autonomic balance, thereby restoring the efferent arm of the cholinergic anti-inflammatory pathway.
The relationship between binding of ligands to the pattern recognition AGE receptor (RAGE) and inflammation, gene expression, oxidative and nitrosative stress, and damage to the macro- and microvasculature is not entirely clear. Elevated levels of glucose bind to proteins and form AGEs, which bind to RAGEs. RAGE signaling activates NADPH oxidase and production of reactive oxygen species (ROS). Increased ROS increases advanced oxidation protein products (AOPPs), more AGEs, and AGE-modification of oxidized LDLs (oxLDLs). Furthermore, increased ROS may deplete glutathione, thereby suppressing glyoxalase I activity, a mechanism favoring further AGE accumulation. AGEs, AOPPs, macrophage glycoprotein (MAC-1), and AGE-oxLDL ligands of RAGE sustain stimulation of RAGE, and these processes, together with increased ROS, activate key transcription factors, such as nuclear factor-κB (NF-κB) and Egr-1, which increase gene transcription factors and activate inflammatory mechanisms. Consequences include increased migration and activation of RAGE-expressing neutrophils, monocytes/macrophages, T-cells, and dendritic cells. This results in the release of the pro-inflammatory RAGE ligands S100/calgranulins and high-mobility group protein box-1 (HMGB1). In this inflammatory environment, further AGEs may be formed as well. Via interaction with RAGE, these ligands magnify activation of NF-κB, Agr-1, and other factors, thereby amplifying cellular stress and tissue damage leading to neurovascular dysfunction. Soluble RAGE (sRAGE) is formed from the cleavage of RAGE by disintegrins, such as ADAM 10, a metalloproteinase, and β- and γ-secretases. sRAGE or a spliced variant (esRAGE) compete for binding of ligands to RAGE, and a deficiency could theoretically initiate the sequence of events activating an inflammatory cascade with an increase in the expression of pro-inflammatory cytokines [E-selectin, endothelin-1 tissue factor, vascular endothelial growth factor, and other pro-inflammatory cytokines (interleukin-6 and tumor necrosis factor-α)] and damage to neurons, kidney, eye, the vasculature, and even bone. Increasing sRAGE or its administration could competitively reduce activation of the AGE/RAGE pathway and it consequences (5).
Vagal chemoreceptors could be activated directly by substances, such as short-chain fatty acids that can be transported across the epithelial barrier to the portal circulation (84), or by paracrine mediators, such as 5-HT, histamine, CCK, ATP, or glucagon-like peptides released by the various mucosal epithelial layer taste cells (85, 86). That vagal mucosal chemoreceptors might be involved in activation of the “microbiome–gut–brain axis” (85), which is substantiated by animal studies where beneficial bacteria were applied to the epithelium at known concentrations and vagal nerve activity was recorded. There is now strong evidence from animal studies that gut microorganisms can activate the vagus nerve and that such activation plays a critical role in mediating effects on the brain and, subsequently, behavior. The anxiogenic effect of orally administered subclinical doses of Campylobacter jejuni on mice was associated with a significant increase in c-Fos expression in neurons bilaterally in the vagal ganglia and activated visceral sensory nuclei in the brainstem. The areas of brainstem activation, the NTS and lateral parabrachial nucleus, participate in neural information processing that ultimately lead to autonomic neuroendocrine and behavioral responses (87). These findings suggest that the influence of the bacteria on autonomic neurotransmission is mediated centrally, likely through histaminergic nerves and the suprachiasmatic nucleus (88). In a pioneering study, intraduodenal injection of a Lactobacillus johnsonii strain increased gastric vagus massed multiunit firing within 15 min of application (88). Given what is known of the vagal anti-inflammatory reflex, it seems plausible that gut microbiota-induced modulation of vagal mediated “periphery to brain” signaling could translate into changes in efferent neural pathways controlling immune responses (89).
Conclusion
Currently, the depth and breath of the gut microbiome and its implications on obesity, the pathogenesis of diabetes, and its role in the autonomic cholinergic anti-inflammatory pathway that influences health and disease remain poorly understood. The microbiota gut–brain axis is a complex, bidirectional communication, which we are currently in the initial stages of understanding. It appears as though there are strong implications that intestinal dysbiosis via regulation of SCFAs, other paracrine mediators, the critical role of the gut microbiota in endotoxemia, and the effects of LPS on neurons are associated with obesity, metabolic syndrome, and diabetes and its complications.
Associated with the intestinal dysbiosis, animal studies have demonstrated an associated chronic low-grade inflammatory state, which is likely to play a contributing, if not a pivotal, role in diabetic-associated disorders. Clearly, the onset of diabetes is heralded by impairment of HRV, dictated by disordered hypothalamic function, which may owe its origin to the gut dysbiosis.
These seminal data offer significant translational opportunities for further research. Clinical studies evaluating the therapeutic and preventive strategies to prevent or alter this dysbiosis are clearly needed and offer potential new pathways for disease management.
Author Contributions
AV and DJ: manuscript preparation and revision and final approval of the version to be published. PP and VN: manuscript preparation and final approval of the version to be published.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
The authors would like to thank Jerry L. Nadler, MD FAHA MACP FACE, for his thoughtful review and comments.
References
1. Amed S, Dean HJ, Panagiotopoulos C, Sellers EA, Hadjiyannakis S, Laubscher TA, et al. Type 2 diabetes, medication-induced diabetes, and monogenic diabetes in Canadian children: a prospective national surveillance study. Diabetes Care (2010) 33:786–91. doi:10.2337/dc09-1013
2. Pinhas-Hamiel O, Zeitler P. The global spread of type 2 diabetes mellitus in children and adolescents. J Pediatr (2005) 146:693–700. doi:10.1016/j.jpeds.2004.12.042
3. Pittenger GL, Taylor-Fishwick D, Vinik AI. A role for islet neogenesis in curing diabetes. Diabetologia (2009) 52:735–8. doi:10.1007/s00125-009-1322-y
4. DeFronzo RA. Banting lecture. From the triumvirate to the ominous octet: a new paradigm for the treatment of type 2 diabetes mellitus. Diabetes (2009) 58:773–95. doi:10.2337/db09-9028
5. Vinik AI. The conductor of the autonomic orchestra. Front Endocrinol (2012) 3:71. doi:10.3389/fendo.2012.00071
6. Vinik A, Maser R, Ziegler D. Autonomic imbalance: prophet of doom or scope for hope? Diabet Med (2011) 28:643–51. doi:10.1111/j.1464-5491.2010.03184.x
7. Parekh PJ, Arusi E, Vinik AI, Johnson DA. The role and influence of gut microbiota in pathogenesis and management of obesity and metabolic syndrome. Front Endocrinol (2014) 5:47. doi:10.3389/fendo.2014.00047
8. Calle MC, Fernandez ML. Inflammation and type 2 diabetes. Diabetes Metab (2012) 38:183–91. doi:10.1016/j.diabet.2011.11.006
9. Moschen AR, Molnar C, Enrich B, Geiger S, Ebenbichler CF, Tilg H. Adipose and liver expression of interleukin (IL)-1 family members in morbid obesity and effects of weight loss. Mol Med (2011) 17:840–5. doi:10.2119/molmed.2010.00108
10. van Greevenbroek MM, Schalkwijk CG, Stehouwer CD. Obesity-associated low-grade inflammation in type 2 diabetes mellitus: causes and consequences. Neth J Med (2013) 71:174–87.
11. Cani PD, Amar J, Iglesias MA, Poggi M, Knauf C, Bastelica D, et al. Metabolic endotoxemia initiates obesity and insulin resistance. Diabetes (2007) 56:1761–72. doi:10.2337/db06-1491
12. Remely M, Aumueller E, Jahn D, Hippe B, Brath H, Haslberger AG. Microbiota and epigenetic regulation of inflammatory mediators in type 2 diabetes and obesity. Benef Microbes (2014) 5:33–43. doi:10.3920/BM2013.006
13. Hooper LV, Stappenbeck TS, Hong CV, Gordon JI. Angiogenins: a new class of microbicidal proteins involved in innate immunity. Nat Immunol (2003) 4(3):269–73. doi:10.1038/ni888
14. Geurts L, Lazarevic V, Derrien M, Everard A, Van RM, Knauf C, et al. Altered gut microbiota and endocannabinoid system tone in obese and diabetic leptin-resistant mice: impact on apelin regulation in adipose tissue. Front Microbiol (2011) 2:149. doi:10.3389/fmicb.2011.00149
15. Geurts L, Neyrinck AM, Delzenne NM, Knauf C, Cani PD. Gut microbiota controls adipose tissue expansion, gut barrier and glucose metabolism: novel insights into molecular targets and interventions using prebiotics. Benef Microbes (2014) 5:3–17. doi:10.3920/BM2012.0065
16. Cani PD, Osto M, Geurts L, Everard A. Involvement of gut microbiota in the development of low-grade inflammation and type 2 diabetes associated with obesity. Gut Microbes (2012) 3:279–88. doi:10.4161/gmic.19625
17. Qin J, Li Y, Cai Z, Li S, Zhu J, Zhang F, et al. A metagenome-wide association study of gut microbiota in type 2 diabetes. Nature (2012) 490:55–60. doi:10.1038/nature11450
18. Karlsson FH, Tremaroli V, Nookaew I, Bergstrom G, Behre CJ, Fagerberg B, et al. Gut metagenome in European women with normal, impaired and diabetic glucose control. Nature (2013) 498:99–103. doi:10.1038/nature12198
19. Fallucca F, Porrata C, Fallucca S, Pianesi M. Influence of diet on gut microbiota, inflammation and type 2 diabetes mellitus. First experience with macrobiotic Ma-Pi 2 diet. Diabetes Metab Res Rev (2014) 30(Suppl 1):48–54. doi:10.1002/dmrr.2518
20. Fallucca F, Fontana L, Fallucca S, Pianesi M. Gut microbiota and Ma-Pi 2 macrobiotic diet in the treatment of type 2 diabetes. World J Diabetes (2015) 6:403–11. doi:10.4239/wjd.v6.i3.403
21. Soare A, Del TR, Roncella E, Khazrai YM, Angeletti S, Dugo L, et al. The effect of macrobiotic Ma-Pi 2 diet on systemic inflammation in patients with type 2 diabetes: a post hoc analysis of the MADIAB trial. BMJ Open Diabetes Res Care (2015) 3:e000079. doi:10.1136/bmjdrc-2014-000079
22. Soare A, Khazrai YM, Del TR, Roncella E, Fontana L, Fallucca S, et al. The effect of the macrobiotic Ma-Pi 2 diet vs. the recommended diet in the management of type 2 diabetes: the randomized controlled MADIAB trial. Nutr Metab (Lond) (2014) 11:39. doi:10.1186/1743-7075-11-39
23. Porrata C, Sanchez J, Correa V, Abuin A, Hernandez-Triana M, Dacosta-Calheiros RV, et al. Ma-pi 2 macrobiotic diet intervention in adults with type 2 diabetes mellitus. MEDICC Rev (2009) 11:29–35.
24. Chen P, Zhang Q, Dang H, Liu X, Tian F, Zhao J, et al. Antidiabetic effect of Lactobacillus casei CCFM0412 on mice with type 2 diabetes induced by a high-fat diet and streptozotocin. Nutrition (2014) 30:1061–8. doi:10.1016/j.nut.2014.03.022
25. Zhang Y, Guo X, Guo J, He Q, Li H, Song Y, et al. Lactobacillus casei reduces susceptibility to type 2 diabetes via microbiota-mediated body chloride ion influx. Sci Rep (2014) 4:5654. doi:10.1038/srep05654
26. Matsuzaki T, Yamazaki R, Hashimoto S, Yokokura T. Antidiabetic effects of an oral administration of Lactobacillus casei in a non-insulin-dependent diabetes mellitus (NIDDM) model using KK-Ay mice. Endocr J (1997) 44:357–65. doi:10.1507/endocrj.44.357
27. Yadav H, Jain S, Sinha PR. Oral administration of dahi containing probiotic Lactobacillus acidophilus and Lactobacillus casei delayed the progression of streptozotocin-induced diabetes in rats. J Dairy Res (2008) 75:189–95. doi:10.1017/S0022029908003129
28. Matsuzaki T, Nagata Y, Kado S, Uchida K, Kato I, Hashimoto S, et al. Prevention of onset in an insulin-dependent diabetes mellitus model, NOD mice, by oral feeding of Lactobacillus casei. APMIS (1997) 105:643–9. doi:10.1111/j.1699-0463.1997.tb05066.x
29. Naito E, Yoshida Y, Makino K, Kounoshi Y, Kunihiro S, Takahashi R, et al. Beneficial effect of oral administration of Lactobacillus casei strain Shirota on insulin resistance in diet-induced obesity mice. J Appl Microbiol (2011) 110:650–7. doi:10.1111/j.1365-2672.2010.04922.x
30. Everard A, Belzer C, Geurts L, Ouwerkerk JP, Druart C, Bindels LB, et al. Cross-talk between Akkermansia muciniphila and intestinal epithelium controls diet-induced obesity. Proc Natl Acad Sci U S A (2013) 110:9066–71. doi:10.1073/pnas.1219451110
31. Santacruz A, Collado MC, Garcia-Valdes L, Segura MT, Martin-Lagos JA, Anjos T, et al. Gut microbiota composition is associated with body weight, weight gain and biochemical parameters in pregnant women. Br J Nutr (2010) 104:83–92. doi:10.1017/S0007114510000176
32. Karlsson CL, Onnerfalt J, Xu J, Molin G, Ahrne S, Thorngren-Jerneck K. The microbiota of the gut in preschool children with normal and excessive body weight. Obesity (Silver Spring) (2012) 20:2257–61. doi:10.1038/oby.2012.110
33. Derrien M, Vaughan EE, Plugge CM, de Vos WM. Akkermansia muciniphila gen. nov., sp. nov., a human intestinal mucin-degrading bacterium. Int J Syst Evol Microbiol (2004) 54:1469–76. doi:10.1099/ijs.0.02873-0
34. Cani PD, Everard A. [Akkermansia muciniphila: a novel target controlling obesity, type 2 diabetes and inflammation?]. Med Sci (Paris) (2014) 30:125–7. doi:10.1051/medsci/20143002003
35. Graessler J, Qin Y, Zhong H, Zhang J, Licinio J, Wong ML, et al. Metagenomic sequencing of the human gut microbiome before and after bariatric surgery in obese patients with type 2 diabetes: correlation with inflammatory and metabolic parameters. Pharmacogenomics J (2013) 13:514–22. doi:10.1038/tpj.2012.43
36. de Vos WM, de Vos EA. Role of the intestinal microbiome in health and disease: from correlation to causation. Nutr Rev (2012) 70(Suppl 1):S45–56. doi:10.1111/j.1753-4887.2012.00505.x
37. Suez J, Korem T, Zeevi D, Zilberman-Schapira G, Thaiss CA, Maza O, et al. Artificial sweeteners induce glucose intolerance by altering the gut microbiota. Nature (2014) 514:181–6. doi:10.1038/nature13793
38. Remely M, Aumueller E, Merold C, Dworzak S, Hippe B, Zanner J, et al. Effects of short chain fatty acid producing bacteria on epigenetic regulation of FFAR3 in type 2 diabetes and obesity. Gene (2014) 537:85–92. doi:10.1016/j.gene.2013.11.081
39. Li M, Wang B, Zhang M, Rantalainen M, Wang S, Zhou H, et al. Symbiotic gut microbes modulate human metabolic phenotypes. Proc Natl Acad Sci U S A (2008) 105:2117–22. doi:10.1073/pnas.0712038105
40. Miquel S, Martin R, Rossi O, Bermudez-Humaran LG, Chatel JM, Sokol H, et al. Faecalibacterium prausnitzii and human intestinal health. Curr Opin Microbiol (2013) 16:255–61. doi:10.1016/j.mib.2013.06.003
41. Greiner T, Backhed F. Effects of the gut microbiota on obesity and glucose homeostasis. Trends Endocrinol Metab (2011) 22:117–23. doi:10.1016/j.tem.2011.01.002
42. Furet JP, Kong LC, Tap J, Poitou C, Basdevant A, Bouillot JL, et al. Differential adaptation of human gut microbiota to bariatric surgery-induced weight loss: links with metabolic and low-grade inflammation markers. Diabetes (2010) 59:3049–57. doi:10.2337/db10-0253
43. Parekh PJ, Balart LA, Johnson DA. The influence of the gut microbiome on obesity, metabolic syndrome and gastrointestinal disease. Clin Transl Gastroenterol (2015) 6:e91. doi:10.1038/ctg.2015.16
44. Quevrain E, Maubert MA, Michon C, Chain F, Marquant R, Tailhades J, et al. Identification of an anti-inflammatory protein from Faecalibacterium prausnitzii, a commensal bacterium deficient in Crohn’s disease. Gut (2016) 65(3):415–25. doi:10.1136/gutjnl-2014-307649
45. Barengolts E. Vitamin D and prebiotics may benefit the intestinal microbacteria and improve glucose homeostasis in prediabetes and type 2 diabetes. Endocr Pract (2013) 19:497–510. doi:10.4158/EP12263.RA
46. Panwar H, Rashmi HM, Batish VK, Grover S. Probiotics as potential biotherapeutics in the management of type 2 diabetes – prospects and perspectives. Diabetes Metab Res Rev (2013) 29:103–12. doi:10.1002/dmrr.2376
47. Delzenne NM, Kok N. Effects of fructans-type prebiotics on lipid metabolism. Am J Clin Nutr (2001) 73:456S–8S.
48. Delzenne NM, Neyrinck AM, Backhed F, Cani PD. Targeting gut microbiota in obesity: effects of prebiotics and probiotics. Nat Rev Endocrinol (2011) 7:639–46. doi:10.1038/nrendo.2011.126
49. Human Microbiome Project Consortium. Structure, function and diversity of the healthy human microbiome. Nature (2012) 486:207–14. doi:10.1038/nature11234
50. Ley RE, Turnbaugh PJ, Klein S, Gordon JI. Microbial ecology: human gut microbes associated with obesity. Nature (2006) 444:1022–3. doi:10.1038/4441022a
51. Everard A, Lazarevic V, Derrien M, Girard M, Muccioli GG, Neyrinck AM, et al. Responses of gut microbiota and glucose and lipid metabolism to prebiotics in genetic obese and diet-induced leptin-resistant mice. Diabetes (2011) 60:2775–86. doi:10.2337/db11-0227
52. Cani PD, Knauf C, Iglesias MA, Drucker DJ, Delzenne NM, Burcelin R. Improvement of glucose tolerance and hepatic insulin sensitivity by oligofructose requires a functional glucagon-like peptide 1 receptor. Diabetes (2006) 55:1484–90. doi:10.2337/db05-1360
53. Wu X, Ma C, Han L, Nawaz M, Gao F, Zhang X, et al. Molecular characterisation of the faecal microbiota in patients with type II diabetes. Curr Microbiol (2010) 61:69–78. doi:10.1007/s00284-010-9582-9
54. Cani PD, Lecourt E, Dewulf EM, Sohet FM, Pachikian BD, Naslain D, et al. Gut microbiota fermentation of prebiotics increases satietogenic and incretin gut peptide production with consequences for appetite sensation and glucose response after a meal. Am J Clin Nutr (2009) 90:1236–43. doi:10.3945/ajcn.2009.28095
55. Cani PD, Dewever C, Delzenne NM. Inulin-type fructans modulate gastrointestinal peptides involved in appetite regulation (glucagon-like peptide-1 and ghrelin) in rats. Br J Nutr (2004) 92:521–6. doi:10.1079/BJN20041225
56. Cani PD, Neyrinck AM, Maton N, Delzenne NM. Oligofructose promotes satiety in rats fed a high-fat diet: involvement of glucagon-like Peptide-1. Obes Res (2005) 13:1000–7. doi:10.1038/oby.2005.117
57. Cani PD, Possemiers S, Van de Wiele T, Guiot Y, Everard A, Rottier O, et al. Changes in gut microbiota control inflammation in obese mice through a mechanism involving GLP-2-driven improvement of gut permeability. Gut (2009) 58:1091–103. doi:10.1136/gut.2008.165886
58. Ejtahed HS, Mohtadi-Nia J, Homayouni-Rad A, Niafar M, Asghari-Jafarabadi M, Mofid V, et al. Effect of probiotic yogurt containing Lactobacillus acidophilus and Bifidobacterium lactis on lipid profile in individuals with type 2 diabetes mellitus. J Dairy Sci (2011) 94:3288–94. doi:10.3168/jds.2010-4128
59. Tabuchi M, Ozaki M, Tamura A, Yamada N, Ishida T, Hosoda M, et al. Antidiabetic effect of Lactobacillus GG in streptozotocin-induced diabetic rats. Biosci Biotechnol Biochem (2003) 67:1421–4. doi:10.1271/bbb.67.1421
60. Calcinaro F, Dionisi S, Marinaro M, Candeloro P, Bonato V, Marzotti S, et al. Oral probiotic administration induces interleukin-10 production and prevents spontaneous autoimmune diabetes in the non-obese diabetic mouse. Diabetologia (2005) 48:1565–75. doi:10.1007/s00125-005-1831-2
61. Ma X, Hua J, Li Z. Probiotics improve high fat diet-induced hepatic steatosis and insulin resistance by increasing hepatic NKT cells. J Hepatol (2008) 49:821–30. doi:10.1016/j.jhep.2008.05.025
62. Amar J, Chabo C, Waget A, Klopp P, Vachoux C, Bermudez-Humaran LG, et al. Intestinal mucosal adherence and translocation of commensal bacteria at the early onset of type 2 diabetes: molecular mechanisms and probiotic treatment. EMBO Mol Med (2011) 3:559–72. doi:10.1002/emmm.201100159
63. Gomes AC, Bueno AA, de Souza RG, Mota JF. Gut microbiota, probiotics and diabetes. Nutr J (2014) 13:60. doi:10.1186/1475-2891-13-60
64. Maritim AC, Sanders RA, Watkins JB III. Diabetes, oxidative stress, and antioxidants: a review. J Biochem Mol Toxicol (2003) 17:24–38. doi:10.1002/jbt.10058
65. Edwards JF, Casellini CM, Parson HK, Obrosova IG, Yorek M, Vinik AI. Role of peroxynitrite in the development of diabetic peripheral neuropathy. Diabetes Care (2015) 38:e100–1. doi:10.2337/dc14-2918
66. Lieb DC, Parson HK, Mamikunian G, Vinik AI. Cardiac autonomic imbalance in newly diagnosed and established diabetes is associated with markers of adipose tissue inflammation. Exp Diabetes Res (2012) 2012:878760. doi:10.1155/2012/878760
67. Harris M, Cowie C, Stern M, Boyko E, Reiber G, Bennett P. Diabetes in America. Bethesda, MD: National Institutes of Health (1995). p. 233–57.
68. Mohamadshahi M, Veissi M, Haidari F, Javid AZ, Mohammadi F, Shirbeigi E. Effects of probiotic yogurt consumption on lipid profile in type 2 diabetic patients: a randomized controlled clinical trial. J Res Med Sci (2014) 19:531–6.
69. Vinik AI, Maser RE, Ziegler D. Neuropathy: the crystal ball for cardiovascular disease? Diabetes Care (2010) 33:1688–90. doi:10.2337/dc10-0745
70. Kootte RS, Vrieze A, Holleman F, Dallinga-Thie GM, Zoetendal EG, de Vos WM, et al. The therapeutic potential of manipulating gut microbiota in obesity and type 2 diabetes mellitus. Diabetes Obes Metab (2012) 14:112–20. doi:10.1111/j.1463-1326.2011.01483.x
71. Ridaura VK, Faith JJ, Rey FE, Cheng J, Duncan AE, Kau AL, et al. Gut microbiota from twins discordant for obesity modulate metabolism in mice. Science (2013) 341:1241214. doi:10.1126/science.1241214
72. Vrieze A, Van NE, Holleman F, Salojarvi J, Kootte RS, Bartelsman JF, et al. Transfer of intestinal microbiota from lean donors increases insulin sensitivity in individuals with metabolic syndrome. Gastroenterology (2012) 143:913–6. doi:10.1053/j.gastro.2012.06.031
73. Borovikova LV, Ivanova S, Zhang M, Yang H, Botchkina GI, Watkins LR, et al. Vagus nerve stimulation attenuates the systemic inflammatory response to endotoxin. Nature (2000) 405:458–62. doi:10.1038/35013070
75. Saravia F, Homo-Delarche F. Is innervation an early target in autoimmune diabetes? Trends Immunol (2003) 24:574–9. doi:10.1016/j.it.2003.09.010
76. Razavi R, Chan Y, Afifiyan FN, Liu XJ, Wan X, Yantha J, et al. TRPV1+ sensory neurons control beta cell stress and islet inflammation in autoimmune diabetes. Cell (2006) 127:1123–35. doi:10.1016/j.cell.2006.10.038
77. Watkins LR, Goehler LE, Relton JK, Tartaglia N, Silbert L, Martin D, et al. Blockade of interleukin-1 induced hyperthermia by subdiaphragmatic vagotomy: evidence for vagal mediation of immune-brain communication. Neurosci Lett (1995) 183:27–31. doi:10.1016/0304-3940(94)11105-R
78. Hansen MK, O’Connor KA, Goehler LE, Watkins LR, Maier SF. The contribution of the vagus nerve in interleukin-1beta-induced fever is dependent on dose. Am J Physiol Regul Integr Comp Physiol (2001) 280:R929–34.
79. Goehler LE, Relton JK, Dripps D, Kiechle R, Tartaglia N, Maier SF, et al. Vagal paraganglia bind biotinylated interleukin-1 receptor antagonist: a possible mechanism for immune-to-brain communication. Brain Res Bull (1997) 43:357–64. doi:10.1016/S0361-9230(97)00020-8
80. Paolisso G, Manzella D, Montano N, Gambardella A, Varricchio M. Plasma leptin concentrations and cardiac autonomic nervous system in healthy subjects with different body weights. J Clin Endocrinol Metab (2000) 85:1810–4. doi:10.1210/jcem.85.5.6511
81. Wakabayashi S, Aso Y. Adiponectin concentrations in sera from patients with type 2 diabetes are negatively associated with sympathovagal balance as evaluated by power spectral analysis of heart rate variation. Diabetes Care (2004) 27:2392–7. doi:10.2337/diacare.27.10.2392
82. Bernik TR, Friedman SG, Ochani M, DiRaimo R, Ulloa L, Yang H, et al. Pharmacological stimulation of the cholinergic antiinflammatory pathway. J Exp Med (2002) 195:781–8. doi:10.1084/jem.20011714
83. Van Maanen MA, Vervoordeldonk MJ, Tak PP. The cholinergic anti-inflammatory pathway: towards innovative treatment of rheumatoid arthritis. Nat Rev Rheumatol (2009) 5:229–32. doi:10.1038/nrrheum.2009.31
84. Hara H, Haga S, Aoyama Y, Kiriyama S. Short-chain fatty acids suppress cholesterol synthesis in rat liver and intestine. J Nutr (1999) 129:942–8.
85. Raybould HE. Gut chemosensing: interactions between gut endocrine cells and visceral afferents. Auton Neurosci (2010) 153:41–6. doi:10.1016/j.autneu.2009.07.007
86. Bertrand PP. The cornucopia of intestinal chemosensory transduction. Front Neurosci (2009) 3:48. doi:10.3389/neuro.21.003.2009
87. Goehler LE, Gaykema RP, Opitz N, Reddaway R, Badr N, Lyte M. Activation in vagal afferents and central autonomic pathways: early responses to intestinal infection with Campylobacter jejuni. Brain Behav Immun (2005) 19:334–44. doi:10.1016/j.bbi.2004.09.002
88. Tanida M, Yamano T, Maeda K, Okumura N, Fukushima Y, Nagai K. Effects of intraduodenal injection of Lactobacillus johnsonii La1 on renal sympathetic nerve activity and blood pressure in urethane-anesthetized rats. Neurosci Lett (2005) 389:109–14. doi:10.1016/j.neulet.2005.07.036
Keywords: diabetes, intestinal microbiome, autonomic nervous system, intestinal dysbiosis, metabolic syndrome, obesity
Citation: Parekh PJ, Nayi VR, Johnson DA and Vinik AI (2016) The Role of Gut Microflora and the Cholinergic Anti-inflammatory Neuroendocrine System in Diabetes Mellitus. Front. Endocrinol. 7:55. doi: 10.3389/fendo.2016.00055
Received: 05 January 2016; Accepted: 18 May 2016;
Published: 08 June 2016
Edited by:
Gaetano Santulli, Columbia University, USAReviewed by:
Agnes Lehuen, INSERM, FranceMartin Gerbert Frasch, University of Washington Seattle, USA
Zhaoping Li, Ronald Reagan UCLA Medical Center, USA
Copyright: © 2016 Parekh, Nayi, Johnson and Vinik. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Aaron I. Vinik, dmluaWthaUBldm1zLmVkdQ==