 Emilie Pastel
Emilie Pastel Jean-Christophe Pointud
Jean-Christophe Pointud Antoine Martinez
Antoine Martinez A. Marie Lefrançois-Martinez
A. Marie Lefrançois-Martinez- 1Diabetes and Obesity Research Group, University of Exeter Medical School, Exeter, UK
- 2CNRS, UMR 6293/INSERM U1103, Génétique, Reproduction et Développement, Clermont Université, Aubière, France
Aldose reductase (AKR1B) proteins are monomeric enzymes, belonging to the aldo-keto reductase (AKR) superfamily. They perform oxidoreduction of carbonyl groups from a wide variety of substrates, such as aliphatic and aromatic aldehydes or ketones. Due to the involvement of human aldose reductases in pathologies, such as diabetic complications and cancer, AKR1B subgroup enzymatic properties have been extensively characterized. However, the issue of AKR1B function in non-pathologic conditions remains poorly resolved. Adrenal activities generated large amount of harmful aldehydes from lipid peroxidation and steroidogenesis, including 4-hydroxynonenal (4-HNE) and isocaproaldehyde (4-methylpentanal), which can both be reduced by AKR1B proteins. More recently, some AKR1B isoforms have been shown to be endowed with prostaglandin F synthase (PGFS) activity, suggesting that, in addition to possible scavenger function, they could instigate paracrine signals. Interestingly, the adrenal gland is one of the major sites for human and murine AKR1B expression, suggesting that their detoxifying/signaling activity could be specifically required for the correct handling of adrenal function. Moreover, chronic effects of ACTH result in a coordinated regulation of genes encoding the steroidogenic enzymes and some AKR1B isoforms. This review presents the molecular mechanisms accounting for the adrenal-specific expression of some AKR1B genes. Using data from recent mouse genetic models, we will try to connect their enzymatic properties and regulation with adrenal functions.
Introduction
The pituitary adrenocorticotropic hormone is the main regulator of adrenal steroidogenesis acting through the cAMP-dependent protein kinase (PKA) signaling pathway. The fixation of ACTH second messenger, cAMP, on PKA regulatory subunits, leads to the release of catalytic subunits that in turn phosphorylate several targets, including transcription factors, such as the CCAAT enhancer-binding protein (C/EBP) and the cAMP response element-binding protein (CREB). Acting coordinately with tissue-specific factors such as the steroidogenic factor 1 (SF1), they stimulate expression of genes encoding enzymes and proteins involved in cholesterol metabolism, mobilization, and transport. In the adrenal cortex, steroidogenesis activation results in the generation of large amount of lipid aldehydes i.e., isocaproaldehyde (4-methylpentanal) produced by the CYP11A1 cholesterol side-chain cleavage and 4-hydroxynonenal (4-HNE), whose harmfulness has to be supported by coordinately regulated detoxifying enzymes. We and other groups have previously observed that the adrenal gland is one of the main sites of expression of both murine and human AKR1B proteins (1–3). Aldose reductases are cytosolic monomeric enzymes, belonging to the aldo-keto reductase (AKR) superfamily. This superfamily encompasses more than 150 NAD(P)(H)-dependent oxidoreductases distributed in all prokaryotic and eukaryotic kingdoms, including yeast, plant, invertebrates, and vertebrates. They catalyze the reduction of carbonyl groups from a wide variety of substrates, such as aliphatic and aromatic aldehydes, ketones, keto prostaglandins, ketosteroids, and xenobiotics. Based on sequence identity, these proteins are divided in 15 families termed AKR1–AKR15, each family having less than 40% amino acid sequence identity with the others (4–6).
Among the AKR1 family, the aldose reductase subgroup-designated AKR family 1 member B (AKR1B) is one of the most characterized because of its involvement in human diseases, such as diabetic complications resulting from the ability of the former AKR family 1 member B1 (AKR1B1) to reduce glucose into sorbitol in a NADPH + H+-dependent manner during hyperglycemia. In addition to glucose conversion, AKR1B proteins display multiple other activities, including reduction of aldehyde group of by-products derived from lipid peroxidation or steroid synthesis, retinoids, xenobiotics, and prostaglandins (1, 7–9). The AKR1B subfamily includes proteins sharing a high degree of similarity (i.e., more than 65% of identity; Table 1). They are organized in two subgroups based on their ability to reduce glucose: aldose reductases (AR; AKR1B1–6) and aldose reductase-like proteins (ARLP; Akr1b7–19), respectively (4, 10–12). Their structure, enzymatic properties, and substrate specificities have been the subject of many studies (1, 5, 7, 13–17), emphasizing that in addition to their high percentage of identity, they also display redundant substrate specificities and overlapping expression patterns. These potential redundancies, then, complicate study of their distinct biological functions in specific physiological or pathological processes. Analysis of murine genetic models and identification of the mechanisms regulating their expression are the necessary steps to complete our understanding in AKR1Bs biological function.
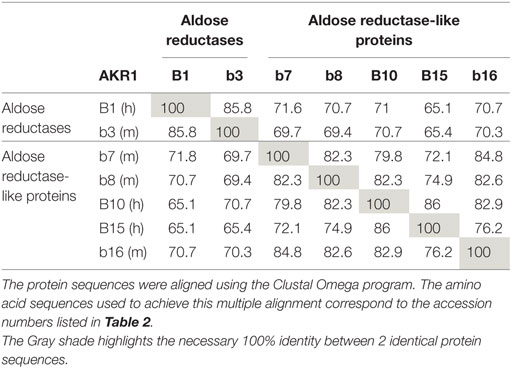
Table 1. Comparison of protein sequence identity (%) between human (h) and murine (m) AKR1B proteins.
This review will provide an updated integrative view on specific regulations of human and murine aldose reductase genes with enzymatic and functional data in the adrenal gland physiology [further information on AKR1Bs in other endocrine functions is reviewed in Ref. (18)]. Since several studies allowed identification of some murine and human aldose reductase genes as orthologs, common features will be presented for each corresponding pair, and individual isoform specificities will be discussed.
Human and Murine AKR1B Gene Synopsis
Human AKR1B Genes
Three human AKR1B genes organized in tandem on chromosome 7q33–35 have been identified (Table 2; Figure 1): AKR1B1 [human aldose reductase (19)], AKR1B10 [also designated as HSI reductase: human small intestine reductase (1, 7)], and AKR1B15 (20). AKR1B1 seems to be ubiquitously expressed, whereas AKR1B10 expression was only reported in small intestine, colon, liver, thymus, and adrenal gland (1, 7). AKR1B15 gene was recently characterized and identified as closely related to the AKR1B1 and AKR1B10 cluster on chromosome 7 (Figure 1). AKR1B15 undergoes alternative splicing, giving rise to two protein isoforms, designated as AK1R1B15.1 and AKR1B15.2, expressed in thyroid gland and testis, respectively, and both in adipose tissue and placenta. AKR1B15.1 transcript encodes a putative protein sharing 68 and 91% sequence identity with AKR1B1 and AKR1B10, respectively (21). Both AKR1B15 transcripts were absent from human adrenal (20).
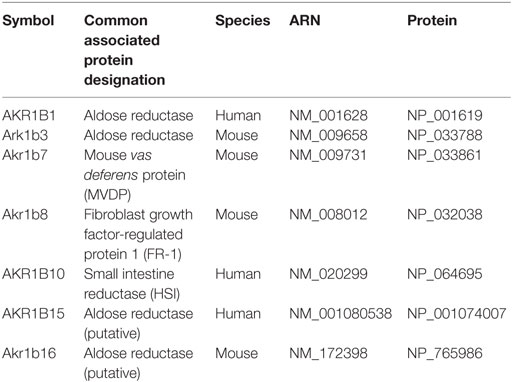
Table 2. Human and murine members of the aldo-keto reductase B1 subgroup (AKR1B).
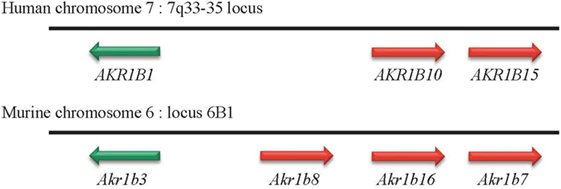
Figure 1. Genomic organization of AKR1B genes in humans and mice. In human and mouse genomes, genes encoding aldose reductase are located on chromosomes 7 and 6, respectively. Whatever the case, these genes are organized in tandem.
Murine Akr1b Genes
Four murine Akr1b genes have been described: Akr1b3 (murine aldose reductase), Akr1b7 [previously named MVDP: mouse vas deferens protein (22)], Akr1b8 [previously named FR-1: fibroblast growth factor (FGF)-related protein (23)], and Akr1b16 (21) (Table 2). Murine aldose reductase genes are located on chromosome 6 (locus 6B1), and their tandem arrangement suggests (as for the three human AKR1Bs) that these four genes arise from an ancestral gene duplication event (10, 12) (Figure 1). Several studies had allowed identification of murine Akr1b3, Akr1b8 as the orthologs of the human AKR1B1 and AKR1B10, respectively. However, this phylogenetic analysis between human and mouse AR has some limits and will be commented below. AKR1B1, Akr1b3, and Akr1b16 are rather ubiquitously expressed (11, 21), whereas Akr1b7 and Akr1b8 exhibit a restricted tissue distribution. Indeed, Akr1b7 is detected in vas deferens, adrenal glands, gonads, intestine, white adipose tissue, eye, liver, and kidney (2, 22, 24–26) and Akr1b8 in testis, heart, adrenal glands, intestine, and liver (2, 11, 23).
AKR1B in Adrenals: Between Detoxification and Paracrine Signaling
Akr1b3/AKR1B1: Expression Pattern and Relevant Functions
In studies using murine adrenal cell lines (Y1 adrenocortical cells and MPC862L chromaffin cells), we found that Akr1b3 protein accumulates in both adrenal cortex and medulla. Moreover, in vivo and ex vivo hormonal manipulations demonstrated that unlike the other murine Akr1b7 and Akr1b8 isoforms, Akr1b3 is expressed in the whole gland (27). Finally, cAMP stimulation failed to modulate Akr1b3 expression in Y1 cell line, confirming that Akr1b3 was insensitive to ACTH signaling (28) (Table 3).
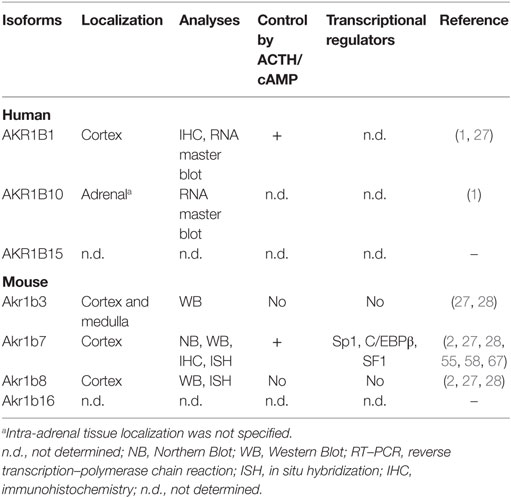
Table 3. Localization and regulation of AKR1B in adrenal gland.
Considering their enzymatic properties and expression levels in murine adrenal cortex, Akr1b7 and Akr1b8 are considered as the main isocaproaldehyde reductase and 4-HNE reductase, respectively, while Akr1b3 could rather participate in the elimination of these toxic compounds in basal physiological conditions (27–29). Moreover, Akr1b3 also displays 9-,11-endoperoxide reductase activity that, when coupled to COX-1 (cyclooxygenase type 1), allows prostaglandin F2α (PGF2α) synthesis in adrenal cortex and medulla (see below).
Despite all these evidences upon Akr1b3 involvement in both lipid aldehyde detoxification and PGF2α synthesis, in vivo Akr1b3 gene invalidation (Akr1b3−/− mice) did not highlight any phenotype related to adrenal gland (30, 31). The lack of adrenal dysfunction in Akr1b3−/− mice may result from the redundancy of enzymatic properties carried by the other murine isoforms expressed in the gland (Akr1b7 and Akr1b8) that could then compensate Akr1b3 loss.
In human adrenal gland, AKR1B1 transcripts have been initially detected using RNA master blot (1) (Table 3). Thereafter, using immunohistochemistry, we confirmed those results and demonstrated that AKR1B1 expression pattern is restricted to the cortex of adrenal gland (27). Treatment of the human adrenocortical tumor cells NC1-H295 with forskolin (adenylyl cyclase inducer) allowed us to suggest that similar to the murine isoform Akr1b7, AKR1B1 expression was sensitive to ACTH (32) (Table 3). The molecular mechanisms and cis-acting elements responsible for ACTH/cAMP responsiveness of AKR1B1 gene have not been investigated to date (Figure 2). Analysis of AKR1B1 expression in stress-related disorders was not explored to date. Moreover, analysis of adrenal samples from Cushing’s disease (ACTH-producing pituitary tumor) revealed unchanged mRNA levels of AKR1B1 gene (32).
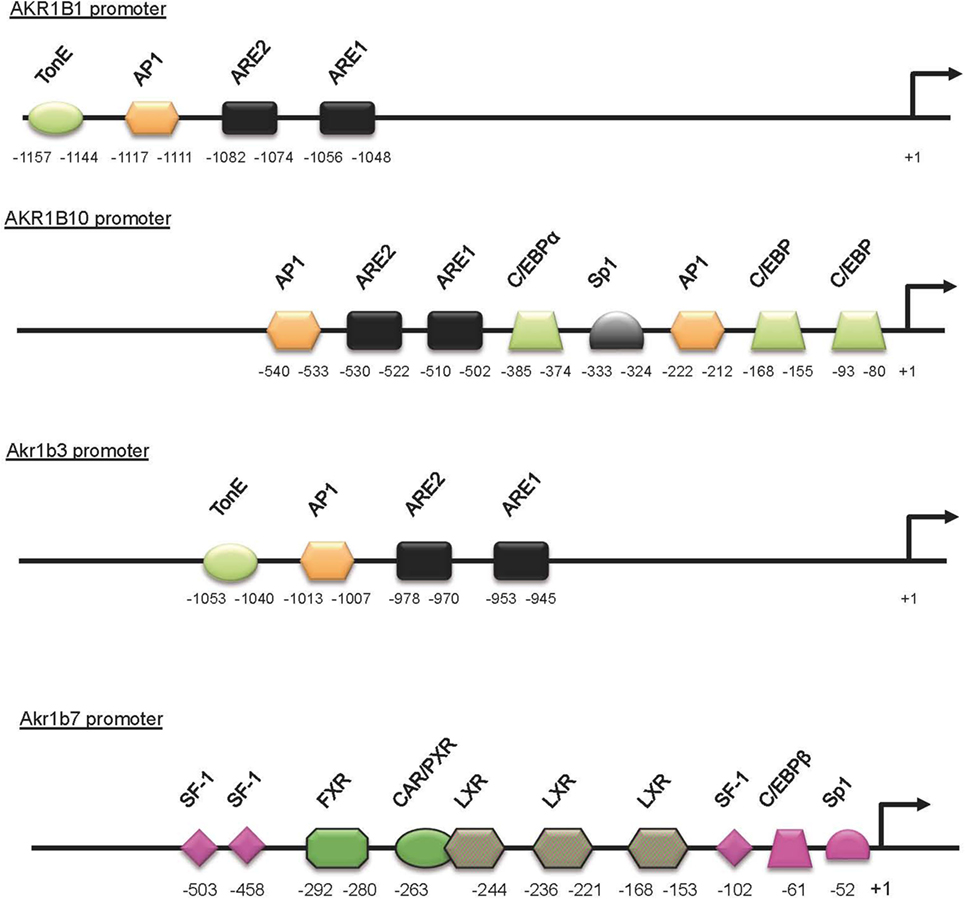
Figure 2. Schematic representation of the AKR1B1, AKR1B10, Akr1b3, and Akr1b7 promoters. Cis- and trans-acting factors shown to be involved in stress responsiveness are indicated. The DNA-binding site for transcription factors and nuclear receptors required for the Akr1b7-specific expression in adrenal gland (pink boxes) are shown. LXR-binding sites are involved in both adrenal and intestine Akr1b7 expression. TonE, tonicity response element; AP1, activator protein 1 binding site; ARE, antioxidant response element; C/EBP, CCAAT enhancer binding protein binding site; Sp1, selective promoter factor 1 binding site.
Based on its enzymatic properties, AKR1B1 has long been considered as the sole isocaproaldehyde reductase in the human adrenal gland (33) (Figure 3A; Table 4). Interestingly, unlike murine Akr1b7 isoform, NADPH-dependent isocaproaldehyde reductase activity carried by AKR1B1 was inhibited by tolrestat, a potent and specific aldose reductase inhibitor belonging to the carboxylic acids group of AR inhibitors (13, 29, 33). We demonstrated that AKR1B1 was also able to convert PGH2 into PGF2α (34) (Figure 3C; Table 4). This 9-,11-endoperoxide reductase activity is also strictly NADPH-dependent and inhibited by tolrestat.
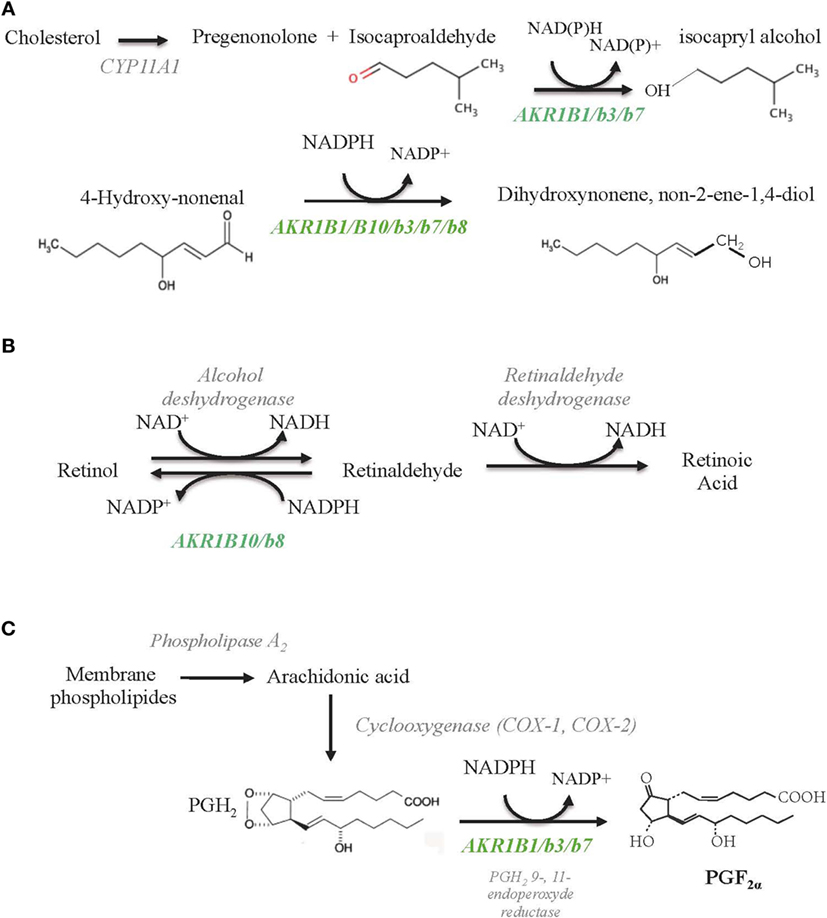
Figure 3. Schematic diagrams of enzymatic reactions illustrating AKR1B involvement toward lipid aldehyde detoxification (A), retinoids (B), and prostaglandins (C) metabolism. All the indicated substrates and enzymatic activities are supposed to coexist in the adrenal glands, but their relative importance in adrenal function remains unknown so far. AKR1B isoforms endowed with each of these activities are indicated in green.
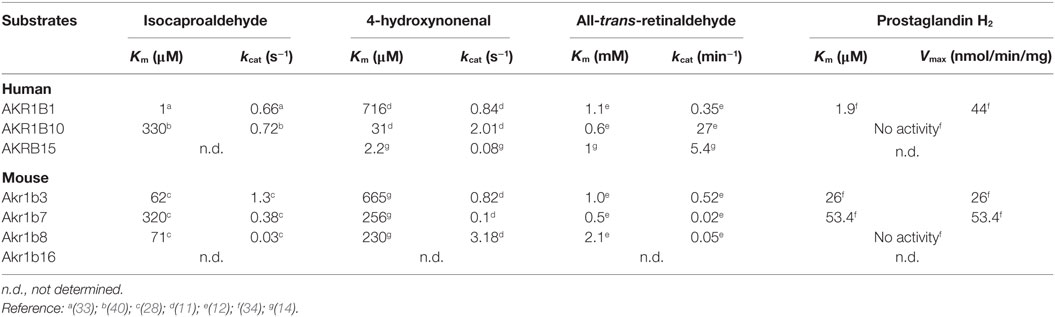
Table 4. Kinetic constants of AKR1B toward isocaproaldehyde, 4-hydroxynonenal, retinaldehyde, and prostaglandin H2.
We observed that in the human adrenal gland, AKR1B1 and the cAMP-inducible COX-2 isoform were co-localized in steroidogenic cortical cells (27) (Table 3). Then, we can consider that human adrenal cortex has the potential to produce PGF2α in response to ACTH surge. The shared properties of human AKR1B1 and mouse Akr1b7, such as hormonal regulation and reductase activity toward common substrates, prompted us to postulate that they can be considered as functional orthologs at least in the adrenal cortex (32). Moreover, increased levels of AKR1B1 transcripts in human were observed in adrenocortical adenomas harboring glucocorticoid autonomous hypersecretion (32). The possibility that AKR1B1 prostaglandin F synthase (PGFS) activity could participate in an intra-adrenal feedback loop between endocrine activities of cortex and medulla in human adrenal gland remains to be explored.
Given the high expression of AKR1B1 in the adrenal cortex, we evaluated alterations in its expression in association with human adrenal disorders. The relative abundance of AKR1B1 mRNA was decreased in adrenocortical carcinomas (ACC) when compared to benign tumors, Cushing’s hyperplasia, or normal adrenals (32). These data were reinforced by de Reyniès et al., who demonstrated that decreased AKR1B1 expression was associated with malignancy using an unsupervised clustering analysis of the human adrenal tumors transcriptome (35). This identified AKR1B1 as a potential negative marker for adrenocortical malignancy.
Akr1b8/AKR1B10: Phylogeny, Regulation, and Enzymatic Specificities
Akr1b8 mRNA was initially detected in both fetal and adult murine adrenal cortex but remained undetected in the medulla by in situ hybridization (2). In fibroblasts as well as in adrenocortical Y1 cell line, Akr1b8 was previously shown to be controlled by the FGF and phorbol myristate acetate (PMA) (23, 28). On the contrary, dexamethasone-induced ACTH suppression did not influence Akr1b8 mRNA and protein accumulation (27), suggesting that its biological function did not seem to be related to the ACTH-dependent steroidogenic activity present in adrenocortical cells (Table 3). Interestingly, in digestive tract organs, such as liver and small intestine, Akr1b8 gene was recently showed to be a target of NF-E2-related factor2 (Nrf2), which mediates transcriptional response to oxidative stress by binding to antioxidant response element (ARE) sites (36). As expected, Akr1b8 expression was downregulated in digestive tract from Nrf2 knockout mice. However, no adrenal phenotype was described in these mice, and neither oxidative stress nor Nrf2 have been involved in adrenal Akr1b8 expression so far.
Previous studies demonstrated that some AKR1B proteins showed efficient catalytic activity for the reduction of the abundant and highly reactive lipid-derived aldehyde 4-hydroxy-2-nonenal and the phospholipid aldehydes (17). In particular, Akr1b8 displays the most efficient phospholipid aldehyde and HNE-reductase activity in mouse tissues (17, 28, 37) (Figure 3; Table 4). All these enzymatic data suggest that in the adrenal cortex, Akr1b8 isoform could be devoted to detoxify aldehyde lipids abundantly accumulated in this tissue (38). Furthermore, expression of an antisense RNA directed against Akr1b7 suppressed isocaproaldehyde reductase activity in adrenocortical Y1 cells without any alteration of Akr1b8 protein accumulation (29). In view of its enzymatic features and constitutive expression, Akr1b8 is unlikely to be the principal isocaproaldehyde reductase in the adrenocortical gland (28). Akr1b8 gene disruption in mice led to reduced lipid synthesis and diminished proliferation of colonic epithelial cells but had no evident effect on general appearance, body weight, and reproduction. However, in vivo adrenal Akr1b8 physiological role remains to be examined since Akr1b8 gene disruption first report did not notice evident effect on the adrenal physiology (39).
Whether Akr1b8 and human AKR1B10 gene can be considered as ortholog is still a matter of debate since they share high sequence identity, and proteins display several close structural and enzymatic properties (11) (Tables 1 and 4). In contrast to Akr1b8, AKR1B10 gene expression is not controlled by FGF (12). Although AKR1B10 mRNA was initially detected in adrenal glands using a human RNA Master Blot, to date, there is no more information available on its in situ localization and transcriptional control in this organ (1). Moreover, AKR1B10 gene expression pattern only partially overlaps that of Akr1b8, since AKR1B10 transcripts are absent from heart, lung, or testis (7, 11).
Comparative studies demonstrated that AKR1B10 exhibits higher 4-HNE-reductase activity than AKR1B1, while lower than the murine Akr1b8 (11, 16). Ex vivo studies revealed that both human AKR1B1 and -B10 also share the ability to reduce isocaproaldehyde (1, 40). Nevertheless, in a comparative enzymatic study, Hara and colleagues showed that AKR1B1 had a more effective isocaproaldehyde reductase activity than AKR1B10, suggesting that in human steroidogenic organs, the latter was unlikely to play a major role in the detoxification of steroidogenic by-products (41).
The AKR superfamily has been added as a novel group of cytosolic enzymes that could contribute to retinoid–redox conversion. Based on their cofactor specificity (NADPH), AKR work in the reductive direction (42). Retinol (vitamin A) and its derivatives, retinaldehyde and retinoic acid (RA), are essential for the growth and maintenance of many tissues. RA is a key molecule in the development of different vertebrate organs by promoting cell differentiation and apoptosis. The control of retinaldehyde levels is essential in the regulation of RA synthesis and therefore of its signaling role. Once synthesized from β-carotene through the β-carotene 15,15′ monooxygenase 1 (BCO1), retinaldehyde has two alternative fates, its irreversible oxidation to RA (metabolism fate) by the aldehyde dehydrogenases (ADH) or its reduction back to retinol (storage fate) by the retinaldehyde reductase activity of AKR (43) (Figure 3B; Table 4). Comparative in vitro enzymatic studies on murine and human AKRs have fairly evidenced that among AKR1B proteins, AKR1B10 is so far the only retinaldehyde reductase with the highest kcat value for the retinaldehyde reduction (11, 12, 44). Ex vivo AKR1B10 overexpression in different cell systems demonstrated its contribution in increasing retinol production (45, 46). In rodent, previous data evidenced that normal adrenal gland may function as an important site of retinoic acid synthesis involving class I- and IV-ADH, thus furthering retinaldehyde metabolism rather than its storage through the AKR activity (47). According to its expression, whether the well-established retinaldehyde reductase activity of AKR1B10 is operated in normal human adrenal physiology remains to be explored.
AKR1B10 expression was initially characterized in hepatocellular carcinoma and subsequently found to be altered by tumorigenesis process in several other organs (7, 48–50). Moreover, AKR1B10 expression was associated with smoker’s non-small cell lung carcinomas (48) and was suggested to be involved in drug resistance (51). A putative mechanism by which the activity of AKR1B enzymes could promote tumor growth is the conversion of retinaldehyde to retinol resulting in RA deprivation and blockage of its differentiating effect, promoting cell proliferation and fostering tumorigenesis (43). Furthermore, recent studies have shown that in breast cancer cells, AKR1B10 associates with acetyl-CoA carboxylase-alpha (ACCA), the rate-limiting enzyme of de novo synthesis of long-chain fatty acids, and blocks its ubiquitination and proteasome degradation. Long-chain fatty acids are the building blocks of biomembranes and the precursor of lipid second messengers, playing a critical role in cell growth and proliferation (52). The AKR1B10-mediated regulation on ACCA stability represents a novel regulatory mechanism, in which AKR1B10 promotes cell survival via modulating lipid synthesis, mitochondrial function, oxidative stress, and carbonyl levels (53).
Adrenocortical carcinomas are very aggressive and rare malignant tumors with poor prognosis (54). Microarray analysis was used to seek molecular predictors of malignancy and survival in a large cohort of unilateral adrenocortical tumors (http://www.ebi.ac.uk/arrayexpress, experiment E-TABM-311). Unsupervised clustering analysis allowed robust discrimination of malignant and benign tumors. On the basis of this analysis, AKR1B10 expression was not found to be associated with the ACC group (35).
Akr1b7: Expression Profile, Detoxification Function, and Paracrine Action
High levels of Akr1b7 transcripts were initially observed by in situ hybridization in fetal and adult murine adrenal cortex but were undetectable in the medulla (2). We confirmed these results by immunohistochemistry experiments, which allowed us to further restrict major Akr1b7 expression to the zona fasciculata (55). In vivo, ACTH suppression with dexamethasone treatment resulted in a marked decrease of Akr1b7 mRNA levels that were restored when the treated mice were injected with exogenous ACTH. This ACTH/cAMP-induced Akr1b7 transcription was blocked by a PKA inhibitor (H89) in the murine adrenocortical ATC and Y1 cell lines (55, 56).
In the adrenal gland, basal and ACTH-induced expressions of Akr1b7 gene depend on three SF1 response element (SFRE) and on other cis-elements located in the upstream promoter region (Figure 3). Using transgenic mice and transfection experiments, we characterized a cryptic SFRE 102 bp upstream of the transcription start site that supports basal expression of Akr1b7 in the adrenal cortex. Among the two other SFREs identified further upstream, the site at −458 was a bona fide SFRE, essential for both basal and cAMP-stimulated promoter activity. The last SFRE, an Sp1, and C/EBPβ binding sites, respectively, localized at positions −503, −52, and −61 are all involved in cAMP responsiveness (57, 58).
AKR1Bs are capable to handle the large amount of isocaproaldehyde, a toxic by-product coming from the cholesterol side-chain cleavage during the initial step of steroid biosynthesis. Furthermore, isocaproaldehyde accumulation decreased viability of Y1 cells (29). Although Akr1b3, Akr1b7, and Akr1b8 all were able to reduce isocaproaldehyde, the two former seemed to be the more efficient reductases for this substrate (28). However, the silencing of Akr1b7 gene was sufficient to abolish the cAMP-induced isocaproaldehyde reductase activity in Y1 cells. Therefore, Akr1b7 was the main enzyme in charge of isocaproaldehyde detoxification in the adrenal cortex (29). Altogether, these data showed that in the adrenal cortex, ACTH not only controls expression of enzymes synthetizing steroids but also of proteins scavenging toxic by-products derived from steroidogenesis.
Madore et al. established that the bovine 20α-hydroxysteroid dehydrogenase, AKR1B5 was responsible for PGF2α synthesis in the endometrium (59). Thereafter, we demonstrated by ex vivo studies, that AKR1B1, Akr1b3, and Akr1b7 were also able to reduce PGH2 into PGF2α, whereas Akr1b8 and AKR1B10 were devoid of this PGF2α synthase activity (Table 4). Due to their recent discovery, this 9-,11-endoperoxide reductase activity has not been investigated yet for Akr1b16 and AKR1B15. Moreover, their enzymatic constants suggested that AKR1B1, Akr1b3, and Akr1b7 had a higher 9-,11-endoperoxide reductase activity than the other PGF synthases already described (34). Prostaglandins are paracrine/autocrine signal molecules produced from a common precursor, PGH2, which is derived from arachidonic acid by COX-1 or COX-2. Unlike COX-1, which is a constitutively expressed enzyme, COX-2 is not expressed in most organs under basal conditions but can be stimulated by inflammation and various mitogenic factors (60). Following these observations, we carefully examined the PGF2α biosynthetic pathway in the adrenal gland (27).
Prostaglandin F2α was produced by both cortical (steroidogenic cells) and medullary (chromaffin cells) tissue of the adrenal gland. In primary adrenocortical cell culture, PGF2α release was induced 2.5-fold by ACTH treatment. This secretion was correlated with ACTH responsiveness of both COX-2 and Akr1b7. Using ex vivo gain- and loss-of-function strategies, we demonstrated the pivotal role of Akr1b7 in ACTH-induced PGF2α release, and it is functionally coupled with COX-2. In the adrenal medulla in which Akr1b7 was not expressed, PGF2α was produced from the coordinated activities of Akr1b3 and COX-1. Adrenal expression of PGF2α-specific receptor (FP) was restricted to the chromaffin cells, suggesting that both autocrine and paracrine mechanisms (within the medulla and between steroidogenic and medulla cells, respectively) were relaying PGF2α action. In agreement with this hypothesis, we demonstrated that PGF2α repressed both basal and glucocorticoid-induced dopamine release in the chromaffin cell line MPC862L. Comparison of the PGF2α-responsiveness of isolated cells and whole adrenal tissue cultures showed that PGF2α-mediated repression of glucocorticoid release is an indirect mechanism relying on a decrease in catecholamine secretion, which in turn decreased cortical steroidogenesis.
These functional data led us to propose an intra-adrenal feedback loop in which adrenal endocrine activities are regulated through the involvement of AKRs [Figure 4 and Ref. (18)]. Surprisingly, however, the absence of Akr1b7 in vivo did not affect basal adrenocortical function as illustrated by normal glucocorticoid plasma levels in Akr1b7−/− mice (61). Indeed, these mice displayed an obese phenotype that did not rely on adrenal dysfunction but on the lack of Akr1b7-dependent production of PGF2α within the stromal vascular adipose tissue (3, 61). Adrenal expression of Akr1b3 and b8 is not affected in knockout mice and since they all share redundant enzymatic activities regarding detoxification of lipid aldehydes (see Akr1b3/AKR1B1: Expression Pattern and Relevant Functions and Akr1b8/AKR1B10: Phylogeny, Regulation, and Enzymatic Specificities), the remaining isoforms can compensate the absence of Akr1b7 at least in basal conditions. Importantly, Akr1b7 is the only one out of the three adrenal isoforms to be ACTH-responsive (27) and also the most abundantly expressed (3). Taken together, these hallmarks would predict that physiological importance of Akr1b enzymes in adrenal function should be rather explored under stress conditions during which scavenging capacity of constitutive (and less abundant) isoforms should be exhausted.
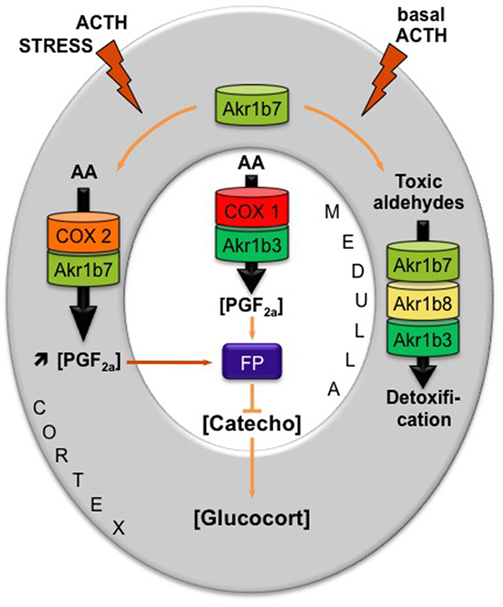
Figure 4. Proposed model integrating dual functions of aldose reductases in the regulation of mouse adrenal endocrine functions [adapted from Ref. (18)]. AKR1B family is endowed with enzymatic activities that at least ensure two metabolic functions: the production of PGF2α and the detoxification of lipid aldehydes. Arachidonic acid (AA) is metabolized into PGH2, the precursor of all prostanoids, by COX enzymes and then converted into PGF2α by the 9-,11-endoperoxyde reductase activity of PGF synthases of the AKR1B family. AKR1B are also capable to reduce toxic lipid aldehydes resulting from the particularly high prooxidant activities of P450 cytochromes in adrenocortical cells (i.e., isocaproaldehyde and 4-HNE). The mechanism integrating these dual functions was the following: (1) In basal conditions, PGF2α is constitutively secreted by chromaffin cells (by the coupling of COX-1 and Akr1b3), thus regulating catecholamine production and also limiting their paracrine action on steroidogenesis. (2) During a stress situation, ACTH transiently induces COX-2 and Akr1b7 expression, which results in PGF2α production inside the cortex. PGF2α produced in the cortex then represses catecholamine release by the medulla via a paracrine action on its FP receptor. Decreased catecholamine release in turn reduces the effect of ACTH on glucocorticoids production (27). After the stress response has ended, COX-2 returns to undetectable levels. The coupling between Akr1b7 and COX-2 does not take place. Then, Akr1b7 together with Akr1b8 and Akr1b3 function only as cortical detoxifying enzymes of the harmful aldehydes produced under chronic/basal stimulation of steroidogenesis. Catecho, catecholamine; Glucocort, glucocorticoids.
Future Directions
Fighting against oxidative stress is a challenging but mandatory task for adrenocortical cells. Indeed, P450 cytochrome systems involved in steroidogenesis, and in particular glucocorticoid production, contribute very significantly to oxidative stress by cellular reactive oxygen species (ROS) production (62). The redox imbalance due to excessive ROS production can cause adrenal damage that may progress to severe insufficiency, including familial glucocorticoid deficiency (FGD). Therefore, adrenal cortex is well supplied in antioxidant defense genes encoding enzymes of the superoxide dismutase (SOD), glutathione peroxidase (GPX), and peroxiredoxin (PRDX) families [for review, see Ref. (63)]. Since the precursor works of Feige’s group showing the ACTH responsiveness of SOD2 expression, the expected coordinated regulation of antioxidant enzymatic systems and P450s systems producing prooxidant by-products has been somewhat neglected (64). Accordingly, AKR1B enzymes family may be considered as antioxidant defense genes. Among these, ACTH-responsive ones, e.g., Akr1b7 and SOD2 genes, could participate in the adaptive response of antioxidant systems of adrenal cortex under stress conditions. Disturbance in redox homeostasis was the most recently discovered cause of FGD and mutations in NNT gene (nicotinamide nucleotide transhydrogenase) account for about 10% of cases (65). NNT ensures mitochondrial NADPH supply that is essential to ROS detoxification enzymatic systems. Then, it would be interesting to know whether adrenal-specific deficit in AKR1B enzymes could contribute to cortical damage or adrenal insufficiency in mice carrying a spontaneous Nnt mutation (66). This could provide the proof of principle for studying the physiological contribution of AKR1B family in detoxifying function in steroidogenic organs and beyond.
Author Contributions
All authors listed have made substantial, direct, and intellectual contribution to the work and approved it for publication.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Funding
This work was supported by funds from the Centre National de la Recherche Scientifique, Clermont Université, Institut National de la Santé et de la Recherche Médicale and grant from the Région Auvergne.
References
1. Hyndman DJ, Flynn TG. Sequence and expression levels in human tissues of a new member of the aldo-keto reductase family. Biochim Biophys Acta (1998) 1399:198–202. doi:10.1016/S0167-4781(98)00109-2
2. Lau ET, Cao D, Lin C, Chung SK, Chung SS. Tissue-specific expression of two aldose reductase-like genes in mice: abundant expression of mouse vas deferens protein and fibroblast growth factor-regulated protein in the adrenal gland. Biochem J (1995) 312(Pt 2):609–15. doi:10.1042/bj3120609
3. Pastel E, Pointud JC, Loubeau G, Dani C, Slim K, Martin G, et al. Aldose reductases influence prostaglandin F2α levels and adipocyte differentiation in male mouse and human species. Endocrinology (2015) 156:1671–84. doi:10.1210/en.2014-1750
4. Hyndman D, Bauman DR, Heredia VV, Penning TM. The aldo-keto reductase superfamily homepage. Chem Biol Interact (2003) 14(3–144):621–31. doi:10.1016/S0009-2797(02)00193-X
5. Jez JM, Bennett MJ, Schlegel BP, Lewis M, Penning TM. Comparative anatomy of the aldo-keto reductase superfamily. Biochem J (1997) 326(Pt 3):625–36. doi:10.1042/bj3260625
6. Jez JM, Flynn TG, Penning TM. A new nomenclature for the aldo-keto reductase superfamily. Biochem Pharmacol (1997) 54:639–47. doi:10.1016/S0006-2952(97)84253-0
7. Cao D, Fan ST, Chung SS. Identification and characterization of a novel human aldose reductase-like gene. J Biol Chem (1998) 273:11429–35. doi:10.1074/jbc.273.19.11429
8. Jin Y, Penning TM. Aldo-keto reductases and bioactivation/detoxication. Annu Rev Pharmacol Toxicol (2007) 47:263–92. doi:10.1146/annurev.pharmtox.47.120505.105337
9. Petrash JM. All in the family: aldose reductase and closely related aldo-keto reductases. Cell Mol Life Sci (2004) 61:737–49. doi:10.1007/s00018-003-3402-3
10. Ho HT, Jenkins NA, Copeland NG, Gilbert DJ, Winkles JA, Louie HW, et al. Comparisons of genomic structures and chromosomal locations of the mouse aldose reductase and aldose reductase-like genes. Eur J Biochem (1999) 259:726–30. doi:10.1046/j.1432-1327.1999.00110.x
11. Joshi A, Rajput S, Wang C, Ma J, Cao D. Murine aldo-keto reductase family 1 subfamily B: identification of AKR1B8 as an ortholog of human AKR1B10. Biol Chem (2010) 391:1371–8. doi:10.1515/BC.2010.144
12. Ruiz FX, Moro A, Gallego O, Ardèvol A, Rovira C, Petrash JM, et al. Human and rodent aldo-keto reductases from the AKR1B subfamily and their specificity with retinaldehyde. Chem Biol Interact (2011) 191:199–205. doi:10.1016/j.cbi.2011.02.007
13. Barski OA, Tipparaju SM, Bhatnagar A. The aldo-keto reductase superfamily and its role in drug metabolism and detoxification. Drug Metab Rev (2008) 40:553–624. doi:10.1080/03602530802431439
14. Giménez-Dejoz J, Kolář MH, Ruiz FX, Crespo I, Cousido-Siah A, Podjarny A, et al. Substrate specificity, inhibitor selectivity and structure-function relationships of aldo-keto reductase 1B15: a novel human retinaldehyde reductase. PLoS One (2015) 10:e0134506. doi:10.1371/journal.pone.0134506
15. Penning TM. The aldo-keto reductases (AKRs): overview. Chem Biol Interact (2015) 234:236–46. doi:10.1016/j.cbi.2014.09.024
16. Shen Y, Zhong L, Johnson S, Cao D. Human aldo-keto reductases 1B1 and 1B10: a comparative study on their enzyme activity toward electrophilic carbonyl compounds. Chem Biol Interact (2011) 191:192–8. doi:10.1016/j.cbi.2011.02.004
17. Spite M, Baba SP, Ahmed Y, Barski OA, Nijhawan K, Petrash JM, et al. Substrate specificity and catalytic efficiency of aldo-keto reductases with phospholipid aldehydes. Biochem J (2007) 405:95–105. doi:10.1042/BJ20061743
18. Pastel E, Pointud JC, Volat F, Martinez A, Lefrançois-Martinez AM. Aldo-keto reductases 1B in endocrinology and metabolism. Front Pharmacol (2012) 3:148. doi:10.3389/fphar.2012.00148
19. Bohren KM, Bullock B, Wermuth B, Gabbay KH. The aldo-keto reductase superfamily. cDNAs and deduced amino acid sequences of human aldehyde and aldose reductases. J Biol Chem (1989) 264:9547–51.
20. Weber S, Salabei JK, Möller G, Kremmer E, Bhatnagar A, Adamski J, et al. Aldo-keto Reductase 1B15 (AKR1B15): a mitochondrial human aldo-keto reductase with activity toward steroids and 3-keto-acyl-CoA conjugates. J Biol Chem (2015) 290:6531–45. doi:10.1074/jbc.M114.610121
21. Salabei JK, Li X-P, Petrash JM, Bhatnagar A, Barski OA. Functional expression of novel human and murine AKR1B genes. Chem Biol Interact (2011) 191:177–84. doi:10.1016/j.cbi.2011.01.020
22. Pailhoux EA, Martinez A, Veyssiere GM, Jean CG. Androgen-dependent protein from mouse vas deferens. cDNA cloning and protein homology with the aldo-keto reductase superfamily. J Biol Chem (1990) 265:19932–6.
23. Donohue PJ, Alberts GF, Hampton BS, Winkles JA. A delayed-early gene activated by fibroblast growth factor-1 encodes a protein related to aldose reductase. J Biol Chem (1994) 269:8604–9.
24. Brunskill EW, Sequeira-Lopez MLS, Pentz ES, Lin E, Yu J, Aronow BJ, et al. Genes that confer the identity of the renin cell. J Am Soc Nephrol (2011) 22:2213–25. doi:10.1681/ASN.2011040401
25. Schmidt DR, Schmidt S, Holmstrom SR, Makishima M, Yu RT, Cummins CL, et al. AKR1B7 is induced by the farnesoid X receptor and metabolizes bile acids. J Biol Chem (2011) 286:2425–32. doi:10.1074/jbc.M110.181230
26. Tirard J, Gout J, Lefrancois-Martinez AM, Martinez A, Begeot M, Naville D. A novel inhibitory protein in adipose tissue, the aldo-keto reductase AKR1B7: its role in adipogenesis. Endocrinology (2007) 148:1996–2005. doi:10.1210/en.2006-1707
27. Lambert-Langlais S, Pointud JC, Lefrancois-Martinez AM, Volat F, Manin M, Coudore F, et al. Aldo keto reductase 1B7 and prostaglandin F2alpha are regulators of adrenal endocrine functions. PLoS One (2009) 4:e7309. doi:10.1371/journal.pone.0007309
28. Martinez A, Aigueperse C, Val P, Dussault M, Tournaire C, Berger M, et al. Physiological functions and hormonal regulation of mouse vas deferens protein (AKR1B7) in steroidogenic tissues. Chem Biol Interact (2001) 13(0–132):903–17. doi:10.1016/S0009-2797(00)00244-1
29. Lefrancois-Martinez AM, Tournaire C, Martinez A, Berger M, Daoudal S, Tritsch D, et al. Product of side-chain cleavage of cholesterol, isocaproaldehyde, is an endogenous specific substrate of mouse vas deferens protein, an aldose reductase-like protein in adrenocortical cells. J Biol Chem (1999) 274:32875–80. doi:10.1074/jbc.274.46.32875
30. Aida K, Ikegishi Y, Chen J, Tawata M, Ito S, Maeda S, et al. Disruption of aldose reductase gene (Akr1b1) causes defect in urinary concentrating ability and divalent cation homeostasis. Biochem Biophys Res Commun (2000) 277:281–6. doi:10.1006/bbrc.2000.3648
31. Ho HT, Chung SK, Law JW, Ko BC, Tam SC, Brooks HL, et al. Aldose reductase-deficient mice develop nephrogenic diabetes insipidus. Mol Cell Biol (2000) 20:5840–6. doi:10.1128/MCB.20.16.5840-5846.2000
32. Lefrancois-Martinez AM, Bertherat J, Val P, Tournaire C, Gallo-Payet N, Hyndman D, et al. Decreased expression of cyclic adenosine monophosphate-regulated aldose reductase (AKR1B1) is associated with malignancy in human sporadic adrenocortical tumors. J Clin Endocrinol Metab (2004) 89:3010–9. doi:10.1210/jc.2003-031830
33. Matsuura K, Deyashiki Y, Bunai Y, Ohya I, Hara A. Aldose reductase is a major reductase for isocaproaldehyde, a product of side-chain cleavage of cholesterol, in human and animal adrenal glands. Arch Biochem Biophys (1996) 328:265–71. doi:10.1006/abbi.1996.0172
34. Kabututu Z, Manin M, Pointud JC, Maruyama T, Nagata N, Lambert S, et al. Prostaglandin F2{alpha} synthase activities of aldo-keto reductase 1B1, 1B3 and 1B7. J Biochem (2009) 145:161–8. doi:10.1093/jb/mvn152
35. de Reyniès A, Assié G, Rickman DS, Tissier F, Groussin L, René-Corail F, et al. Gene expression profiling reveals a new classification of adrenocortical tumors and identifies molecular predictors of malignancy and survival. J Clin Oncol (2009) 27:1108–15. doi:10.1200/JCO.2008.18.5678
36. Luo L, Chen Y, Wu D, Shou J, Wang S, Ye J, et al. Butylated hydroxyanisole induces distinct expression patterns of Nrf2 and detoxification enzymes in the liver and small intestine of C57BL/6 mice. Toxicol Appl Pharmacol (2015) 288:339–48. doi:10.1016/j.taap.2015.08.006
37. Srivastava S, Chandra A, Ansari NH, Srivastava SK, Bhatnagar A. Identification of cardiac oxidoreductase(s) involved in the metabolism of the lipid peroxidation-derived aldehyde-4-hydroxynonenal. Biochem J (1998) 329(Pt 3):469–75. doi:10.1042/bj3290469
38. Burczynski JM, Hayes JR, Longhurst PA, Colby HD. Species differences in adrenal lipid peroxidation: role of alpha-tocopherol. Free Radic Biol Med (1999) 26:987–91. doi:10.1016/S0891-5849(98)00289-5
39. Shen Y, Ma J, Yan R, Ling H, Li X, Yang W, et al. Impaired self-renewal and increased colitis and dysplastic lesions in colonic mucosa of AKR1B8-deficient mice. Clin Cancer Res (2015) 21:1466–76. doi:10.1158/1078-0432.CCR-14-2072
40. Martin H-J, Maser E. Role of human aldo-keto-reductase AKR1B10 in the protection against toxic aldehydes. Chem Biol Interact (2009) 178:145–50. doi:10.1016/j.cbi.2008.10.021
41. Endo S, Matsunaga T, Mamiya H, Ohta C, Soda M, Kitade Y, et al. Kinetic studies of AKR1B10, human aldose reductase-like protein: endogenous substrates and inhibition by steroids. Arch Biochem Biophys (2009) 487:1–9. doi:10.1016/j.abb.2009.05.009
42. Crosas B, Cederlund E, Torres D, Jornvall H, Farres J, Pares X. A vertebrate aldo-keto reductase active with retinoids and ethanol. J Biol Chem (2001) 276:19132–40. doi:10.1074/jbc.M010478200
43. Ruiz FX, Porté S, Parés X, Farrés J. Biological role of aldo-keto reductases in retinoic acid biosynthesis and signaling. Front Pharmacol (2012) 3:58. doi:10.3389/fphar.2012.00058
44. Crosas B, Hyndman DJ, Gallego O, Martras S, Parés X, Flynn TG, et al. Human aldose reductase and human small intestine aldose reductase are efficient retinal reductases: consequences for retinoid metabolism. Biochem J (2003) 373:973–9. doi:10.1042/BJ20021818
45. Gallego O, Ruiz FX, Ardèvol A, Domínguez M, Alvarez R, de Lera AR, et al. Structural basis for the high all-trans-retinaldehyde reductase activity of the tumor marker AKR1B10. Proc Natl Acad Sci U S A (2007) 104:20764–9. doi:10.1073/pnas.0705659105
46. Wang R, Wang G, Ricard MJ, Ferris B, Strulovici-Barel Y, Salit J, et al. Smoking-induced upregulation of AKR1B10 expression in the airway epithelium of healthy individuals. Chest (2010) 138:1402–10. doi:10.1378/chest.09-2634
47. Haselbeck RJ, Ang HL, Deltour L, Duester G. Retinoic acid and alcohol/retinol dehydrogenase in the mouse adrenal gland: a potential endocrine source of retinoic acid during development. Endocrinology (1997) 138:3035–41. doi:10.1210/endo.138.7.5274
48. Fukumoto S, Yamauchi N, Moriguchi H, Hippo Y, Watanabe A, Shibahara J, et al. Overexpression of the aldo-keto reductase family protein AKR1B10 is highly correlated with smokers’ non-small cell lung carcinomas. Clin Cancer Res (2005) 11:1776–85. doi:10.1158/1078-0432.CCR-04-1238
49. Rajkumar T, Vijayalakshmi N, Gopal G, Sabitha K, Shirley S, Raja UM, et al. Identification and validation of genes involved in gastric tumorigenesis. Cancer Cell Int (2010) 10:45. doi:10.1186/1475-2867-10-45
50. Yoshitake H, Takahashi M, Ishikawa H, Nojima M, Iwanari H, Watanabe A, et al. Aldo-keto reductase family 1, member B10 in uterine carcinomas: a potential risk factor of recurrence after surgical therapy in cervical cancer. Int J Gynecol Cancer (2007) 17:1300–6. doi:10.1111/j.1525-1438.2007.00932.x
51. Matsunaga T, Wada Y, Endo S, Soda M, El-Kabbani O, Hara A. Aldo-keto reductase 1B10 and its role in proliferation capacity of drug-resistant cancers. Front Pharmacol (2012) 3:5. doi:10.3389/fphar.2012.00005
52. Ma J, Yan R, Zu X, Cheng JM, Rao K, Liao DF, et al. Aldo-keto reductase family 1 B10 affects fatty acid synthesis by regulating the stability of acetyl-CoA carboxylase-alpha in breast cancer cells. J Biol Chem (2008) 283:3418–23. doi:10.1074/jbc.M707650200
53. Wang C, Yan R, Luo D, Watabe K, Liao DF, Cao D. Aldo-keto reductase family 1 member B10 promotes cell survival by regulating lipid synthesis and eliminating carbonyls. J Biol Chem (2009) 284:26742–8. doi:10.1074/jbc.M109.022897
54. Libè R, Fratticci A, Bertherat J. Adrenocortical cancer: pathophysiology and clinical management. Endocr Relat Cancer (2007) 14:13–28. doi:10.1677/erc.1.01130
55. Aigueperse C, Martinez A, Lefrancois-Martinez AM, Veyssiere G, Jean CI. Cyclic AMP regulates expression of the gene coding for a mouse vas deferens protein related to the aldo-keto reductase superfamily in human and murine adrenocortical cells. J Endocrinol (1999) 160:147–54. doi:10.1677/joe.0.1600147
56. Ragazzon B, Lefrancois-Martinez AM, Val P, Sahut-Barnola I, Tournaire C, Chambon C, et al. Adrenocorticotropin-dependent changes in SF-1/DAX-1 ratio influence steroidogenic genes expression in a novel model of glucocorticoid-producing adrenocortical cell lines derived from targeted tumorigenesis. Endocrinology (2006) 147:1805–18. doi:10.1210/en.2005-1279
57. Martinez A, Val P, Sahut-Barnola I, Aigueperse C, Veyssiere G, Lefrancois-Martinez AM. Steroidogenic factor-1 controls the aldose reductase akr1b7 gene promoter in transgenic mice through an atypical binding site. Endocrinology (2003) 144:2111–20. doi:10.1210/en.2002-220825
58. Val P, Aigueperse C, Ragazzon B, Veyssiere G, Lefrancois-Martinez AM, Martinez A. Adrenocorticotropin/3′,5′-cyclic AMP-mediated transcription of the scavenger akr1-b7 gene in adrenocortical cells is dependent on three functionally distinct steroidogenic factor-1-responsive elements. Endocrinology (2004) 145:508–18. doi:10.1210/en.2003-1093
59. Madore E, Harvey N, Parent J, Chapdelaine P, Arosh JA, Fortier MA. An aldose reductase with 20 alpha-hydroxysteroid dehydrogenase activity is most likely the enzyme responsible for the production of prostaglandin f2 alpha in the bovine endometrium. J Biol Chem (2003) 278:11205–12. doi:10.1074/jbc.M208318200
60. Ramsay RG, Ciznadija D, Vanevski M, Mantamadiotis T. Transcriptional regulation of cyclo-oxygenase expression: three pillars of control. Int J Immunopathol Pharmacol (2003) 16:59–67.
61. Volat FE, Pointud JC, Pastel E, Morio B, Sion B, Hamard G, et al. Depressed levels of prostaglandin F2α in mice lacking Akr1b7 increase basal adiposity and predispose to diet-induced obesity. Diabetes (2012) 61:2796–806. doi:10.2337/db11-1297
62. Rapoport R, Sklan D, Hanukoglu I. Electron leakage from the adrenal cortex mitochondrial P450scc and P450c11 systems: NADPH and steroid dependence. Arch Biochem Biophys (1995) 317:412–6. doi:10.1006/abbi.1995.1182
63. Prasad R, Kowalczyk JC, Meimaridou E, Storr HL, Metherell LA. Oxidative stress and adrenocortical insufficiency. J Endocrinol (2014) 221:R63–73. doi:10.1530/JOE-13-0346
64. Chinn AM, Ciais D, Bailly S, Chambaz E, LaMarre J, Feige JJ. Identification of two novel ACTH-responsive genes encoding manganese-dependent superoxide dismutase (SOD2) and the zinc finger protein TIS11b [tetradecanoyl phorbol acetate (TPA)-inducible sequence 11b]. Mol Endocrinol (2002) 16:1417–27. doi:10.1210/mend.16.6.0844
65. Meimaridou E, Kowalczyk J, Guasti L, Hughes CR, Wagner F, Frommolt P, et al. Mutations in NNT encoding nicotinamide nucleotide transhydrogenase cause familial glucocorticoid deficiency. Nat Genet (2012) 44:740–2. doi:10.1038/ng.2299
66. Toye AA, Lippiat JD, Proks P, Shimomura K, Bentley L, Hugill A, et al. A genetic and physiological study of impaired glucose homeostasis control in C57BL/6J mice. Diabetologia (2005) 48:675–86. doi:10.1007/s00125-005-1680-z
67. Aigueperse C, Val P, Pacot C, Darne C, Lalli E, Sassone-Corsi P, et al. SF-1 (steroidogenic factor-1), C/EBPbeta (CCAAT/enhancer binding protein), and ubiquitous transcription factors NF1 (nuclear factor 1) and Sp1 (selective promoter factor 1) are required for regulation of the mouse aldose reductase-like gene (AKR1B7) expression in adrenocortical cells. Mol Endocrinol (2001) 15:93–111. doi:10.1210/mend.15.1.0577
Keywords: aldose reductase, adrenal physiology, AKR1B
Citation: Pastel E, Pointud J-C, Martinez A and Lefrançois-Martinez AM (2016) Aldo-Keto Reductases 1B in Adrenal Cortex Physiology. Front. Endocrinol. 7:97. doi: 10.3389/fendo.2016.00097
Received: 03 May 2016; Accepted: 06 July 2016;
Published: 22 July 2016
Edited by:
James A. Carr, Texas Tech University, USAReviewed by:
Tullio Florio, University of Genoa, ItalyTamas Kozicz, Radboud University Nijmegen, Netherlands
Copyright: © 2016 Pastel, Pointud, Martinez and Lefrançois-Martinez. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: A. Marie Lefrançois-Martinez, YS1tYXJpZS5sZWZyYW5jb2lzLW1hcnRpbmV6QHVuaXYtYnBjbGVybW9udC5mcg==