 Iacopo Gesmundo1,2
Iacopo Gesmundo1,2 Tania Villanova1,2
Tania Villanova1,2 Eleonora Gargantini1,2Emanuela Arvat3
Eleonora Gargantini1,2Emanuela Arvat3 Ezio Ghigo2
Ezio Ghigo2 Riccarda Granata1,2*
Riccarda Granata1,2*
- 1Laboratory of Molecular and Cellular Endocrinology, Department of Medical Sciences, University of Torino, Torino, Italy
- 2Department of Medical Sciences, Division of Endocrinology, Diabetes and Metabolism, University of Torino, Torino, Italy
- 3Department of Medical Sciences, Division of Oncological Endocrinology, University of Torino, Torino, Italy
Glucocorticoid receptor (GR) activation has been shown to reduce adult hippocampal progenitor cell proliferation and neurogenesis. By contrast, mineralocorticoid receptor (MR) signaling is associated with neuronal survival in the dentate gyrus of the hippocampus, and impairment of hippocampal MR has been linked to pathological conditions, such as depression or neurodegenerative disorders. Here, we aimed to further clarify the protective role of MR in adult hippocampal neurons by studying the survival and proliferative effects of the highly potent MR agonist fludrocortisone (Fludro) in adult rat hippocampal progenitor cells (AHPs), along with the associated signaling mechanisms. Fludro, which upregulated MR but not GR expression, increased survival and proliferation and prevented apoptosis in AHPs cultured in growth factor-deprived medium. These effects were blunted by the MR antagonist spironolactone and by high doses of the GR agonist dexamethasone. Moreover, they involved signaling through cAMP/protein kinase A (PKA)/cAMP response element-binding protein, phosphoinositide 3-kinase (PI3K)/Akt and its downstream targets glycogen synthase kinase-3β (GSK-3β) and mammalian target of rapamycin. Furthermore, Fludro attenuated the detrimental effects of amyloid-β peptide 1–42 (Aβ1–42) on cell survival, proliferation, and apoptosis in AHPs, and increased the phosphorylation of both PI3K/Akt and GSK-3β, which was reduced by Aβ1–42. Finally, Fludro blocked Aβ1–42-induced hyperphosphorylation of Tau protein, which is a main feature of Alzheimer’s disease. Overall, these results are the first to show the protective and proliferative role of Fludro in AHPs, suggesting the potential therapeutic importance of targeting MR for increasing hippocampal neurogenesis and for treating neurodegenerative diseases.
Introduction
The hypothalamic–pituitary–adrenal (HPA) axis is the endocrine center of the stress system, consisting of corticotrophin-releasing hormone (CRH), pituitary adrenocorticotropic hormone (ACTH), and adrenal corticosteroid release. At the central level, corticosteroids influence neuronal plasticity and excitability, neurogenesis and neuronal death, as well as neuroendocrine control and behavioral responses (1). Their actions in the brain are mediated by glucocorticoid receptors (GRs) and mineralocorticoid receptors (MRs), which act in the cell nuclei as activators of gene transcription factors. MR expression is particularly high in the hippocampus and other limbic regions, where it co-localizes with GRs, which bind corticosteroid with a 10-fold lower affinity (3). GRs, in turn, are expressed throughout the brain, in neurons and glial cells, and are most abundant in hypothalamic neurons and corticotroph cells (2). Due to the differences in affinity, MR is always occupied by corticosterone, even at basal levels of HPA axis activity; however, higher hormone levels, after stress or circadian peak, progressively saturate the GR (3). Recently, rapid non-genomic effects have been demonstrated for MR and GR, which act as low-affinity membrane-associated receptor variants in response to rising corticosteroid levels (4).
Adult neurogenesis, which mainly occurs in the hippocampal subgranular zone of the dentate gyrus, is a tightly coordinated process involving proliferation, migration, and differentiation of progenitor cells. Numerous signal transduction mechanisms have been shown involved in hippocampal synaptic plasticity, memory formation, and neuronal cell proliferation and survival. These include cAMP/protein kinase A (PKA) and cAMP response element-binding protein (CREB) signaling, which play a central role in different steps of adult neurogenesis (5–7), and the phosphatidylinositol 3-kinase (PI3K)/Akt pathway, a key regulator of hippocampal progenitor cell proliferation and survival, also required for the mitogenic effects of several growth factors (8–10). Interestingly, PI3K/Akt was found to promote CREB phosphorylation, suggesting cross-talk mechanisms between these pathways (9). In addition, the downstream effectors of PI3K/Akt, glycogen synthase kinase-3β (GSK-3β) and mammalian target of rapamycin (mTOR) have been found involved in hippocampal synaptic plasticity and neuronal regeneration, and their signaling has been shown to be impaired in neurodegenerative disorders, including Alzheimer’s disease (AD) (11, 12).
Adult neurogenesis is also regulated by a variety of factors, including environmental factors, stress, hormones, and drugs (13, 14). Among the different hormones, elevation of glucocorticoids or administration of exogenous glucocorticoids has been shown to decrease progenitor cell proliferation and hippocampal neurogenesis (15–21), and adrenalectomy-induced depletion of glucocorticoids was found to attenuate these detrimental effects (22). On the other hand, several studies have shown that, differently from GR, MR activation is involved in hippocampal neuronal survival (4, 23). Indeed, deletion of MR in mice was found to reduce granule cell neurogenesis (24) and to impair learning ability (25), whereas MR overexpression increased differentiation and survival of embryonic stem (ES) cell-derived neurons (26); furthermore, forebrain MR overexpression enhanced memory, reduced anxiety, and attenuated neuronal loss in mouse cerebral ischemia (27). In addition, the natural MR agonist aldosterone was shown to counteract apoptotic hippocampal cell death induced by the GR agonist dexamethasone (DEX) (28) and to enhance neurogenesis in adrenalectomized rats (29). In humans, reduced MR expression and/or MR antagonism were found to be detrimental for the central functions and to affect HPA axis activity, suggesting that hippocampal MR, more than GR, might have clinical relevance in pathological conditions, such as depression, anxiety, or neurodegenerative diseases (30, 31).
Interestingly, different studies have shown impairment of the HPA axis and high glucocorticoid levels in animal models of AD (32, 33). Indeed, AD is an irreversible, progressive age-related neurodegenerative disorder that slowly destroys memory and thinking skills, and the hippocampus is one of the first brain regions to be damaged. Furthermore, it is characterized by accumulation of insoluble extraneuronal senile plaques, mainly composed of amyloid-β peptide (Aβ) and intraneuronal deposits of neurofibrillary tangles (NFTs), formed by hyperphosphorylated Tau protein, which cause massive neuronal death (34, 35).
Although a role for glucocorticoids and GR has been suggested in the progression of AD (33), to date, the direct role of MR agonists on Aβ-induced toxicity has not been investigated and, based on the survival effects in neurons, it can be hypothesized an MR-induced protection against Aβ peptide-induced detrimental effects in hippocampal cells.
The synthetic potent and selective MR agonist fludrocortisone (Fludro) has been previously found to display beneficial effects on executive function and memory in young depressed patients, as well as younger and older healthy individuals (36, 37); however, its role on neuronal cell survival has never been assessed. Therefore, based on the foregoing, the present study aimed to determine whether Fludro exerts survival and proliferative actions in adult rat hippocampal progenitor cells (AHPs), in stress conditions, such as growth factor deprivation, and Aβ-induced toxicity. In addition to the MR agonist, the role of the MR selective antagonist spironolactone (Spiro) and of the GR agonist DEX was also investigated, together with the underlying signaling pathways.
Materials and Methods
Reagents
Fludrocortisone, Spiro, DEX, β-amyloid fragment (1–42), wortmannin, rapamycin, 3-[4,5-dimethylthiazol-2-yl]-2,5-diphenyl tetrazolium bromide (MTT), Stemline neural stem cell medium, polyornithine, bovine serum albumin (BSA), and primers for RT-PCR were from Sigma-Aldrich (Milano, Italy). Human b-FGF, penicillin, streptomycin, fungizone, and trypsin were from Life Technologies, Inc. (Invitrogen, Milano, Italy). Quantikine immunoassay caspase-3 Colorimetric kit was provided by Assay Designs, Bologna, Italy. KT5720 was from Biomol Research Laboratory Inc. (DBA, Italy). Rabbit polyclonal antibodies P-Akt (Ser473), P-GSK-3β (Ser9), P-CREB (Ser133), and P-p70S6K (Thr389); mouse monoclonal antibody for P-Tau (Ser396) (PHF13) was from Cell Signalling Technology (Euroclone SpA, Milano, Italy). Total antibodies were from Abcam (Cambridge, UK). RT-PCR and Real-Time PCR reagents were from Life Technologies, Inc. (Invitrogen, Milano, Italy). Primers for RT-PCR were from TibMolBiol (Genova, Italy).
Cell Culture and Treatments
The clonal population of AHP cells was a kind gift from Prof. Jorgen Isgaard (Laboratory of Experimental Endocrinology, Sahlgrenska Academy, University of Gothenburg, Sweden). The cells were isolated and cultured as previously described (10, 38). Briefly, AHPs were grown in polyornithine poly-coated flasks or wells. For normal proliferating conditions, they were cultured in Stemline neural stem cell medium, supplemented with 20 ng/ml of human b-FGF [normal medium (NM)] at 37°C in a 5% CO2 humidified atmosphere. For experimental conditions, the cells were switched to Dulbecco’s Modified Eagle’s medium (DMEM)/F12 (Sigma-Aldrich, Milano, Italy), supplemented with 0.1% BSA without growth factors and b-FGF for 12 h (control medium, c), then replaced with the same fresh medium, in either absence or presence of the different stimuli for further 24 h. For Aβ1–42 treatments, AHPs were maintained in control medium for 24 h and after removal of the medium they were incubated for the indicated times with Aβ1–42 and either with or without Fludro.
Cell Survival and Proliferation
Cells were seeded in NM in polyornithine-coated 96-well plates at a cell concentration of 5 × 103 cells/well. After 48 h, the medium was changed to control medium and cells were treated with different stimuli for further 24 h. Cell survival was assessed by MTT assay. The cells were incubated with 1 mg/ml MTT for approximately 1 h, then the medium was aspirated and the formazan product solubilized with 100 μl dimethyl sulfoxide (DMSO). Cell viability was assessed by spectrophotometry at 570 nm absorbance using the LT-4000 microplate reader (Euroclone, Milano, Italy). Cell proliferation was assessed using the 5-bromo-2-deoxyuridine (BrdU) incorporation ELISA (Roche Diagnostic SpA, Milano, Italy). Briefly, cells were incubated with BrdU labeling solution for 2 h at 37°C. After removal of the labeling solution, they were fixed, denatured, and incubated for 90 min with anti-BrdU antibody conjugate, which was subsequently removed by rinsing three times. Finally, the cells were incubated in substrate solution at room temperature and proliferation assessed by colorimetric detection at 450 nm absorbance using the LT-4000 microplate reader (Euroclone, Milano, Italy).
Caspase-3 Activity
Caspase-3 activity was assessed by Caspase-3 Colorimetric kit (Assay Designs, Bologna, Italy) in cell lysates, according to the manufacturer’s instruction. Caspase-3 activity was assessed by colorimetric detection at 450 nm absorbance using the LT-4000 microplate reader (Euroclone, Milano, Italy).
cAMP Assay
Cells were seeded in NM in polyornithine-coated 6-well dishes at a concentration of 2 × 105 cells. After 48 h, the medium was changed to control medium and the cells were incubated in the presence of 100 μM of 3-isobutyl-1-methylxanthine (IBMX) and with or without Fludro 1 μM at the indicated time. cAMP was measured from cell lysates using the Cyclic AMP Assay (R&D System, Space Srl, Milano, Italy), as previously described (10). Briefly after incubations, the medium was removed and the cells lysed with ice-cold 0.1 N HCl. Cell lysates were centrifuged for 10 min at 800 rpm, and cAMP in the cell lysates was measured according to the manufacturer’s instructions.
RT-PCR and Real-Time PCR
Total RNA extraction from AHP cells and reverse transcription to cDNA from 3 μg RNA were performed as previously described (39). Nine microliters of cDNA were amplified by standard polymerase chain reaction (PCR) in a 50 μl volume using AmpliTaq Gold Polymerase in a GeneAmp PCR System (Perkin Elmer, Milano, Italy) as described (10). The following primer pairs were used for both RT-PCR and real time PCR: MR (40), forward 5′-TACGACAATTCCAAGCCCGACACC-3′, reverse 5′-TACCTTGGCCCACTTCACGACCTG-3′ (99 bp) (NM_013131). GR (40), forward 5′-AGGGGAGGGGGAGCGTAATGG-3, reverse 5′-CCTCTGCTGCTTGGAATCTGC-3′ (119 bp) (AY293740); rat 18S rRNA, forward 5′-GTGGAGCGATTTGTCTGGTT-3′, and reverse 5′-CGCTGAGCCAGTTCAGTGTA-3′(X01117). Amplification for 18S rRNA subunit was used as internal control. For real-time PCR, cDNA was treated with DNA-free DNAse (LifeTech, Monza, Italy). Real-time PCR was performed with 50 ng cDNA, 100 nmol/L of each primer and the IQ-SYBR-green mastermix (Bio-Rad, Milano, Italy) using the ABI-Prism 7300 (Applied Biosystems).
Western Blot Analysis
Immunoblot analysis was performed as previously described (10, 39). Proteins (50 μg) were resolved in 11% SDS-PAGE and transferred to a nitrocellulose membrane; after blocking with 1% BSA in Tris-buffered saline with 0.1% Tween for 2 h at room temperature, membranes were incubated overnight at 4°C with the specific antibody (P-CREB, P-Akt, P-GSK-3β, P-p70S6K, and P-Tau) (dilution 1:1000). Blots were reprobed with the respective total antibodies for normalization. Immunoreactive proteins were visualized using horseradish peroxidase-conjugated goat anti-mouse or goat anti-rabbit (1:5000) by enhanced chemiluminescence using ChemiDoc XRS (Bio-Rad, Milano, Italy) and densitometric analysis performed with Quantity One software (Bio-Rad).
Statistical Analysis
Data are expressed as means ± SE. Results were analyzed using two-tailed Student t-test or two-way ANOVA followed by Tukey honestly significant difference for post-ANOVA comparisons (GraphPad Prism 5.0 Software, San Diego, CA, USA). Significance was established when P < 0.05.
Results
MR and GR Expression in AHP Cells
MR and GR mRNAs were found expressed in AHPs, as assessed by RT-PCR (Figure 1A). Moreover, real-time PCR analysis showed that treatment with Fludro (1 μM) for 24 h strongly increased MR gene expression in AHPs (Figure 1B), whereas no variation was observed for GR (Figure 1C).
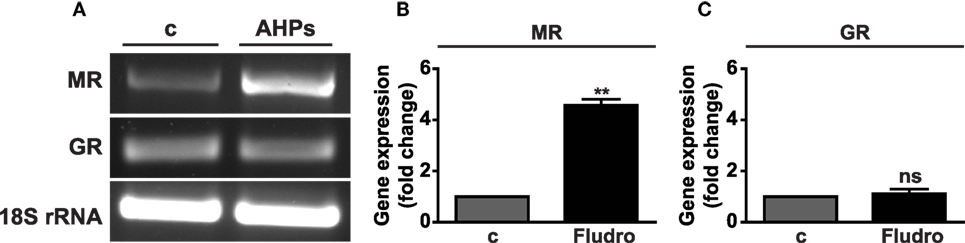
Figure 1. MR and GR mRNA expression in AHPs. (A) Representative images of MR and GR mRNA assessed by RT-PCR. Rat brain was used as positive control (c); 18S rRNA served as internal control. MR (B) and GR (C) gene expression assessed by real-time PCR in AHPs cultured in growth factor-deprived medium (c, control), in either absence or presence of Fludro (1 μM) for 24 h. mRNA expression was normalized to 18S rRNA and reported as fold increase vs. control. Results are the mean ± SEM of at least three independent experiments, each performed in duplicate. **P < 0.01 vs. c; ns, not significant.
Fludro Promotes Survival and Proliferation of AHPs and Prevents Apoptosis
Once ascertained MR and GR expression, we sought to determine the biological effects induced by different concentrations (0.01–2 μM) of the potent synthetic MR agonist Fludro in AHPs, which were cultured in growth factor-deprived medium (referred to as control medium). As previously reported (10, 38), cell survival and proliferation were reduced in control, with respect to growth factor-containing medium (NM) (Figures 2A,B). However, cell survival and proliferation progressively increased in AHPs treated with Fludro, with respect to NM. Fludro displayed these effects at the highest concentrations tested, namely from 0.5 μM and particularly, at 1 and 2 μM (Figures 2A,B). We next determined the potential antiapoptotic role of the MR agonist and observed that, at the same concentrations, Fludro strongly reduced serum starvation-induced apoptosis to levels that were even below those of control, as assessed by reduction of caspase-3 activity (Figure 2C). Therefore, following these results, 1 μM was selected as the best Fludro concentration for the subsequent experiments.
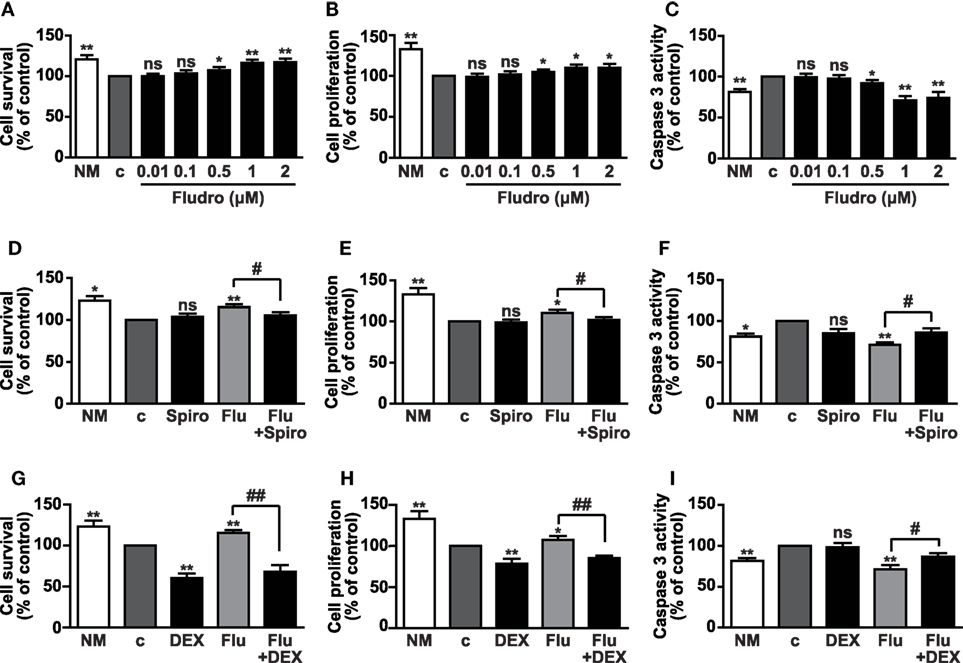
Figure 2. Fludro survival, proliferative, and antiapoptotic effects in AHPs. The cells were cultured in either normal medium (NM) or in growth factor-deprived medium (c, control medium) for 12 h and then for further 24 h with Fludro alone, at the indicated concentrations (A–C), or with 1 μM Fludro (Flu) with or without either 1 μM Spiro (D–F) or 5 μM DEX (G–I). Cell survival and proliferation were assessed by MTT and BrdU incorporation assays, respectively, and apoptosis by caspase-3 activity. Results, expressed as percentage of control, are the mean ± SE of three independent experiments. *P < 0.05, **P < 0.01 vs. c; #P < 0.05, ##P < 0.01; ns, not significant.
To next determine the involvement of MR in the effects of Fludro, AHPs were treated with Fludro, either alone or in combination with the MR selective antagonist Spiro (41). Fludro survival, proliferative, and antiapoptotic actions were found reduced by cotreatment with Spiro (Figures 2D–F). Interestingly, the protective effects of Fludro were also abolished by high concentrations (5 μM) (4) of the GR agonist DEX (Figures 2G–I), which was previously shown to inhibit the proliferation of hippocampal progenitors (17, 42).
Taken together, these results suggest that Fludro elicits protective effects in AHPs, which are counteracted by either selective MR antagonists or high concentrations of GR agonists.
Fludro Survival and Proliferative Effects Involve Signaling Through cAMP/Protein PKA and CREB
Activation of the cAMP/PKA/CREB cascade plays an important role in the regulation of adult neurogenesis (6, 7). Furthermore, elevation of cAMP levels and activation of CREB by the MR agonist aldosterone has been previously demonstrated in porcine coronary artery vascular smooth muscle cells through non-genomic effects (43), as well as the crosstalk between cAMP and aldosterone signaling in human hepatoma cell lines (44). Therefore, we next investigated whether Fludro-induced survival and proliferation would imply activation of the cAMP/PKA/CREB pathway. cAMP levels were found elevated by Fludro (1 μM), peaking at 15 min and being still significantly increased at 90 min with respect to untreated cells. The adenylyl cyclase activator forskolin was used as positive control and strongly increased cAMP levels, as expected (Figure 3A). Fludro also promoted the phosphorylation of CREB on serine 133, which peaked at 15 min and progressively returned to basal levels at 90 min (Figure 3B). The specific PKA inhibitor KT5720 abolished Fludro-induced survival and proliferation in cells cultured in growth factor-deprived medium, further suggesting the involvement of PKA/CREB signaling in the effects of the MR agonist (Figures 3C,D).
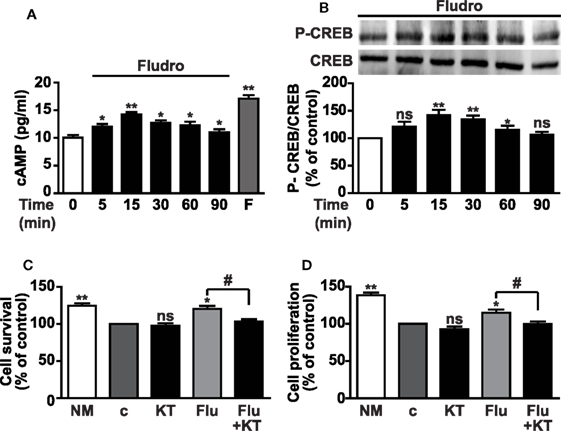
Figure 3. Involvement of cAMP/PKA/CREB signaling in Fludro survival and proliferative effects. (A) Intracellular cAMP levels in growth factor-deprived AHPs cultured for the indicated times with 1 μM Fludro, in the presence of phosphodiesterase inhibitor IBMX (100 μM), that was added 30 min before stimulation. Forskolin (F) (50 μM for 2 min) was used as positive control. Results are the mean ± SE of three independent experiments (*P < 0.05, **P < 0.01 vs. time 0). (B) CREB phosphorylation on serine 133 (top panel) evaluated by Western blot in lysates from AHPs incubated with Fludro (1 μM) for the indicated times. Equal protein loading was determined by reprobing with total CREB antibody (bottom panel). Blots are representative of three independent experiments. Graphs show the densitometric analysis of phosphorylated proteins normalized to total proteins and reported as percentage of basal (*P < 0.05 and **P < 0.01 vs. time 0; ns, not significant). (C,D) Cell survival and proliferation assessed by MTT assay and BrdU, respectively, in AHP cells cultured in normal medium (NM) or in growth factor-deprived medium (c, control medium) for 12 h, and then for further 24 h with 0.5 μM KT5720 (KT), either alone or with 1 μM Fludro (Flu). Data, expressed as percentage of control, are the mean ± SE (n = 3); *P < 0.05, **P < 0.01 vs. c; #P < 0.05; ns, not significant.
Phosphorylation of PI3K/Akt, GSK-3β, and mTOR Is Required for Fludro Survival, Proliferative, and Antiapoptotic Effects
Activation of the PI3K/Akt pathway has been shown to play a pivotal role in both survival and proliferation of adult hippocampal progenitors, also induced by different growth factors (5, 9, 10). Furthermore, PI3K/Akt-mediated inactivation of GSK-3β through phosphorylation on Ser9 residue is crucial for hippocampal neurogenesis and neuronal survival (11). Here, Fludro-treated AHPs showed a time-dependent increase of Akt phosphorylation, which peaked at 30 min and decreased thereafter, although being still significantly elevated at 60 and 90 min compared to basal (Figure 4A). Similarly, Fludro strongly and time-dependently increased GSK-3β phosphorylation at Ser9, which was maintained up to 90 min after stimulation (Figure 4B). The role of Fludro was also assessed on activation of the other Akt effector mTOR, (12). Specifically, we studied the effect of Fludro on the mTOR target p70S6K and found a time-dependent increase of its phosphorylation at Thr389, compared to basal (Figure 4C). Next, to further assess the role of Akt and mTOR in Fludro effects, AHPs were treated with the specific inhibitors of these pathways. Fludro survival, proliferative, and antiapoptotic actions were found reduced by either the Akt inhibitor wortmannin (Figures 4D–F) or the mTOR inhibitor rapamycin (Figures 4G–I), whereas these compounds alone were inactive. Overall, these results suggest that Fludro-induced protection in AHPs requires activation of PI3K/Akt and mTOR, as well as inactivation of GSK-3β.
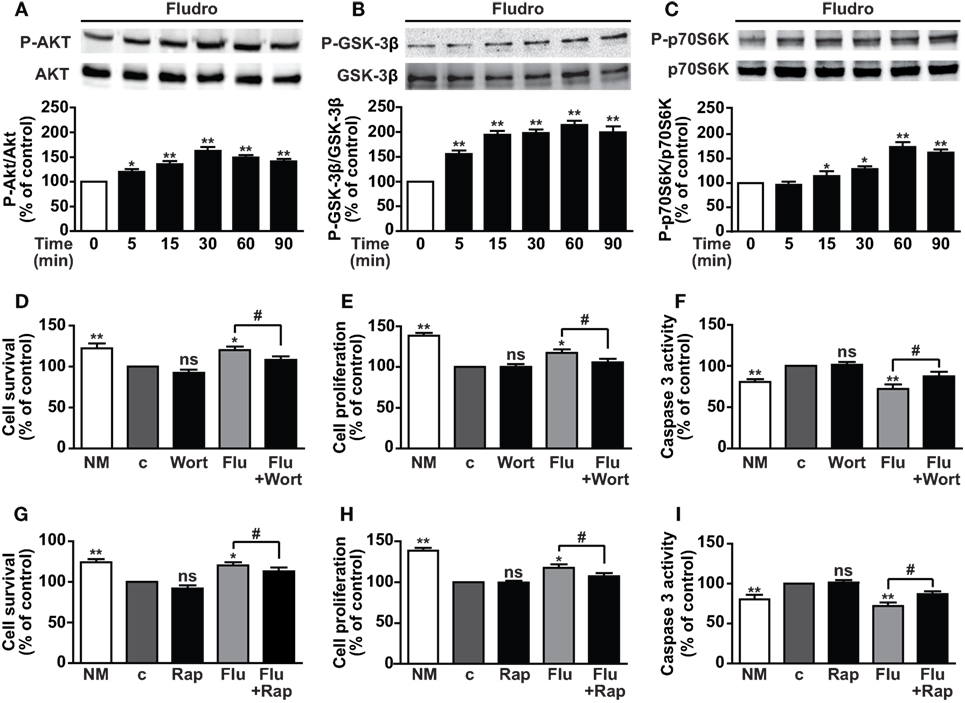
Figure 4. Involvement of PI3K/Akt, GSK-3β, and mTOR/p70S6K signaling in Fludro effects. (A) Akt, (B) GSK-3β, and (C) p70S6K phosphorylation evaluated by Western blot in AHPs treated with 1 μM Fludro for the indicated times (upper panels). Blots, each representative of three independent experiments, were reprobed with total antibodies for normalization (lower panels). Graphs show phosphorylated proteins normalized to total proteins and reported as percentage of basal (*P < 0.05 and **P < 0.01 vs. time 0). (D–I) Cell survival, proliferation, and apoptosis assessed by MTT, BrdU, and caspase-3 activity, respectively, in AHPs cultured in normal medium (NM) or in growth factor-deprived medium (c, control medium) for 12 h, and then for further 24 h with either wortmannin (Wort, 100 nM) (D–F) or rapamycin (Rap, 50 nM) (G–I), both alone or with 1 μM Fludro (Flu). Data, expressed as percentage of c, are the mean ± SE of four independent experiments. *P < 0.05, **P < 0.01 vs. c; #P < 0.05; ns, not significant.
Fludro Counteracts the Detrimental Effects of Aβ Peptide 1–42
Aβ peptides exert toxic effects in a variety of neural cell types by reducing survival and proliferation. Indeed, abnormal Aβ peptide 1–42 (Aβ1–42) deposition is one of the main hallmarks of AD and contributes to neuronal apoptotic cell death in the hippocampus and cerebral cortex. Moreover, Aβ1–42 plays a critical role in the hyperphosphorylation of Tau protein and the consequent formation of NFTs (34, 35). Fludro-induced survival and proliferative effects observed in growth factor-deprived conditions led us to hypothesize a protective role of the MR agonist against Aβ1–42-induced cell death. In AHPs treated for 24 h with 1 μM Aβ1–42, whose concentration was selected on the basis of our previous findings (10), coincubation with Fludro attenuated the detrimental effect of the peptide by increasing survival and proliferation and restoring apoptosis to control levels (Figures 5A–C). Consistent with the previous results, Fludro, which per se promoted both Akt and GSK-3β phosphorylation, also counteracted Aβ1–42-induced reduction of Akt and GSK-3β phosphorylation (Figures 5D,E). Next, the effect of Fludro was examined on Tau protein, whose hyperphosphorylation by GSK-3β is involved in the formation of NFTs and the pathogenesis of AD (35, 45). Figure 5F shows that Fludro alone had no effect with respect to control; however, it completely blocked Aβ1–42-induced phosphorylation of Tau. Collectively, these findings suggest a protective role for Fludro against Aβ1–42-induced toxicity in hippocampal progenitors.
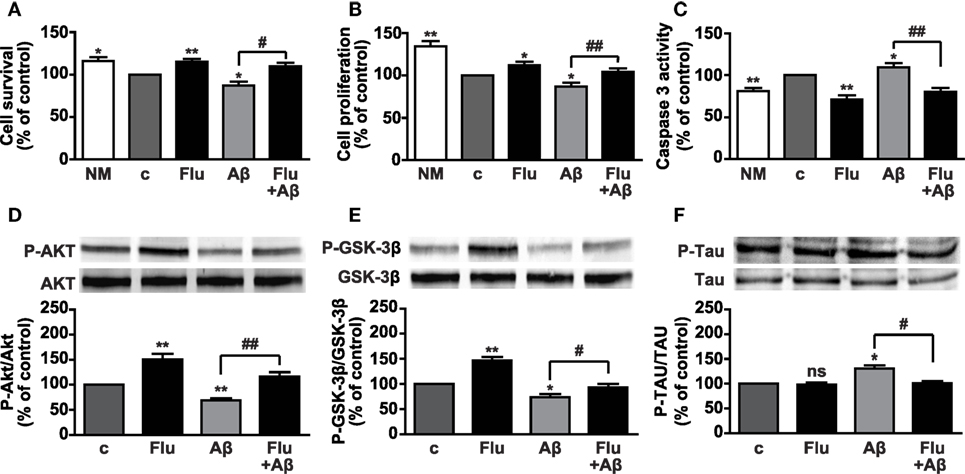
Figure 5. Survival effects of Fludro against Aβ1–42-induced toxicity in AHPs. (A–C) Cell survival, proliferation, and apoptosis in cells treated for 24 h in control medium (c) with or without Aβ1–42 (1 μM) and in either absence or presence of Fludro (1 μM). Data, expressed as percentage of c, are the mean ± SE of four replicates. *P < 0.05 and **P < 0.01 vs. c; #P < 0.05 and ##P < 0.01; ns, not significant. (D) Akt, (E) GSK-3β, and (F) Tau phosphorylation (top panels) assessed by Western blot in cells treated with Aβ1–42 (1 μM) and Fludro (1 μM), either alone or in combination. The cells were pretreated with Fludro for 30 min and then treated with Aβ1–42 for 60 min. Blots were reprobed with total antibodies for normalization (lower panels). Graphs show phosphorylated proteins normalized to total proteins and reported as percentage of basal. *P < 0.05 and **P < 0.01 vs. c; #P < 0.05 and ##P < 0.01; ns, not significant (n = 3).
Discussion
This study is the first to describe the survival, proliferative, and antiapoptotic effects of the MR agonist Fludro in adult rat hippocampal progenitors. Fludro counteracted the detrimental effects of growth factor deprivation through mechanisms involving activation of the cAMP/PKA/CREB signaling and PI3K/Akt and mTOR pathways, as well as inactivation of GSK-3β. Furthermore, Fludro reduced the negative effects of Aβ1–42 on AHP survival, proliferation, and apoptosis through Akt and GSK-3β-mediated signaling and inhibition of Aβ1–42-induced hyperphosphorylation of Tau protein.
Adult hippocampal neurogenesis comprises proliferation and differentiation of progenitors, survival and maturation of new neurons, and their integration into neuronal circuits. Defects in neurogenesis cause memory impairment and cognitive deficits and have been found to be critical events in aging and neurodegenerative diseases, including AD (13, 14). Both MR and GR are abundantly expressed in the limbic areas, where they display quite opposite effects (1, 2). Indeed, MRs play an essential role in the neuroendocrine and behavioral responses to stress and in cognitive functions, and are involved in the integrity and stability of neuronal networks (4, 23). On the other hand, activation of GR by high levels of glucocorticoids promotes neuronal death through cell cycle arrest and induction of apoptosis (17, 20, 21, 46). We show here expression of both MR and GR in AHPs and interestingly, MR, but not GR, mRNA expression was found increased by treatment with Fludro, suggesting a specific role for MR in mediating Fludro survival and proliferative effects. This assumption was also bolstered by the results showing that the MR antagonist Spiro was able to counteract the actions of Fludro in AHPs. Furthermore, previous studies have demonstrated that MR overexpression in vitro was associated with increased survival of rat primary cortical neurons, as well as neuroprotection in rat hippocampus and decreased sensitivity to stress (47). In addition, exposure to chronic stressors was previously found to downregulate hippocampal MR, leading to a reduced MR:GR ratio, and decreased hippocampal MRs have been associated with enhanced stress-induced HPA axis activity (48). Interestingly, antidepressant administration increased MR expression (49); furthermore mice with conditional forebrain-specific MR overexpression showed an attenuated HPA axis response to stress (50) and MR overexpression was recently shown to confer resilience to the effects of chronic stress on hippocampus-dependent function and structural plasticity (51). Moreover, murine ES cells that overexpressed human MR and were induced to differentiate into mature neurons showed increased neuron survival and increased ratio of anti- vs. proapoptotic molecules (26). Therefore, in our study increased MR expression may be a mechanism whereby Fludro promotes cell survival and counteracts apoptosis induced by growth factor deprivation.
Although the neuroprotective actions of MR and of natural MR agonists, such as aldosterone have been described (4, 23, 26–28), to the best of our knowledge, this is the first study showing the protective role of Fludro in hippocampal progenitors. We and others have recently reported that high doses of Fludro display inhibitory effects on the HPA axis in humans, likely through binding to hippocampal MR (52–54), whereas MR antagonism increased HPA axis activity (31). Accordingly, Fludro also enhanced efficacy of antidepressants and improved memory and executive functions in young depressed patients (48). These findings led us to hypothesize that the highly selective MR agonist Fludro, similarly to the natural agonist aldosterone, would display protective action in adult hippocampal progenitors. Indeed, we show here that Fludro counteracted the effects of growth factor deprivation in AHPs by promoting cell survival and proliferation, and reducing apoptosis. To date, very few studies have described the effects of Fludro on cells; among these, Fludro was found to stimulated cell viability in bladder cancer cell lines (55), whereas in neurons, it increased nerve growth factor (NGF)-induced neurite outgrowth in a neuronal model derived from PC12 pheochromocytoma line (56). Conversely, at variance with the previously observed survival actions of MR, a recent study demonstrated Fludro-induced neuron damage in pyramidal cells of the hippocampal CA3 region, which expressed only MR and not GR (57).
Herein, we show that the survival effects of Fludro in AHPs were attenuated not only by the MR antagonist Spiro, suggesting MR specificity, but also by the GR agonist DEX. In fact, as previously demonstrated in hippocampal cells (17), DEX alone strongly reduced cell survival and proliferation in growth factor-deprived AHPs, whereas it had no effect on apoptosis. Fludro was likely unable to counteract such a strong death response to GR activation; in addition, DEX also reduced the antiapoptotic effect of Fludro, although having no proapoptotic effect on its own. These results suggest that upon stress conditions, such as high DEX levels, MR response is unable to attenuate the detrimental effects of GR.
The limitation of the present study is that the experiments have been performed using a single cell type. However, the adult rat hippocampus-derived progenitors, employed herein, were originally isolated from the rat hippocampal dentate gyrus and are a well-established cell model, which have been thoroughly studied with regard to their proliferative capacities and lineage stability. Most importantly, they can generate the three main lineages of the central nervous system (8, 38, 58).
In addition to the classical long-lasting genomic mechanisms regulating gene transcription, MRs have been recently shown to influence cellular physiology through rapid non-genomic pathways (59). These are represented by low-affinity membrane receptors that are activated when corticosteroid levels rise, for example, shortly after stress exposure, and are most likely located on the presynaptic terminal in hippocampal neurons; moreover, their effects were found to be blocked by Spiro (4, 60). Furthermore, aldosterone has been shown to induce a rapid increase of cAMP levels and to promote CREB phosphorylation in vascular smooth muscle cells through non-genomic mechanisms; in this study, however, these effects were not blocked by the MR antagonist Spiro or by inhibitors of transcription and protein synthesis (43). Moreover, cAMP potentiated the activation of transcription by aldosterone of a GRE-containing promoter in different cell types (44). In line with these studies, we show here that Fludro rapidly increased cAMP in AHPs and also promoted the phosphorylation of CREB, suggesting activation of non-genomic pathways. Furthermore, Fludro-induced survival and proliferation was reduced by the PKA inhibitor KT5720, further supporting the implication of cAMP/PKA/CREB signaling, which is recognized as a central player in different steps of adult neurogenesis, including hippocampal cell survival, maturation, and integration of new neurons (6, 7).
Recently, rapid non-genomic effects of glucocorticoids were also observed in hippocampal neurons, where corticosterone promoted the activation of both PKA and PI3K/Akt (61); however, the role of MR on activation of survival signaling pathways is still quite unknown. Furthermore, the importance of PI3K/Akt activity on proliferation and survival of adult hippocampal progenitors (8, 9), as well as its association with CREB phosphorylation, has been previously described (9). Interestingly, we show here rapid Fludro-induced phosphorylation of Akt in AHPs; moreover, Akt inhibition by wortmannin reduced Fludro survival, proliferative, and antiapoptotic effects. Similarly, Fludro rapidly increased the phosphorylation of the PI3K/Akt downstream targets GSK-3β and mTOR/p70S6K, which resulted in their inactivation and activation, respectively. GSK-3β, a key component of the Wnt pathway, plays a crucial role in progenitor cell differentiation and survival during neurogenesis, as well as in neuronal survival (11). Furthermore, impaired GSK-3β activity has been associated with psychiatric disorders and neurodegenerative diseases, including AD (45). Interestingly, as for Akt, Fludro survival and mitogenic actions were reduced in the presence of the mTOR inhibitor rapamycin, suggesting rapamycin sensitive mTOR complex 1 (mTORC1)-mediated mechanisms. Accordingly, mTOR/p70S6K pathway has been linked to synaptic plasticity and neurogenesis, and mTOR signaling impairment has been related to neurodegenerative disorders, including autism and AD (12). Therefore, on the basis of the protective effects observed in growth factor-deprived conditions and the signaling pathways involved, we next sought to determine whether Fludro would protect AHPs from the detrimental effects of Aβ1–42 peptide. Indeed, Aβ1–42 is a main responsible for neuronal death, memory, and cognitive impairment in AD (34, 35). Furthermore, different studies have described the correlation between high glucocorticoid levels or dysregulation in the corticosteroid system and AD (32, 33). In fact, in both in vitro and in vivo models, stress-levels of glucocorticoids were found to increase Aβ formation and Tau accumulation, through GR-mediated mechanisms (62). Moreover, elevated glucocorticoid levels increased susceptibility of cholinergic neurons to Aβ1–42-mediated toxicity in vivo (63) and in hippocampal neuronal cultures (64). Conversely, CRH, which initiates neuroendocrine responses to stress by activating the HPA axis, was found to reduce cell death caused by an Aβ peptide in primary hippocampal and cortical neurons, an effect that was blocked by a CRH receptor antagonist and by an inhibitor of PKA (65). However, so far the direct effect of MR agonists on Aβ-induced toxicity in hippocampal cells has never been investigated. Here, we show for the first time that Fludro counteracts the detrimental effects of Aβ1–42 in AHPs, by increasing cell survival and proliferation and reducing apoptosis. Fludro also increased both Akt and GSK-3β phosphorylation, that was reduced by Aβ1–42, and restored Aβ1–42-induced phosphorylation of Tau to basal levels.
Overall, these findings are the first to show the protective and proliferative role of Fludro in adult hippocampal progenitors exposed to different stress stimuli, including those characteristics of neurodegenerative diseases. These effects likely involved both genomic and non-genomic mechanisms, as suggested by the activation of specific signaling pathways. Therefore, the results of this study further support the previously observed neuroprotective role of MR and suggest that MR agonists may represent a possible therapeutic strategy for reducing hippocampal cell loss, improving neurogenesis and for approaching neurodegenerative diseases, such as AD.
Author Contributions
IG performed the experiments, analyzed the results, and prepared the paper draft; TV and E.Garg. contributed to the experiments and results’ analysis; EA and EG contributed to the conception of the work and critically revised the manuscript; RG designed the study, supervised the work and the results’ analysis, and wrote the paper.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We are grateful to Dr. Fabio Settanni for its technical support and to Prof. Jörgen Isgaard for kindly providing AHP cells. We also thank the Neuroscience Institute of Torino and the Italian Group of Neuroendocrine Sciences (GiSNe).
Funding
This work was supported by Ministero dell’Istruzione, dell’Università e della Ricerca Scientifica e Tecnologica (Italian Ministry of Instruction and Research) (PRIN 2010 B5B2NL to EG), by Ex-60% 2012, University of Torino (to EG) and by CRT 2015 (2015/2573) (to RG).
Abbreviations
Aβ, amyloid-beta peptide; AD, Alzheimer’s disease; AHP, adult rat hippocampal progenitor cells; BrdU, 5-bromo-2-deoxyuridine; BSA, bovine serum albumin; cAMP, cyclic AMP; CREB, cAMP response element-binding protein; DEX, dexamethasone; DMEM, Dulbecco’s Modified Eagle’s medium; ELISA, enzyme-linked immunosorbent assay; ERK1/2, extracellular signal-regulated protein kinases 1 and 2; FGF, fibroblast growth factor; Fludro, fludrocortisone; GSK-3β, glycogen synthase kinase-3 beta; IBMX, 3-isobutyl-1-methylxanthine; mTOR, mammalian target of rapamycin; MTT, 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide; NFTs, neurofibrillary tangles; NM, normal medium; p70S6K, p70 ribosomal S6 kinase; PI3K, phosphatidylinositol 3-kinase; PKA, protein kinase A; RT-PCR, reverse transcriptase-polymerase chain reaction; Spiro, spironolactone.
References
1. De Kloet ER, Vreugdenhil E, Oitzl MS, Joels M. Brain corticosteroid receptor balance in health and disease. Endocr Rev (1998) 19:269–301. doi:10.1210/edrv.19.3.0331
2. Van Eekelen JA, Jiang W, De Kloet ER, Bohn MC. Distribution of the mineralocorticoid and the glucocorticoid receptor mRNAs in the rat hippocampus. J Neurosci Res (1988) 21:88–94. doi:10.1002/jnr.490210113
3. Arriza JL, Simerly RB, Swanson LW, Evans RM. The neuronal mineralocorticoid receptor as a mediator of glucocorticoid response. Neuron (1988) 1:887–900. doi:10.1016/0896-6273(88)90136-5
4. Joels M, Karst H, Derijk R, De Kloet ER. The coming out of the brain mineralocorticoid receptor. Trends Neurosci (2008) 31:1–7. doi:10.1016/j.tins.2007.10.005
5. Dudek H, Datta SR, Franke TF, Birnbaum MJ, Yao R, Cooper GM, et al. Regulation of neuronal survival by the serine-threonine protein kinase Akt. Science (1997) 275:661–5. doi:10.1126/science.275.5300.661
6. Nakagawa S, Kim JE, Lee R, Malberg JE, Chen J, Steffen C, et al. Regulation of neurogenesis in adult mouse hippocampus by cAMP and the cAMP response element-binding protein. J Neurosci (2002) 22:3673–82.
7. Merz K, Herold S, Lie DC. CREB in adult neurogenesis – master and partner in the development of adult-born neurons? Eur J Neurosci (2011) 33:1078–86. doi:10.1111/j.1460-9568.2011.07606.x
8. Aberg MA, Aberg ND, Palmer TD, Alborn AM, Carlsson-Skwirut C, Bang P, et al. IGF-I has a direct proliferative effect in adult hippocampal progenitor cells. Mol Cell Neurosci (2003) 24:23–40. doi:10.1016/S1044-7431(03)00082-4
9. Peltier J, O’neill A, Schaffer DV. PI3K/Akt and CREB regulate adult neural hippocampal progenitor proliferation and differentiation. Dev Neurobiol (2007) 67:1348–61. doi:10.1002/dneu.20506
10. Gargantini E, Lazzari L, Settanni F, Taliano M, Trovato L, Gesmundo I, et al. Obestatin promotes proliferation and survival of adult hippocampal progenitors and reduces amyloid-beta-induced toxicity. Mol Cell Endocrinol (2016) 422:18–30. doi:10.1016/j.mce.2015.11.008
11. Hur EM, Zhou FQ. GSK3 signalling in neural development. Nat Rev Neurosci (2010) 11:539–51. doi:10.1038/nrn2870
12. Takei N, Nawa H. mTOR signaling and its roles in normal and abnormal brain development. Front Mol Neurosci (2014) 7:28. doi:10.3389/fnmol.2014.00028
13. Spalding KL, Bergmann O, Alkass K, Bernard S, Salehpour M, Huttner HB, et al. Dynamics of hippocampal neurogenesis in adult humans. Cell (2013) 153:1219–27. doi:10.1016/j.cell.2013.05.002
14. Aimone JB, Li Y, Lee SW, Clemenson GD, Deng W, Gage FH. Regulation and function of adult neurogenesis: from genes to cognition. Physiol Rev (2014) 94:991–1026. doi:10.1152/physrev.00004.2014
15. Cameron HA, Gould E. Adult neurogenesis is regulated by adrenal steroids in the dentate gyrus. Neuroscience (1994) 61:203–9. doi:10.1016/0306-4522(94)90224-0
16. Gould E, McEwen BS, Tanapat P, Galea LA, Fuchs E. Neurogenesis in the dentate gyrus of the adult tree shrew is regulated by psychosocial stress and NMDA receptor activation. J Neurosci (1997) 17:2492–8.
17. Kim JB, Ju JY, Kim JH, Kim TY, Yang BH, Lee YS, et al. Dexamethasone inhibits proliferation of adult hippocampal neurogenesis in vivo and in vitro. Brain Res (2004) 1027:1–10. doi:10.1016/j.brainres.2004.07.093
18. Murray F, Smith DW, Hutson PH. Chronic low dose corticosterone exposure decreased hippocampal cell proliferation, volume and induced anxiety and depression like behaviours in mice. Eur J Pharmacol (2008) 583:115–27. doi:10.1016/j.ejphar.2008.01.014
19. David DJ, Samuels BA, Rainer Q, Wang JW, Marsteller D, Mendez I, et al. Neurogenesis-dependent and -independent effects of fluoxetine in an animal model of anxiety/depression. Neuron (2009) 62:479–93. doi:10.1016/j.neuron.2009.04.017
20. Brummelte S, Galea LAM. Chronic high corticosterone reduces neurogenesis in the dentate gyrus of adult male and female rats. Neuroscience (2010) 168:680–90. doi:10.1016/j.neuroscience.2010.04.023
21. Garza JC, Guo M, Zhang W, Lu XY. Leptin restores adult hippocampal neurogenesis in a chronic unpredictable stress model of depression and reverses glucocorticoid-induced inhibition of GSK-3beta/beta-catenin signaling. Mol Psychiatry (2011) 17:790–808. doi:10.1038/mp.2011.161
22. Tanapat P, Hastings NB, Rydel TA, Galea LAM, Gould E. Exposure to fox odor inhibits cell proliferation in the hippocampus of adult rats via an adrenal hormone-dependent mechanism. JComp Neurol (2001) 437:496–504. doi:10.1002/cne.1297
23. Gomez-Sanchez CE, Gomez-Sanchez EP. The protective side of the mineralocorticoid receptor. Endocrinology (2012) 153:1565–7. doi:10.1210/en.2011-2184
24. Gass P, Kretz O, Wolfer DP, Berger S, Tronche F, Reichardt HM, et al. Genetic disruption of mineralocorticoid receptor leads to impaired neurogenesis and granule cell degeneration in the hippocampus of adult mice. EMBO Rep (2000) 1:447–51. doi:10.1093/embo-reports/kvd088
25. Berger S, Wolfer DP, Selbach O, Alter H, Erdmann G, Reichardt HM, et al. Loss of the limbic mineralocorticoid receptor impairs behavioral plasticity. Proc Natl Acad Sci U S A (2006) 103:195–200. doi:10.1073/pnas.0503878102
26. Munier M, Law F, Meduri G, Le Menuet D, Lombes M. Mineralocorticoid receptor overexpression facilitates differentiation and promotes survival of embryonic stem cell-derived neurons. Endocrinology (2012) 153:1330–40. doi:10.1210/en.2011-1436
27. Lai M, Horsburgh K, Bae SE, Carter RN, Stenvers DJ, Fowler JH, et al. Forebrain mineralocorticoid receptor overexpression enhances memory, reduces anxiety and attenuates neuronal loss in cerebral ischaemia. Eur J Neurosci (2007) 25:1832–42. doi:10.1111/j.1460-9568.2007.05427.x
28. Crochemore C, Lu J, Wu Y, Liposits Z, Sousa N, Holsboer F, et al. Direct targeting of hippocampal neurons for apoptosis by glucocorticoids is reversible by mineralocorticoid receptor activation. Mol Psychiatry (2005) 10:790–8. doi:10.1038/sj.mp.4001679
29. Fischer AK, Von Rosenstiel P, Fuchs E, Goula D, Almeida OF, Czeh B. The prototypic mineralocorticoid receptor agonist aldosterone influences neurogenesis in the dentate gyrus of the adrenalectomized rat. Brain Res (2002) 947:290–3. doi:10.1016/S0006-8993(02)03042-1
30. Derijk RH, Wust S, Meijer OC, Zennaro MC, Federenko IS, Hellhammer DH, et al. A common polymorphism in the mineralocorticoid receptor modulates stress responsiveness. J Clin Endocrinol Metab (2006) 91:5083–9. doi:10.1210/jc.2006-0915
31. Berardelli R, Karamouzis I, Marinazzo E, Prats E, Picu A, Giordano R, et al. Effect of acute and prolonged mineralocorticoid receptor blockade on spontaneous and stimulated hypothalamic-pituitary-adrenal axis in humans. Eur J Endocrinol (2010) 162:1067–74. doi:10.1530/EJE-09-1076
32. Joshi YB, Chu J, Pratico D. Stress hormone leads to memory deficits and altered tau phosphorylation in a model of Alzheimer’s disease. J Alzheimers Dis (2012) 31:167–76. doi:10.3233/JAD-2012-120328
33. Brureau A, Zussy C, Delair B, Ogier C, Ixart G, Maurice T, et al. Deregulation of hypothalamic-pituitary-adrenal axis functions in an Alzheimer’s disease rat model. Neurobiol Aging (2013) 34:1426–39. doi:10.1016/j.neurobiolaging.2012.11.015
34. Benilova I, Karran E, De Strooper B. The toxic Abeta oligomer and Alzheimer’s disease: an emperor in need of clothes. Nat Neurosci (2012) 15:349–57. doi:10.1038/nn.3028
35. De Strooper B, Karran E. The cellular phase of Alzheimer’s disease. Cell (2016) 164:603–15. doi:10.1016/j.cell.2015.12.056
36. Hinkelmann K, Wingenfeld K, Kuehl LK, Fleischer J, Heuser I, Wiedemann K, et al. Stimulation of the mineralocorticoid receptor improves memory in young and elderly healthy individuals. Neurobiol Aging (2015) 36:919–24. doi:10.1016/j.neurobiolaging.2014.09.008
37. Otte C, Wingenfeld K, Kuehl LK, Kaczmarczyk M, Richter S, Quante A, et al. Mineralocorticoid receptor stimulation improves cognitive function and decreases cortisol secretion in depressed patients and healthy individuals. Neuropsychopharmacology (2015) 40:386–93. doi:10.1038/npp.2014.181
38. Johansson I, Destefanis S, Aberg ND, Aberg MA, Blomgren K, Zhu C, et al. Proliferative and protective effects of growth hormone secretagogues on adult rat hippocampal progenitor cells. Endocrinology (2008) 149:2191–9. doi:10.1210/en.2007-0733
39. Gallo D, Gesmundo I, Trovato L, Pera G, Gargantini E, Minetto MA, et al. GH-releasing hormone promotes survival and prevents TNF-alpha-induced apoptosis and atrophy in C2C12 myotubes. Endocrinology (2015) 156:3239–52. doi:10.1210/EN.2015-1098
40. Marini F, Pozzato C, Andreetta V, Jansson B, Arban R, Domenici E, et al. Single exposure to social defeat increases corticotropin-releasing factor and glucocorticoid receptor mRNA expression in rat hippocampus. Brain Res (2006) 1067:25–35. doi:10.1016/j.brainres.2005.10.002
41. Kagawa CM. Blocking the renal electrolyte effects of mineralocorticoids with an orally active steroidal spirolactone. Endocrinology (1960) 67:125–32. doi:10.1210/endo-67-1-125
42. Gould E, Cameron HA, Daniels DC, Woolley CS, McEwen BS. Adrenal hormones suppress cell division in the adult rat dentate gyrus. J Neurosci (1992) 12:3642–50.
43. Christ M, Gunther A, Heck M, Schmidt BM, Falkenstein E, Wehling M. Aldosterone, not estradiol, is the physiological agonist for rapid increases in cAMP in vascular smooth muscle cells. Circulation (1999) 99:1485–91. doi:10.1161/01.CIR.99.11.1485
44. Massaad C, Houard N, Lombes M, Barouki R. Modulation of human mineralocorticoid receptor function by protein kinase A. Mol Endocrinol (1999) 13:57–65. doi:10.1210/mend.13.1.0226
45. Llorens-Martin M, Jurado J, Hernandez F, Avila J. GSK-3beta, a pivotal kinase in Alzheimer disease. Front Mol Neurosci (2014) 7:46. doi:10.3389/fnmol.2014.00046
46. Crochemore C, Michaelidis TM, Fischer D, Loeffler JP, Almeida OF. Enhancement of p53 activity and inhibition of neural cell proliferation by glucocorticoid receptor activation. FASEB J (2002) 16:761–70. doi:10.1096/fj.01-0577com
47. Macleod MR, Johansson IM, Soderstrom I, Lai M, Gido G, Wieloch T, et al. Mineralocorticoid receptor expression and increased survival following neuronal injury. Eur J Neurosci (2003) 17:1549–55. doi:10.1046/j.1460-9568.2003.02587.x
48. De Kloet ER, Otte C, Kumsta R, Kok L, Hillegers MH, Hasselmann H, et al. STRESS and DEPRESSION a crucial role of the mineralocorticoid receptor. J Neuroendocrinol (2016). doi:10.1111/jne.12379
49. Reul JM, Stec I, Soder M, Holsboer F. Chronic treatment of rats with the antidepressant amitriptyline attenuates the activity of the hypothalamic-pituitary-adrenocortical system. Endocrinology (1993) 133:312–20. doi:10.1210/endo.133.1.8391426
50. Rozeboom AM, Akil H, Seasholtz AF. Mineralocorticoid receptor overexpression in forebrain decreases anxiety-like behavior and alters the stress response in mice. Proc Natl Acad Sci U S A (2007) 104:4688–93. doi:10.1073/pnas.0606067104
51. Kanatsou S, Fearey BC, Kuil LE, Lucassen PJ, Harris AP, Seckl JR, et al. Overexpression of mineralocorticoid receptors partially prevents chronic stress-induced reductions in hippocampal memory and structural plasticity. PLoS One (2015) 10:e0142012. doi:10.1371/journal.pone.0142012
52. Otte C, Jahn H, Yassouridis A, Arlt J, Stober N, Maass P, et al. The mineralocorticoid receptor agonist, fludrocortisone, inhibits pituitary-adrenal activity in humans after pre-treatment with metyrapone. Life Sci (2003) 73:1835–45. doi:10.1016/S0024-3205(03)00513-7
53. Buckley TM, Mullen BC, Schatzberg AF. The acute effects of a mineralocorticoid receptor (MR) agonist on nocturnal hypothalamic-adrenal-pituitary (HPA) axis activity in healthy controls. Psychoneuroendocrinology (2007) 32:859–64. doi:10.1016/j.psyneuen.2007.05.016
54. Karamouzis I, Berardelli R, Marinazzo E, D’angelo V, Zinna D, Minetto MA, et al. The acute effect of fludrocortisone on basal and hCRH-stimulated hypothalamic – pituitary – adrenal (HPA) axis in humans. Pituitary (2013) 16:378–85. doi:10.1007/s11102-012-0435-3
55. Ishiguro H, Kawahara T, Zheng Y, Kashiwagi E, Li Y, Miyamoto H. Differential regulation of bladder cancer growth by various glucocorticoids: corticosterone and prednisone inhibit cell invasion without promoting cell proliferation or reducing cisplatin cytotoxicity. Cancer Chemother Pharmacol (2014) 74:249–55. doi:10.1007/s00280-014-2496-7
56. Yeyeodu ST, Witherspoon SM, Gilyazova N, Ibeanu GC. A rapid, inexpensive high throughput screen method for neurite outgrowth. Curr Chem Genomics (2010) 4:74–83. doi:10.2174/1875397301004010074
57. Sasaki K, Yoshizaki F. Investigation into hippocampal nerve cell damage through the mineralocorticoid receptor in mice. Mol Med Rep (2015) 12:7211–20. doi:10.3892/mmr.2015.4406
58. Palmer TD, Markakis EA, Willhoite AR, Safar F, Gage FH. Fibroblast growth factor-2 activates a latent neurogenic program in neural stem cells from diverse regions of the adult CNS. J Neurosci (1999) 19:8487–97.
59. Karst H, Berger S, Turiault M, Tronche F, Schutz G, Joels M. Mineralocorticoid receptors are indispensable for nongenomic modulation of hippocampal glutamate transmission by corticosterone. Proc Natl Acad Sci U S A (2005) 102:19204–7. doi:10.1073/pnas.0507572102
60. Olijslagers JE, De Kloet ER, Elgersma Y, Van Woerden GM, Joels M, Karst H. Rapid changes in hippocampal CA1 pyramidal cell function via pre- as well as postsynaptic membrane mineralocorticoid receptors. Eur J Neurosci (2008) 27:2542–50. doi:10.1111/j.1460-9568.2008.06220.x
61. Yang S, Roselli F, Patchev AV, Yu S, Almeida OF. Non-receptor-tyrosine kinases integrate fast glucocorticoid signaling in hippocampal neurons. J Biol Chem (2013) 288:23725–39. doi:10.1074/jbc.M113.470146
62. Green KN, Billings LM, Roozendaal B, McGaugh JL, Laferla FM. Glucocorticoids increase amyloid-beta and tau pathology in a mouse model of Alzheimer’s disease. J Neurosci (2006) 26:9047–56. doi:10.1523/JNEUROSCI.2797-06.2006
63. Abraham I, Harkany T, Horvath KM, Veenema AH, Penke B, Nyakas C, et al. Chronic corticosterone administration dose-dependently modulates A beta((1-42))- and NMDA-induced neurodegeneration in rat magnocellular nucleus basalis. J Neuroendocrinol (2000) 12:486–94. doi:10.1046/j.1365-2826.2000.00475.x
64. Goodman YD, Bruce AJ, Cheng B, Mattson MP. Estrogens attenuate and corticosterone exacerbates excitotoxicity, oxidative injury, and amyloid beta-peptide toxicity in hippocampal neurons. J Neurochem (1996) 66:1836–44. doi:10.1046/j.1471-4159.1996.66051836.x
Keywords: mineralocorticoid receptor agonists, glucocorticoid receptor agonists, hippocampal progenitors, hippocampal progenitor cell proliferation and survival, amyloid beta peptide
Citation: Gesmundo I, Villanova T, Gargantini E, Arvat E, Ghigo E and Granata R (2016) The Mineralocorticoid Agonist Fludrocortisone Promotes Survival and Proliferation of Adult Hippocampal Progenitors. Front. Endocrinol. 7:66. doi: 10.3389/fendo.2016.00066
Received: 15 April 2016; Accepted: 01 June 2016;
Published: 16 June 2016
Edited by:
Pierrette Gaudreau, Centre hospitalier de l’Université de Montréal, CanadaReviewed by:
Gábor B. Makara, Hungarian Academy of Sciences, HungaryBenno Roozendaal, Radboud University Nijmegen Medical Centre, Netherlands
Copyright: © 2016 Gesmundo, Villanova, Gargantini, Arvat, Ghigo and Granata. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Riccarda Granata, cmljY2FyZGEuZ3JhbmF0YUB1bml0by5pdA==