 Eric Seidel
Eric Seidel Ute I. Scholl
Ute I. Scholl- Department of Nephrology, University Hospital Düsseldorf, Heinrich Heine University, Düsseldorf, Germany
The adrenal cortex is a major site of steroid hormone production. Two hormones are of particular importance: aldosterone, which is produced in the zona glomerulosa in response to volume depletion and hyperkalemia, and cortisol, which is produced in the zona fasciculata in response to stress. In both cases, acute stimulation leads to increased hormone production, and chronic stimulation causes hyperplasia of the respective zone. Aldosterone- and cortisol-producing adenomas (APAs and CPAs) are benign tumors of the adrenal cortex that cause excess hormone production, leading to primary aldosteronism and Cushing’s syndrome, respectively. About 40% of the APAs carry somatic heterozygous gain-of-function mutations in the K+ channel KCNJ5. These mutations lead to sodium permeability, depolarization, activation of voltage-gated Ca2+ channels, and Ca2+ influx. Mutations in the Na+/K+-ATPase subunit ATP1A1 and the plasma membrane Ca2+-ATPase ATP2B3 similarly cause Na+ or H+ permeability and depolarization, whereas mutations in the Ca2+ channel CACNA1D directly lead to increased calcium influx. One in three CPAs carries a recurrent gain-of-function mutation (L206R) in the PRKACA gene, encoding the catalytic subunit of PKA. This mutation causes constitutive PKA activity by abolishing the binding of the inhibitory regulatory subunit to the catalytic subunit. These mutations activate pathways that are relatively specific to the respective cell type (glomerulosa versus fasciculata), and there is little overlap in mutation spectrum between APAs and CPAs, but co-secretion of both hormones can occur. Mutations in CTNNB1 (beta-catenin) and GNAS (Gsα) are exceptions, as they can cause both APAs and CPAs through pathways that are incompletely understood.
Introduction
Adrenal masses are common tumors in humans. Adrenal incidentalomas may be found in more than 4% of computed tomography series (1), and about 7% are malignant (2). Among hormone-producing lesions, besides pheochromocytomas, cortisol-producing and aldosterone-producing adenomas (CPAs and APAs) of the adrenal cortex are frequently diagnosed (2). Aldosterone and cortisol are physiologically synthesized in the two outer layers of the adrenal cortex (zonae glomerulosa and fasciculata, respectively) from their common precursor cholesterol. The two main stimuli of aldosterone production are angiotensin II (ATII) and hyperkalemia. ATII levels rise in states of volume depletion, via activation of the renin–angiotensin system. Binding of ATII to the AT1 receptor, a G protein-coupled receptor in the glomerulosa membrane, leads to the inhibition of potassium channels, depolarization and activation of voltage-gated calcium channels, and the release of calcium from intracellular stores (Figure 1). Other factors that physiologically regulate aldosterone release in concert with ATII and K+ are corticotropin (ACTH, stimulatory) and atrial natriuretic peptide (ANP, inhibitory) (3). Binding of aldosterone to the mineralocorticoid receptor leads to the increased activity of downstream effectors, such as the Na+/K+-ATPase or the epithelial sodium channel (ENaC) (4). The increased activity of these pumps and channels in kidney and intestine causes increased sodium and water reabsorption and an increase in systemic blood pressure.
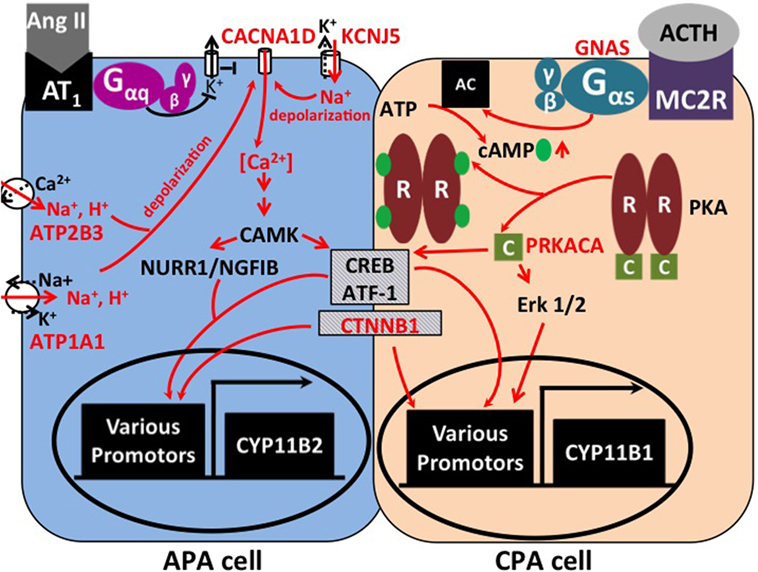
Figure 1. Signaling pathways affected by mutations in APAs and CPAs. In zona glomerulosa, binding of angiotensin II (AngII) to its receptor inhibits potassium channels via G protein signaling. This leads to depolarization and opening of voltage-gated calcium channels. Increased intracellular calcium results in the activation of Ca2+/calmodulin-dependent protein kinase (CAMK) and the activation of transcription factors, such as NURR1/NGFIB, CREB, and ATF-1. As a consequence, genes involved in proliferation and aldosterone production (e.g., aldosterone synthase, CYP11B2) are activated (5). Mutations in KCNJ5, ATP1A1, and ATP2B3 lead to abnormal permeability for sodium or protons, which causes cellular depolarization and activation of the same pathways. Similarly, mutations in the calcium channel gene CACNA1D lead to increased calcium influx. In the zona fasciculata, binding of corticotropin (ACTH) to the melanocortin receptor (MC2R) causes activation of adenylate cyclase (AC) by the Gαs subunit (encoded by GNAS). Binding of cAMP to the regulatory subunit (“R”) of protein kinase A (PKA) leads to release of the catalytic subunit (“C,” encoded by PRKACA) from the complex. Transcription factors CREB, ATF-1, and Erk 1/2 cause increased expression of genes involved in proliferation and cortisol production, such as 11β-hydroxylase (CYP11B1). Hypercortisolism can occur due to activating mutations in GNAS and PRKACA. Activating mutations in β-catenin (CTNNB1) are found in both APAs and CPAs; the underlying mechanisms are incompletely understood.
Cortisol is released from the zona fasciculata upon stimulation by pituitary ACTH, in response to stress. ACTH binds to the melanocortin receptor 2, a G protein-coupled receptor, which activates adenylate cyclase (6). As a result, cAMP is produced, which binds to the regulatory subunit of protein kinase A (PKA), causing release of its catalytic subunit. The catalytic subunit then phosphorylates target proteins, such as CREB and ATF, which lead to cortisol production and proliferation (7) (Figure 1).
Cortisol influences a variety of biological processes, including skeletal growth, immune response, glucose and lipid metabolism, cognition, and reproduction (8–10).
Cortisol-producing adenomas and APAs feature the compelling combination of both hormone production and proliferation, suggesting that they carry genetic changes that activate both processes. Such changes have been identified over the past 5 years through exome sequencing. Comparing DNA sequences from tumor specimens and corresponding normal tissue (such as blood or adjacent tissue) can reveal tumor-specific (somatic) mutations, which are candidates for disease causation (11). This review will discuss recent genetic discoveries in APAs and CPAs and the underlying pathways.
KCNJ5 Mutations in Primary Aldosteronism
Primary aldosteronism (PA) features autonomous production of aldosterone from the adrenal gland and accounts for about 10% of hypertension in referral centers. The two most common causes are APAs and bilateral adrenal hyperplasia. Other causes, such as unilateral hyperplasia, malignant tumors, or familial hyperaldosteronism, are rare (12–16).
In the first exome sequencing study of APAs, Choi et al. analyzed four tumors and corresponding blood samples (11). This revealed only two to three somatic mutations per tumor. One gene (KCNJ5) was mutated in two tumors, with one tumor carrying a heterozygous G151R mutation, and the second carrying a heterozygous L168R mutation. By Sanger sequencing, these two mutations were found in 6 of 18 additional APAs. KCNJ5 encodes an inward rectifier potassium channel, Kir3.4, or GIRK4. The G151 and L168 residues are located within or close to the selectivity filter of the channel (17), which allows only potassium, but not the smaller sodium ions, to pass through the channel. This suggested an effect of the variants on potassium selectivity. Accordingly, by electrophysiology, mutant channels were found to be permeable to sodium and cause cellular depolarization. These effects were inferred to contribute to aldosterone production and proliferation through the activation of voltage-gated calcium channels and calcium entry (3, 11) (Figure 1). Additional support for the notion that KCNJ5 mutations are sufficient to cause aldosterone production and proliferation came from the discovery of heterozygous germ line KCNJ5 mutations in families with early-onset PA and massive bilateral adrenal hyperplasia (11, 18–20). The high frequency of KCNJ5 mutations in APAs (about 35% in European cohorts, more than 60% in Asian cohorts) has subsequently been confirmed in large cohorts (21–29) (Table 1). A higher prevalence in Asian cohorts may be due to selection bias; individuals with KCNJ5 mutations tend to have a more florid presentation at least in some cohorts. Interestingly, KCNJ5 mutations are more prevalent in females than in males, which could account for the higher overall prevalence of APAs in females, a finding that remains unexplained. In vitro studies in the aldosterone-producing human adrenocortical cancer cell line HAC15 have demonstrated that gain-of-function mutations in KCNJ5 lead to increased expression of aldosterone synthase and increased aldosterone production (30–32). Lastly, a recent study confirmed the role of CYP11B2 transcriptional regulators NURR1 and ATF2 in mutant KCNJ5-induced aldosterone production (33) (Figure 1).
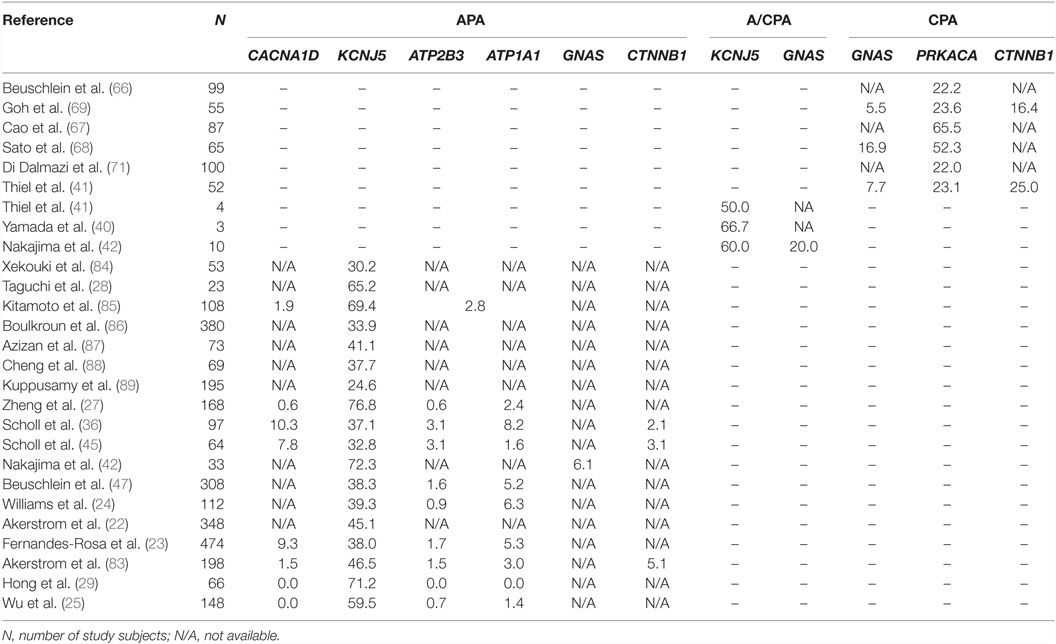
Table 1. Mutation frequencies in APAs, A/CPAs, and CPAs.
KCNJ5 Mutations and Glucocorticoids
Interestingly, tumors with KCNJ5 mutations tend to be larger than other tumors and have fasciculata-like features by histopathology and gene expression analysis, which may have implications for the radiological diagnosis of these tumors (34–36). Another line of evidence pointing to a more fasciculata-like or mixed glomerulosa–fasciculata phenotype of KCNJ5-positive APAs is the finding that heterologous expression of a KCNJ5 variant in HAC15 cells causes not only upregulation of CYP11B2 expression but also increased expression of CYP11B1 and synthesis of hybrid steroids 18-hydroxycorticsol and 18-oxocortisol, as well as corticosterone (31, 33). This raises the question whether KCNJ5-positive APAs produce clinically relevant amounts of glucocorticoids. Interestingly, hypersecretion of cortisol and aldosterone are not mutually exclusive in adrenal adenomas, and cases of aldosterone and cortisol co-secreting adenomas (A/CPAs) have been reported (37–40). This phenotype may be underdiagnosed due to incomplete screening for subclinical Cushing’s syndrome (CS) in patients with APAs; many of these patients will not receive dexamethasone suppression tests. Yamada et al. reported three female patients with hypertension and hypokalemia who were diagnosed with A/CPAs. Two had KCNJ5 mutations (G151R and L168R) (40). Thiel et al. reported KCNJ5 mutations (G151R and L168R) in two of four A/CPAs, and no mutations in PRKACA, ATP1A1, ATP2B3, and CACNA1D were found (41). Lastly, Nakajima et al. demonstrated KCNJ5 mutations in 6 of 10 A/CPAs (42). This suggests that KCNJ5 mutations may cause excess secretion of not only aldosterone but also glucocorticoids, leading to PA with discrete features of CS. Potential explanations include the overlapping role of transcriptional regulators CREB and ATF in the regulation of both aldosterone and cortisol production (Figure 1) as well as a potential role of Ca2+ in cAMP formation (43).
In summary, KCNJ5 mutations have been extensively studied in the context of PA. However, the physiological role of KCNJ5 in human adrenal glomerulosa remains largely undetermined, and animal studies have been hampered by extremely low or absent expression of kcnj5 in rodents (44).
CACNA1D Mutations in Primary Aldosteronism
The gene with the second highest somatic mutation burden in APAs is CACNA1D, with frequencies of about 8–11% described in the initial exome sequencing studies and similar findings in a large follow-up study (23, 35, 45) (Table 1). Similar to KCNJ5 mutations, CACNA1D mutations are heterozygous. However, mutations are more scattered throughout the protein. CACNA1D encodes an L-type voltage-gated calcium channel (CaV1.3). Mutant CACNA1D channels show activation at more hyperpolarized membrane potentials and, in some cases, reduced channel inactivation compared to wild-type channels (45). In line with the notion that these effects will lead to increased calcium entry, expression of mutant CACNA1D channels causes increased aldosterone production in the adrenocortical cancer cell line H295R (46). Again, similar to KCNJ5 variants, additional evidence for a role of CACNA1D in PA came from the discovery of germ line variants at the same positions found to be mutated in tumors (45). Among 100 unrelated subjects with early-onset PA and hypertension, two carried de novo mutations in CACNA1D. Interestingly, these subjects had a multi-organ phenotype, including primary aldosteronism, seizures, and neurologic abnormalities (PASNA) (45). The discovery of mutations in calcium channels as a cause of PA may suggest that specific calcium channel blockers could be useful in patients carrying such mutations (46).
ATPase Mutations in Primary Aldosteronism
Additional somatic mutations in APAs without corresponding germ line mutations have been identified. Beuschlein et al. first described heterozygous or hemizygous somatic mutations in the ATP1A1 and ATP2B3 genes in 5.2 and 1.6% of APAs, respectively. ATP1A1 encodes a sodium/potassium ATPase subunit, whereas ATP2B3 encodes the plasma membrane calcium ATPase. Mutations in both ATPases cluster within the M4 helix, again suggesting a gain-of-function mechanism (47). Azizan et al. subsequently demonstrated that ATP1A1 mutations cause an ouabain-sensitive, voltage-dependent inward Na+ or H+ current, respectively. Heterologous expression of mutant ATP1A1 in human adrenocortical H295R cells led to increased aldosterone production and CYP11B2 expression levels (35), consistent with a role of mutant ATP1A1 in cellular depolarization and activation of voltage-gated calcium channels, as with mutated KCNJ5. Similarly, a mutation in ATP2B3 was shown to induce a pathological Na+ permeability, with increased intracellular Ca2+ levels and aldosterone production in H295R cells (48).
Specific Features and Origin of CACNA1D- and Atpase-Mutant APAs
Azizan and colleagues first suggested an association of CACNA1D and ATP1A1 mutations with a glomerulosa-like phenotype (35), whereas other groups reported mixed histological phenotypes (23, 36). Glomerulosa-like features in CACNA1D and ATP1A1-positive tumors could suggest that these tumors are derived from zona glomerulosa cells. Indeed, Nishimoto et al. recently studied 42 normal adrenal glands from kidney donors and identified so-called aldosterone-producing cell clusters (APCCs), nests of cells just below the adrenal capsule that feature high expression of aldosterone synthase and protrude into cortisol-producing cells (49). Remarkably, targeted next-generation sequencing of DNA from 23 APCCs identified known somatic CACNA1D mutations in six cases and known somatic ATP1A1 mutations in two cases, suggesting that APCCs may represent precursors of a subtype of APAs. These results also support the presence of APCCs and potentially subclinical PA in a substantial number of apparently healthy individuals, which is interesting, given that prior clinical studies identified a higher risk of developing hypertension in individuals with increased aldosterone levels within the physiologic range (50). No somatic KCNJ5 mutations were identified in APCCs, suggesting that APAs carrying such mutations may arise from cells of the zona fasciculata or may grow more rapidly, with precursors evading detection in apparently healthy individuals.
Investigations of Multinodular Tumors
Even though the classical presentation of aldosterone-producing adenoma is that of a uninodular lesion, many cases feature associated hyperplasia or multiple secondary nodules, many of which do not show increased expression of aldosterone synthase. Investigations of individual nodules revealed the presence of characteristic APA mutations in aldosterone-producing nodules, whereas non-producing nodules do not carry such mutations (51). Some individuals carry different aldosterone-driver mutations in different nodules, suggesting that independent mutation events account for the development of multiple nodules (52, 53). Whether germ line susceptibility variants promote the formation of multiple tumors remain to be determined. Interestingly, some adenomas appear to show intra-tumoral heterogeneity, indicating that the somatic events underlying APA formation can also occur in the context of preexisting nodules (53). This has led to the proposal of a two-hit model of adenoma development, with one hit being responsible for proliferation and another hit causing hormone production (54). However, the rarity of such findings and the absence of second hits explaining proliferation in the exomes of tumors carrying aldosterone-driver mutations suggest that APA driver mutations alone are sufficient to cause proliferation and hormone production in the majority of APAs.
CACNA1H Mutations in Familial Hyperaldosteronism
One additional ion channel gene implicated in PA to date has been found to be mutated in the germ line only, but not in APAs. A novel germ line heterozygous variant in the CACNA1H gene (M1549V) was found in 5 of 40 unrelated subjects with PA and hypertension diagnosed at age 10 or below (55). Microscopic glomerulosa hyperplasia without macroscopic enlargement was demonstrated in one subject who had undergone unilateral adrenalectomy, suggesting a limited proliferative effect of the variant. CACNA1H encodes the low-voltage-activated T-type calcium channel CaV3.2 (56). CaV3.2 has been hypothesized to be responsible for fine adjustments in the aldosterone production when activated by small changes in potassium or ATII levels and appears to be necessary for glomerulosa membrane potential oscillations (55, 57, 58). The observed M1549V variant causes impaired channel inactivation and a slight shift of activation to more hyperpolarized potentials (55), as well as increased CYP11B2 expression (59), suggesting a pathophysiology similar to that of CACNA1D variants.
Somatic Mutations in Adrenal Cushing’s Syndrome
Cushing’s syndrome features hypercortisolism and is associated with a plethora of signs and symptoms, including weight gain, hypertension, diabetes mellitus, lethargy, acne, depression, hirsutism, and increased mortality (60, 61). CPAs are less frequent than ACTH-secreting pituitary tumors (62), but still account for up to 10% of endogenous CS (60, 63, 64). Somatic PRKAR1A loss-of-function mutations were identified as a cause of sporadic CPAs in a hypothesis-driven approach (65).
Following the description of somatic mutations in PA, using exome sequencing, four groups independently identified somatic mutations in the PRKACA gene as a cause of CS (66–69). PRKACA encodes the catalytic subunit of protein kinase A involved in the regulation of adrenal cortisol production (see Introduction and Figure 1). Beuschlein and colleagues sequenced the exomes of 10 CPAs and identified heterozygous somatic PRKACA mutations in eight, with a frequency of 37% in the entire cohort of CPAs associated with overt CS. No PRKACA variants were found in CPAs associated with subclinical CS, APAs, or inactive adenomas, and the presence of PRKACA variants was associated with a more severe phenotype (66). All but one tumor carried a single variant, L206R, suggesting a gain-of-function effect. L206 is located in the highly conserved interaction site between the regulatory and the catalytic subunits of PKA, and binding of the regulatory subunit at this position prevents substrate phosphorylation. Molecular modeling and functional analysis of PKA activity suggested that the L206R mutation would lead to a steric hindrance and prevent inhibition of catalytic activity by the regulatory subunit (70). Somatic PRKACA variants other than L206R are exceedingly rare (71). Further support for the causative role of increased PKA activity in CS came from the discovery of germ line PRKACA duplications in subjects with bilateral adrenal hyperplasia and CS (66).
These results were confirmed in independent cohorts. Cao et al. reported an L205R variant (equivalent to L206R in the initial report) in the PRKACA gene in 27 of 39 CPAs. Further, two GNAS (Gαs) mutations and a CTNNB1 (β-catenin) mutation were found (see below) (67). Sato et al. screened tumors of 65 patients with ACTH-independent CS. They identified PRKACAL206R mutations in 52.3% and GNAS mutations in 16.9% of the tumors. In addition, they provided evidence of an association of PRKACAL206R with smaller tumor size and a more severe phenotype (68). Lastly, Goh et al. reported a PRKACAL206R mutation in 24% of CPAs (35% of cases with overt CS). They also reported CTNNB1 mutations in 16% and GNAS mutations in 6% of tumors (69). Similar results were found in additional cohorts (39, 41, 71) (Table 1). Functionally, L206R has been shown to enhance the phosphorylation of PKA downstream effectors CREB and ATF in cell culture and tumor tissue samples (69) (Figure 1).
Mutations in GNAS and CTNNB1 in Cushing’s Syndrome and Primary Aldosteronism
Mutations in GNAS have long been known to inhibit GTPase activity of the Gαs subunit and thereby cause constitutive Gαs activation, abnormal cAMP signaling, endocrine hyperfunction, and tumor formation; postzygotic GNAS mutations are found in McCune–Albright syndrome, which can be associated with CS (72, 73). The discovery of mutually exclusive somatic gain-of-function mutations of PRKACA and GNAS in CPAs (see above) has further demonstrated that increased cAMP signaling is sufficient to cause tumorigenesis and cortisol hypersecretion. However, somewhat unexpectedly, given the absence of PRKACA mutations in APAs, GNAS variants were also reported in A/CPAs in two instances (42). On a molecular level, given the accessory role of ACTH in stimulating aldosterone secretion, increased cAMP signaling may play a role.
CTNNB1 encodes β-catenin of the Wnt/β–catenin pathway, which is known to play an important role in adrenocortical development and cancer (74). Activating mutations are not only found in benign and malignant adrenal tumors (75) but also in tumors of other organs. Such mutations prevent β-catenin degradation and cause proliferation. Even though such events have been shown to trigger benign aldosterone-secreting and cortisol-secreting tumor development as well as malignancy in a mouse model and human tissue samples (36, 45, 69, 75–77), the exact mechanisms underlying hormone secretion in CTNNB1 positive tumors remain to be determined.
In this context, a common pathway of PRKACA, GNAS, and CTNNB1 has been suggested (78). However, it has been shown that GNAS and CTNNB1 mutations are not always mutually exclusive in CPAs, and that mutations of CTNNB1 are also present in non-secreting adrenal tumors (79). A recent study described an association with pregnancy in two of three cases with APAs and CTNNB1 mutations and suggested that the manifestation may be mediated by CTNNB1-induced LHCGR expression and increased LH levels in pregnancy (80). However, the absence of an association with pregnancy in previously described female cases (81), the high prevalence of LHCGR overexpression in APAs (82), and the finding of CTNNB1 mutations in male individuals with APAs (83) suggest a role of additional factors.
Conclusion and Open Questions
Taken together, the recent findings on the genetic causes of APAs and CPAs suggest that both result from gain-of-function mutations that concurrently lead to excess hormone hypersecretion and increased proliferation. In most cases, a single mutation is apparently sufficient for tumor formation and hormone hypersecretion. There is little overlap between CPAs and APAs in terms of the mutational spectrum. While CPAs often carry mutations that lead to increased intracellular cAMP levels, mutations known to cause APAs mostly affect intracellular calcium signaling. Overlapping roles in the function of transcription factors ATF and CREB in glomerulosa and fasciculata function, as well as overlapping roles of signaling downstream of ACTH and calcium in cortisol and aldosterone synthesis, may explain the presence of KCNJ5 and GNAS mutations in tumors secreting both cortisol and aldosterone (Figure 1). Open questions include the determinants of the histological phenotype of APAs with certain mutations, the molecular pathways involved in proliferation of both APAs and CPAs, potential additional factors that drive hormone production in tumors with CTNNB1 mutations, and the pathogenesis of tumors without mutations in known driver genes. In summary, despite significant progress over the past few years, the pathophysiology behind CPAs and APAs has not been fully unraveled. Distinct and common molecular switches appear to exist in both disorders.
Author Contributions
All authors listed have made substantial, direct, and intellectual contribution to the work and approved it for publication.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Funding
This work was funded by a grant of the Ministry of Innovation, Science and Research of the State of North Rhine-Westphalia (NRW Rückkehrerprogramm to US).
References
1. Bovio S, Cataldi A, Reimondo G, Sperone P, Novello S, Berruti A, et al. Prevalence of adrenal incidentaloma in a contemporary computerized tomography series. J Endocrinol Invest (2006) 29:298–302. doi:10.1007/BF03344099
2. Young WF Jr. Clinical practice. The incidentally discovered adrenal mass. N Engl J Med (2007) 356:601–10. doi:10.1056/NEJMcp065470
3. Spat A, Hunyady L. Control of aldosterone secretion: a model for convergence in cellular signaling pathways. Physiol Rev (2004) 84:489–539. doi:10.1152/physrev.00030.2003
4. Masilamani S, Kim GH, Mitchell C, Wade JB, Knepper MA. Aldosterone-mediated regulation of ENaC alpha, beta, and gamma subunit proteins in rat kidney. J Clin Invest (1999) 104:R19–23. doi:10.1172/JCI7840
5. Bandulik S, Tauber P, Penton D, Schweda F, Tegtmeier I, Sterner C, et al. Severe hyperaldosteronism in neonatal Task3 potassium channel knockout mice is associated with activation of the intraadrenal renin-angiotensin system. Endocrinology (2013) 154:2712–22. doi:10.1210/en.2013-1101
6. Simpson ER, Waterman MR. Regulation of the synthesis of steroidogenic enzymes in adrenal cortical cells by ACTH. Annu Rev Physiol (1988) 50:427–40. doi:10.1146/annurev.ph.50.030188.002235
7. Rosenberg D, Groussin L, Jullian E, Perlemoine K, Bertagna X, Bertherat J. Role of the PKA-regulated transcription factor CREB in development and tumorigenesis of endocrine tissues. Ann N Y Acad Sci (2002) 968:65–74. doi:10.1111/j.1749-6632.2002.tb04327.x
8. Oakley RH, Cidlowski JA. Cellular processing of the glucocorticoid receptor gene and protein: new mechanisms for generating tissue-specific actions of glucocorticoids. J Biol Chem (2011) 286:3177–84. doi:10.1074/jbc.R110.179325
9. Sapolsky RM, Romero LM, Munck AU. How do glucocorticoids influence stress responses? Integrating permissive, suppressive, stimulatory, and preparative actions. Endocr Rev (2000) 21:55–89. doi:10.1210/er.21.1.55
10. Barnes PJ. Anti-inflammatory actions of glucocorticoids: molecular mechanisms. Clin Sci (1998) 94:557–72. doi:10.1042/cs0940557
11. Choi M, Scholl UI, Yue P, Bjorklund P, Zhao B, Nelson-Williams C, et al. K+ channel mutations in adrenal aldosterone-producing adenomas and hereditary hypertension. Science (2011) 331:768–72. doi:10.1126/science.1198785
12. Conn JW. Presidential address. I. Painting background. II. Primary aldosteronism, a new clinical syndrome. J Lab Clin Med (1955) 45:3–17.
13. Stowasser M, Gordon RD, Rutherford JC, Nikwan NZ, Daunt N, Slater GJ. Diagnosis and management of primary aldosteronism. J Renin Angiotensin Aldosterone Syst (2001) 2:156–69. doi:10.3317/jraas.2001.022
14. Mulatero P, Dluhy RG, Giacchetti G, Boscaro M, Veglio F, Stewart PM. Diagnosis of primary aldosteronism: from screening to subtype differentiation. Trends Endocrinol Metab (2005) 16:114–9. doi:10.1016/j.tem.2005.02.007
15. Rossi GP, Bernini G, Caliumi C, Desideri G, Fabris B, Ferri C, et al. A prospective study of the prevalence of primary aldosteronism in 1,125 hypertensive patients. J Am Coll Cardiol (2006) 48:2293–300. doi:10.1016/j.jacc.2006.07.059
16. Korah HE, Scholl UI. An update on familial hyperaldosteronism. Horm Metab Res (2015) 47:941–6. doi:10.1055/s-0035-1564166
17. Heginbotham L, Lu Z, Abramson T, MacKinnon R. Mutations in the K+ channel signature sequence. Biophys J (1994) 66:1061–7. doi:10.1016/S0006-3495(94)80887-2
18. Scholl UI, Nelson-Williams C, Yue P, Grekin R, Wyatt RJ, Dillon MJ, et al. Hypertension with or without adrenal hyperplasia due to different inherited mutations in the potassium channel KCNJ5. Proc Natl Acad Sci U S A (2012) 109:2533–8. doi:10.1073/pnas.1121407109
19. Charmandari E, Sertedaki A, Kino T, Merakou C, Hoffman DA, Hatch MM, et al. A novel point mutation in the KCNJ5 gene causing primary hyperaldosteronism and early-onset autosomal dominant hypertension. J Clin Endocrinol Metab (2012) 97:E1532–9. doi:10.1210/jc.2012-1334
20. Monticone S, Bandulik S, Stindl J, Zilbermint M, Dedov I, Mulatero P, et al. A case of severe hyperaldosteronism caused by a de novo mutation affecting a critical salt bridge Kir3.4 residue. J Clin Endocrinol Metab (2015) 100:E114–8. doi:10.1210/jc.2014-3636
21. Lenzini L, Rossitto G, Maiolino G, Letizia C, Funder JW, Rossi GP. A meta-analysis of somatic KCNJ5 K channel mutations in 1636 patients with an aldosterone-producing adenoma. J Clin Endocrinol Metab (2015) 100:E1089–95. doi:10.1210/jc.2015-2149
22. Akerstrom T, Crona J, Delgado Verdugo A, Starker LF, Cupisti K, Willenberg HS, et al. Comprehensive re-sequencing of adrenal aldosterone producing lesions reveal three somatic mutations near the KCNJ5 potassium channel selectivity filter. PLoS One (2012) 7:e41926. doi:10.1371/journal.pone.0041926
23. Fernandes-Rosa FL, Williams TA, Riester A, Steichen O, Beuschlein F, Boulkroun S, et al. Genetic spectrum and clinical correlates of somatic mutations in aldosterone-producing adenoma. Hypertension (2014) 64:354–61. doi:10.1161/HYPERTENSIONAHA.114.03419
24. Williams TA, Monticone S, Schack VR, Stindl J, Burrello J, Buffolo F, et al. Somatic ATP1A1, ATP2B3, and KCNJ5 mutations in aldosterone-producing adenomas. Hypertension (2014) 63:188–95. doi:10.1161/HYPERTENSIONAHA.113.01733
25. Wu VC, Huang KH, Peng KY, Tsai YC, Wu CH, Wang SM, et al. Prevalence and clinical correlates of somatic mutation in aldosterone producing adenoma-Taiwanese population. Sci Rep (2015) 5:11396. doi:10.1038/srep11396
26. Wang B, Li X, Zhang X, Ma X, Chen L, Zhang Y, et al. Prevalence and characterization of somatic mutations in chinese aldosterone-producing adenoma patients. Medicine (Baltimore) (2015) 94:e708. doi:10.1097/MD.0000000000000708
27. Zheng FF, Zhu LM, Nie AF, Li XY, Lin JR, Zhang K, et al. Clinical characteristics of somatic mutations in Chinese patients with aldosterone-producing adenoma. Hypertension (2015) 65:622–8. doi:10.1161/HYPERTENSIONAHA.114.03346
28. Taguchi R, Yamada M, Nakajima Y, Satoh T, Hashimoto K, Shibusawa N, et al. Expression and mutations of KCNJ5 mRNA in Japanese patients with aldosterone-producing adenomas. J Clin Endocrinol Metab (2012) 97:1311–9. doi:10.1210/jc.2011-2885
29. Hong AR, Kim JH, Song YS, Lee KE, Seo SH, Seong MW, et al. Genetics of aldosterone-producing adenoma in Korean patients. PLoS One (2016) 11:e0147590. doi:10.1371/journal.pone.0147590
30. Monticone S, Hattangady NG, Nishimoto K, Mantero F, Rubin B, Cicala MV, et al. Effect of KCNJ5 mutations on gene expression in aldosterone-producing adenomas and adrenocortical cells. J Clin Endocrinol Metab (2012) 97:E1567–72. doi:10.1210/jc.2011-3132
31. Oki K, Plonczynski MW, Lam ML, Gomez-Sanchez EP, Gomez-Sanchez CE. The potassium channel, Kir3.4 participates in angiotensin II-stimulated aldosterone production by a human adrenocortical cell line. Endocrinology (2012) 153:4328–35. doi:10.1210/en.2012-1241
32. Oki K, Plonczynski MW, Luis Lam M, Gomez-Sanchez EP, Gomez-Sanchez CE. Potassium channel mutant KCNJ5 T158A expression in HAC-15 cells increases aldosterone synthesis. Endocrinology (2012) 153:1774–82. doi:10.1210/en.2011-1733
33. Hattangady NG, Karashima S, Yuan L, Ponce-Balbuena D, Jalife J, Gomez-Sanchez CE, et al. Mutated KCNJ5 activates the acute and chronic regulatory steps in aldosterone production. J Mol Endocrinol (2016). doi:10.1530/JME-15-0324
34. Azizan EA, Lam BY, Newhouse SJ, Zhou J, Kuc RE, Clarke J, et al. Microarray, qPCR, and KCNJ5 sequencing of aldosterone-producing adenomas reveal differences in genotype and phenotype between zona glomerulosa- and zona fasciculata-like tumors. J Clin Endocrinol Metab (2012) 97:E819–29. doi:10.1210/jc.2011-2965
35. Azizan EA, Poulsen H, Tuluc P, Zhou J, Clausen MV, Lieb A, et al. Somatic mutations in ATP1A1 and CACNA1D underlie a common subtype of adrenal hypertension. Nat Genet (2013) 45:1055–60. doi:10.1038/ng.2716
36. Scholl UI, Healy JM, Thiel A, Fonseca AL, Brown TC, Kunstman JW, et al. Novel somatic mutations in primary hyperaldosteronism are related to the clinical, radiological and pathological phenotype. Clin Endocrinol (Oxf) (2015) 83:779–89. doi:10.1111/cen.12873
37. Adachi J, Hirai Y, Terui K, Nakano T, Fukuda Y, Suda T, et al. A report of 7 cases of adrenal tumors secreting both cortisol and aldosterone. Intern Med (2003) 42:714–8. doi:10.2169/internalmedicine.42.714
38. Willenberg HS, Spath M, Maser-Gluth C, Engers R, Anlauf M, Dekomien G, et al. Sporadic solitary aldosterone- and cortisol-co-secreting adenomas: endocrine, histological and genetic findings in a subtype of primary aldosteronism. Hypertens Res (2010) 33:467–72. doi:10.1038/hr.2010.18
39. Nakajima Y, Okamura T, Gohko T, Satoh T, Hashimoto K, Shibusawa N, et al. Somatic mutations of the catalytic subunit of cyclic AMP-dependent protein kinase (PRKACA) gene in Japanese patients with several adrenal adenomas secreting cortisol [Rapid Communication]. Endocr J (2014) 61:825–32. doi:10.1507/endocrj.EJ14-0282
40. Yamada M, Nakajima Y, Taguchi R, Okamura T, Ishii S, Tomaru T, et al. KCNJ5 mutations in aldosterone- and cortisol-co-secreting adrenal adenomas [Rapid Communication]. Endocr J (2012) 59:735–41. doi:10.1507/endocrj.EJ12-0247
41. Thiel A, Reis AC, Haase M, Goh G, Schott M, Willenberg HS, et al. PRKACA mutations in cortisol-producing adenomas and adrenal hyperplasia: a single-center study of 60 cases. Eur J Endocrinol (2015) 172:677–85. doi:10.1530/EJE-14-1113
42. Nakajima Y, Okamura T, Horiguchi K, Gohko T, Miyamoto T, Satoh T, et al. GNAS mutations in adrenal aldosterone-producing adenomas [Rapid Communication]. Endocr J (2016) 63:199–204. doi:10.1507/endocrj.EJ15-0642
43. Spät A, Hunyady L, Szanda G. Signaling interactions in the adrenal cortex. Front Endocrinol (Lausanne) (2016) 7:17. doi:10.3389/fendo.2016.00017
44. Chen AX, Nishimoto K, Nanba K, Rainey WE. Potassium channels related to primary aldosteronism: expression similarities and differences between human and rat adrenals. Mol Cell Endocrinol (2015) 417:141–8. doi:10.1016/j.mce.2015.09.011
45. Scholl UI, Goh G, Stolting G, de Oliveira RC, Choi M, Overton JD, et al. Somatic and germline CACNA1D calcium channel mutations in aldosterone-producing adenomas and primary aldosteronism. Nat Genet (2013) 45:1050–4. doi:10.1038/ng.2695
46. Xie CB, Haris Shaikh L, Garg S, Tanriver G, Teo AE, Zhou J, et al. Regulation of aldosterone secretion by Cav1.3. Sci Rep (2016) 6:24697. doi:10.1038/srep24697
47. Beuschlein F, Boulkroun S, Osswald A, Wieland T, Nielsen HN, Lichtenauer UD, et al. Somatic mutations in ATP1A1 and ATP2B3 lead to aldosterone-producing adenomas and secondary hypertension. Nat Genet (2013) 45:440–4. doi:10.1038/ng.2550
48. Tauber P, Aichinger B, Christ C, Stindl J, Rhayem Y, Beuschlein F, et al. Cellular pathophysiology of an adrenal adenoma-associated mutant of the plasma membrane Ca2+-ATPase ATP2B3. Endocrinology (2016) 157(6):2489–99. doi:10.1210/en.2015-2029
49. Nishimoto K, Tomlins SA, Kuick R, Cani AK, Giordano TJ, Hovelson DH, et al. Aldosterone-stimulating somatic gene mutations are common in normal adrenal glands. Proc Natl Acad Sci U S A (2015) 112:E4591–9. doi:10.1073/pnas.1505529112
50. Vasan RS, Evans JC, Larson MG, Wilson PW, Meigs JB, Rifai N, et al. Serum aldosterone and the incidence of hypertension in nonhypertensive persons. N Engl J Med (2004) 351:33–41. doi:10.1056/NEJMoa033263
51. Dekkers T, ter Meer M, Lenders JW, Hermus AR, Schultze Kool L, Langenhuijsen JF, et al. Adrenal nodularity and somatic mutations in primary aldosteronism: one node is the culprit? J Clin Endocrinol Metab (2014) 99:E1341–51. doi:10.1210/jc.2013-4255
52. Fernandes-Rosa FL, Giscos-Douriez I, Amar L, Gomez-Sanchez CE, Meatchi T, Boulkroun S, et al. Different somatic mutations in multinodular adrenals with aldosterone-producing adenoma. Hypertension (2015) 66:1014–22. doi:10.1161/HYPERTENSIONAHA.115.05993
53. Nanba K, Chen AX, Omata K, Vinco M, Giordano TJ, Else T, et al. Molecular heterogeneity in aldosterone-producing adenomas. J Clin Endocrinol Metab (2016) 101:999–1007. doi:10.1210/jc.2015-3239
54. Lalli E, Barhanin J, Zennaro MC, Warth R. Local control of aldosterone production and primary aldosteronism. Trends Endocrinol Metab (2016) 27:123–31. doi:10.1016/j.tem.2016.01.003
55. Scholl UI, Stölting G, Nelson-Williams C, Vichot AA, Choi M, Loring E, et al. Recurrent gain of function mutation in calcium channel CACNA1H causes early-onset hypertension with primary aldosteronism. Elife (2015) 4:e06315. doi:10.7554/eLife.06315
56. Perez-Reyes E. Molecular physiology of low-voltage-activated t-type calcium channels. Physiol Rev (2003) 83:117–61. doi:10.1152/physrev.00018.2002
57. Felizola SJ, Maekawa T, Nakamura Y, Satoh F, Ono Y, Kikuchi K, et al. Voltage-gated calcium channels in the human adrenal and primary aldosteronism. J Steroid Biochem Mol Biol (2014) 144 Pt B:410–6. doi:10.1016/j.jsbmb.2014.08.012
58. Hu C, Rusin CG, Tan Z, Guagliardo NA, Barrett PQ. Zona glomerulosa cells of the mouse adrenal cortex are intrinsic electrical oscillators. J Clin Invest (2012) 122:2046–53. doi:10.1172/JCI61996
59. Reimer EN, Walenda G, Seidel E, Scholl UI. CACNA1HM1549V mutant calcium channel causes autonomous aldosterone production in HAC15 cells and is inhibited by Mibefradil. Endocrinology (2016). doi:10.1210/en.2016-1170
60. Newell-Price J, Trainer P, Besser M, Grossman A. The diagnosis and differential diagnosis of Cushing’s syndrome and pseudo-Cushing’s states. Endocr Rev (1998) 19:647–72. doi:10.1210/edrv.19.5.0346
61. Lindholm J, Juul S, Jorgensen JO, Astrup J, Bjerre P, Feldt-Rasmussen U, et al. Incidence and late prognosis of Cushing’s syndrome: a population-based study. J Clin Endocrinol Metab (2001) 86:117–23. doi:10.1210/jc.86.1.117
62. Nieman LK, Biller BM, Findling JW, Newell-Price J, Savage MO, Stewart PM, et al. The diagnosis of Cushing’s syndrome: an Endocrine Society Clinical Practice Guideline. J Clin Endocrinol Metab (2008) 93:1526–40. doi:10.1210/jc.2008-0125
63. Newell-Price J, Bertagna X, Grossman AB, Nieman LK. Cushing’s syndrome. Lancet (2006) 367:1605–17. doi:10.1016/S0140-6736(06)68699-6
65. Bertherat J, Groussin L, Sandrini F, Matyakhina L, Bei T, Stergiopoulos S, et al. Molecular and functional analysis of PRKAR1A and its locus (17q22-24) in sporadic adrenocortical tumors: 17q losses, somatic mutations, and protein kinase A expression and activity. Cancer Res (2003) 63:5308–19. doi:10.1016/s0889-8529(05)70113-4
66. Beuschlein F, Fassnacht M, Assie G, Calebiro D, Stratakis CA, Osswald A, et al. Constitutive activation of PKA catalytic subunit in adrenal Cushing’s syndrome. N Engl J Med (2014) 370:1019–28. doi:10.1056/NEJMoa1310359
67. Cao Y, He M, Gao Z, Peng Y, Li Y, Li L, et al. Activating hotspot L205R mutation in PRKACA and adrenal Cushing’s syndrome. Science (2014) 344:913–7. doi:10.1126/science.1249480
68. Sato Y, Maekawa S, Ishii R, Sanada M, Morikawa T, Shiraishi Y, et al. Recurrent somatic mutations underlie corticotropin-independent Cushing’s syndrome. Science (2014) 344:917–20. doi:10.1126/science.1252328
69. Goh G, Scholl UI, Healy JM, Choi M, Prasad ML, Nelson-Williams C, et al. Recurrent activating mutation in PRKACA in cortisol-producing adrenal tumors. Nat Genet (2014) 46:613–7. doi:10.1038/ng.2956
70. Calebiro D, Hannawacker A, Lyga S, Bathon K, Zabel U, Ronchi C, et al. PKA catalytic subunit mutations in adrenocortical Cushing’s adenoma impair association with the regulatory subunit. Nat Commun (2014) 5:5680. doi:10.1038/ncomms6680
71. Di Dalmazi G, Kisker C, Calebiro D, Mannelli M, Canu L, Arnaldi G, et al. Novel somatic mutations in the catalytic subunit of the protein kinase A as a cause of adrenal Cushing’s syndrome: a European multicentric study. J Clin Endocrinol Metab (2014) 99:E2093–100. doi:10.1210/jc.2014-2152
72. Landis CA, Masters SB, Spada A, Pace AM, Bourne HR, Vallar L. GTPase inhibiting mutations activate the alpha chain of Gs and stimulate adenylyl cyclase in human pituitary tumours. Nature (1989) 340:692–6. doi:10.1038/340692a0
73. Weinstein LS, Shenker A, Gejman PV, Merino MJ, Friedman E, Spiegel AM. Activating mutations of the stimulatory G protein in the McCune-Albright syndrome. N Engl J Med (1991) 325:1688–95. doi:10.1056/NEJM199112123252403
74. El Wakil A, Lalli E. The Wnt/beta-catenin pathway in adrenocortical development and cancer. Mol Cell Endocrinol (2011) 332:32–7. doi:10.1016/j.mce.2010.11.014
75. Tadjine M, Lampron A, Ouadi L, Bourdeau I. Frequent mutations of beta-catenin gene in sporadic secreting adrenocortical adenomas. Clin Endocrinol (Oxf) (2008) 68:264–70. doi:10.1111/j.1365-2265.2007.03033.x
76. Berthon A, Sahut-Barnola I, Lambert-Langlais S, de Joussineau C, Damon-Soubeyrand C, Louiset E, et al. Constitutive beta-catenin activation induces adrenal hyperplasia and promotes adrenal cancer development. Hum Mol Genet (2010) 19:1561–76. doi:10.1093/hmg/ddq029
77. Gaujoux S, Tissier F, Groussin L, Libe R, Ragazzon B, Launay P, et al. Wnt/beta-catenin and 3′,5′-cyclic adenosine 5′-monophosphate/protein kinase A signaling pathways alterations and somatic beta-catenin gene mutations in the progression of adrenocortical tumors. J Clin Endocrinol Metab (2008) 93:4135–40. doi:10.1210/jc.2008-0631
78. Wilson CH, McIntyre RE, Arends MJ, Adams DJ. The activating mutation R201C in GNAS promotes intestinal tumourigenesis in Apc(Min/+) mice through activation of Wnt and ERK1/2 MAPK pathways. Oncogene (2010) 29:4567–75. doi:10.1038/onc.2010.202
79. Bonnet S, Gaujoux S, Launay P, Baudry C, Chokri I, Ragazzon B, et al. Wnt/beta-catenin pathway activation in adrenocortical adenomas is frequently due to somatic CTNNB1-activating mutations, which are associated with larger and nonsecreting tumors: a study in cortisol-secreting and -nonsecreting tumors. J Clin Endocrinol Metab (2011) 96:E419–26. doi:10.1210/jc.2010-1885
80. Teo AE, Garg S, Haris Shaikh L, Zhou J, Karet Frankl FE, Gurnell M, et al. Pregnancy, primary aldosteronism, and adrenal CTNNB1 mutations. N Engl J Med (2015) 373(15):1429–36. doi:10.1056/NEJMoa1504869
81. Murtha TD, Carling T, Scholl UI. Pregnancy, primary aldosteronism, and somatic CTNNB1 mutations. N Engl J Med (2016) 374:1492–3. doi:10.1056/NEJMc1514508
82. Teo AED, Garg S, Haris Shaikh L, Zhou J, Karet Frankl FE, Gurnell M, et al. Pregnancy, primary aldosteronism, and adrenal CTNNB1 mutations. N Engl J Med (2015) 373:1429–36. doi:10.1056/NEJMoa1504869
83. Akerstrom T, Maharjan R, Sven Willenberg H, Cupisti K, Ip J, Moser A, et al. Activating mutations in CTNNB1 in aldosterone producing adenomas. Sci Rep (2016) 6:19546. doi:10.1038/srep19546
84. Xekouki P, Hatch MM, Lin L, Rodrigo de A, Azevedo M, de la Luz Sierra M, et al. KCNJ5 mutations in the National Institutes of Health cohort of patients with primary hyperaldosteronism: an infrequent genetic cause of Conn’ syndrome. Endocr Relat Cancer (2012) 19:255–60. doi:10.1530/ERC-12-0022
85. Kitamoto T, Suematsu S, Matsuzawa Y, Saito J, Omura M, Nishikawa T. Comparison of cardiovascular complications in patients with and without KCNJ5 gene mutations harboring aldosterone-producing adenomas. J Atheroscler Thromb (2015) 22:191–200. doi:10.5551/jat.24455
86. Boulkroun S, Beuschlein F, Rossi GP, Golib-Dzib JF, Fischer E, Amar L, et al. Prevalence, clinical, and molecular correlates of KCNJ5 mutations in primary aldosteronism. Hypertension (2012) 59:592–8. doi:10.1161/HYPERTENSIONAHA.111.186478
87. Azizan EA, Murthy M, Stowasser M, Gordon R, Kowalski B, Xu S, et al. Somatic mutations affecting the selectivity filter of KCNJ5 are frequent in 2 large unselected collections of adrenal aldosteronomas. Hypertension (2012) 59:587–91. doi:10.1161/HYPERTENSIONAHA.111.186239
88. Cheng CJ, Sung CC, Wu ST, Lin YC, Sytwu HK, Huang CL, et al. Novel KCNJ5 mutations in sporadic aldosterone-producing adenoma reduce Kir3.4 membrane abundance. J Clin Endocrinol Metab (2015) 100:E155–63. doi:10.1210/jc.2014-3009
Keywords: KCNJ5, CACNA1D, ATP1A1, ATP2B3, CTNNB1
Citation: Seidel E and Scholl UI (2016) Intracellular Molecular Differences in Aldosterone- Compared to Cortisol-Secreting Adrenal Cortical Adenomas. Front. Endocrinol. 7:75. doi: 10.3389/fendo.2016.00075
Received: 30 April 2016; Accepted: 14 June 2016;
Published: 27 June 2016
Edited by:
Andre Lacroix, Centre hospitalier de l’Université de Montréal, CanadaReviewed by:
Nils Lambrecht, University of California Irvine, USAGuido Di Dalmazi, Medizinische Klinik und Poliklinik IV, Germany
Copyright: © 2016 Seidel and Scholl. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Ute I. Scholl, dXRlLnNjaG9sbEBtZWQudW5pLWR1ZXNzZWxkb3JmLmRl