 Hervé Lefebvre
Hervé Lefebvre Michaël Thomas1,2
Michaël Thomas1,2 Céline Duparc
Céline Duparc Estelle Louiset
Estelle Louiset- 1U982, Laboratory of Neuronal and Neuroendocrine Differentiation and Communication, INSERM, Institute for Research and Innovation in Biomedicine, Mont-Saint-Aignan, France
- 2Normandie Université, UNIROUEN, Rouen, France
- 3Department of Endocrinology, Diabetes and Metabolic Diseases, University Hospital of Rouen, Rouen, France
- 4U1016, INSERM, Institut Cochin, Paris, France
- 5Department of Endocrinology and Metabolic Diseases, Hôpital Cochin, Assistance Publique-Hôpitaux de Paris, Paris, France
In the normal human adrenal gland, steroid secretion is regulated by a complex network of autocrine/paracrine interactions involving bioactive signals released by endothelial cells, nerve terminals, chromaffin cells, immunocompetent cells, and adrenocortical cells themselves. ACTH can be locally produced by medullary chromaffin cells and is, therefore, a major mediator of the corticomedullary functional interplay. Plasma ACTH also triggers the release of angiogenic and vasoactive agents from adrenocortical cells and adrenal mast cells and, thus, indirectly regulates steroid production through modulation of the adrenal blood flow. Adrenocortical neoplasms associated with steroid hypersecretion exhibit molecular and cellular defects that tend to reinforce the influence of paracrine regulatory loops on corticosteroidogenesis. Especially, ACTH has been found to be abnormally synthesized in bilateral macronodular adrenal hyperplasia responsible for hypercortisolism. In these tissues, ACTH is detected in a subpopulation of adrenocortical cells that express gonadal markers. This observation suggests that ectopic production of ACTH may result from impaired embryogenesis leading to abnormal maturation of the adrenogonadal primordium. Globally, the current literature indicates that ACTH is a major player in the autocrine/paracrine processes occurring in the adrenal gland in both physiological and pathological conditions.
Introduction
The adrenal cortex is a heterogeneous tissue that not only contains steroidogenic cells but also hosts various cell types that are able to locally release a wide variety of bioactive signals. This histological organization results in a complex interactive network that participates in the regulation of both basal and ACTH-induced corticosteroidogenesis. The intracortical sources of regulatory factors include chromaffin cells arranged in cords or islets, nerve fibers originating from extraadrenal neurones or cell bodies located in the adrenal medulla, cells of the immune system, including lymphocytes, macrophages/monocytes and mast cells, endothelial cells, and adipocytes. These autocrine/paracrine mechanisms have been extensively reviewed during the past years (1–5). However, their physiological role, especially their exact contribution to the regulation of corticosteroid synthesis remains a matter of debate although some data indicate that intraadrenal regulatory signals may mediate parts of the biological effects of ACTH on the adrenal cortex. Interestingly, adrenocortical neoplasms associated with steroid hypersecretion exhibit molecular and cellular defects that tend to reinforce the potency of paracrine factors to activate corticosteroidogenesis. For instance, abnormal expression of ACTH has been reported in adrenocortical cells in both tumors and hyperplasias responsible for hypercortisolism (6–11).
In the present review article, we have updated the current knowledge on the role of ACTH in the cell-to-cell communication processes occurring in the adrenal cortex in both physiological and pathological conditions. We will also discuss the potential interest of the intraadrenal ACTH regulatory loop for the clinical management of patients with primary adrenal excess of corticosteroids.
Effect of ACTH on the Adrenal Vasculature
The adrenal cortex is a richly vascularized organ. This extensive vasculature is essential for delivery of tropic hormone and steroid hormones precursors to the gland and secretion of mature hormones into the blood flow. Furthermore, the establishment of such dense vascular network ensures that every adrenocortical cell is in contact with at least one endothelial cell (12, 13). This remarkable histological organization allows paracrine regulation of adrenocortical cells by endothelial cells through release of endothelins, adrenomedullin, nitric oxide, and prostacyclin (14). For instance, Rossi et al. have shown that endothelin-1 released by endothelial cells is an important regulator of aldosterone secretion, and may then indirectly influence arterial blood pressure (15, 16). The release of endothelin by adrenocortical sinusoids is thought to mediate the modulation of adrenal steroidogenesis by the adrenal blood (17). Interestingly, ACTH appears able to both act on development and maintenance of the adrenal vasculature and regulate the adrenal blood flow (18, 19), influencing thus steroid production through an indirect effect in addition to its intrinsic steroidogenic action on adrenocortical cells (Figure 1).
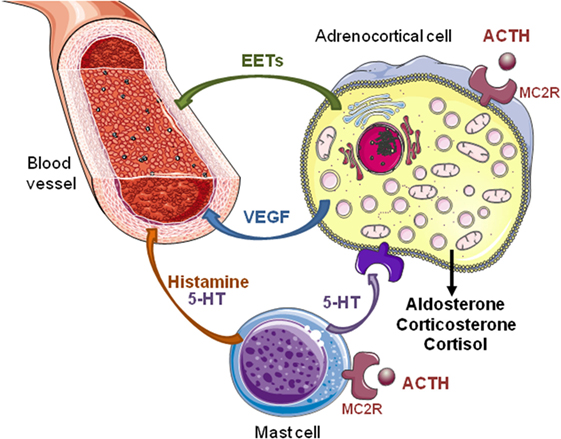
Figure 1. Direct and indirect effects of ACTH on steroidogenesis in the adrenal gland. ACTH exerts a direct stimulatory effect on adrenocortical cells via melanocortin type 2 receptors (MC2R). Activation of MC2R increases secretion of steroid hormones, vascular endothelial growth factor (VEGF), and epoxyeicosatrienoic acids (EETs). Angiogenesis induced by VEGF, an endothelial cell-specific mitogen factor, allows adrenal tissue growth in response to ACTH. Increase in adrenal blood flow elicited by EETs, which are potent vasorelaxant agents, favors steroid production. In rats, ACTH also activates adrenal mast cells inducing local release of histamine and serotonin (5-HT). These factors modulate the tonicity of adrenal arterioles and, thus, the adrenal blood flow that indirectly influences the steroidogenesis. In addition, 5-HT stimulates steroid secretion through activation of different 5-HT receptor subtypes expressed by normal and tumor human adrenocortical cells.
The modulation of adrenal angiogenesis by ACTH may involve several bioactive signals. Thrombospondins (TSPs) represent a wide family of extracellular proteins consisting of five members, TSP1–5, which can bind multiple cell surface molecules, including heparin sulfate proteoglycans, low-density lipoprotein receptor-related protein, integrins, CD36, and CD47 (20). Owing to the great diversity of their binding partners, TSPs are involved in various biological processes, such as cell adhesion, spreading and migration, and angiogenesis (20, 21). In this respect, TSPs are known to inhibit angiogenesis by preventing migration of capillary endothelial cells. Interestingly, adrenocortical cells release high amounts of TSP2 in response to ACTH (22), suggesting that TSP2 may mediate some of the biological actions of ACTH in the adrenal cortex, especially centripetal adrenocortical cell migration, which is a fundamental process in the dynamic organization and remodeling of the adrenal cortex (23). However, the physiological role of TSP2, which is primarily expressed in zona glomerulosa and zona fasciculata, remains unclear since TSP2-null mice exhibit no alteration in corticosteroid secretion or adrenal development (24). In addition, syndromes of ACTH excess, including Cushing’s disease and 21-hydroxylase deficiency, are associated with adrenocortical hyperplasia, a process that underlies active angiogenesis. Conversely, several observations indicate that vascular endothelial growth factor (VEGF) plays a pivotal role in the trophic effects of ACTH on the adrenal vasculature (25). VEGF is a widely expressed cytokine that acts as an endothelial cell-specific mitogen and angiogenic factor. In the bovine adrenal gland, VEGF is expressed in zona glomerulosa and zona fasciculata cells and its release is stimulated by ACTH (26). Upregulation of VEGF by ACTH has also been reported in human adrenal (27). The effect of ACTH on adrenal VEGF production involves transcription-independent mechanisms, including stabilization of VEGF mRNA by the HuR protein (28). Conversely, ACTH suppression by dexamethasone in mice results in progressive decrease of VEGF expression in adrenocortical cells and regression of the vascular network (29). Interestingly, ACTH also stimulates VEGF expression in human fetal adrenocortical cells, suggesting that VEGF is an important mediator of the trophic action of ACTH during the adrenal development (30). Consistently, an increase in VEGF expression in adrenocortical cells has been noticed in the regenerating adrenal cortex in rats (31, 32). It is noteworthy that, in parallel to its effect on adrenal vasculature development via local synthesis of VEGF, ACTH also favors adrenal tissue growth through an antiapoptotic action on adrenocortical cells (29) and stimulation of synthesis of growth factors (30). Finally, VEGF may be involved in the capacity of ACTH to induce endothelial fenestration, a phenomenon that favors cell-to-cell interactions (26, 33).
The mechanism by which ACTH modulates the adrenal blood flow is obviously not univocal. Adrenocortical cells themselves are able to release vasorelaxant agents in response to ACTH. These compounds include metabolites of arachidonic acid, such as epoxyeicosatrienoic acids (EETs) (34). In addition, in the rat adrenal gland, capsular mast cells modulate the tonicity of adrenal arterioles and, thus, the adrenal blood flow through local release of histamine and serotonin (5-hydroxytryptamine; 5-HT) (17, 35). Interestingly, rat adrenal mast cells are sensitive to the action of ACTH, suggesting that they may represent an important intermediate in the effect of corticotropin on the adrenal blood flow (35). In humans, the presence of mast cells has been reported in both the subcapsular region of the normal adrenal gland (36) and various types of adrenocortical tumors, including deoxycorticosterone-secreting tumors, aldosterone-producing adenomas (APAs), and adrenocortical carcinomas (37–39). However, there is currently no data reported in the literature indicating that human adrenal mast cells are target cells for ACTH.
Indirect Effects of ACTH on Corticosteroidogenesis through Adrenocortical Cell Secretory Products
Adrenocortical cells are important sources of bioactive compounds that are able to modulate steroidogenesis through autocrine/paracrine actions. In man, it is doubtful that corticosteroid may affect their own secretion in physiological conditions. Conversely, the adrenal cortex is known to express the diverse components of the renin–angiotensin system, leading to local synthesis of angiotensin II and potential autocrine/paracrine stimulation of aldosterone secretion (40). Interestingly, renin production, which primarily occurs in zona glomerulosa cells, is stimulated by ACTH and reduced by hypophysectomy or dexamethasone administration (41, 42). This observation may explain why plasma renin levels are usually not suppressed in patients with overt ACTH-dependent hypercortisolism (43, 44). In fact, it is conceivable that, in this condition, ACTH-induced adrenal renin secretion may compensate inhibition of renal renin synthesis by hypervolemia and hypertension secondary to cortisol excess.
Adrenocortical cells also synthesize and release various cytokines, such as interleukin-1 (IL-1), IL-3, IL-6, and tumor necrosis factor-α (TNF- α) (1). These signals may influence the steroidogenic and mitogenic activities of adrenocortical cells via autocrine/paracrine processes (5). Indeed, ACTH has been shown to modulate cytokine production by adrenocortical cells, either positively (IL-6) or negatively (TNF-α) (45, 46). Intraadrenal cytokines may, thus, represent important effectors of ACTH capable of potentiating or attenuating its action on adrenocortical cells. However, there is currently no evidence that the impact of ACTH on adrenal cytokine synthesis may be involved in the pathogenesis of ACTH-dependent adrenal hyperplasia and/or hypercortisolism.
Several types of adrenocortical neoplasms are associated with illicit neuroendocrine differentiation of adrenocortical cells (47). This is especially true for APAs that have been shown to express synaptophysin, neuronal cell adhesion molecule, neuron specific enolase, and SV2 (48, 49). In addition, we have observed that APA cells may also abnormally synthesize 5-HT that is able to stimulate aldosterone secretion through activation of the overexpressed serotonergic type 4 (5-HT4) receptor (50, 51). Because APA cells also express high amounts of the ACTH receptor, i.e., the melanocortin receptor type 2 (MC2R) (51–53), it is conceivable that locally produced 5-HT may act as an amplifier of the stimulatory effect of ACTH on aldosterone secretion by APA tissues.
Inhibins and activin are dimeric peptides belonging to the TGF-β family. Inhibins are formed by combination of the α-subunit encoded by INHA and A or B isoform of the β-subunit, encoded by INHBA and INHBB, respectively. Alternatively, activin is a homodimer composed of two β-subunits. The action of activin is mediated by its specific receptors type I and II, and the intracellular proteins SMAD. Inhibins counteract the biological effects of activin by antagonizing activin type II receptor and formation of an inactive complex with the TGFβ type III receptor β-glycan. Adrenocortical cells are able to express both α and β subunits (54–56). In particular, the α-inhibin is expressed in the zona reticularis under the positive control of ACTH, whereas β-subunits are mainly present in the outer cortex. Both activin receptors and the inhibin co-receptor β-glycan are also detected in the adrenal cortex (55, 56). It has been demonstrated that ACTH stimulates secretion of inhibin A and B, without modifying production of activin A (55). These data indicate that ACTH also controls corticosteroidogenesis through modulation of the intraadrenal activins/inhibins ratio.
Indirect Effects of ACTH on Corticosteroidogenesis through Non-Steroidogenic Adrenal Cells
The adrenal gland is surrounded by adipose tissue and its cortical region contains adipocytes either isolated or arranged in small islets (57). Like cells of the immune system, adipocytes release a wide panel of cytokines, suggesting that they could influence the adrenocortical function through a cell-to-cell communication process. For instance, adipocytes have been shown to activate aldosterone release by secreting soluble bioactive factors, which have not been yet characterized (57–59). Conversely, leptin exerts an inhibitory action on ACTH-induced corticosteroid secretion in human adrenocortical cells without affecting their viability and proliferation (60–63). It is possible that peri- and intraadrenal adipocytes may be controlled by ACTH and, thus, constitute a relay in the action of the hormone on the adrenal cortex. In support of this hypothesis, it has been reported that murine adipocyte cell lines and immortalized adipocytes express the MC2R, and ACTH regulate adipocyte functions in these models (64–66). In addition, patients with congenital adrenal hyperplasia due to 21-hydroxylase deficiency, a condition which is associated with chronically high plasma ACTH levels, can present with adrenal myelolipoma (67–69), suggesting a role of ACTH in the development of lipomatous tissue inclusions in the adrenal glands. However, in contrast to the observations made in murine cells, human mature adipocytes only express low levels of the MC2R and ACTH does not influence lipolysis in the mature human adipose tissue (70).
Paracrine Control of Adrenal Steroidogenesis by Intraadrenal ACTH
In the normal adrenal gland, chromaffin cells release detectable amounts of ACTH that is a major mediator in the corticomedullary functional interaction (71, 72) (Figure 2). This secretory process can be activated by corticotropin-releasing hormone (CRH) that is expressed in the adrenal medullary tissue (11). As chromaffin cells are also regulated by splanchnic nerves and proinflammatory cytokines, it seems possible that they may be important intermediates in the activation of the adrenal cortex during stress and inflammation. However, the physiological role of the paracrine control of corticosteroidogenesis by adrenomedullary ACTH remains unclear (3).
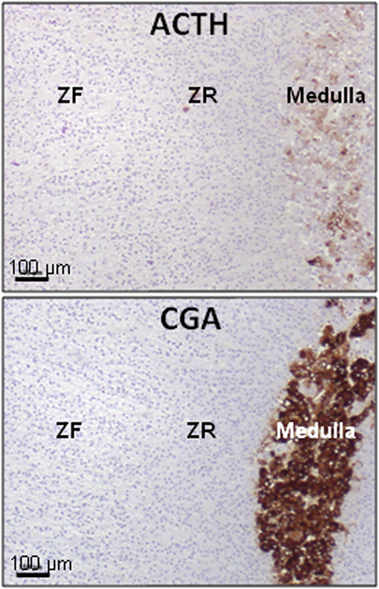
Figure 2. Presence of ACTH in the normal human adrenal. ACTH immunoreactivity is exclusively located in some chromaffin cells (high panel), identified as chromogranin A (CGA) immunoreactive cells (low panel), in the medulla. ACTH was detected by using antibodies against the N-terminal region of the peptide. ZF, zona fasciculata; ZR, zona reticularis. Illustration of the data published in Ref. (9).
Conversely, clinicopathological studies have shown that paracrine interactions involving ACTH produced by chromaffin cells may play a role in the pathogenesis of hypercortisolism. Pheochromocytoma can occasionally produce ACTH resulting in ACTH-dependent Cushing’s syndrome (73–75). Hypercortisolism seems to be mainly the consequence of the endocrine corticotropic action of ACTH whose plasma levels are typically elevated in this situation (76). Nevertheless, this mechanism may not be exclusive. In fact, hyperplasia of the adrenal cortex adjacent to pheochromocytoma has also been observed, indicating that ACTH originating from the pheochromocytoma tissue can stimulate adrenocortical cells in a paracrine manner (11, 77–80). Such a histological pattern is close to what is observed in corticomedullary mixed tumors that are composed of intermingled adrenocortical and pheochromocytoma tissues (81–84). These rare tumors are sometimes associated with hypercortisolism, suggesting that pheochromocytes release paracrine signals capable of activating glucocorticoid synthesis (81, 84). ACTH could be one of them although it is not excluded that catecholamines may exert a stimulatory action on cortisol production through illicit expression of adrenergic receptors in tumor adrenocortical cells (85, 86).
Alternatively, the occurrence of ectopic production of ACTH in the adrenal cortex has already been reported. Very rarely, ACTH-positive cells in the adrenocortical tissue can reveal adrenal micrometastases of an ACTH-secreting cancer. In the published cases, plasma ACTH levels were strongly elevated, as a result of ACTH secretion by the primary tumor (87–89). More surprisingly, a subpopulation of adrenocortical cells has been shown to produce detectable amounts of ACTH in various types of adrenal neoplasms. A first case of adrenocortical cortisol-secreting adenoma associated with production of ACTH by tumor cells, has been described in 2001 by Hiroi et al. (6). Illicit synthesis of ACTH was considered to result from abnormal pituitary differentiation of the tissue as witnessed by co-expression of 17-hydroxylase and pituitary homeobox factor-1 mRNAs by adrenocortical cells (6). At the ultrastructural level, tumor cells exhibited characteristics of both steroidogenic cells and neuroendocrine cells, and the tumor was, thus, referred to as an adrenocortical–pituitary hybrid adenoma. Bilateral macronodular adrenal hyperplasia (BMAH), a rare cause of primary adrenal hypercortisolism, has also been found to contain ACTH-producing cells. This observation was first reported by Pereira et al. who also noticed that ACTH-positive cells were labeled by antibodies to chromogranin A (CGA), suggesting that these cells may correspond to intracortical chromaffin cells (90). Subsequently, several teams reported expression of proopiomelanocortin (POMC) and ACTH in groups of adrenocortical cells in isolated BMAH cases (8, 10, 91) (Figure 3). In one case, it could be shown that ACTH-positive cells also expressed 17-hydroxylase but were negative for pituitary corticotroph markers (8). The role of ACTH in the pathogenesis of BMAH has been more extensively investigated in a large series of 30 cases (9). Adrenal hyperplasia samples expressed POMC mRNA at variable levels. Proconvertase 1, a protease implicated in the maturation of POMC into ACTH, was also visualized in clusters of adrenal cells, indicating that ACTH could be generated from POMC in the BMAH tissues. In agreement with this finding, ACTH immunoreactivity was detected, as previously noticed, in some adrenocortical cells either isolated or arranged in small groups disseminated in the tissues. The presence of corticotropin can also be seen in some chromaffin cells of the adrenal medulla. Surprisingly, adrenocortical ACTH-positive cells also displayed characteristics of steroidogenic cells, such as the presence of numerous lipid inclusions and several markers of steroidogenic differentiation, including steroidogenic factor 1 (SF1), 17-hydroxylase, and the HDL-cholesterol receptor scavenger receptor B1 (SRB1). It could be, thus, concluded that they constitute a subcategory of adrenocortical steroidogenic cells characterized by an unusual capacity to synthesize ACTH. BMAH specimens were found to express very low levels of T-pit [a transduction factor which controls pituitary corticotrophs differentiation (92)], confirming that ectopic synthesis of ACTH in adrenocortical cells does not result from illicit corticotropic-like differentiation of the latter (9) but may rather be considered as an additional feature of neuroendocrine differentiation of the hyperplastic tissues (47, 93, 94). Interestingly, ACTH-containing cells were also positive for gonadal markers like the gonadal marker insulin-like 3 (INSL3), exhibiting thus a pseudo-gonadal phenotype (8, 9, 95, 96). This observation is concordant with expression of POMC and synthesis of ACTH previously reported in testicular Leydig cells and ovarian granulosa cells (97, 98). Because the adrenal glands and gonads both originate from a same tissue precursor, the adrenogonadal primordium, it is likely that the occurrence of these pseudo-gonadal cells in BMAH tissues may be the consequence of altered embryogenesis, explaining the bilaterality of the lesions.
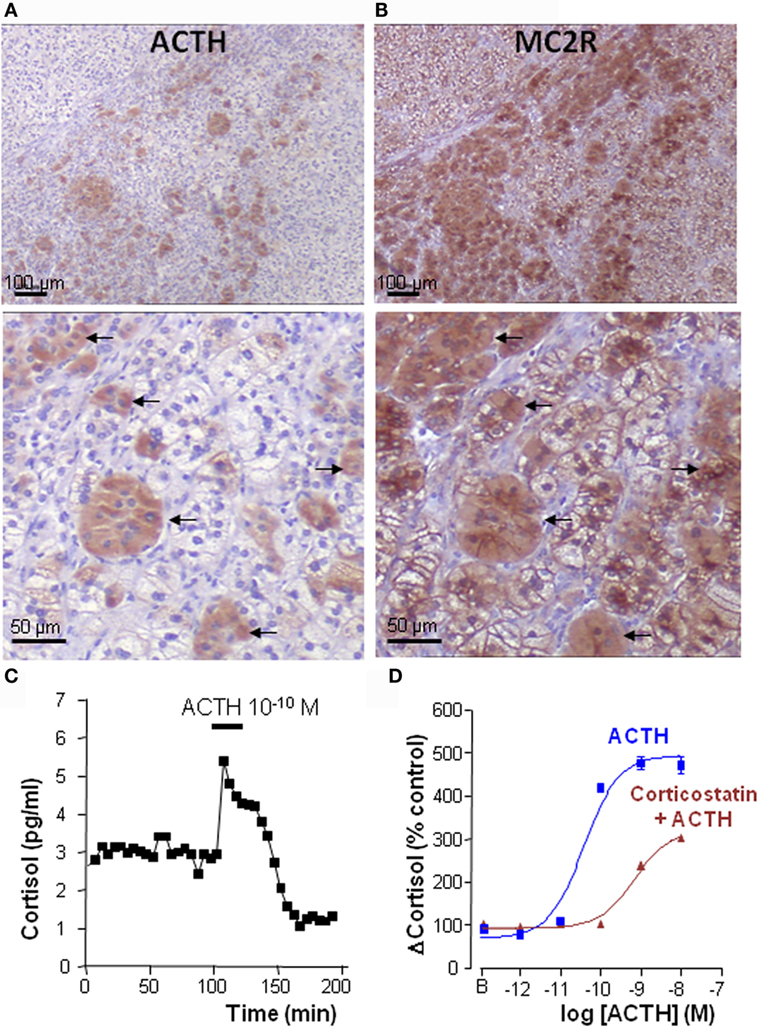
Figure 3. Intraadrenal ACTH regulation of cortisol secretion in bilateral macronodular adrenal hyperplasia (BMAH). (A,B) Consecutive sections of a BMAH tissue labeled with ACTH and MC2R antibodies. (A) Heterogeneous distribution of ACTH-producing cell clusters in BMAH. (B) The ACTH receptor MC2R is highly expressed in ACTH-producing cells (arrows) and in their vicinity. (C) ACTH sensitivity of BMAH tissues. Application of exogenous ACTH stimulated cortisol secretion by perifused BMAH explants. (D) Corticostatin, a MC2R antagonist, inhibited in vitro the cortisol response of cultured BMAH cells to ACTH. Illustration of the data published in Ref. (9).
Perifusion studies revealed that ACTH is released by BMAH tissues in vitro in a pulsatile mode (9), in agreement with older clinical studies showing a clear pulsatility of cortisol secretion in patients with BMAH (99). The ectopic secretion of ACTH by the hyperplastic adrenal glands has also been detected in vivo in two patients through adrenal vein catheterization (9). Taken collectively, these data suggested that, in BMAH tissues, intraadrenal ACTH may exert an autocrine/paracrine action to stimulate cortisol secretion, supplying therefore circulating ACTH that is suppressed by cortisol excess. This hypothesis was supported by statistical analyses showing positive correlations between ACTH and cortisol levels in BMAH culture medium. In addition, basal plasma cortisol concentrations measured in vivo were positively correlated with both the levels of POMC mRNA and the ACTH histological score in the tissues. Most importantly, MC2R antagonists, such as corticostatin and ACTH (7–38), were found to inhibit in vitro spontaneous and ACTH-evoked cortisol secretion by BMAH explants (9) (Figure 3). Clinical studies with MC2R antagonists are now mandatory to confirm that cortisol production is actually dependent on intraadrenal ACTH in patients with BMAH.
Bilateral macronodular adrenal hyperplasia tissues also constitute an interesting model for the study of the regulation of MC2R expression in the adrenal cortex. MC2R mRNA is globally underexpressed in BMAH samples versus normal adrenals (100). In fact, we could observe that, at variance with the normal adrenal cortex that is diffusely labeled by anti-MC2R antibodies, BMAH explants exhibit heterogeneous distribution of the receptor that appears highly expressed in the vicinity of clusters of ACTH-producing cells and more weakly at distance (Figure 3). Indeed, as previously established in the normal adrenal gland (101–103), MC2R seems to be upregulated by ACTH in BMAH tissues. In fact, MC2R mRNA levels were positively correlated with POMC mRNA rates and MC2R-like immunoreactivity was principally visualized in the vicinity of ACTH-positive cells (9). Interestingly, ACTH-producing cells were also found to express the receptor, suggesting that intraadrenal ACTH possibly exerts autocrine actions in BMAH.
All these data indicate that intraadrenal ACTH plays a pivotal role in the pathogenesis of hypercortisolism associated with BMAH. Deciphering the mode of regulation of ACTH production by BMAHs is, thus, essential for the comprehension of the pathophysiology of the disease. At variance with pituitary ACTH, intraadrenal ACTH does not seem to be regulated by cortisol, as suggested by the lack of action of dexamethasone and the glucocorticoid receptor antagonist RU486 on ACTH release by BMAH explants (9). Conversely, we noticed that ligands of various membrane receptors that are known to be abnormally expressed by BMAH cells, i.e., 5-HT, LH/hCG, and glucose-dependent insulinotropic peptide (GIP), are able to activate ACTH production from BMAH tissues in vitro (9). This surprising finding indicated that activation of illicit membrane receptors may stimulate cortisol production via two mechanisms, including a direct effect on corticosteroidogenesis, as previously shown in BMAH cell culture (93, 104), and an indirect action mediated by ACTH secretion (9). Consistently, MC2R antagonists were found to partially inhibit in vitro the cortisol response evoked by GIP in perifused BMAH samples. Taken together, these results suggest that intraadrenal ACTH may be regarded as a common intermediate and amplifier of the action of several illicit membrane receptors in BMAH tissues. Targeting the MC2R with specific antagonists may, thus, represent an efficient strategy for the treatment of BMAH-associated hypercortisolism. The pathophysiology links between intraadrenal ACTH and abnormally expressed receptors may be more complex and could form a complete auto-amplification loop in the hyperplastic tissues. In fact, although the presence of the LH and GIP receptors in adrenocortical cells may be simply considered as features of pseudo-gonadal differentiation of BMAH tissues, overexpression of some membrane receptors, such as 5-HT receptors, may result from local production from ACTH. This hypothesis is supported by intriguing observations recently performed in another type of adrenal hyperplasia associated with hypercortisolism, namely primary pigmented adrenocortical disease (PPNAD). The disease is caused in most patients by germline inactivating mutations that affect the PRKAR1A gene (105), resulting in constitutive activation of protein kinase A (PKA) in adrenocortical cells (106). Since MC2R are positively coupled to the cAMP/PKA pathway, it can be considered that PRKAR1A mutations partly mimic the action of ACTH on adrenal steroidogenic cells. Interestingly, PPNAD have been found to overexpress several types of 5-HT receptors, including the 5-HT4 receptor (107). It seems, thus, conceivable that abnormal expression of 5-HT receptors in BMAH tissues may result from exposure of adrenocortical cells to locally produced ACTH whose release is subsequently increased by the 5-HT signaling pathway.
There is now no doubt that BMAH is a genetically determined condition, ARMC5 being a major susceptibility gene of the disease (108). It is now well established that the development of macronodular adrenal hyperplasia requires inactivation of the two ARMC5 alleles, respectively, by the first germline mutation and a secondary somatic genetic event. Surprisingly, inactivation of ARMC5 expression in the human adrenocortical cell line H295R, which reproduces the molecular defects observed in adrenocortical cells of patients with BMAH, results in a decrease in expression of steroidogenic enzymes (108, 109). It seems, thus, that a second line event is necessary for the emergence of hypercortisolism. We hypothesize that ARMC5 mutations may alter differentiation and/or separation of the adrenogonadal primordium leading to the presence of pseudo-gonadal cells in the adrenal areas. Progressive expression of POMC and ACTH by these cells may then result in cortisol hypersecretion. Because illicit expression of ACTH has also been observed in non-ARMC5-mutated adrenal hyperplasias, it seems that abnormal differentiation of adrenocortical cells is a frequent histological feature in BMAH tissues whatever the causative firstline genetic defect (Figure 4).
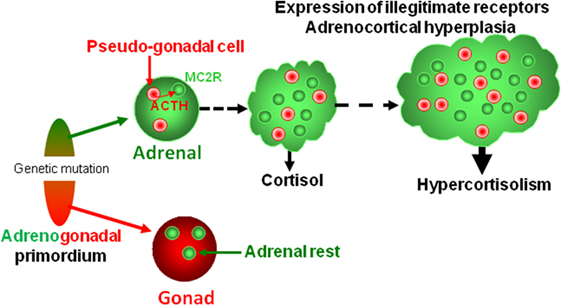
Figure 4. Putative pathophysiological mechanism responsible for bilateral macronodular adrenal hyperplasia. Both gonads and adrenals originate from the adrenogonadal primordium. It can be speculated that the causative genetic mutations may alter differentiation and/or separation of the adrenogonadal primordium leading to the presence of pseudo-gonadal cells in the adrenals. Secretion of ACTH by intraadrenal pseudo-gonadal cells may progressively stimulate, via activation of MC2R, both cortisol secretion and growth of the adrenocortical tissue, leading to bilateral adrenal hyperplasia associated with hypercortisolism. In parallel, sustained activation of the PKA pathway consecutive to activation of MC2R by intraadrenal ACTH may activate expression of some illegitimate receptors. Activation of steroidogenesis by ligands of illegitimate receptors further reinforces cortisol hypersecretion through both an intrinsic stimulatory action and an indirect effect via local release of ACTH.
Conclusion
In addition to its well-known action on adrenocortical cells to promote steroidogenesis through activation of the MC2R, ACTH exerts multiple effects through the paracrine communication processes that occur in the adrenal gland in both physiological and pathophysiological conditions. ACTH can also be abnormally produced in adrenal neoplasms in which the hormone acts, thus, as an autocrine/paracrine factor to activate steroid secretion. This new pathophysiological concept opens novel avenues for the development of original pharmacological treatments of primary adrenal syndromes of steroid excess.
Author Contributions
HL, MT, and EL: wrote the paper. CD, EL: acquisition, analysis, and interpretation of data. EL: designed the figures. JB: revised the paper and gave intellectual contribution.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
This work was supported by Institut National de la Santé et de la Recherche Médicale Unité 982, the Centre Hospitalier Universitaire de Rouen, the Assistance Publique-Hôpitaux de Paris and the Conseil Régional de Normandie, the Société Française d’Endocrinologie (SFE).
References
1. Bornstein SR, Rutkowski H, Vrezas I. Cytokines and steroidogenesis. Mol Cell Endocrinol (2004) 215:135–41. doi:10.1016/j.mce.2003.11.022
2. Ehrhart-Bornstein M, Hinson JP, Bornstein SR, Scherbaum WA, Vinson GP. Intraadrenal interactions in the regulation of adrenocortical steroidogenesis. Endocr Rev (1998) 19:101–43. doi:10.1210/edrv.19.2.0326
3. Haase M, Willenberg HS, Bornstein SR. Update on the corticomedullary interaction in the adrenal gland. Endocr Dev (2011) 20:28–37. doi:10.1159/000321211
4. Nussdorfer GG. Paracrine control of adrenal cortical function by medullary chromaffin cells. Pharmacol Rev (1996) 48:495–530.
5. Nussdorfer GG, Mazzocchi G. Immune-endocrine interactions in the mammalian adrenal gland: facts and hypotheses. Int Rev Cytol (1998) 183:143–84. doi:10.1016/S0074-7696(08)60144-8
6. Hiroi N, Chrousos GP, Kohn B, Lafferty A, Abu-Asab M, Bonat S, et al. Adrenocortical-pituitary hybrid tumor causing Cushing’s syndrome. J Clin Endocrinol Metab (2001) 86:2631–7. doi:10.1210/jcem.86.6.7590
7. Iwata M, Oki Y, Okazawa T, Ishizawa S, Taka C, Yamazaki K, et al. A rare case of adrenocorticotropic hormone (ACTH)-independent macroadrenal hyperplasia showing ectopic production of ACTH. Intern Med (2012) 51:2181–7. doi:10.2169/internalmedicine.51.7547
8. Lefebvre H, Duparc C, Chartrel N, Jegou S, Pellerin A, Laquerriere A, et al. Intraadrenal adrenocorticotropin production in a case of bilateral macronodular adrenal hyperplasia causing Cushing’s syndrome. J Clin Endocrinol Metab (2003) 88:3035–42. doi:10.1210/jc.2002-030014
9. Louiset E, Duparc C, Young J, Renouf S, Tetsi Nomigni M, Boutelet I, et al. Intraadrenal corticotropin in bilateral macronodular adrenal hyperplasia. N Engl J Med (2013) 369:2115–25. doi:10.1056/NEJMoa1215245
10. Mazzuco TL, Thomas M, Martinie M, Cherradi N, Sturm N, Feige J-J, et al. Cellular and molecular abnormalities of a macronodular adrenal hyperplasia causing beta-blocker-sensitive Cushing’s syndrome. Arq Bras Endocrinol Metabol (2007) 51:1452–62. doi:10.1590/S0004-27302007000900007
11. Vrezas I, Willenberg HS, Mansmann G, Hiroi N, Fritzen R, Bornstein SR. Ectopic adrenocorticotropin (ACTH) and corticotropin-releasing hormone (CRH) production in the adrenal gland: basic and clinical aspects. Microsc Res Tech (2003) 61:308–14. doi:10.1002/jemt.10340
12. Kikuta A, Murakami T. Microcirculation of the rat adrenal gland: a scanning electron microscope study of vascular casts. Am J Anat (1982) 164:19–28. doi:10.1002/aja.1001640103
13. Sasano H, Ohashi Y, Suzuki T, Nagura H. Vascularity in human adrenal cortex. Mod Pathol (1998) 11:329–33.
14. Hinson JP, Kapas S. The role of endothelial cell products in the regulation of adrenocortical function: actions of endothelin, nitric oxide, adrenomedullin and PAMP. Horm Metab Res (1998) 30:334–40. doi:10.1055/s-2007-978894
15. Rossi G, Albertin G, Belloni A, Zanin L, Biasolo MA, Prayer-Galetti T, et al. Gene expression, localization, and characterization of endothelin A and B receptors in the human adrenal cortex. J Clin Invest (1994) 94:1226–34. doi:10.1172/JCI117440
16. Rossi GP, Andreis PG, Colonna S, Albertin G, Aragona F, Belloni AS, et al. Endothelin-1[1-31]: a novel autocrine-paracrine regulator of human adrenal cortex secretion and growth. J Clin Endocrinol Metab (2002) 87:322–8. doi:10.1210/jcem.87.1.8134
17. Hinson JP, Vinson GP, Kapas S, Teja R. The relationship between adrenal vascular events and steroid secretion: the role of mast cells and endothelin. J Steroid Biochem Mol Biol (1991) 40:381–9. doi:10.1016/0960-0760(91)90205-J
18. Ansurudeen I, Kopprasch S, Ehrhart-Bornstein M, Bornstein SR, Willenberg HS. Endothelial cell-mediated regulation of aldosterone release from human adrenocortical cells. Mol Cell Endocrinol (2007) 26(5–266):150–6. doi:10.1016/j.mce.2006.12.011
19. Ansurudeen I, Kopf PG, Gauthier KM, Bornstein SR, Cowley AW, Campbell WB. Aldosterone secretagogues increase adrenal blood flow in male rats. Endocrinology (2014) 155:127–32. doi:10.1210/en.2013-1532
20. Adams JC, Lawler J. The thrombospondins. Cold Spring Harb Perspect Biol (2011) 3:a009712. doi:10.1101/cshperspect.a009712
21. Stenina-Adognravi O. Thrombospondins: old players, new games. Curr Opin Lipidol (2013) 24:401–9. doi:10.1097/MOL.0b013e3283642912
22. Danik M, Chinn AM, Lafeuillade B, Keramidas M, Aguesse-Germon S, Penhoat A, et al. Bovine thrombospondin-2: complete complementary deoxyribonucleic acid sequence and immunolocalization in the external zones of the adrenal cortex. Endocrinology (1999) 140:2771–80. doi:10.1210/endo.140.6.6835
23. Pihlajoki M, Dörner J, Cochran RS, Heikinheimo M, Wilson DB. Adrenocortical zonation, renewal, and remodeling. Front Endocrinol (2015) 6:27. doi:10.3389/fendo.2015.00027
24. Kyriakides TR, Zhu YH, Yang Z, Bornstein P. The distribution of the matricellular protein thrombospondin 2 in tissues of embryonic and adult mice. J Histochem Cytochem (1998) 46:1007–15. doi:10.1177/002215549804600904
25. Thomas M, Keramidas M, Monchaux E, Feige J-J. Role of adrenocorticotropic hormone in the development and maintenance of the adrenal cortical vasculature. Microsc Res Tech (2003) 61:247–51. doi:10.1002/jemt.10333
26. Gaillard I, Keramidas M, Liakos P, Vilgrain I, Feige JJ, Vittet D. ACTH-regulated expression of vascular endothelial growth factor in the adult bovine adrenal cortex: a possible role in the maintenance of the microvasculature. J Cell Physiol (2000) 185:226–34. doi:10.1002/1097-4652(200011)185:2<226::AID-JCP7>3.0.CO;2-E
27. Heikkilä P, Arola J, Voutilainen R, Salmenkivi K, Kahri AI, Liu J. Expression of vascular endothelial growth factor in human adrenals. Endocr Res (2000) 26:867–71. doi:10.3109/07435800009048610
28. Cherradi N, Lejczak C, Desroches-Castan A, Feige J-J. Antagonistic functions of tetradecanoyl phorbol acetate-inducible-sequence 11b and HuR in the hormonal regulation of vascular endothelial growth factor messenger ribonucleic acid stability by adrenocorticotropin. Mol Endocrinol (2006) 20:916–30. doi:10.1210/me.2005-0121
29. Thomas M, Keramidas M, Monchaux E, Feige J-J. Dual hormonal regulation of endocrine tissue mass and vasculature by adrenocorticotropin in the adrenal cortex. Endocrinology (2004) 145:4320–9. doi:10.1210/en.2004-0179
30. Shifren JL, Mesiano S, Taylor RN, Ferrara N, Jaffe RB. Corticotropin regulates vascular endothelial growth factor expression in human fetal adrenal cortical cells. J Clin Endocrinol Metab (1998) 83:1342–7. doi:10.1210/jcem.83.4.4730
31. Albertin G, Rucinski M, Carraro G, Forneris M, Andreis P, Malendowicz LK, et al. Adrenomedullin and vascular endothelium growth factor genes are overexpressed in the regenerating rat adrenal cortex, and AM and VEGF reciprocally enhance their mRNA expression in cultured rat adrenocortical cells. Int J Mol Med (2005) 16:431–5. doi:10.3892/ijmm.16.3.431
32. Taniguchi A, Tajima T, Nonomura K, Shinohara N, Mikami A, Koyanagi T. Expression of vascular endothelial growth factor and its receptors Flk-1 and Flt-1 during the regeneration of autotransplanted adrenal cortex in the adrenalectomized rat. J Urol (2004) 171:2445–9. doi:10.1097/01.ju.0000127755.87490.37
33. Apkarian RP, Curtis JC. Hormonal regulation of capillary fenestrae in the rat adrenal cortex: quantitative studies using objective lens staging scanning electron microscopy. Scan Electron Microsc (1986) Pt 4:1381–93.
34. Zhang DX, Gauthier KM, Falck JR, Siddam A, Campbell WB. Steroid-producing cells regulate arterial tone of adrenal cortical arteries. Endocrinology (2007) 148:3569–76. doi:10.1210/en.2007-0169
35. Hinson JP, Vinson GP, Pudney J, Whitehouse BJ. Adrenal mast cells modulate vascular and secretory responses in the intact adrenal gland of the rat. J Endocrinol (1989) 121:253–60. doi:10.1677/joe.0.1210253
36. Lefebvre H, Contesse V, Delarue C, Feuilloley M, Hery F, Grise P, et al. Serotonin-induced stimulation of cortisol secretion from human adrenocortical tissue is mediated through activation of a serotonin4 receptor subtype. Neuroscience (1992) 47:999–1007. doi:10.1016/0306-4522(92)90047-6
37. Aiba M, Iri H, Suzuki H, Kageyama K, Kawai T, Abe O, et al. Numerous mast cells in an 11-deoxycorticosterone-producing adrenocortical tumor. Histologic evaluation of benignancy and comparison with mast cell distribution in adrenal glands and neoplastic counterparts of 67 surgical specimens. Arch Pathol Lab Med (1985) 109:357–60.
38. Duparc C, Moreau L, Dzib JFG, Boyer H-G, Tetsi Nomigni M, Boutelet I, et al. Mast cell hyperplasia is associated with aldosterone hypersecretion in a subset of aldosterone-producing adenomas. J Clin Endocrinol Metab (2015) 100:E550–60. doi:10.1210/jc.2014-3660
39. Louiset E, Isvi K, Gasc JM, Duparc C, Cauliez B, Laquerrière A, et al. Ectopic expression of serotonin7 receptors in an adrenocortical carcinoma co-secreting renin and cortisol. Endocr Relat Cancer (2008) 15:1025–34. doi:10.1677/ERC-08-0085
40. Peters J. Local renin-angiotensin systems in the adrenal gland. Peptides (2012) 34:427–32. doi:10.1016/j.peptides.2012.01.023
41. Baba K, Doi Y, Franco-Saenz R, Mulrow PJ. Mechanisms by which nephrectomy stimulates adrenal renin. Hypertension (1986) 8:997–1002. doi:10.1161/01.HYP.8.11.997
42. Doi Y, Atarashi K, Franco-Saenz R, Mulrow PJ. Effect of changes in sodium or potassium balance, and nephrectomy, on adrenal renin and aldosterone concentrations. Hypertension (1984) 6:I124–9. doi:10.1161/01.HYP.6.2_Pt_2.I124
43. Cui J, Dou J, Yang G, Zang L, Jin N, Chen K, et al. The changes in renin-angiotensin-aldosterone-system in different subtypes of Cushing’s syndrome. Zhonghua Nei Ke Za Zhi (2015) 54:618–22. doi:10.3760/cma.j.issn.0578-1426.2015.07.010
44. Luton JP, Vidal-Trecan G, Mouveroux F, Bricaire H. Hypertension and Cushing’s disease. Ann Med Interne (1983) 134:203–8.
45. Barney M, Call GB, McIlmoil CJ, Husein OF, Adams A, Balls AG, et al. Stimulation by interleukin-6 and inhibition by tumor necrosis factor of cortisol release from bovine adrenal zona fasciculata cells through their receptors. Endocrine (2000) 13:369–77. doi:10.1385/ENDO:13:3:369
46. Judd AM, MacLeod RM. Differential release of tumor necrosis factor and IL-6 from adrenal zona glomerulosa cells in vitro. Am J Physiol (1995) 268:E114–20.
47. Lefebvre H, Prévost G, Louiset E. Autocrine/paracrine regulatory mechanisms in adrenocortical neoplasms responsible for primary adrenal hypercorticism. Eur J Endocrinol (2013) 169:R115–38. doi:10.1530/EJE-13-0308
48. Caroccia B, Fassina A, Seccia TM, Recarti C, Petrelli L, Belloni AS, et al. Isolation of human adrenocortical aldosterone-producing cells by a novel immunomagnetic beads method. Endocrinology (2010) 151:1375–80. doi:10.1210/en.2009-1243
49. Li Q, Johansson H, Kjellman M, Grimelius L. Neuroendocrine differentiation and nerves in human adrenal cortex and cortical lesions. APMIS (1998) 106:807–17. doi:10.1111/j.1699-0463.1998.tb00227.x
50. Cartier D, Jégou S, Parmentier F, Lihrmann I, Louiset E, Kuhn J-M, et al. Expression profile of serotonin4 (5-HT4) receptors in adrenocortical aldosterone-producing adenomas. Eur J Endocrinol (2005) 153:939–47. doi:10.1530/eje.1.02051
51. Ye P, Mariniello B, Mantero F, Shibata H, Rainey WE. G-protein-coupled receptors in aldosterone-producing adenomas: a potential cause of hyperaldosteronism. J Endocrinol (2007) 195:39–48. doi:10.1677/JOE-07-0037
52. Murakami M, Yoshimoto T, Nakabayashi K, Tsuchiya K, Minami I, Bouchi R, et al. Integration of transcriptome and methylome analysis of aldosterone-producing adenomas. Eur J Endocrinol (2015) 173:185–95. doi:10.1530/EJE-15-0148
53. Zwermann O, Suttmann Y, Bidlingmaier M, Beuschlein F, Reincke M. Screening for membrane hormone receptor expression in primary aldosteronism. Eur J Endocrinol (2009) 160:443–51. doi:10.1530/EJE-08-0711
54. Arola J, Liu J, Heikkilä P, Voutilainen R, Kahri A. Expression of inhibin alpha in the human adrenal gland and adrenocortical tumors. Endocr Res (1998) 24:865–7. doi:10.3109/07435809809032699
55. Vänttinen T, Kuulasmaa T, Liu J, Voutilainen R. Expression of activin/inhibin receptor and binding protein genes and regulation of activin/inhibin peptide secretion in human adrenocortical cells. J Clin Endocrinol Metab (2002) 87:4257–63. doi:10.1210/jc.2002-020460
56. Vänttinen T, Liu J, Kuulasmaa T, Kivinen P, Voutilainen R. Expression of activin/inhibin signaling components in the human adrenal gland and the effects of activins and inhibins on adrenocortical steroidogenesis and apoptosis. J Endocrinol (2003) 178:479–89. doi:10.1677/joe.0.1780479
57. Ehrhart-Bornstein M, Lamounier-Zepter V, Schraven A, Langenbach J, Willenberg HS, Barthel A, et al. Human adipocytes secrete mineralocorticoid-releasing factors. Proc Natl Acad Sci U S A (2003) 100:14211–6. doi:10.1073/pnas.2336140100
58. Krug AW, Vleugels K, Schinner S, Lamounier-Zepter V, Ziegler CG, Bornstein SR, et al. Human adipocytes induce an ERK1/2 MAP kinases-mediated upregulation of steroidogenic acute regulatory protein (StAR) and an angiotensin II-sensitization in human adrenocortical cells. Int J Obes (Lond) (2007) 31:1605–16. doi:10.1038/sj.ijo.0803642
59. Schinner S, Willenberg HS, Krause D, Schott M, Lamounier-Zepter V, Krug AW, et al. Adipocyte-derived products induce the transcription of the StAR promoter and stimulate aldosterone and cortisol secretion from adrenocortical cells through the Wnt-signaling pathway. Int J Obes (Lond) (2007) 31:864–70. doi:10.1038/sj.ijo.0803508
60. Glasow A, Bornstein SR. Leptin and the adrenal gland. Eur J Clin Invest (2000) 30(Suppl 3):39–45. doi:10.1046/j.1365-2362.2000.0300s3039.x
61. Glasow A, Haidan A, Hilbers U, Breidert M, Gillespie J, Scherbaum WA, et al. Expression of Ob receptor in normal human adrenals: differential regulation of adrenocortical and adrenomedullary function by leptin. J Clin Endocrinol Metab (1998) 83:4459–66. doi:10.1210/jcem.83.12.5337
62. Pralong FP, Roduit R, Waeber G, Castillo E, Mosimann F, Thorens B, et al. Leptin inhibits directly glucocorticoid secretion by normal human and rat adrenal gland. Endocrinology (1998) 139:4264–8. doi:10.1210/endo.139.10.6254
63. Szücs N, Varga I, Jakab C, Patócs A, Gláz E, Tóth M, et al. Leptin inhibits cortisol and corticosterone secretion in pathologic human adrenocortical cells. Pituitary (2001) 4:71–7. doi:10.1023/A:1012990928218
64. Iwen KAH, Senyaman O, Schwartz A, Drenckhan M, Meier B, Hadaschik D, et al. Melanocortin crosstalk with adipose functions: ACTH directly induces insulin resistance, promotes a pro-inflammatory adipokine profile and stimulates UCP-1 in adipocytes. J Endocrinol (2008) 196:465–72. doi:10.1677/JOE-07-0299
65. Jun D-J, Na K-Y, Kim W, Kwak D, Kwon E-J, Yoon JH, et al. Melanocortins induce interleukin 6 gene expression and secretion through melanocortin receptors 2 and 5 in 3T3-L1 adipocytes. J Mol Endocrinol (2010) 44:225–36. doi:10.1677/JME-09-0161
66. Kim NS, Kim Y-J, Cho SY, Lee TR, Kim SH. Transcriptional activation of melanocortin 2 receptor accessory protein by PPARγ in adipocytes. Biochem Biophys Res Commun (2013) 439:401–6. doi:10.1016/j.bbrc.2013.08.061
67. German-Mena E, Zibari GB, Levine SN. Adrenal myelolipomas in patients with congenital adrenal hyperplasia: review of the literature and a case report. Endocr Pract (2011) 17:441–7. doi:10.4158/EP10340.RA
68. Nermoen I, Rørvik J, Holmedal SH, Hykkerud DL, Fougner KJ, Svartberg J, et al. High frequency of adrenal myelolipomas and testicular adrenal rest tumours in adult Norwegian patients with classical congenital adrenal hyperplasia because of 21-hydroxylase deficiency. Clin Endocrinol (Oxf) (2011) 75:753–9. doi:10.1111/j.1365-2265.2011.04151.x
69. Almeida MQ, Kaupert LC, Brito LP, Lerario AM, Mariani BMP, Ribeiro M, et al. Increased expression of ACTH (MC2R) and androgen (AR) receptors in giant bilateral myelolipomas from patients with congenital adrenal hyperplasia. BMC Endocr Disord (2014) 14:42. doi:10.1186/1472-6823-14-42
70. Kiwaki K, Levine JA. Differential effects of adrenocorticotropic hormone on human and mouse adipose tissue. J Comp Physiol B (2003) 173:675–8. doi:10.1007/s00360-003-0377-1
71. Ehrhart-Bornstein M, Haidan A, Alesci S, Bornstein SR. Neurotransmitters and neuropeptides in the differential regulation of steroidogenesis in adrenocortical-chromaffin co-cultures. Endocr Res (2000) 26:833–42. doi:10.3109/07435800009048606
72. Suda T, Tomori N, Tozawa F, Demura H, Shizume K, Mouri T, et al. Immunoreactive corticotropin and corticotropin-releasing factor in human hypothalamus, adrenal, lung cancer, and pheochromocytoma. J Clin Endocrinol Metab (1984) 58:919–24. doi:10.1210/jcem-58-5-919
73. Ballav C, Naziat A, Mihai R, Karavitaki N, Ansorge O, Grossman AB. Mini-review: pheochromocytomas causing the ectopic ACTH syndrome. Endocrine (2012) 42:69–73. doi:10.1007/s12020-012-9646-7
74. Cohade C, Broussaud S, Louiset E, Bennet A, Huyghe E, Caron P. Ectopic Cushing’s syndrome due to a pheochromocytoma: a new case in the post-partum and review of literature. Gynecol Endocrinol (2009) 25:624–7. doi:10.1080/09513590903015411
75. Oh HC, Koh J-M, Kim MS, Park JY, Shong YK, Lee K-U, et al. A case of ACTH-producing pheochromocytoma associated with pregnancy. Endocr J (2003) 50:739–44. doi:10.1507/endocrj.50.739
76. Newell-Price J, Bertagna X, Grossman AB, Nieman LK. Cushing’s syndrome. Lancet (2006) 367:1605–17. doi:10.1016/S0140-6736(06)68699-6
77. Erem C, Hacihasanoglu A, Ersöz HO, Reis AK, Calik A, Ukinç K, et al. Pheochromocytoma combined with pre-clinical Cushing’s syndrome in the same adrenal gland. J Endocrinol Invest (2005) 28:561–5. doi:10.1007/BF03347247
78. Inoue J, Oishi S, Naomi S, Umeda T, Sato T. Pheochromocytoma associated with adrenocortical adenoma: case report and literature review. Endocrinol Jpn (1986) 33:67–74. doi:10.1507/endocrj1954.33.67
79. Takizawa N, Muguruma K, Sasano H. Pheochromocytoma and subclinical Cushing’s syndrome with focal adrenocortical hyperplasia. Int J Urol (2011) 18:548–9. doi:10.1111/j.1442-2042.2011.02759.x
80. Yoshioka M, Saito H, Kawakami Y, Mineyama H, Sekiya M. Adrenomedullary hyperplasia associated with cortisol producing adenoma. Endocr J (1993) 40:467–71. doi:10.1507/endocrj.40.467
81. Alexandraki KI, Michail OP, Nonni A, Diamantis D, Giannopoulou I, Kaltsas GA, et al. Corticomedullary mixed adrenal tumor: case report and literature review. Endocr J (2009) 56:817–24. doi:10.1507/endocrj.K09E-010
82. Kaneko T, Matsushima H, Homma Y. Dopamine-secreting corticomedullary mixed tumor of the adrenal gland. Int J Urol (2012) 19(12):1123–4. doi:10.1111/j.1442-2042.2012.03107.x
83. Lau SK, Chu PG, Weiss LM. Mixed cortical adenoma and composite pheochromocytoma-ganglioneuroma: an unusual corticomedullary tumor of the adrenal gland. Ann Diagn Pathol (2011) 15:185–9. doi:10.1016/j.anndiagpath.2010.02.005
84. Wieneke JA, Thompson LD, Heffess CS. Corticomedullary mixed tumor of the adrenal gland. Ann Diagn Pathol (2001) 5:304–8. doi:10.1053/adpa.2001.28297
85. Hirata Y, Uchihashi M, Sueoka S, Matsukura S, Fujita T. Presence of ectopic beta-adrenergic receptors on human adrenocortical cortisol-producing adenomas. J Clin Endocrinol Metab (1981) 53:953–7. doi:10.1210/jcem-53-5-953
86. Lacroix A, Tremblay J, Rousseau G, Bouvier M, Hamet P. Propranolol therapy for ectopic beta-adrenergic receptors in adrenal Cushing’s syndrome. N Engl J Med (1997) 337:1429–34. doi:10.1056/NEJM199711133372004
87. Guha P, Sahai SS, Sarkar D, Sardar P, Mandal B, Das BK, et al. Gastric adenocarcinoma associated with lymphangitis carcinomatosa, adrenal metastasis with ectopic ACTH syndrome and malignant common bile duct stricture. Arab J Gastroenterol (2012) 13:31–4. doi:10.1016/j.ajg.2011.07.010
88. Sand M, Uecker S, Bechara FG, Gelos M, Sand D, Wiese TH, et al. Simultaneous ectopic adrenocorticotropic hormone syndrome and adrenal metastasis of a medullary thyroid carcinoma causing paraneoplastic Cushing’s syndrome. Int Semin Surg Oncol (2007) 4:15. doi:10.1186/1477-7800-4-15
89. Satoh H, Saito R, Hisata S, Shiihara J, Taniuchi S, Nakamura Y, et al. An ectopic ACTH-producing small cell lung carcinoma associated with enhanced corticosteroid biosynthesis in the peritumoral areas of adrenal metastasis. Lung Cancer (2012) 76:486–90. doi:10.1016/j.lungcan.2011.12.007
90. Pereira MAA, Araújo RS, Bisi H. Síndrome de Cushing Associada à Hiperplasia Macronodular das Adrenais. Apresentação de um Caso e Revisão da Literatura. Arq Bras Endocrinol Metab (2001) 45:619–27. doi:10.1590/S0004-27302001000600015
91. Mijnhout GS, Danner SA, van de Goot FRW, van Dam EWCM. Macronodular adrenocortical hyperplasia in a postmenopausal woman. Neth J Med (2004) 62:454–5.
92. Pulichino A-M, Vallette-Kasic S, Tsai JP-Y, Couture C, Gauthier Y, Drouin J. Tpit determines alternate fates during pituitary cell differentiation. Genes Dev (2003) 17:738–47. doi:10.1101/gad.1065703
93. Bertherat J, Contesse V, Louiset E, Barrande G, Duparc C, Groussin L, et al. In vivo and in vitro screening for illegitimate receptors in adrenocorticotropin-independent macronodular adrenal hyperplasia causing Cushing’s syndrome: identification of two cases of gonadotropin/gastric inhibitory polypeptide-dependent hypercortisolism. J Clin Endocrinol Metab (2005) 90:1302–10. doi:10.1210/jc.2004-1256
94. Louiset E, Contesse V, Groussin L, Cartier D, Duparc C, Barrande G, et al. Expression of serotonin7 receptor and coupling of ectopic receptors to protein kinase A and ionic currents in adrenocorticotropin-independent macronodular adrenal hyperplasia causing Cushing’s syndrome. J Clin Endocrinol Metab (2006) 91:4578–86. doi:10.1210/jc.2006-0538
95. Ivell R, Heng K, Anand-Ivell R. Insulin-like factor 3 and the HPG axis in the male. Front Endocrinol (2014) 5:6. doi:10.3389/fendo.2014.00006
96. Pelusi C, Fanelli F, Pariali M, Zanotti L, Gambineri A, Pasquali R. Parallel variations of insulin-like peptide 3 (INSL3) and antimüllerian hormone (AMH) in women with the polycystic ovary syndrome according to menstrual cycle pattern. J Clin Endocrinol Metab (2013) 98:E1575–82. doi:10.1210/jc.2013-1107
97. de Keyzer Y, Lenne F, Massias JF, Vieau D, Luton JP, Kahn A, et al. Pituitary-like proopiomelanocortin transcripts in human Leydig cell tumors. J Clin Invest (1990) 86:871–7. doi:10.1172/JCI114787
98. Lacaze-Masmonteil T, de Keyzer Y, Luton JP, Kahn A, Bertagna X. Characterization of proopiomelanocortin transcripts in human nonpituitary tissues. Proc Natl Acad Sci U S A (1987) 84:7261–5. doi:10.1073/pnas.84.20.7261
99. van Aken MO, Pereira AM, van Thiel SW, van den Berg G, Frölich M, Veldhuis JD, et al. Irregular and frequent cortisol secretory episodes with preserved diurnal rhythmicity in primary adrenal Cushing’s syndrome. J Clin Endocrinol Metab (2005) 90:1570–7. doi:10.1210/jc.2004-1281
100. Assie G, Louiset E, Sturm N, René-Corail F, Groussin L, Bertherat J, et al. Systematic analysis of G protein-coupled receptor gene expression in adrenocorticotropin-independent macronodular adrenocortical hyperplasia identifies novel targets for pharmacological control of adrenal Cushing’s syndrome. J Clin Endocrinol Metab (2010) 95:E253–62. doi:10.1210/jc.2009-2281
101. Lebrethon MC, Naville D, Begeot M, Saez JM. Regulation of corticotropin receptor number and messenger RNA in cultured human adrenocortical cells by corticotropin and angiotensin II. J Clin Invest (1994) 93:1828–33. doi:10.1172/JCI117168
102. Mountjoy KG, Bird IM, Rainey WE, Cone RD. ACTH induces up-regulation of ACTH receptor mRNA in mouse and human adrenocortical cell lines. Mol Cell Endocrinol (1994) 99:R17–20. doi:10.1016/0303-7207(94)90160-0
103. Xing Y, Parker CR, Edwards M, Rainey WE. ACTH is a potent regulator of gene expression in human adrenal cells. J Mol Endocrinol (2010) 45:59–68. doi:10.1677/JME-10-0006
104. Lacroix A, Bourdeau I, Lampron A, Mazzuco TL, Tremblay J, Hamet P. Aberrant G-protein coupled receptor expression in relation to adrenocortical overfunction. Clin Endocrinol (Oxf) (2010) 73:1–15. doi:10.1111/j.1365-2265.2009.03689.x
105. Stratakis CA, Boikos SA. Genetics of adrenal tumors associated with Cushing’s syndrome: a new classification for bilateral adrenocortical hyperplasias. Nat Clin Pract Endocrinol Metab (2007) 3:748–57. doi:10.1038/ncpendmet0648
106. Groussin L, Kirschner LS, Vincent-Dejean C, Perlemoine K, Jullian E, Delemer B, et al. Molecular analysis of the cyclic AMP-dependent protein kinase A (PKA) regulatory subunit 1A (PRKAR1A) gene in patients with Carney complex and primary pigmented nodular adrenocortical disease (PPNAD) reveals novel mutations and clues for pathophysiology: augmented PKA signaling is associated with adrenal tumorigenesis in PPNAD. Am J Hum Genet (2002) 71:1433–42. doi:10.1086/344579
107. Bram Z, Renouf S, Duparc C, Ragazzon B, Libe R, Martinez A, et al. Activation of the PKA pathway triggers formation of an illicit serotonergic regulatory loop in primary pigmented nodular adrenal disease (PPNAD) tissues associated with Cushing’s syndrome. Endocrine Abstracts 29 OC7.2. Florence, Italy: (2012).
108. Assié G, Libé R, Espiard S, Rizk-Rabin M, Guimier A, Luscap W, et al. ARMC5 mutations in macronodular adrenal hyperplasia with Cushing’s syndrome. N Engl J Med (2013) 369:2105–14. doi:10.1056/NEJMoa1304603
Keywords: ACTH, aldosterone, cortisol, Cushing’s syndrome, aldosterone-producing adenoma, hyperplasia, adrenocortical cells, VEGF
Citation: Lefebvre H, Thomas M, Duparc C, Bertherat J and Louiset E (2016) Role of ACTH in the Interactive/Paracrine Regulation of Adrenal Steroid Secretion in Physiological and Pathophysiological Conditions. Front. Endocrinol. 7:98. doi: 10.3389/fendo.2016.00098
Received: 30 March 2016; Accepted: 07 July 2016;
Published: 20 July 2016
Edited by:
Nicole Gallo-Payet, Université de Sherbrooke, CanadaReviewed by:
Francisco Gaytán, University of Córdoba, SpainAndre Lacroix, Université de Montréal, Canada
Duarte L. Pignatelli, Institute of Molecular Pathology and Immunology of the University of Porto, Portugal
Copyright: © 2016 Lefebvre, Thomas, Duparc, Bertherat and Louiset. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Hervé Lefebvre, aGVydmUubGVmZWJ2cmVAY2h1LXJvdWVuLmZy