 Vimal Selvaraj
Vimal Selvaraj Lan N. Tu
Lan N. Tu Douglas M. Stocco
Douglas M. Stocco- 1Department of Animal Science, Cornell University, Ithaca, NY, USA
- 2Department of Cell Biology and Biochemistry, Texas Tech University Health Sciences Center, Lubbock, TX, USA
A commentary on
Conditional Steroidogenic Cell-Targeted Deletion of TSPO Unveils a Crucial Role in Viability and Hormone-Dependent Steroid Formation
by Fan J, Campioli E, Midzak A, Culty M, Papadopoulos V. Proc. Natl. Acad. Sci. U S A (2015) 112:7261–7266. doi: 10.1073/pnas.1502670112
Recent reports on Leydig cell-specific Tspo conditional knockout TspocΔ/Δ mice (1), viable global Tspo knockout (Tspo−/−) mice from two independent laboratories (2, 3), and clones of CRISPR/Cas9-mediated Tspo-deleted MA-10 Leydig cells (MA-10TspoΔ/Δ) (4) established that TSPO is not essential for steroid hormone biosynthesis or viability [reviewed in Ref. (5, 6)]. These reports refuted 25 years of dogma that described TSPO as a mitochondrial cholesterol transport protein, indispensable for steroidogenesis. In response, the research group involved in most of the early studies linking TSPO and steroidogenesis investigated Leydig cell-specific and adrenocortical cell-specific TspocΔ/Δ mice (7) and presented results that seem to repudiate the recent findings and revive the old model. In this commentary, we would like to point out that interpretations made in the manuscript by Fan et al. (7) are seriously flawed.
TSPO Deletion Does Not Affect Viability
In Fan et al. (7), it was observed that use of Amhr2cre/+ knock-in mice (8) to generate Leydig cell-specific TspocΔ/Δ mice resulted in low Mendelian ratios for homozygous cre positive mice (HO: Amhr2cre/+TspocΔ/Δ). This was interpreted as partial preimplantation embryo loss, and the authors concluded that TSPO is crucial for viability. This is a fundamental mistake because both Amhr2 and Tspo are in the same chromosome 15 and, therefore, cannot assort independently. The Tspo and Amhr2 genetic positions are just 18.18 cM apart, and the probability for chromosomal crossover between the two loci to get HO mice is calculated as 7.6% [based on Haldane (9), other estimates are similar]. Therefore, the low rate of 4.4% HO mice observed by Fan et al. (7) is anticipated and represents the precise biological value due to linkage of the two loci and is certainly not an indication of embryonic lethality (Figure 1) [Note: Actual values are not identical to calculated numbers because of differences in mouse strain, secondary regulation, specific chromosome structures, and interference at crossover sites]. Expectation of 25% HO mice based on classical Mendelian principles is not applicable in this context, and the interpretation made on this basis is highly inaccurate.
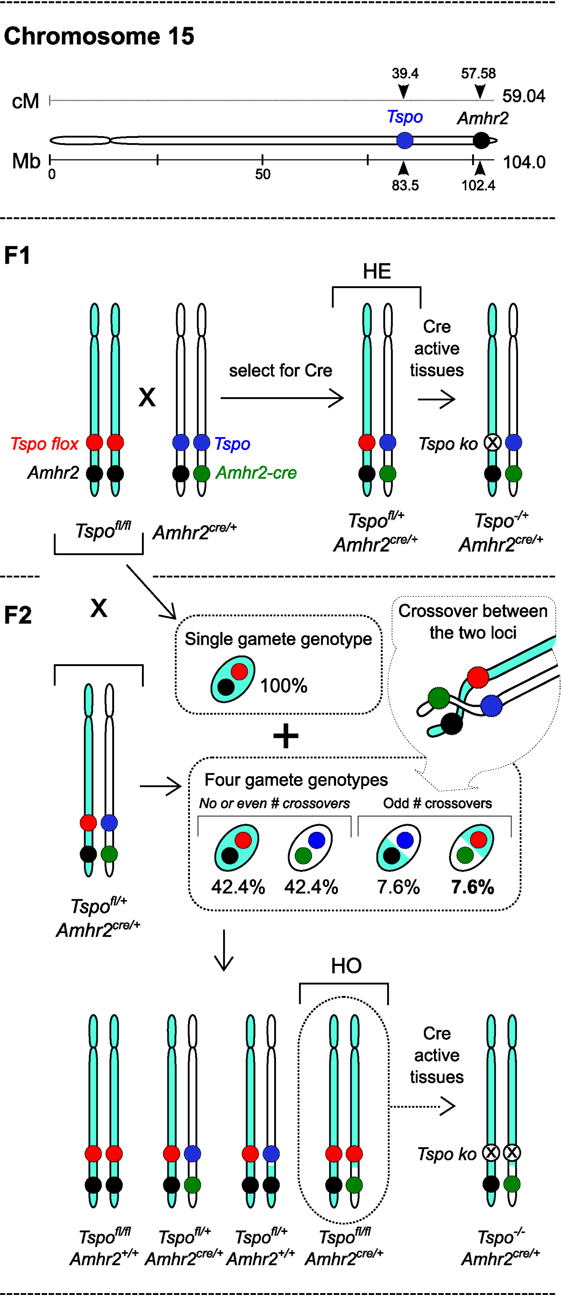
Figure 1. “Embryonic lethality” based on a miscalculation: inheritance of the Tspo and Amhr2 gene loci in chromosome 15 with respective floxed and knock-in cre alleles. Murine chromosome 15 showing the physical positions (in megabases – Mb) and genetic positions (in centimorgans – cM) of Tspo and Amhr2 genes (drawn to scale). F1: breeding between Tspofl/flAmhr2+/+ and Tspo+/+Amhr2Cre/+ can produce cre positive heterozygous Tspofl/+Amhr2Cre/+ (HE) offspring and Tspofl/+Amhr2+/+ offspring (not shown). With tissue-specific cre drivers, there is recombination in cre expressing cells leading to Tspo−/+Amhr2Cre/+ HE mono-allelic knockout cells. F2: HE mice are backcrossed with Tspofl/flAmhr2+/+ mice in order to generate Tspofl/fl, Amhr2Cre/+ (HO) mice. The Tspofl/flAmhr2+/+ mice produce only one gamete genotype, whereas HE mice produce four gamete genotypes, the ratio of which depends on the frequency of odd number of crossovers that occur between the Tspo and Amhr2 loci, and not through independent assortment, because they are in the same chromosome. Consequently, the gamete genotype necessary for generating HO offspring occurs at a calculated frequency of 7.6%. This is much lower than the classic Mendelian ratio of 25% because Tspo and Amhr2 gene loci are closely spaced in chromosome 15, and are, therefore, linked and inherited together at a high frequency. The experimental value of 4.4% observed by Fan et al. (7), is anticipated and indicates the biological value for that particular mouse strain. Therefore, interpretation of embryonic lethality in HO mice based on an incorrect expectation of 25% by Fan et al. (7) is seriously flawed.
To explain their proposed case of embryonic lethality, Fan et al. (7) proposed that the Amhr2cre/+ knock-in mice used to generate gonadal cell type-specific conditional deletions in more than 90 publications (MGI ID: 3042214), in their particular case, could induce global Tspo deletions. The justification provided was published microarray datasets that seemed to detect (with inconsistencies) an increase in Amhr2 transcription in 2-cell embryos. There was no primary data in the manuscript validating this assertion that appears highly unlikely. Even if it were to occur, global Tspo deletions would not affect the linkage and rate of HO mice as described above. In previous studies that used cre on different chromosomes, Mendelian ratios were observed during generation of viable global Tspo−/− mice with similar mouse backgrounds (2, 3).
TSPO is Not Involved in Steroidogenesis
In Fan et al. (7), the Nr5a1creTspocΔ/Δ mice showed expected Tspo deletions in Leydig cells and the adrenal cortex. Testosterone production (both baseline and after induction using hCG) was not affected, consistent with the previous report (1). Although the authors note this as “surprising,” the significance of this observation as indication that TSPO was not involved in mitochondrial cholesterol import in Leydig cells was disregarded. It is not our intention to criticize, but we are indeed under obligation to point out that the proclaimed landmark in vitro studies of Tspo disruption (10) and Tspo knockdown (11) used as foundations for asserting TSPO link to steroidogenesis were performed only using Leydig cells by this same research group. These in vitro results have since not been reproducible both in vitro (2, 4) and in vivo (1, 2) and is now also invalidated by Fan et al. (7) without an explanation.
In Nr5a1creTspocΔ/Δ mice, baseline corticosterone levels were also unaffected, consistent with the previous report (2). However, when induced with ACTH, circulating corticosterone did not increase in both heterozygous (TspocΔ/+) and homozygous (TspocΔ/Δ) deletions of Tspo compared to Tspofl/fl controls, an observation that had no correlation to TSPO expression levels and was in contrast to the previous report (2). This inconsistency and differences observed with regard to lipid accumulation and changes to Lhcgr and Scarb1 expression in Nr5a1creTspocΔ/Δ adrenals (7) may be linked to parallel findings showing that TSPO can affect lipid metabolism in cells (12). Although additional work is necessary, by attempting to provide explanations based on the unsubstantiated conjecture that TSPO is an essential cholesterol transport protein for steroidogenesis, the authors have missed an excellent opportunity to advance understanding of TSPO function.
Concluding Remarks
Global Tspo−/− mice are viable (2, 3) and are an excellent tool for investigating TSPO function in health and disease. Evidence refuting TSPO link to steroidogenesis is not based only on reports in Tspo−/− mice (2, 3). Recent studies, using Tspo-deficient isolated mitochondria (3), in vitro Tspo knockdown (2), and CRISPR/Cas9-mediated Tspo knockout in cell lines (4), have all highlighted problems with reproducibility of prior work. The serious misinterpretations made in Fan et al. (7) are negatively impacting research progress across multiple fields and distracting from pursuit of the core function of TSPO.
Author Contributions
All authors listed have made substantial, direct, and intellectual contribution to the work and approved it for publication.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1. Morohaku K, Pelton SH, Daugherty DJ, Butler WR, Deng W, Selvaraj V. Translocator protein/peripheral benzodiazepine receptor is not required for steroid hormone biosynthesis. Endocrinology (2014) 155:89–97. doi:10.1210/en.2013-1556
2. Tu LN, Morohaku K, Manna PR, Pelton SH, Butler WR, Stocco DM, et al. Peripheral benzodiazepine receptor/translocator protein global knock-out mice are viable with no effects on steroid hormone biosynthesis. J Biol Chem (2014) 289:27444–54. doi:10.1074/jbc.M114.578286
3. Banati RB, Middleton RJ, Chan R, Hatty CR, Wai-Ying Kam W, Quin C, et al. Positron emission tomography and functional characterization of a complete PBR/TSPO knockout. Nat Commun (2014) 5:5452. doi:10.1038/ncomms6452
4. Tu LN, Zhao AH, Stocco DM, Selvaraj V. PK11195 effect on steroidogenesis is not mediated through the translocator protein (TSPO). Endocrinology (2015) 156:1033–9. doi:10.1210/en.2014-1707
5. Selvaraj V, Stocco DM, Tu LN. Minireview: translocator protein (TSPO) and steroidogenesis: a reappraisal. Mol Endocrinol (2015) 29:490–501. doi:10.1210/me.2015-1033
6. Selvaraj V, Stocco DM. The changing landscape in translocator protein (TSPO) function. Trends Endocrinol Metab (2015) 26:341–8. doi:10.1016/j.tem.2015.02.007
7. Fan J, Campioli E, Midzak A, Culty M, Papadopoulos V. Conditional steroidogenic cell-targeted deletion of TSPO unveils a crucial role in viability and hormone-dependent steroid formation. Proc Natl Acad Sci U S A (2015) 112:7261–6. doi:10.1073/pnas.1502670112
8. Jamin SP, Arango NA, Mishina Y, Hanks MC, Behringer RR. Requirement of Bmpr1a for Mullerian duct regression during male sexual development. Nat Genet (2002) 32:408–10. doi:10.1038/ng1003
9. Haldane JBS. The combination of linkage values, and the calculation of distance between the loci of linked factors. J Genet (1919) 8:299–309.
10. Papadopoulos V, Amri H, Li H, Boujrad N, Vidic B, Garnier M. Targeted disruption of the peripheral-type benzodiazepine receptor gene inhibits steroidogenesis in the R2C Leydig tumor cell line. J Biol Chem (1997) 272:32129–35. doi:10.1074/jbc.272.51.32129
11. Hauet T, Yao ZX, Bose HS, Wall CT, Han Z, Li W, et al. Peripheral-type benzodiazepine receptor-mediated action of steroidogenic acute regulatory protein on cholesterol entry into Leydig cell mitochondria. Mol Endocrinol (2005) 19:540–54. doi:10.1210/me.2004-0307
Keywords: translocator protein TSPO, steroid biosynthesis, mitochondria, adrenal cortex, Leydig cells, cholesterol, embryonic lethality, lipid metabolism
Citation: Selvaraj V, Tu LN and Stocco DM (2016) Crucial Role Reported for TSPO in Viability and Steroidogenesis is a Misconception. Commentary: Conditional Steroidogenic Cell-Targeted Deletion of TSPO Unveils a Crucial Role in Viability and Hormone-Dependent Steroid Formation. Front. Endocrinol. 7:91. doi: 10.3389/fendo.2016.00091
Received: 01 June 2016; Accepted: 29 June 2016;
Published: 18 July 2016
Edited by:
Pascale Crepieux, Centre National de la Recherche Scientifique, FranceReviewed by:
Alfredo Ulloa-Aguirre, Universidad Nacional Autónoma de México, MexicoDavid H. Volle, INSERM, France
Copyright: © 2016 Selvaraj, Tu and Stocco. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Vimal Selvaraj, dnM4OEBjb3JuZWxsLmVkdQ==