 Aurélia E. Lewis
Aurélia E. Lewis Reidun Aesoy2
Reidun Aesoy2 Marit Bakke
Marit Bakke- 1Department of Molecular Biology, University of Bergen, Bergen, Norway
- 2Department of Biomedicine, University of Bergen, Bergen, Norway
Adrenocorticotropic hormone regulates adrenal steroidogenesis mainly via the intracellular signaling molecule cAMP. The effects of cAMP are principally relayed by activating protein kinase A (PKA) and the more recently discovered exchange proteins directly activated by cAMP 1 and 2 (EPAC1 and EPAC2). While the intracellular roles of PKA have been extensively studied in steroidogenic tissues, those of EPACs are only emerging. EPAC1 and EPAC2 are encoded by the genes RAPGEF3 and RAPGEF4, respectively. Whereas EPAC1 is ubiquitously expressed, the expression of EPAC2 is more restricted, and typically found in endocrine tissues. Alternative promoter usage of RAPGEF4 gives rise to three different isoforms of EPAC2 that vary in their N-termini (EPAC2A, EPAC2B, and EPAC2C) and that exhibit distinct expression patterns. EPAC2A is expressed in the brain and pancreas, EPAC2B in steroidogenic cells of the adrenal gland and testis, and EPAC2C has until now only been found in the liver. In this review, we discuss current knowledge on EPAC expression and function with focus on the known roles of EPAC in adrenal gland physiology.
Introduction
In response to stress, the hypothalamic–pituitary–adrenal (HPA) axis is activated. Parvocellular neurons, located in the paraventricular nucleus of the hypothalamus release corticotrophin-releasing factor (CRF), which is transported to the anterior pituitary where it stimulates the corticotropes by binding to type I CRF receptors (1). In response to CRF, these cells release the prohormone pro-opiomelanocortin (POMC) (2). Through posttranscriptional modifications, the inert POMC is converted to biologically active peptides, including adrenocorticotropic hormone (ACTH) (3). ACTH enters the systemic circulation, binds to specific receptors located on the surface of adrenocortical cells, and stimulates the production of adrenocorticosteroid hormones, including cortisol, aldosterone, and adrenal androgens (4). Steroid hormones are produced from the same precursor, cholesterol, by a set of cytochrome P450 steroid hydroxylases (CYP11A1, CYP11B1 and CYP11B2, CYP17 and CYP21) and the steroid dehydrogenase 3βHSD (5). The enzymes are differentially expressed in the three zones of the adrenal cortex (zona glomerulosa, zona fasciculata, and zona reticularis) giving rise to zone-specific hormone production. In humans, the primary source of cholesterol for steroid hormone production is low density lipoprotein (LDL), which is imported via the LDL receptor (LDLR) from the blood stream. Once cholesterol enters the cell, hormone-sensitive lipase (HSL) converts it to free cholesterol substrate (6). Free cholesterol is then delivered to the inner mitochondrial membrane by the actions of steroidogenic acute regulatory protein (StAR) and cholesterol-binding proteins. CYP11A1 (or P450 cholesterol side chain cleavage) catalyses the first and rate-limiting enzymatic step in the biosynthesis of all steroid hormones, the conversion of cholesterol to pregnenolone (7, 8). Pregnenolone can be further converted into different hormone intermediates in the endoplasmic reticulum and the final production of cortisol and aldosterone occurs in mitochondria within the zona fasciculata and zona glomerulosa, respectively (5).
In the adrenal cortex, ACTH coordinates the biosynthesis of steroid hormones via the second messenger cAMP. In response to ACTH binding to its receptor, conformational changes induce the release of G-proteins, which then activate membrane bound adenylyl cyclases (ACs). Upon activation, ACs generates cAMP from ATP (Figure 1A). cAMP relays ACTH-mediated functions via the activation of the serine–threonine kinase cAMP-dependent protein kinase A (PKA) or the exchange proteins directly activated by cAMP (EPAC1 and 2 also named cAMP-regulated guanine nucleotide exchange factors (cAMP-GEFs) I and II). Following cAMP activation, PKA and EPAC transmit signals differently. PKA phosphorylates numerous substrates, while EPACs act as guanine exchange factors (GEFs) catalyzing the conversion of the small GTPases Rap1 and Rap2 from an inactive (GDP-bound) to active form [guanine triphosphate (GTP)-bound] (9, 10). While cAMP signaling by PKA in steroidogenic cells has been intensely investigated, the roles of EPAC are only beginning to emerge. This review summarizes our current knowledge of EPAC2 in tissues of the hypothalamus–pituitary–adrenal axis.
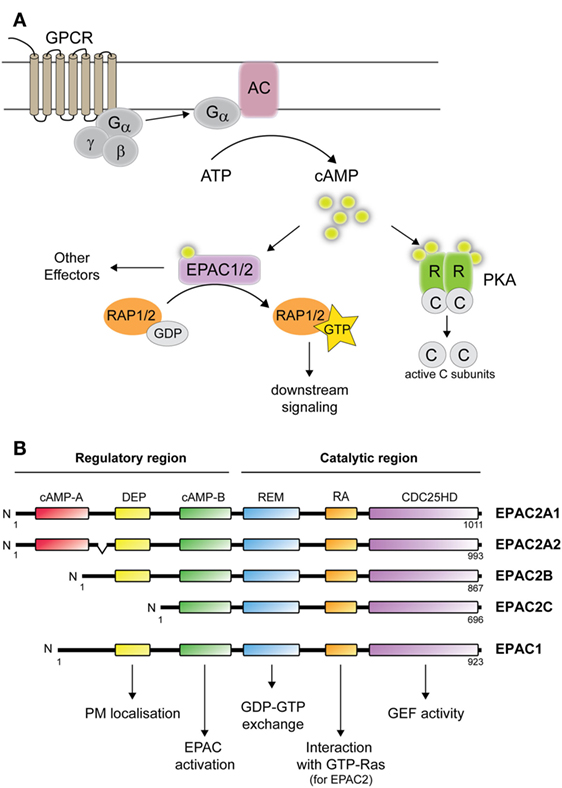
Figure 1. cAMP-mediated signaling and EPAC1 and 2 isoforms. (A) cAMP signaling: following ligand binding of G-protein-coupled receptor, the membrane bound adenylate cyclase (AC) is activated, and generate cAMP from ATP. cAMP subsequently activates PKA and/or EPAC1/2. Binding of cAMP to PKA causes the release of the catalytic subunits, which phosphorylate a variety of targets. Binding of cAMP to EPAC leads to guanosine diphosphate (GDP) to guanine triphosphate (GTP) exchange on Rap1 or Rap2. (B) Domain structure illustration of EPAC1 and EPAC2 isoforms consisting of: cAMP-binding domains A and B, disheveled, Egl-10, pleckstrin (DEP) domain, Ras-exchange motif (REM), Ras association (RA) domain, and CDC25-homology domain (HD). The protein structure of EPAC2A2 is shown in accordance to EPAC2A1. Functional roles of each domain are also indicated. PM, plasma membrane; GEF, guanine nucleotide exchange factor activity.
Structure and Function of EPAC Isoforms
The identification of EPACs was reported in 1998 by two separate groups using different approaches. EPAC1 was discovered by de Rooij and colleagues in a database search for proteins containing cAMP-binding domains with the ultimate goal to explain the PKA-independent cAMP-induced activation of Rap1 (9). The same year, Kawasaki et al. identified both EPAC1 and EPAC2 in a screen for brain-specific genes with cAMP-binding motifs (11). In recent years, it has become evident that the EPAC proteins play essential roles in many biological processes, in some where EPAC and PKA collaborate to achieve a common biological response and in some where the two cAMP effectors have separate functions [reviewed in Ref. (12–14)].
Expression of EPAC Isoforms
The EPAC proteins are encoded by two different genes: RAPGEF3 (EPAC1) and RAPGEF4 (EPAC2), which both give rise to multiple transcripts. Three transcripts are produced from RAPGEF3, but only variant 1, encoding EPAC1, has been studied (13). EPAC1 is relatively ubiquitously expressed (9, 11). Specific parts of the brain, thyroid gland, proximal tubules of the kidney, ovary, and skeletal muscle express the highest levels of EPAC1, but lower levels of EPAC1 have been found in virtually all tissues examined, as well as in hematopoietic cells (15). Studies on murine tissues suggest that EPAC1, as well as EPAC2, mRNA expression is also regulated at different stages of development in embryos and after birth (16).
At the transcript level, four different isoforms arise from RAPGEF4, termed EPAC2A1, EPAC2A2, EPAC2B, and EPAC2C (Figure 1B) (17–20). The EPAC2A2 transcript might be specifically expressed in the brain (20), but the existence of a corresponding protein is yet to be demonstrated. Thus, potential biological roles for this isoform remain unknown. By contrast, the three other mRNAs are known to give rise to the proteins EPAC2A, EPAC2B, and EPAC2C. EPAC2A was the initial EPAC2 isoform identified (11), and is expressed predominantly in brain (with high levels in the cerebral cortex, hippocampus, habenula, cerebellum, and hypothalamus), pituitary, and endocrine pancreas (11, 17, 20). The EPAC2B isoform was identified by the group of Dr. Seino during their efforts in developing an EPAC2 knockout model (19). While confirming deletion of EPAC2A, they discovered the presence of a shorter transcript in the adrenal gland that lacked the N-terminal cAMP-binding domain [(19); Figure 1B]. Until now, EPAC2B expression has only been demonstrated in the adrenal gland (19–21), in the Leydig cell-derived cell line MA10 (21) and in endocrine pancreas (20). The physiological roles of EPAC2B appear to diverge from those of EPAC2A since EPAC2B is not able to substitute for EPAC2A in cellular assays monitoring insulin secretion (19). The shortest EPAC2 isoform, EPAC2C, was also identified by the group of Dr. Seino (17, 18) and has so far only been found in the liver, presumably solely in hepatocytes (18, 20). The strict tissue-specific expression of the different EPAC2 isoforms is controlled, at least in part, by DNA methylation. Detailed analyses of the EPAC2 gene have led to the identification of alternative promoters for the different isoforms (18, 20) and bisulfite sequencing demonstrated that the methylation status of the different promoters nearly perfectly mimics their activity and the expression pattern of the corresponding isoform (20).
Structure and Activation of EPAC Proteins
EPACs are multidomain proteins consisting of two main parts, i.e., an N-terminal regulatory region and a C-terminal catalytic region (Figure 1B). The regulatory region is built up by a cAMP-binding domain and a disheveled, Egl-10, pleckstrin (DEP) domain. EPAC2A contains two cAMP-binding domains, cAMP-A and cAMP-B, while EPAC1 has only one such domain. The domain structure of EPAC2B is similar to EPAC1 and lacks the first cAMP-A binding domain, while EPAC2C lacks both the cAMP-A and DEP domains (18, 19). The N-terminal cAMP-A domain in EPAC2A binds cAMP with low affinity and is not believed to be important for cAMP-induced activation (10). Instead, this domain appears to be important for the localization of EPAC2A near the plasma membrane (19). The DEP domain, by its ability to interact with phosphatidic acid, is also important in targeting EPAC to the plasma membrane upon activation by cAMP (22). The catalytic region consists of a CDC25 homology domain (CDC25HD) that catalyzes Rap1 activation, a Ras-exchange motif (REM) domain, and a Ras association (RA) domain (23). The regulatory regions of EPAC1 and EPAC2 function as inhibitors of the C-terminal GEF domain in the absence of cAMP. Binding of cAMP induces a conformational change that opens the catalytic CDC25HD domain from auto-inhibitory restraints and thereby permits GTP loading of Rap (10, 24–26). Both EPAC1 and EPAC2 contain potential RA domains, but only EPAC2 has been shown to interact with Ras-GTP, which contributes in recruiting EPAC2 to the plasma membrane. The enrichment of EPAC2 on the membrane through Ras binding is crucial for EPAC2-mediated Rap1 activation (27, 28). In addition to the plasma membrane, other subcellular localizations have been observed, such as the perinuclear region, nuclear membranes, and mitochondria for EPAC1 [reviewed in Ref. (29)] and the Golgi apparatus and the nucleus for EPAC2B (21).
Physiological Roles of EPAC
EPACs regulate a multitude of cAMP-mediated cellular processes in many different tissues [extensively reviewed in Ref. (23)], including the formation of cell–cell adhesion (24, 30–33), cell proliferation (34, 35), differentiation (36, 37), cell survival (38), ion channels regulation (39–41), and Ca2+-mediated signaling (23, 42, 43). The development of EPAC knockout mice models has led to a better insight into the biological functions of these proteins. In spite of the involvement of EPAC in multiple cellular pathways, mice lacking EPAC1, EPAC2, or both EPAC1 and EPAC2 do not show gross developmental or reproductive abnormalities. However recent studies have revealed that EPAC1−/− and EPAC2−/− mice display various phenotypes in response to stress or other challenges. For example, mice lacking EPAC1 or EPAC2 exhibit impaired glucose tolerance and dysfunctional insulin secretion after glucose challenge when compared with their wild-type littermates (44–46). Double knockout mice of both EPACs in the forebrain showed defects in long-term potentiation, spatial learning, and social interactions (47), whereas knocking out only EPAC2 is sufficient to induce social interactions impairment (48). Loss of EPAC2 also causes defects in memory retrieval in a fear condition paradigm (49). Interestingly, single nucleotide polymorphisms within the gene encoding EPAC2 have been linked to autism. Screening of 48 autistic individuals for mutations in the RAPGEF4 gene showed that four rare missense mutations may be a cause of autism (50). In spine synapses, these mutations alter the protein function of EPAC2 by affecting its Rap-GEF activity, the synaptic protein distribution and spine morphology (51). Activation of EPAC2 results in shrinkage of dendritic spine size as well as increased motility and turnover of the spines, thereby contributing to the plasticity of brain circuits (51, 52). Mice lacking EPAC1 or EPAC2 also present with phenotypes in the heart. Deletion of EPAC1 causes a mild decrease in basal cardiac functions, but more interestingly protects mice hearts from various stressors, such as arrhythmogenic stress (53). However, partly in contrast to this study, deletion of EPAC1 was reported to have no effect in cardiac function (54). Instead, loss of EPAC2 was shown to protect against β-adrenergic receptor-dependent arrhythmia (54). Most of the biological functions aforementioned have been attributed retrospectively to EPAC1 and EPAC2A due to their expression pattern. The roles of EPAC2B and EPAC2C, which were discovered later, are overall less studied. The potential roles of EPAC2B are discussed in chapter 2. In the liver, EPAC2C has been shown to suppress apoptosis and iNOS expression and activity in hepatocytes (55). EPAC2C may also control bile acid-stimulated canalicular formation in the liver (56).
EPAC in the HPA Axis
EPAC2A is expressed in the hypothalamus and pituitary gland and EPAC2B in the adrenal gland. cAMP is known to be an essential regulator at all levels in the HPA axis (57) and, here, we review how EPAC2 is emerging to contribute to cAMP-mediated actions.
EPAC in the Hypothalamus and the Pituitary
In response to various stress factors, the expression of CRF is stimulated in the PVN of the hypothalamus. CRF synthesis is dependent in part upon the neuropeptide pituitary adenylate cyclase-activating polypeptide (PACAP) and a subsequent elevation of cAMP (57, 58). So far, only PKA-dependent signaling has been reported to relay the stimulatory effect of cAMP on CRF expression. PKA inhibition was indeed shown to prevent binding of cAMP response element (CRE)-binding protein (CREB) on the CRF gene promoter and to inhibit transcriptional activation (59). EPAC has not been studied in PACAP-mediated actions in the PVN. However, EPAC was shown to mediate the effects of PACAP on long-term depression of synaptic transmission in the hippocampus through Rap in murine hippocampal slices (60), and in mice deleted for both Epac1 and Epac2 (47). These studies may, therefore, suggest a potential contribution of EPAC in PACAP-responsive PVN. In the suprachiasmatic nuclei of the hypothalamus, EPAC has been associated with leptin signaling (61) and the regulation of factors involved in setting circadian rhythms in Ref. (62). In noradrenergic neurons isolated from locus coeruleus (LC) in culture, EPAC, but not PKA, was shown to be involved in mediating the actions of cAMP (63). Thus, upon CRF binding to type-1 CRF receptor, LC neurons differentiate into norepinephrine-producing neurons via the activation of cAMP–EPAC–ERK/MAPK pathway, by potentiating brain-derived neurotrophic factor-stimulated synaptic plasticity via tyrosine kinase B signaling (63). Since LC neurons innervate PVN neurons, EPAC may, hence, indirectly stimulate the secretion of CRF from the hypothalamus [reviewed in Ref. (64)].
EPAC2A is also the dominant isoform expressed in the pituitary (19), but we still have very limited information about the potential roles of EPAC2 in this gland. Experiments in AtT20 pituitary cells demonstrated that EPAC, presumably EPAC2A, acts as a mediator of CRF1-induced signaling in corticotropes (65) and in HEK-293 overexpressing CRH-R2β cells (66). Upon activation of the CRH receptor, EPAC2A is involved in cAMP-mediated induction of ERK signaling (65), a pathway previously reported to induce POMC transcription in a PKA-independent manner (67). In addition, CRF receptor activation via Gq is known to signal to phospholipase Cɛ (PLCɛ) and, hence, inositol (1,4,5) triphosphate (IP3) to induce calcium stores mobilization that contributes to ACTH secretion (1, 2, 68). Considering that the activation of PLCɛ by EPAC–Rap2 has been reported to activate Ca2+ release and the secretion of hormones in different tissues (23), EPAC may also contribute to the secretion of ACTH in corticotropes.
EPAC in Adrenal Physiology
Role of EPAC versus PKA in Steroidogenesis
In the adrenal cortex, PKA is undoubtedly the major mediator by which cAMP regulates steroidogenesis (69–71). Once PKA is activated, both an acute and a chronic response occur, which contribute to increased steroid hormone synthesis. During the acute response, PKA phosphorylates HSL, which converts cholesterol esters to free cholesterol. This rapid response also involves an increase in StAR, which facilitates the movement of cholesterol to the inner mitochondrial membrane where the rate-limiting enzyme CYP11A1 resides (72). The chronic response corresponds to the transcriptional activation of all the other steroidogenic enzymes (73, 74). In a study by Schimmer et al., using microarray technology to investigate the effects of ACTH in mouse adrenal Y1 cells, the involvement of PKA was shown to account for up to 60% of the effects of ACTH on transcription, while only 6% could be assigned to PKC (70). This study clearly validated the dominant role of PKA in steroidogenesis, but left about 34% of the ACTH effects to be independent of PKA and PKC. In addition, another study had pointed to the importance of cAMP signaling, mediated independently of PKA, for aldosterone production in the adrenal zona glomerulosa (75). These findings suggest a role for EPAC in the regulation of adrenal function. The specific expression of the EPAC2B isoform in steroidogenic cells (19, 21) also points to roles for EPAC-dependent signaling in these cells. We, therefore, systematically assessed the involvement of PKA versus EPAC in steroidogenesis using cell permeable cAMP analogs specific for PKA and EPAC1/2 (N6-benzoyl-cAMP and 8-p-chlorophenylthio-2-O-methyl-cAMP) in adrenocortical cell lines (21). Our study demonstrated that PKA, and not EPAC2B, is the essential cAMP-induced regulator of factors involved in steroid hormone production (such as StAR, CYP11A1, and CYP17) as well as for the biosynthesis of cortisol and aldosterone. The role of EPAC2 was also studied in bovine zona fasciculata, expressing high levels of EPAC2 mRNA (76), using the same EPAC-specific cAMP analog (77). Although this analog induced cortisol biosynthesis, a non-hydrolyzable EPAC activator had no effect. The study concluded that metabolites of the hydrolyzable EPAC-specific analog induced the increase in cortisol observed in a cAMP-independent manner. Although seemingly opposite results were obtained with the same EPAC activating compound, these two studies indicate that EPAC is not important for cortisol production. cAMP rapidly induces the transcription factor nerve growth factor-induced clone B (NGFI-B), a regulator of several steroid hydroxylase genes (78–80). In adrenocortical cells in culture, we also found that NGFI-B-induction by cAMP is mediated by PKA and not by EPAC (21). Current investigations on HPA axis regulation in mouse knockout models will provide insights into the potential roles for EPAC1/2 in this neuroendocrine system.
EPAC2B Contributes to Cytoskeletal Remodeling in the Adrenal Cortex
In adrenocortical cells in culture, cAMP characteristically induces changes in cell shape and a concomitant reorganization of F-actin microfilaments [reviewed in Ref. (81)]. Furthermore, cytoskeletal reorganization has been shown to contribute to steroidogenic hormone production by allowing the correct positioning of lipid droplets, the ER and mitochondria where cholesterol and its metabolites are transported and metabolized (82–84). While using PKA- and EPAC-specific agonists to study their effect on steroidogenesis in adrenocortical cell lines, we observed that the activation of both cAMP effectors contributed to cell rounding and the reorganization of F-actin fibers (21). Considering that PKA activation contributes to the regulation and expression of many enzymes necessary for steroidogenesis, the additional effect mediated by PKA on F-actin remodeling would, hence, correlate well with enzymatic outputs. By contrast, the effects of EPAC on F-actin remodeling are not correlated to steroidogenesis and may, therefore, contribute to other aspects of adrenal physiology. In line with this, we also found that activation of EPAC2B induced a marked decrease in migration (21). This finding implies that EPAC2B plays a role in cell motility and this suggests wider implications, such as adrenal cancer cell invasion. Although EPAC2 has so far not been implicated in cancer development, several studies have demonstrated roles, albeit contradictory, for EPAC1 in cell migration and metastasis (85). The molecular mechanisms implicated include, at least, an increase of Ca2+ release mediated by PLC-IP3 promoting actin remodelling and cell migration of melanoma cells (86) as well as integrin activation important for cell migration and metastasis of pancreatic cancer cells (87). EPAC2B may, hence, act in the same way as EPAC1 in the adrenal gland.
Conclusion
Since the discovery of EPACs in 1998, our understanding of cAMP-induced signaling and its roles in physiological processes has changed dramatically. Important initial in vitro studies on EPAC paved the way for current phenotypic analyses of genetic mouse models lacking EPAC in single or double knockouts. Based on these gene knockout models an important picture has emerged, namely that although deletion of EPAC does not cause gross defects in mice kept at standard protected conditions in the animal facility, exposure to stressful situations provoke significant phenotypes. EPAC2 is expressed along the HPA axis, and it is interesting to note that whereas the hypothalamus and pituitary specifically express EPAC2A, the adrenal cortex expresses solely the EPAC2B isoform. While EPAC has been shown to mediate potential roles in the hypothalamus, putative functions in the PVN are yet to be determined. At the hypophyseal level, EPAC2A has been implicated in the regulation of POMC expression, and in the adrenal cortex, EPAC2B affects the migration of adrenocortical cells in culture. The generation of spatial and temporal conditional gene knockout models is now required to pinpoint the specific roles of the different EPAC isoforms during development and adult life. Moreover, the ongoing efforts to develop isoform-specific agonists and antagonists hold great promise for insights into isoform-specific functions. Such compounds will also be important potential new drugs to treat diseases in which EPAC plays a role. Several studies do indeed suggest that EPACs are promising drug targets (88), giving hope that small molecules targeting EPACs will serve as useful treatments in the future.
Author Contributions
AEL, RA, and MB wrote parts of the manuscript. AL coordinated editing.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1. Hauger RL, Risbrough V, Oakley RH, Olivares-Reyes JA, Dautzenberg FM. Role of CRF receptor signaling in stress vulnerability, anxiety, and depression. Ann N Y Acad Sci (2009) 1179:120–43. doi:10.1111/j.1749-6632.2009.05011.x
2. Stojilkovic SS, Tabak J, Bertram R. Ion channels and signaling in the pituitary gland. Endocr Rev (2010) 31:845–915. doi:10.1210/er.2010-0005
3. Smith AI, Funder JW. Proopiomelanocortin processing in the pituitary, central nervous system, and peripheral tissues. Endocr Rev (1988) 9:159–79. doi:10.1210/edrv-9-1-159
4. Gallo-Payet N. Adrenal and extra-adrenal functions of ACTH. J Mol Endocrinol (2016). doi:10.1530/JME-15-0257
5. Payne AH, Hales DB. Overview of steroidogenic enzymes in the pathway from cholesterol to active steroid hormones. Endocr Rev (2004) 25:947–70. doi:10.1210/er.2003-0030
6. Kraemer FB, Shen WJ. Hormone-sensitive lipase: control of intracellular tri-(di-)acylglycerol and cholesteryl ester hydrolysis. J Lipid Res (2002) 43:1585–94. doi:10.1194/jlr.R200009-JLR200
7. Ishimura K, Fujita H. Light and electron microscopic immunohistochemistry of the localization of adrenal steroidogenic enzymes. Microsc Res Tech (1997) 36:445–53. doi:10.1002/(SICI)1097-0029(19970315)36:6<445::AID-JEMT2>3.0.CO;2-H
8. Miller WL, Bose HS. Early steps in steroidogenesis: intracellular cholesterol trafficking. J Lipid Res (2011) 52:2111–35. doi:10.1194/jlr.R016675
9. de Rooij J, Zwartkruis FJ, Verheijen MH, Cool RH, Nijman SM, Wittinghofer A, et al. Epac is a Rap1 guanine-nucleotide-exchange factor directly activated by cyclic AMP. Nature (1998) 396:474–7. doi:10.1038/24884
10. de Rooij J, Rehmann H, van Triest M, Cool RH, Wittinghofer A, Bos JL. Mechanism of regulation of the Epac family of cAMP-dependent RapGEFs. J Biol Chem (2000) 275:20829–36. doi:10.1074/jbc.M001113200
11. Kawasaki H, Springett GM, Mochizuki N, Toki S, Nakaya M, Matsuda M, et al. A family of cAMP-binding proteins that directly activate Rap1. Science (1998) 282:2275–9. doi:10.1126/science.282.5397.2275
12. Almahariq M, Mei FC, Cheng X. Cyclic AMP sensor EPAC proteins and energy homeostasis. Trends Endocrinol Metab (2014) 25:60–71. doi:10.1016/j.tem.2013.10.004
13. Banerjee U, Cheng X. Exchange protein directly activated by cAMP encoded by the mammalian rapgef3 gene: structure, function and therapeutics. Gene (2015) 570:157–67. doi:10.1016/j.gene.2015.06.063
14. Sugawara K, Shibasaki T, Takahashi H, Seino S. Structure and functional roles of Epac2 (Rapgef4). Gene (2016) 575:577–83. doi:10.1016/j.gene.2015.09.029
15. Tiwari S, Felekkis K, Moon EY, Flies A, Sherr DH, Lerner A. Among circulating hematopoietic cells, B-CLL uniquely expresses functional EPAC1, but EPAC1-mediated Rap1 activation does not account for PDE4 inhibitor-induced apoptosis. Blood (2004) 103:2661–7. doi:10.1182/blood-2003-06-2154
16. Ulucan C, Wang X, Baljinnyam E, Bai Y, Okumura S, Sato M, et al. Developmental changes in gene expression of Epac and its upregulation in myocardial hypertrophy. Am J Physiol Heart Circ Physiol (2007) 293:H1662–72. doi:10.1152/ajpheart.00159.2007
17. Ozaki N, Shibasaki T, Kashima Y, Miki T, Takahashi K, Ueno H, et al. cAMP-GEFII is a direct target of cAMP in regulated exocytosis. Nat Cell Biol (2000) 2:805–11. doi:10.1038/35041046
18. Ueno H, Shibasaki T, Iwanaga T, Takahashi K, Yokoyama Y, Liu LM, et al. Characterization of the gene EPAC2: structure, chromosomal localization, tissue expression, and identification of the liver-specific isoform. Genomics (2001) 78:91–8. doi:10.1006/geno.2001.6641
19. Niimura M, Miki T, Shibasaki T, Fujimoto W, Iwanaga T, Seino S. Critical role of the N-terminal cyclic AMP-binding domain of Epac2 in its subcellular localization and function. J Cell Physiol (2009) 219:652–8. doi:10.1002/jcp.21709
20. Hoivik EA, Witsoe SL, Bergheim IR, Xu Y, Jakobsson I, Tengholm A, et al. DNA methylation of alternative promoters directs tissue specific expression of Epac2 isoforms. PLoS One (2013) 8:e67925. doi:10.1371/journal.pone.0067925
21. Aumo L, Rusten M, Mellgren G, Bakke M, Lewis AE. Functional roles of protein kinase A (PKA) and exchange protein directly activated by 3’,5’-cyclic adenosine 5’-monophosphate (cAMP) 2 (EPAC2) in cAMP-mediated actions in adrenocortical cells. Endocrinology (2010) 151:2151–61. doi:10.1210/en.2009-1139
22. Consonni SV, Gloerich M, Spanjaard E, Bos JL. cAMP regulates DEP domain-mediated binding of the guanine nucleotide exchange factor Epac1 to phosphatidic acid at the plasma membrane. Proc Natl Acad Sci U S A (2012) 109:3814–9. doi:10.1073/pnas.1117599109
23. Schmidt M, Dekker FJ, Maarsingh H. Exchange protein directly activated by cAMP (epac): a multidomain cAMP mediator in the regulation of diverse biological functions. Pharmacol Rev (2013) 65:670–709. doi:10.1124/pr.110.003707
24. Rehmann H, Prakash B, Wolf E, Rueppel A, De Rooij J, Bos JL, et al. Structure and regulation of the cAMP-binding domains of Epac2. Nat Struct Biol (2003) 10:26–32. doi:10.1038/nsb878
25. Rehmann H, Arias-Palomo E, Hadders MA, Schwede F, Llorca O, Bos JL. Structure of Epac2 in complex with a cyclic AMP analogue and RAP1B. Nature (2008) 455:124–7. doi:10.1038/nature07187
26. Rehmann H, Das J, Knipscheer P, Wittinghofer A, Bos JL. Structure of the cyclic-AMP-responsive exchange factor Epac2 in its auto-inhibited state. Nature (2006) 439:625–8. doi:10.1038/nature04468
27. Li Y, Asuri S, Rebhun JF, Castro AF, Paranavitana NC, Quilliam LA. The RAP1 guanine nucleotide exchange factor Epac2 couples cyclic AMP and Ras signals at the plasma membrane. J Biol Chem (2006) 281:2506–14. doi:10.1074/jbc.M508165200
28. Liu C, Takahashi M, Li Y, Song S, Dillon TJ, Shinde U, et al. Ras is required for the cyclic AMP-dependent activation of Rap1 via Epac2. Mol Cell Biol (2008) 28:7109–25. doi:10.1128/MCB.01060-08
29. Grandoch M, Roscioni SS, Schmidt M. The role of Epac proteins, novel cAMP mediators, in the regulation of immune, lung and neuronal function. Br J Pharmacol (2010) 159:265–84. doi:10.1111/j.1476-5381.2009.00458.x
30. Rangarajan S, Enserink JM, Kuiperij HB, de Rooij J, Price LS, Schwede F, et al. Cyclic AMP induces integrin-mediated cell adhesion through Epac and Rap1 upon stimulation of the beta 2-adrenergic receptor. J Cell Biol (2003) 160:487–93. doi:10.1083/jcb.200209105
31. Bos JL. Epac: a new cAMP target and new avenues in cAMP research. Nat Rev Mol Cell Biol (2003) 4:733–8. doi:10.1038/nrm1197
32. Fukuhara S, Sakurai A, Sano H, Yamagishi A, Somekawa S, Takakura N, et al. Cyclic AMP potentiates vascular endothelial cadherin-mediated cell-cell contact to enhance endothelial barrier function through an Epac-Rap1 signaling pathway. Mol Cell Biol (2005) 25:136–46. doi:10.1128/MCB.25.1.136-146.2005
33. Lorenowicz MJ, Fernandez-Borja M, Kooistra MR, Bos JL, Hordijk PL. PKA and Epac1 regulate endothelial integrity and migration through parallel and independent pathways. Eur J Cell Biol (2008) 87:779–92. doi:10.1016/j.ejcb.2008.05.004
34. Aivatiadou E, Ripolone M, Brunetti F, Berruti G. cAMP-Epac2-mediated activation of Rap1 in developing male germ cells: RA-RhoGAP as a possible direct down-stream effector. Mol Reprod Dev (2009) 76:407–16. doi:10.1002/mrd.20963
35. Hochbaum D, Hong K, Barila G, Ribeiro-Neto F, Altschuler DL. Epac, in synergy with cAMP-dependent protein kinase (PKA), is required for cAMP-mediated mitogenesis. J Biol Chem (2008) 283:4464–8. doi:10.1074/jbc.C700171200
36. Bryn T, Mahic M, Enserink JM, Schwede F, Aandahl EM, Tasken K. The cyclic AMP-Epac1-Rap1 pathway is dissociated from regulation of effector functions in monocytes but acquires immunoregulatory function in mature macrophages. J Immunol (2006) 176:7361–70. doi:10.4049/jimmunol.176.12.7361
37. Petersen RK, Madsen L, Pedersen LM, Hallenborg P, Hagland H, Viste K, et al. Cyclic AMP (cAMP)-mediated stimulation of adipocyte differentiation requires the synergistic action of Epac- and cAMP-dependent protein kinase-dependent processes. Mol Cell Biol (2008) 28:3804–16. doi:10.1128/MCB.00709-07
38. Cullen K, McCool J, Anwer MS, Webster CR. Activation of cAMP-guanine exchange factor (cAMP-GEF) confers protein kinase A independent protection from hepatocyte apoptosis. Am J Physiol Gastrointest Liver Physiol (2004) 2:G334–43. doi:10.1152/ajpgi.00517.2003
39. Honegger KJ, Capuano P, Winter C, Bacic D, Stange G, Wagner CA, et al. Regulation of sodium-proton exchanger isoform 3 (NHE3) by PKA and exchange protein directly activated by cAMP (EPAC). Proc Natl Acad Sci U S A (2006) 103:803–8. doi:10.1073/pnas.0503562103
40. Tengholm A. Cyclic AMP dynamics in the pancreatic beta-cell. Ups J Med Sci (2012) 117:355–69. doi:10.3109/03009734.2012.724732
41. Zhang CL, Katoh M, Shibasaki T, Minami K, Sunaga Y, Takahashi H, et al. The cAMP sensor Epac2 is a direct target of antidiabetic sulfonylurea drugs. Science (2009) 325:607–10. doi:10.1126/science.1172256
42. Kang G, Chepurny OG, Malester B, Rindler MJ, Rehmann H, Bos JL, et al. cAMP sensor Epac as a determinant of ATP-sensitive potassium channel activity in human pancreatic beta cells and rat INS-1 cells. J Physiol (2006) 573:595–609. doi:10.1113/jphysiol.2006.107391
43. Morel E, Marcantoni A, Gastineau M, Birkedal R, Rochais F, Garnier A, et al. cAMP-binding protein Epac induces cardiomyocyte hypertrophy. Circ Res (2005) 97:1296–304. doi:10.1161/01.RES.0000194325.31359.86
44. Shibasaki T, Takahashi H, Miki T, Sunaga Y, Matsumura K, Yamanaka M, et al. Essential role of Epac2/Rap1 signaling in regulation of insulin granule dynamics by cAMP. Proc Natl Acad Sci U S A (2007) 104:19333–8. doi:10.1073/pnas.0707054104
45. Kai AK, Lam AK, Chen Y, Tai AC, Zhang X, Lai AK, et al. Exchange protein activated by cAMP 1 (Epac1)-deficient mice develop beta-cell dysfunction and metabolic syndrome. FASEB J (2013) 27:4122–35. doi:10.1096/fj.13-230433
46. Song WJ, Mondal P, Li Y, Lee SE, Hussain MA. Pancreatic beta-cell response to increased metabolic demand and to pharmacologic secretagogues requires EPAC2A. Diabetes (2013) 62:2796–807. doi:10.2337/db12-1394
47. Yang Y, Shu X, Liu D, Shang Y, Wu Y, Pei L, et al. Impairs learning and social interactions via aberrant regulation of miR-124 and Zif268 translation. Neuron (2012) 73:774–88. doi:10.1016/j.neuron.2012.02.003
48. Srivastava DP, Jones KA, Woolfrey KM, Burgdorf J, Russell TA, Kalmbach A, et al. Social, communication, and cortical structural impairments in Epac2-deficient mice. J Neurosci (2012) 32:11864–11878.
49. Ostroveanu A, van der Zee EA, Eisel UL, Schmidt M, Nijholt IM. Exchange protein activated by cyclic AMP 2 (Epac2) plays a specific and time-limited role in memory retrieval. Hippocampus (2010) 20:1018–26. doi:10.1002/hipo.20700
50. Bacchelli E, Blasi F, Biondolillo M, Lamb JA, Bonora E, Barnby G, et al. Screening of nine candidate genes for autism on chromosome 2q reveals rare nonsynonymous variants in the cAMP-GEFII gene. Mol Psychiatry (2003) 8:916–24. doi:10.1038/sj.mp.4001340
51. Woolfrey KM, Srivastava DP, Photowala H, Yamashita M, Barbolina MV, Cahill ME, et al. Epac2 induces synapse remodeling and depression and its disease-associated forms alter spines. Nat Neurosci (2009) 12:1275–84. doi:10.1038/nn.2386
52. Penzes P, Woolfrey KM, Srivastava DP. Epac2-mediated dendritic spine remodeling: implications for disease. Mol Cell Neurosci (2011) 46:368–80. doi:10.1016/j.mcn.2010.11.008
53. Okumura S, Fujita T, Cai W, Jin M, Namekata I, Mototani Y, et al. Epac1-dependent phospholamban phosphorylation mediates the cardiac response to stresses. J Clin Invest (2014) 124:2785–801. doi:10.1172/JCI64784
54. Pereira L, Cheng H, Lao DH, Na L, van Oort RJ, Brown JH, et al. Epac2 mediates cardiac beta1-adrenergic-dependent sarcoplasmic reticulum Ca2+ leak and arrhythmia. Circulation (2013) 127:913–22. doi:10.1161/CIRCULATIONAHA.12.148619
55. Zhang B, Nweze I, Lakshmanan J, Harbrecht BG. Activation of a cyclic amp-guanine exchange factor in hepatocytes decreases nitric oxide synthase expression. Shock (2013) 39:70–6. doi:10.1097/SHK.0b013e3182760530
56. Fu D, Wakabayashi Y, Lippincott-Schwartz J, Arias IM. Bile acid stimulates hepatocyte polarization through a cAMP-Epac-MEK-LKB1-AMPK pathway. Proc Natl Acad Sci U S A (2011) 108:1403–8. doi:10.1073/pnas.1018376108
57. Kageyama K, Tamasawa N, Suda T. Signal transduction in the hypothalamic corticotropin-releasing factor system and its clinical implications. Stress (2011) 14:357–67. doi:10.3109/10253890.2010.536279
58. Kageyama K, Suda T. Regulatory mechanisms underlying corticotropin-releasing factor gene expression in the hypothalamus. Endocr J (2009) 56:335–44. doi:10.1507/endocrj.K09E-075
59. Agarwal A, Halvorson LM, Legradi G. Pituitary adenylate cyclase-activating polypeptide (PACAP) mimics neuroendocrine and behavioral manifestations of stress: evidence for PKA-mediated expression of the corticotropin-releasing hormone (CRH) gene. Brain Res Mol Brain Res (2005) 138:45–57. doi:10.1016/j.molbrainres.2005.03.016
60. Ster J, de Bock F, Bertaso F, Abitbol K, Daniel H, Bockaert J, et al. Epac mediates PACAP-dependent long-term depression in the hippocampus. J Physiol (2009) 587:101–13. doi:10.1113/jphysiol.2008.157461
61. Fukuda M, Williams KW, Gautron L, Elmquist JK. Induction of leptin resistance by activation of cAMP-Epac signaling. Cell Metab (2011) 13:331–9. doi:10.1016/j.cmet.2011.01.016
62. O’Neill JS, Maywood ES, Chesham JE, Takahashi JS, Hastings MH. cAMP-dependent signaling as a core component of the mammalian circadian pacemaker. Science (2008) 320:949–53. doi:10.1126/science.1152506
63. Traver S, Marien M, Martin E, Hirsch EC, Michel PP. The phenotypic differentiation of locus ceruleus noradrenergic neurons mediated by brain-derived neurotrophic factor is enhanced by corticotropin releasing factor through the activation of a cAMP-dependent signaling pathway. Mol Pharmacol (2006) 70:30–40. doi:10.1124/mol.106.022715
64. Dunn AJ, Swiergiel AH. The role of corticotropin-releasing factor and noradrenaline in stress-related responses, and the inter-relationships between the two systems. Eur J Pharmacol (2008) 583:186–93. doi:10.1016/j.ejphar.2007.11.069
65. Van Kolen K, Dautzenberg FM, Verstraeten K, Royaux I, De Hoogt R, Gutknecht E, et al. Corticotropin releasing factor-induced ERK phosphorylation in AtT20 cells occurs via a cAMP-dependent mechanism requiring EPAC2. Neuropharmacology (2010) 58:135–44. doi:10.1016/j.neuropharm.2009.06.022
66. Markovic D, Punn A, Lehnert H, Grammatopoulos DK. Molecular determinants and feedback circuits regulating type 2 CRH receptor signal integration. Biochim Biophys Acta (2011) 1813:896–907. doi:10.1016/j.bbamcr.2011.02.005
67. Kovalovsky D, Refojo D, Liberman AC, Hochbaum D, Pereda MP, Coso OA, et al. Activation and induction of NUR77/NURR1 in corticotrophs by CRH/cAMP: involvement of calcium, protein kinase A, and MAPK pathways. Mol Endocrinol (2002) 16:1638–51. doi:10.1210/mend.16.7.0863
68. Gutknecht E, Van der Linden I, Van Kolen K, Verhoeven KF, Vauquelin G, Dautzenberg FM. Molecular mechanisms of corticotropin-releasing factor receptor-induced calcium signaling. Mol Pharmacol (2009) 75:648–57. doi:10.1124/mol.108.050427
69. Clark BJ, Ranganathan V, Combs R. Post-translational regulation of steroidogenic acute regulatory protein by cAMP-dependent protein kinase A. Endocr Res (2000) 26:681–9. doi:10.3109/07435800009048587
70. Schimmer BP, Cordova M, Cheng H, Tsao A, Goryachev AB, Schimmer AD, et al. Global profiles of gene expression induced by adrenocorticotropin in Y1 mouse adrenal cells. Endocrinology (2006) 147:2357–67. doi:10.1210/en.2005-1526
71. Aesoy R, Mellgren G, Morohashi K, Lund J. Activation of cAMP-dependent protein kinase increases the protein level of steroidogenic factor-1. Endocrinology (2002) 143:295–303. doi:10.1210/en.143.1.295
72. Stocco DM. The role of the StAR protein in steroidogenesis: challenges for the future. J Endocrinol (2000) 164:247–53. doi:10.1677/joe.0.1640247
73. Sewer MB, Waterman MR. ACTH modulation of transcription factors responsible for steroid hydroxylase gene expression in the adrenal cortex. Microsc Res Tech (2003) 61:300–7. doi:10.1002/jemt.10339
74. Sewer MB, Waterman MR. Adrenocorticotropin/cyclic adenosine 3’,5’-monophosphate-mediated transcription of the human CYP17 gene in the adrenal cortex is dependent on phosphatase activity. Endocrinology (2002) 143:1769–77. doi:10.1210/endo.143.5.8820
75. Gambaryan S, Butt E, Tas P, Smolenski A, Allolio B, Walter U. Regulation of aldosterone production from zona glomerulosa cells by ANG II and cAMP: evidence for PKA-independent activation of CaMK by cAMP. Am J Physiol Endocrinol Metab (2006) 290:E423–33. doi:10.1152/ajpendo.00128.2005
76. Liu H, Enyeart JA, Enyeart JJ. ACTH inhibits bTREK-1 K+ channels through multiple cAMP-dependent signaling pathways. J Gen Physiol (2008) 132:279–94. doi:10.1085/jgp.200810003
77. Enyeart JA, Enyeart JJ. Metabolites of an Epac-selective cAMP analog induce cortisol synthesis by adrenocortical cells through a cAMP-independent pathway. PLoS One (2009) 4:e6088. doi:10.1371/journal.pone.0006088
78. Bassett MH, Suzuki T, Sasano H, De Vries CJ, Jimenez PT, Carr BR, et al. The orphan nuclear receptor NGFIB regulates transcription of 3beta-hydroxysteroid dehydrogenase. implications for the control of adrenal functional zonation. J Biol Chem (2004) 279:37622–30. doi:10.1074/jbc.M405431200
79. Wilson TE, Mouw AR, Weaver CA, Milbrandt J, Parker KL. The orphan nuclear receptor NGFI-B regulates expression of the gene encoding steroid 21-hydroxylase. Mol Cell Biol (1993) 13:861–8. doi:10.1128/MCB.13.2.861
80. Zhang P, Mellon SH. Multiple orphan nuclear receptors converge to regulate rat P450c17 gene transcription: novel mechanisms for orphan nuclear receptor action. Mol Endocrinol (1997) 11:891–904. doi:10.1210/mend.11.7.9940
81. Gallo-Payet N, Payet MD. Mechanism of action of ACTH: beyond cAMP. Microsc Res Tech (2003) 61:275–87. doi:10.1002/jemt.10337
82. Sewer MB, Li D. Regulation of steroid hormone biosynthesis by the cytoskeleton. Lipids (2008) 43:1109–15. doi:10.1007/s11745-008-3221-2
83. Sewer MB, Li D. Regulation of adrenocortical steroid hormone production by RhoA-diaphanous 1 signaling and the cytoskeleton. Mol Cell Endocrinol (2013) 371:79–86. doi:10.1016/j.mce.2012.11.014
84. Issop L, Rone MB, Papadopoulos V. Organelle plasticity and interactions in cholesterol transport and steroid biosynthesis. Mol Cell Endocrinol (2013) 371:34–46. doi:10.1016/j.mce.2012.12.003
85. Almahariq M, Mei FC, Cheng X. The pleiotropic role of exchange protein directly activated by cAMP 1 (EPAC1) in cancer: implications for therapeutic intervention. Acta Biochim Biophys Sin (Shanghai) (2016) 48:75–81. doi:10.1093/abbs/gmv115
86. Baljinnyam E, De Lorenzo MS, Xie LH, Iwatsubo M, Chen S, Goydos JS, et al. Exchange protein directly activated by cyclic AMP increases melanoma cell migration by a Ca2+-dependent mechanism. Cancer Res (2010) 70:5607–17. doi:10.1158/0008-5472.CAN-10-0056
87. Almahariq M, Chao C, Mei FC, Hellmich MR, Patrikeev I, Motamedi M, et al. Pharmacological inhibition and genetic knockdown of exchange protein directly activated by cAMP 1 reduce pancreatic cancer metastasis in vivo. Mol Pharmacol (2015) 87:142–9. doi:10.1124/mol.114.095158
Keywords: ACTH, steroidogenesis, cyclic AMP, EPAC, HPA axis, adrenal glands, adrenal cortex hormones
Citation: Lewis AE, Aesoy R and Bakke M (2016) Role of EPAC in cAMP-Mediated Actions in Adrenocortical Cells. Front. Endocrinol. 7:63. doi: 10.3389/fendo.2016.00063
Received: 15 March 2016; Accepted: 30 May 2016;
Published: 13 June 2016
Edited by:
Nicole Gallo-Payet, University of Sherbrooke, CanadaReviewed by:
A-Marie Lefrançois Martinez, Université Blaise Pascal, FranceKristof Van Kolen, Janssen Research and Development, Belgium
Copyright: © 2016 Lewis, Aesoy and Bakke. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Aurélia E. Lewis, YXVyZWxpYS5sZXdpc0B1aWIubm8=