 Fady Hannah-Shmouni
Fady Hannah-Shmouni Fabio R. Faucz
Fabio R. Faucz Constantine A. Stratakis
Constantine A. Stratakis- Program on Developmental Endocrinology and Genetics (PDEGEN), Section on Endocrinology and Genetics (SEGEN), National Institute of Child Health and Human Development (NICHD), National Institutes of Health (NIH), Bethesda, MD, USA
Alterations in the cyclic (c)AMP-dependent signaling pathway have been implicated in the majority of benign adrenocortical tumors (ACTs) causing Cushing syndrome (CS). Phosphodiesterases (PDEs) are enzymes that regulate cyclic nucleotide levels, including cyclic adenosine monophosphate (cAMP). Inactivating mutations and other functional variants in PDE11A and PDE8B, two cAMP-binding PDEs, predispose to ACTs. The involvement of these two genes in ACTs was initially revealed by a genome-wide association study in patients with micronodular bilateral adrenocortical hyperplasia. Thereafter, PDE11A or PDE8B genetic variants have been found in other ACTs, including macronodular adrenocortical hyperplasias and cortisol-producing adenomas. In addition, downregulation of PDE11A expression and inactivating variants of the gene have been found in hereditary and sporadic testicular germ cell tumors, as well as in prostatic cancer. PDEs confer an increased risk of ACT formation probably through, primarily, their action on cAMP levels, but other actions might be possible. In this report, we review what is known to date about PDE11A and PDE8B and their involvement in the predisposition to ACTs.
Introduction
The development and function of the endocrine system is highly dependent on second messengers in hormonal signaling. Advances in molecular and genetic studies and appreciation for the impact of second messengers on endocrine physiology and disease sparked important discoveries in signal pathway research especially over the past decades. The first second messenger to be identified was cyclic adenosine monophosphate (cAMP), described in 1958 by Earl Sutherland (1). The production and degradation of cAMP is regulated by adenyl cyclases (AC) and phosphodiesterases (PDEs), respectively (Figure 1) (1, 2). Recent studies have demonstrated a link between genetic alterations in PDEs and increased predisposition of tumor formation, particularly in the prostate, testis, and the adrenal cortex (3, 4). In primary bilateral macronodular adrenocortical hyperplasia (PBMAH), aberrant expression of several non-mutated G-protein-coupled receptors (GPCRs) showed that cAMP signaling could be increased without genetic mutations (5). In primary pigmented nodular adrenocortical disease (PPNAD), germline inactivating mutations of the protein kinase A regulatory subunit type 1 (PRKAR1A)-linked cAMP-dependent protein kinase (PKA) to adrenocortical tumors (ACTs) (6). Indeed, cAMP signaling dysregulation through expression defects or mutations appears to underlie the pathogenesis of most benign ACTs (5, 7).
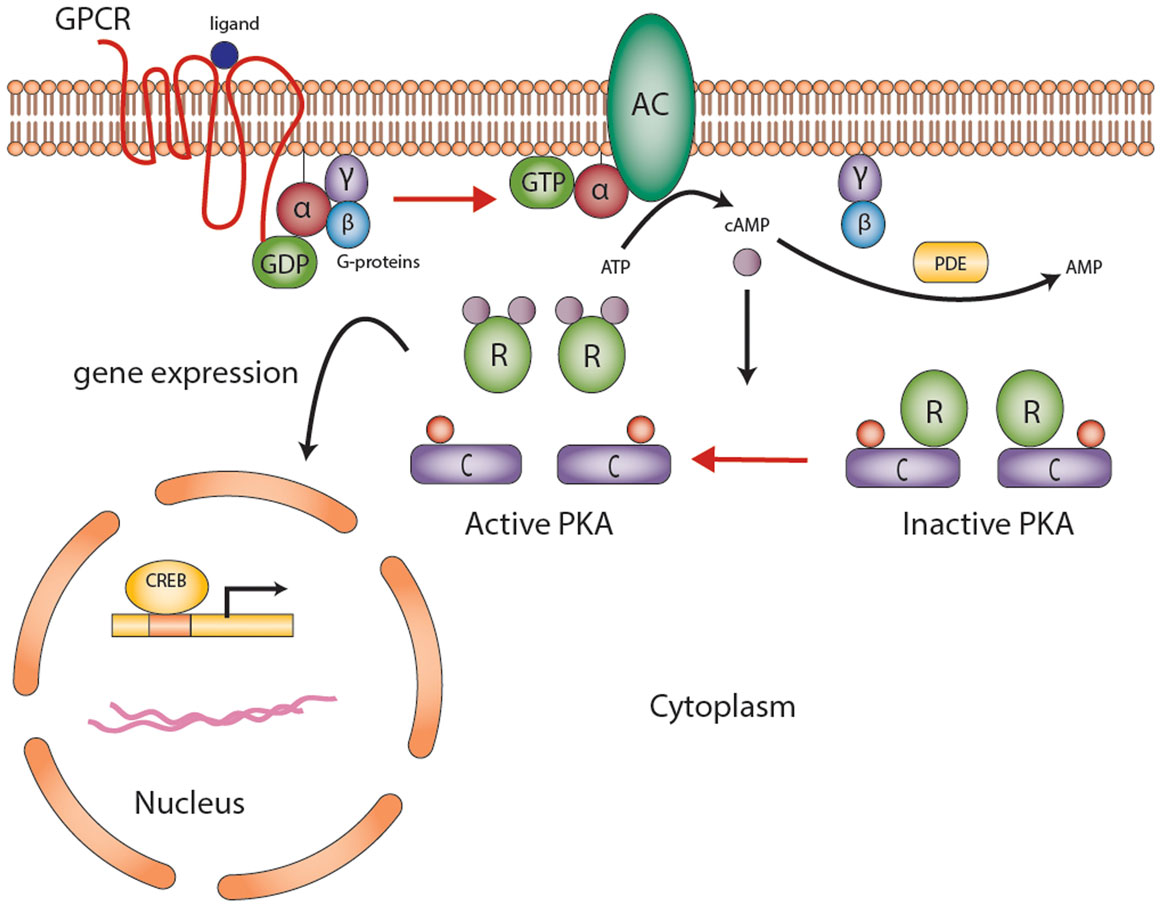
Figure 1. The cyclic AMP-dependent signaling pathway. G protein-coupled receptors (GPCRs) undergo conformational changes in response to various extracellular stimuli. Gsα subunit (G-proteins) exchanges GDP for GTP that activates adenylyl cyclase (AC), converting ATP to cAMP. Elevated cAMP levels, regulated by PDEs (also responsible for degradation of cGMP to GMP), then activate protein kinase A (PKA). PKA consists of a tetramer of two homo- or heterodimers regulatory subunits (R) and two catalytic subunits (C) responsible for the phosphorylation of several enzymes and transcription factors downstream [e.g., cAMP-response element-binding protein (CREB)]. The end product is gene expression to mediate cell growth and differentiation. Abbreviations: α, β, γ, Gs-protein subunits; AC, adenyl cyclase; C, catalytic subunit of protein kinase A; cAMP, cyclic AMP; CREB, cyclic AMP response element-binding protein, a transcription factor; GPCR, G-protein-coupled receptor; PDE, phosphodiesterase; PKA, protein kinase A; R, the regulatory subunits of protein kinase. Courtesy of Stratakis Lab, NICHD, NIH.
Phosphodiesterases exist in over 100 isoforms and are derived from 21 genes separated into 11 PDE gene families (summarized in Table 1) (8–10). PDEs function through the hydrolyzation of cAMP (PDEs isoform 4, 7, and 8) and cyclic guanosine monophosphate (cGMP) (PDEs isoforms 5, 6, and 9) into AMP and GMP, respectively (8, 9). Dual-specificity PDEs (acting on both cAMP and cGMP with varying affinities) include the PDE1, PDE2, PDE3, PDE10, and PDE11 enzymes. All PDEs share a major structural feature; a conserved catalytic domain with about 300 amino acids located near the C-terminal regions, and a variable regulatory domain located in the N-terminal regions. PDEs vary in a number of ways, including a difference in substrate selectivity, tissue distribution, kinetic, and tissue expression (Table 1). The adrenal cortex expresses several isoforms of PDEs, including PDE2A that has been implicated in the downregulation of aldosterone production in adrenal zona glomerulosa cells and the regulation of the adrenocorticotropic hormone (ACTH)-induced increase in intracellular cAMP in the zona fasciculata cells (11).
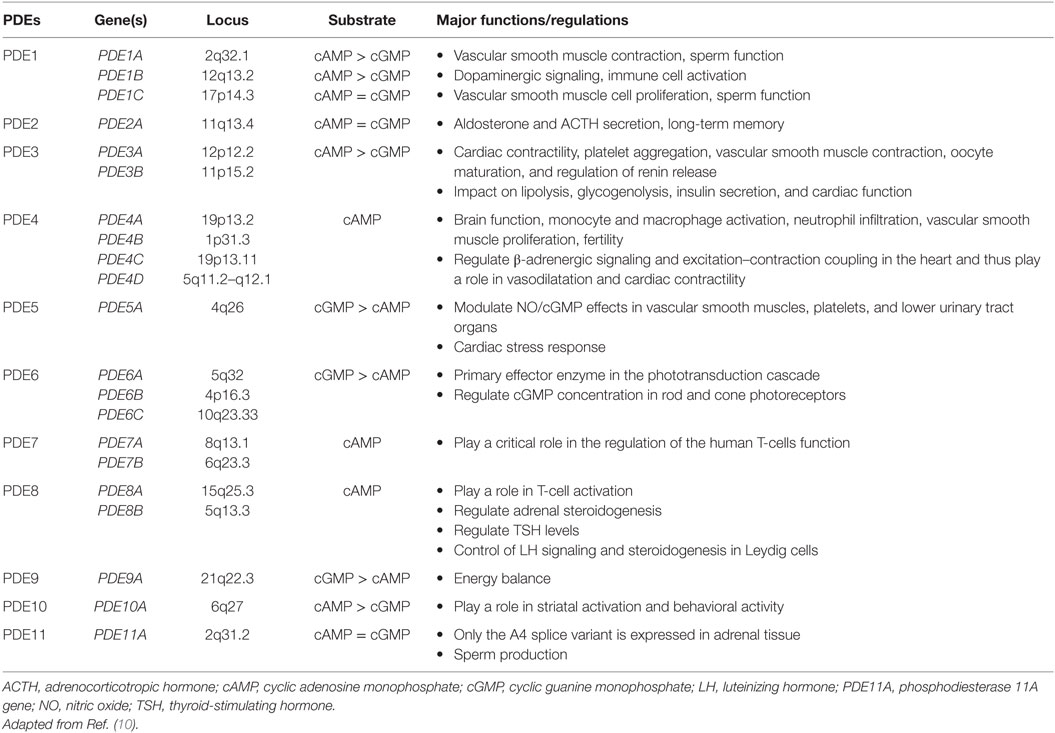
Table 1. Characteristics of phosphodiesterases (PDEs).
Recent studies have demonstrated inactivating mutations and other germline variants in PDE11A and PDE8B in ACT causing Cushing syndrome (CS) (12–17). Here, we present a brief overview of the alterations of PDEs in ACTs. Given the breadth of this topic, we begin with a discussion of the cAMP-dependent signaling pathway in physiology, describe the current classification of ACTs, and then proceed with a discussion of PDE alterations in ACTs.
The cAMP-Dependent Signaling Pathway
Briefly, GPCRs undergo conformational changes in response to various extracellular stimuli, such as ACTH (Figure 1). The first step in cAMP activation in adrenal cortex is the action of ACTH on its seven-transmembrane receptor, ACTHR [e.g., melanocortin 2 receptor (MC2R)]. This activation leads to the dissociation of the Gsα subunit (encoded by the GNAS gene) from the heterotrimeric G-proteins, activation of AC, generation of cAMP, and activating PKA (Figure 1). PKA exists as a tetrameric complex of two regulatory subunits (alpha and beta type 1 or alpha and beta type 2, encoded by PRKAR1A, PRKAR2A, PRKAR1B, and PRKAR2B) and two catalytic subunits (catalytic alpha and catalytic beta, encoded by PRKACA and PRKACB, respectively); the latter subunits are responsible for the phosphorylation of several enzymes and transcription factors downstream, including the cAMP-response element-binding protein (CREB). Abnormalities in some of these genes predispose to the formation of cortisol-producing ACTs (Figure 2) and increased steroid hormone secretion. The latter is often mediated by secondary factors regulated by PKA; in one study, silencing p54(nrb)/NONO expression in H295R human adrenocortical cells decreased the ability of the cells to increase intracellular cAMP production and subsequent cortisol biosynthesis in response to ACTH (18). The expression of multiple PDE isoforms, including PDE2A, PDE3A, PDE3B, PDE4A, PDE4D, and PDE11A, was induced in p54(nrb)/NONO knockdown cells, which suggests that these proteins may be responsible for the splicing and degradation of PDE transcripts (18). Studies in knockout mice (Table 2) have also pointed out to the significance of cAMP signaling-regulating genes in ACT formation.
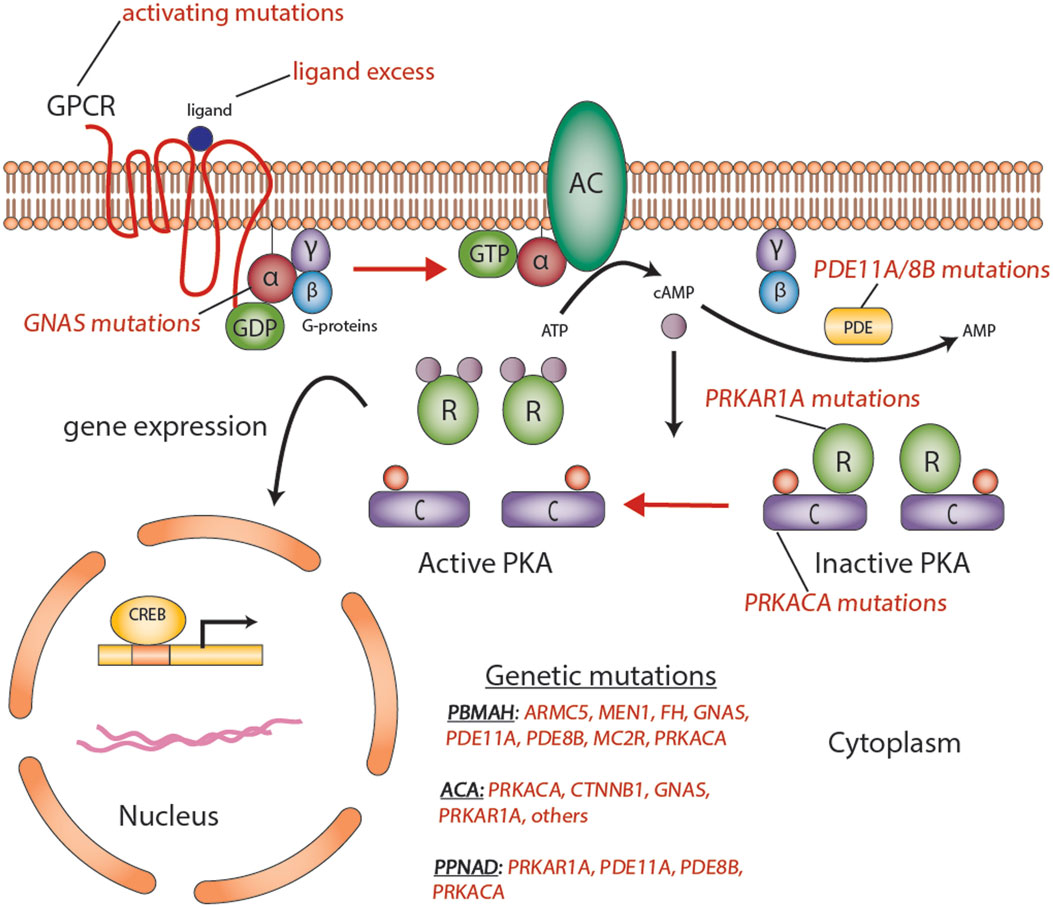
Figure 2. Aberrations in the cyclic AMP-dependent signaling pathway in adrenocortical tumors. Mutations such as activating mutations of GNAS, in McCune–Albright syndrome; inactivating germline mutations in PDE11A in primary pigmented nodular adrenocortical hyperplasia (PPNAD); and inactivating germline mutations of PRKAR1A in Carney’s complex, all predispose to the formation of adrenocortical tumors through a cyclic AMP-dependent process. Abbreviations: α, β, γ, Gs-protein subunits; AC, adenyl cyclase; ACA, adrenocortical adenoma; C, catalytic subunit of protein kinase A; cAMP, cyclic AMP; CREB, cyclic AMP response element-binding protein, a transcription factor; GPCR, G-protein-coupled receptor; PBMAH, bilateral macronodular adrenocortical hyperplasia; PDE, phosphodiesterase; PKA, protein kinase A; R, the regulatory subunits of protein kinase; PPNAD, primary pigmented nodular adrenocortical disease. Courtesy of Stratakis Lab, NICHD, NIH.
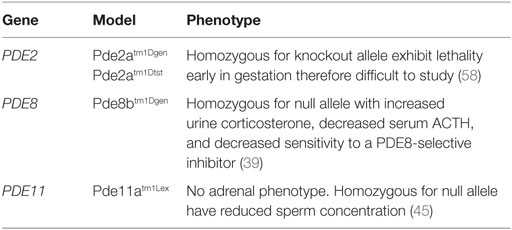
Table 2. Adrenocortical phosphodiesterases mouse models and phenotypes.
A Historical Perspective and Nomenclature of Adrenocortical Tumors
In 1912, Cushing described the pituitary tumors that cause the condition that today bears his name (19). The causes of CS are broadly divided into ACTH-dependent and ACTH-independent disease. In 1984, the first description of Carney complex, a multiple neoplasia syndrome associated with spotty skin pigmentation, cardiac myxomas, pituitary tumors, and CS caused by PPNAD, provided the first insight into the genetic forms of ACT (20). In 1991, Weinstein et al. (21) described Gsα subunit (GNAS) mutations in individuals with McCune-Albright syndrome (MAS); MAS is classically associated with polyostotic fibrous dysplasia, café-au-lait skin spots, and precocious puberty but also with adrenal hyperplasia and/or tumors (21). MAS, albeit rare, appears to be the most frequent cause of CS among ACTH-independent adrenal hyperplasia in the infantile period (22). Kirschner et al. (23, 24) identified the regulatory subunit type 1A (R1α) of PKA (encoded by the PRKAR1A gene on chromosome 17q22-24) as the cause of PPNAD and CNC (25). PPNAD is the most frequent endocrine manifestation in CNC.
In 2006, the first association between PDE mutations and ACT, using a genome-wide association (GWA) study approach, was identified in patients with CS with PPNAD but without GNAS or PRKAR1A mutations (12). Three inactivating mutations in PDE11A were identified initially in patients, predominantly children, with micronodular adrenocortical hyperplasia (iMAD), a rare form of bilateral adrenocortical hyperplasia (BAH) leading to CS. Subsequent studies found other PDE mutations or functional variants, including alterations in PDE8B, in other ACTs, including PBMAH (14–17, 26, 27).
The new genetic findings influenced the diagnostic classification of these ACTs (5). In brief, three major ACTH-independent ACT subtypes exist: bilateral hyperplasias (BAH), adrenocortical adenomas (ACA), and adrenocortical cancer (ACC) (5). PBMAH, a form of BAH, is estimated to affect ~2% of patients with endogenous CS (5). This disease should be distinguished from secondary adrenocortical hyperplasia, which can occur after long-term stimulation by ACTH in Cushing disease (CS in the context of an ACTH-producing pituitary tumor) or ectopic ACTH secretion, predominantly from a neuroendocrine tumor (28). PBMAH is usually sporadic, but familial forms have been described (29). Cortisol-producing adenoma (CPA) is a benign subset of ACAs causing adrenal CS.
Bilateral adrenocortical hyperplasias can be broadly classified on the basis of the size of their nodules into micronodular (<1 cm in diameter) or macrocronodular (>1 cm in diameter) (5). The micronodular subtype is divided into pigmented (c-PPNAD, familial seen in CNC, or isolated, i-PPNAD) and not pigmented (e.g., iMAD) (5). PBMAH is the most common macronodular BAH [previously referred to as massive macronodular adrenocortical disease (MMAD) and ACTH-independent macronodular adrenocortical hyperplasia (AIMAH)] and is largely caused by mutations of the ARMC5 gene (30); several genes have been implicated in other forms of macronodular BAH, including GNAS, APC, and MEN1. Patients with MAS may develop a form of macronodular bilateral adrenal hyperplasia, called primary bimorphic adrenocortical disease (PBAD).
PDEs in Adrenocortical Tumors
Mutations and variants in ACTs that lead to functional abnormalities of cAMP signaling have been reported in the GNAS, PRKAR1A, PDE11A, and PDE8B genes (9). Mutation-negative disease with activation of the cAMP pathway has been reported (31), suggesting that additional genetic (or perhaps epigenetic) “hits” may play a role in the pathogenesis of ACT.
PDE2A
The predominant PDE isoform in adrenal tissue is PDE2A (32). Three PDE2A isoforms exist: PDE2A1, PDE2A2, and PDE2A3, and exhibit higher affinity for cGMP than cAMP (33). PDE2A is implicated in the downregulation of aldosterone production in adrenal zona glomerulosa cells and the regulation of the ACTH-induced increase in intracellular cAMP (11). This ACTH response is described as a rapid and sustained activation of AC followed by a biphasic effect of ACTH on PDE2 activity with an initial and rapid inhibition, followed by a delayed activation (11). In one study, PDE2 involvement was observed to be more important in rat than in human adrenal glomerulosa cells, whereas AC was more stimulated in human than in rat glomerulosa cells (34). Thus, PDE2 activity is involved in the regulation of cAMP accumulation induced by ACTH and suggests that ACTH inhibits this activity. However, no studies to date have reported an association between alterations in PDE2 and ACTs. PDE2A has been shown to be upregulated in beta-catenin (CTNNB1)-mutated ACTs (35). However, it has not been studied in individuals with PPNAD that also have somatic mutations in CTNNB1 (36, 37). Importantly, Pde2a knockout mice do not survive past 17–18 days gestation (38); Pde2a heterozygote mice are not known to develop ACTs or even hypertension.
PDE8
The PDE8 family of proteins includes two genes, PDE8A and PDE8B, which encode for two highly specific enzymes responsible for the highest affinity of the PDEs to degrade cAMP (32). Through negative modulation, these isomers play an important role in adrenal, ovarian, and testicular steroidogenesis (32, 39). Although homologous in structure and function, PDE8B is the major regulator of one or more pools of cAMP in steroidogenesis and carries the highest expression across zona fasciculata compared to other PDEs (14, 32, 39). PDE8A is expressed from a small population of zona fasciculata cells that lie adjacent to zona glomerulosa (32), while PDE8B is expressed throughout the zona fasciculata. The PDE8B locus, like that of other PDEs, is quite complex and encodes multiple isoforms, arising mainly from alternative splicing and displaying tissue-specific expression (32, 40, 41). Tsai et al. (39) demonstrated that Pde8b knockout mice showed elevated urinary corticosterone as a result of adrenal hypersensitivity toward ACTH (39), pointing to PDE8B’s possible role in regulating steroidogenesis. However, the investigators also demonstrated that these mice do not develop adrenal hyperplasia or increased adrenal size.
A GWA study identified a link between the 5q13 locus harboring the PDE8B gene and iMAD (12). A novel missense mutation was found in PDE8B (c.914A > C, p.P305H) in a 2-year-old girl with iMAD where her father carried the same genetic defect with subclinical disease (15); this pattern of an unaffected male passing on the disease to an affected female was also seen in other alterations of PDEs (12, 42). The p.P305H mutation led to higher levels of cAMP when introduced in HEK293 cells (15).
A more recent study of ACTs found several variations in PDE8B: missense substitutions p.H391A, p.P660L, and p.V697I and the c.1365-5G>A splice variant in PDE8B were identified (43). Interestingly, one patient with ACC had both the missense p.R121H and the splice c.1365-5G>A variations, while the other germline PDE8B mutations were found in samples including PBMAH, PPNAD, and secreting and even non-secreting ACAs (43).
Perhaps, the most important recent finding confirming PDE8B’s role in ACT pathogenesis was the genome-wide transcriptomic work by Wilmot Roussel et al. (44). Among over 3000 genes that showed correlation with cortisol secretion in 22 unilateral ACAs (5 non-secreting, 6 subclinical cortisol producing, and 11 cortisol producing), PDE8B showed the strongest positive correlation (44). Accordingly, there was marked increase of the PKA activity to cAMP ratio in secreting adenomas compared to non-secreting adenomas (44).
PDE11A
PDE11A is located on chromosome 2q31.2 and encodes a dual-specificity PDE that degrades both cAMP and cGMP (32). This gene is highly polymorphic in the general population (42) and was the first of the PDEs to be linked with an inherited condition associated with ACTs. Four different transcript variants exist (Table 1), with only the PDE11A4 detected in adrenal tissues (32). Pde11a knockout mice show impaired sperm function and spermatogenesis (45), but no adrenal phenotype has been described for the knock out or heterozygote mouse. Several studies have reported conflicting results with regard to the adrenal expression of PDE11A, suggesting that its expression may only be driven in the diseased adrenal gland (46, 47). The exact role of PDE11A in regulating adrenocortical cAMP levels also remains largely unknown.
Horvath et al. (12) published the first GWA single-nucleotide polymorphism (SNP) association between PDE11A mutations and ACTs in patients with CS from PPNAD or iMAD without known genetic defects (12). Three inactivating mutations in PDE11A were identified. These tumors showed 2q31–2q35 loss of heterozygosity (LOH) and elevated cAMP levels (12), supporting PDE11A’s role in tumor formation. Three of the four patients had PPNAD; a mother and her daughter with the same PDE11A gene mutation, and a third unrelated patient with a different PDE11A mutation, in which the adrenal glands were described as small (largest total adrenal weight = 6.9 g and normal = 8–9 g) with very minor involvement of the superficial cortex evidenced by a few transcapsular cortical extensions into the peri-adrenal fat (48). The fourth patient’s adrenal glands were slightly enlarged owing to hyperplasia of the superficial cortex with a few PPNAD-type nodules in the deep cortex (48). The PDE11A mutation was inherited from her father who had an enlarged right adrenal gland but no CS (48).
In another study, Horvath et al. (42) examined two relatively frequent variants of PDE11A in ACTs and the general population (42). Twelve of 745 controls had these variants, with a lower frequency in patients with ACTs (1.6%; χ2 = 14.62, P < 0.0001). In vitro data demonstrated elevated cAMP levels in HeLa and HEK293 cells, particularly when the p.R804H mutation was studied (42). Another study showed the p.R867G PDE11A gene variant in one patient with familial PBMAH (49). The mechanism by which partially inactivated PDE11A causes adrenocortical overgrowth is largely unclear; the most likely explanation is chronic (albeit modest) elevations of cAMP levels in adrenocortical tissues. Collectively, these experiments suggest that genetic variations in PDE11A may be low-penetrance alleles that occur relatively frequently in the general population and may predispose to the development of ACTs.
The association of PDE11A variants and ACTs was studied further in larger cohorts. Libé et al. (27) examined the role of the PDE11A in a large cohort of ACT, and found an inactivating mutation (p.R307*) in one ACC, with a significant difference between ACC and controls for a polymorphism in exon 6 (p.E421E; OR, 2.1; P = 0.03) (27). Three associated polymorphisms located in intron 10–exon 11–intron 11 were also significant in these tumors (OR, 0.5; P = 0.01) (27). Other variants in the study included 22 germline missense variants (18.8%) in ACA, compared to only 11 missense variants (5.7%) in controls (16 versus 10% in ACC, 19 versus 10% in ACA, and 24 versus 9% in PBMAH; OR, 3.53; P = 0.05) (27). This study suggested a higher frequency of mutations in ACTs, especially PBMAH, when compared to controls. In another study that examined a large cohort of patients with PBMAH, the frequency of all PDE11A variants (e.g., p.D609N or p.M878V) was significantly higher among patients with PBMAH (28%) than controls (7.2%) (P = 5 × 10−5) (17). These variants were also studied in HEK293 cells, where the mutant PDE11A-transfected cells had higher cAMP levels than the wild-type ones (P < 0.05), suggesting that these mutants exhibit diminished cAMP hydrolytic activity (17).
These experiments pointed to several important points about the possible PDE11A’s role in adrenocortical tumorigenesis. First, the spectrum of ACTs varies from benign to malignant. Second, bilateral disease is favored. Third, the allelic losses of the wild-type allele in ACC with missense mutations supports PDE11A role as a tumor suppressor gene. Fourth, PDE11A sequence defects may underlie at least part of the commonly found adrenocortical incidentalomas.
Alterations in PDEs may also be involved in modifying the expression of syndromic diseases associated with ACTs. CNC is caused by PRKAR1A mutations, as discussed earlier (23). In one study of 150 patients with CNC, a higher frequency of PDE11A variants was observed when compared with healthy controls (25.3 versus 6.8%, P < 0.0001), particularly in men (30.8 versus 13%, P = 0.025, PPNAD subgroup) (16). Importantly, these men had a higher incidence of large-cell calcifying Sertoli cell tumors, as well (16). Moreover, simultaneous in vitro inactivation of PRKAR1A and PDE11A by small inhibitory RNA led to increased PKA activity and/or cAMP signaling (16). Thus, it is conceivable that PDE11A-inactivating variants act in concert with other genes in disease predisposition and/or progression.
Aberrations in the cAMP-Dependent Signaling Pathway in Cortisol-Producing Adenomas
Several genetic aberrations in the cAMP-dependent signaling pathway have been implicated in CPA. The most common genetic aberration in CPA is a somatic-activating mutations of PRKACA (c.617A>G/p.L206R) with an estimated incidence of ~42% (86 of 206 tumors studied to date) (50–52), with a predilection to younger patients with overt CS, suggesting a driver mutation role in tumorigenesis (44). Somatic mutations in GNAS were identified in 5–17% of CPA (53). The somatic allelic losses of PRKAR1A were described in 23% of CPA; these tumors were smaller in size and had a paradoxical increase in urinary cortisol levels after dexamethasone suppression (54), due to increased glucocorticoid receptor expression in ACT (55), as often observed in patients with c-PPNAD. Defects in Wnt-signaling have been reported in CPA, with CTNNB1 (p.S45P, p.S45F) in ~23% of cases (56). There are still many unknown genetic defects that lead to CPA formation.
Future Directions
There has been significant progress in PDE-related research over the past two decades. Genetic testing has uncovered several adrenocortical conditions that were linked to aberrations in PDE, often preceded by a long and insidious pre-diagnostic course. This has allowed earlier identification and better management of these lesions. However, there are several unanswered questions. PDE-related research is hampered by inherent (i.e., complexity of their structures, many intracellular interactions, and largely unknown function) and technical issues (e.g., there is lack of specific antibodies for the multiple isoform of each PDE). In addition to improving the characterization of PDE expression and function, future studies should also focus on the characterization of patients with various ACT phenotypes and PDE genotypes.
Preliminary results in using recombinant compounds to activate or inhibit the PDE11A structure may have important implications for drug development. Jäger et al. (47) produced approximately fourfold to fivefold increase in PDE11A-mediated hydrolysis of both cAMP and cGMP, with some degree of PDE11A specificity, with a cGMP analog (Rp-8-pCPTPET-cGMPS) bound to the PDE11A4 GAF domain (47). Furthermore, Ceyhan et al. (57) showed that BC11-28 and BC11-38 (potent and selective PDE11A inhibitors) in both yeast-based and enzyme assays had a >350-fold selectivity for inhibiting PDE11’s cGMP hydrolytic activity versus all other PDEs, while only BC11-38 inhibited PDE11A cAMP hydrolytic activity in H295R cells (57). Thus, a targeted molecular therapy approach for lesions related to defects in PDE may aid in the future management of affected or at risk patients.
Conclusion
Alterations in PDEs that lead to dysregulation of the cAMP-dependent signaling pathway have been linked to the development of ACT. These lesions are usually benign and represent an important group of genetic disorders causing CS. As genetic technology continues to revolutionize the field of endocrine genetics and as we continue to discover novel disease-causing genes on an unprecedented scale, new methods to rapidly assess the functional significance of PDE variants singly, or in combination, will evolve. In this review, we focused our discussion on the various genotypes and phenotypes of ACT due to alterations in PDE, particularly mutations in PDE8B and PDE11A. Although there has been significant progress in PDE-related research over the past two decades, there are as yet unidentified molecular causes for all of these lesions. We hope that one day targeted molecular therapies will replace adrenalectomy as the treatment of choice for these lesions.
Author Contributions
All authors contributed equally to the conception or design of the work; or the acquisition, analysis, or interpretation of data for the work; drafting the work or revising it critically for important intellectual content; final approval of the version to be published; agreement to be accountable for all aspects of the work in ensuring that questions related to the accuracy or integrity of any part of the work are appropriately investigated and resolved.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Funding
This work was supported by the intramural program of the Eunice Kennedy Shriver National Institute of Child Health and Human Development, National Institutes of Health (NIH), protocol HD008920.
Abbreviations
AC, adenyl cyclase; ACTH, adrenocorticotropic hormone; AIMAH, ACTH-independent macronodular adrenal hyperplasia; AMP/ATP, adenosine monophosphate/adenosine triphosphate; Alleles, alternative forms of a gene; cAMP, cyclic adenosine monophosphate; CNC, Carney complex; CS, Cushing syndrome; CREB, cAMP-response element-binding protein; GMP/GDP/GTP, guanosine monophosphate/guanosine diphosphate/guanosine triphosphate; Genes, units of inheritance at specific locations (loci) on a chromosome; GPCRs, G protein-coupled receptors; GWA, genome-wide association; heterozygous, a genotype with two different alleles of a gene for a particular trait; homozygous, a genotype with the same allele of a gene for a particular trait; MMAD, massive macronodular adrenocortical disease; mutations, alteration of genetic material producing a new variation; PBAD, primary bimorphic adrenocortical disease; PBMAH, primary bilateral macronodular adrenocortical hyperplasia; PDEs, phosphodiesterases; phenotype, detectable expression of a genotype; PKA, protein kinase A; PPNAD, primary pigmented micronodular adrenal disease; PRKAR1A, protein kinase A regulatory subunit type 1.
References
1. Rall TW, Sutherland EW. Formation of a cyclic adenine ribonucleotide by tissue particles. J Biol Chem (1958) 232(2):1065–76.
2. Butcher RW, Sutherland EW. Adenosine 3’,5’-phosphate in biological materials. I. Purification and properties of cyclic 3’,5’-nucleotide phosphodiesterase and use of this enzyme to characterize adenosine 3’,5’-phosphate in human urine. J Biol Chem (1962) 237:1244–50.
3. Horvath A, Korde L, Greene MH, Libe R, Osorio P, Faucz FR, et al. Functional phosphodiesterase 11A mutations may modify the risk of familial and bilateral testicular germ cell tumors. Cancer Res (2009) 69(13):5301–6. doi:10.1158/0008-5472.CAN-09-0884
4. de Alexandre RB, Horvath AD, Szarek E, Manning AD, Leal LF, Kardauke F, et al. Phosphodiesterase sequence variants may predispose to prostate cancer. Endocr Relat Cancer (2015) 22(4):519–30. doi:10.1530/ERC-15-0134
5. Stratakis CA, Boikos SA. Genetics of adrenal tumors associated with Cushing’s syndrome: a new classification for bilateral adrenocortical hyperplasias. Nat Clin Pract Endocrinol Metab (2007) 3(11):748–57. doi:10.1038/ncpendmet0648
6. Stratakis CA, Kirschner LS, Carney JA. Clinical and molecular features of the Carney complex: diagnostic criteria and recommendations for patient evaluation. J Clin Endocrinol Metab (2001) 86(9):4041–6. doi:10.1210/jcem.86.9.7903
7. Horvath A, Stratakis CA. Unraveling the molecular basis of micronodular adrenal hyperplasia. Curr Opin Endocrinol Diabetes Obes (2008) 15(3):227–33. doi:10.1097/MED.0b013e3282fe7416
8. Francis SH, Blount MA, Corbin JD. Mammalian cyclic nucleotide phosphodiesterases: molecular mechanisms and physiological functions. Physiol Rev (2011) 91(2):651–90. doi:10.1152/physrev.00030.2010
9. Azevedo MF, Faucz FR, Bimpaki E, Horvath A, Levy I, de Alexandre RB, et al. Clinical and molecular genetics of the phosphodiesterases (PDEs). Endocr Rev (2014) 35(2):195–233. doi:10.1210/er.2013-1053
10. Vezzosi D, Bertherat J. Phosphodiesterases in endocrine physiology and disease. Eur J Endocrinol (2011) 165(2):177–88. doi:10.1530/EJE-10-1123
11. Spiessberger B, Bernhard D, Herrmann S, Feil S, Werner C, Luppa PB, et al. cGMP-dependent protein kinase II and aldosterone secretion. FEBS J (2009) 276(4):1007–13. doi:10.1111/j.1742-4658.2008.06839.x
12. Horvath A, Boikos S, Giatzakis C, Robinson-White A, Groussin L, Griffin KJ, et al. A genome-wide scan identifies mutations in the gene encoding phosphodiesterase 11A4 (PDE11A) in individuals with adrenocortical hyperplasia. Nat Genet (2006) 38(7):794–800. doi:10.1038/ng1809
13. Boikos SA, Horvath A, Heyerdahl S, Stein E, Robinson-White A, Bossis I, et al. Phosphodiesterase 11A expression in the adrenal cortex, primary pigmented nodular adrenocortical disease, and other corticotropin-independent lesions. Horm Metab Res (2008) 40(5):347–53. doi:10.1055/s-2008-1076694
14. Horvath A, Giatzakis C, Tsang K, Greene E, Osorio P, Boikos S, et al. A cAMP-specific phosphodiesterase (PDE8B) that is mutated in adrenal hyperplasia is expressed widely in human and mouse tissues: a novel PDE8B isoform in human adrenal cortex. Eur J Hum Genet (2008) 16(10):1245–53. doi:10.1038/ejhg.2008.85
15. Horvath A, Mericq V, Stratakis CA. Mutation in PDE8B, a cyclic AMP-specific phosphodiesterase in adrenal hyperplasia. N Engl J Med (2008) 358(7):750–2. doi:10.1056/NEJMc0706182
16. Libé R, Horvath A, Vezzosi D, Fratticci A, Coste J, Perlemoine K, et al. Frequent phosphodiesterase 11A gene (PDE11A) defects in patients with Carney complex (CNC) caused by PRKAR1A mutations: PDE11A may contribute to adrenal and testicular tumors in CNC as a modifier of the phenotype. J Clin Endocrinol Metab (2011) 96(1):E208–14. doi:10.1210/jc.2010-1704
17. Vezzosi D, Libé R, Baudry C, Rizk-Rabin M, Horvath A, Levy I, et al. Phosphodiesterase 11A (PDE11A) gene defects in patients with acth-independent macronodular adrenal hyperplasia (AIMAH): functional variants may contribute to genetic susceptibility of bilateral adrenal tumors. J Clin Endocrinol Metab (2012) 97(11):E2063–9. doi:10.1210/jc.2012-2275
18. Lu JY, Sewer MB. p54nrb/NONO regulates cyclic AMP-dependent glucocorticoid production by modulating phosphodiesterase mRNA splicing and degradation. Mol Cell Biol (2015) 35(7):1223–37. doi:10.1128/MCB.00993-14
19. Cushing H. The basophil adenomas of the pituitary body and their clinical manifestations (pituitary basophilism). 1932. Obes Res (1994) 2(5):486–508. doi:10.1002/j.1550-8528.1994.tb00097.x
20. Stratakis CA. Clinical genetics of multiple endocrine neoplasias, Carney complex and related syndromes. J Endocrinol Invest (2001) 24(5):370–83. doi:10.1007/BF03343875
21. Weinstein LS, Shenker A, Gejman PV, Merino MJ, Friedman E, Spiegel AM. Activating mutations of the stimulatory G protein in the McCune-Albright syndrome. N Engl J Med (1991) 325(24):1688–95. doi:10.1056/NEJM199112123252403
22. Kirk JM, Brain CE, Carson DJ, Hyde JC, Grant DB. Cushing’s syndrome caused by nodular adrenal hyperplasia in children with McCune-Albright syndrome. J Pediatr (1999) 134(6):789–92. doi:10.1016/S0022-3476(99)70302-1
23. Kirschner LS, Carney JA, Pack SD, Taymans SE, Giatzakis C, Cho YS, et al. Mutations of the gene encoding the protein kinase A type I-alpha regulatory subunit in patients with the Carney complex. Nat Genet (2000) 26(1):89–92. doi:10.1038/79238
24. Kirschner LS, Sandrini F, Monbo J, Lin JP, Carney JA, Stratakis CA. Genetic heterogeneity and spectrum of mutations of the PRKAR1A gene in patients with the Carney complex. Hum Mol Genet (2000) 9(20):3037–46. doi:10.1093/hmg/9.20.3037
25. Rothenbuhler A, Stratakis CA. Clinical and molecular genetics of Carney complex. Best Pract Res Clin Endocrinol Metab (2010) 24(3):389–99. doi:10.1016/j.beem.2010.03.003
26. Horvath A, Faucz F, Finkielstain GP, Nikita ME, Rothenbuhler A, Almeida M, et al. Haplotype analysis of the promoter region of phosphodiesterase type 8B (PDE8B) in correlation with inactivating PDE8B mutation and the serum thyroid-stimulating hormone levels. Thyroid (2010) 20(4):363–7. doi:10.1089/thy.2009.0260
27. Libé R, Fratticci A, Coste J, Tissier F, Horvath A, Ragazzon B, et al. Phosphodiesterase 11A (PDE11A) and genetic predisposition to adrenocortical tumors. Clin Cancer Res (2008) 14(12):4016–24. doi:10.1158/1078-0432.CCR-08-0106
28. Lacroix A. Heredity and cortisol regulation in bilateral macronodular adrenal hyperplasia. N Engl J Med (2013) 369(22):2147–9. doi:10.1056/NEJMe1312792
29. Lacroix A. ACTH-independent macronodular adrenal hyperplasia. Best Pract Res Clin Endocrinol Metab (2009) 23(2):245–59. doi:10.1016/j.beem.2008.10.011
30. Assié G, Libé R, Espiard S, Rizk-Rabin M, Guimier A, Luscap W, et al. ARMC5 mutations in macronodular adrenal hyperplasia with Cushing’s syndrome. N Engl J Med (2013) 369(22):2105–14. doi:10.1056/NEJMoa1304603
31. Bimpaki EI, Nesterova M, Stratakis CA. Abnormalities of cAMP signaling are present in adrenocortical lesions associated with ACTH-independent Cushing syndrome despite the absence of mutations in known genes. Eur J Endocrinol (2009) 161(1):153–61. doi:10.1530/EJE-09-0027
32. Lakics V, Karran EH, Boess FG. Quantitative comparison of phosphodiesterase mRNA distribution in human brain and peripheral tissues. Neuropharmacology (2010) 59(6):367–74. doi:10.1016/j.neuropharm.2010.05.004
33. Beavo JA, Hardman JG, Sutherland EW. Stimulation of adenosine 3’,5’-monophosphate hydrolysis by guanosine 3’,5’-monophosphate. J Biol Chem (1971) 246(12):3841–6.
34. Côté M, Payet MD, Rousseau E, Guillon G, Gallo-Payet N. Comparative involvement of cyclic nucleotide phosphodiesterases and adenylyl cyclase on adrenocorticotropin-induced increase of cyclic adenosine monophosphate in rat and human glomerulosa cells. Endocrinology (1999) 140(8):3594–601. doi:10.1210/endo.140.8.6889
35. Durand J, Lampron A, Mazzuco TL, Chapman A, Bourdeau I. Characterization of differential gene expression in adrenocortical tumors harboring beta-catenin (CTNNB1) mutations. J Clin Endocrinol Metab (2011) 96(7):E1206–11. doi:10.1210/jc.2010-2143
36. Tadjine M, Lampron A, Ouadi L, Horvath A, Stratakis CA, Bourdeau I. Detection of somatic beta-catenin mutations in primary pigmented nodular adrenocortical disease (PPNAD). Clin Endocrinol (Oxf) (2008) 69(3):367–73. doi:10.1111/j.1365-2265.2008.03273.x
37. Gaujoux S, Tissier F, Groussin L, Libé R, Ragazzon B, Launay P, et al. Wnt/beta-catenin and 3’,5’-cyclic adenosine 5’-monophosphate/protein kinase A signaling pathways alterations and somatic beta-catenin gene mutations in the progression of adrenocortical tumors. J Clin Endocrinol Metab (2008) 93(10):4135–40. doi:10.1210/jc.2008-0631
38. Stephenson DT, Coskran TM, Wilhelms MB, Adamowicz WO, O’Donnell MM, Muravnick KB, et al. Immunohistochemical localization of phosphodiesterase 2A in multiple mammalian species. J Histochem Cytochem (2009) 57(10):933–49. doi:10.1369/jhc.2009.953471
39. Tsai LC, Shimizu-Albergine M, Beavo JA. The high-affinity cAMP-specific phosphodiesterase 8B controls steroidogenesis in the mouse adrenal gland. Mol Pharmacol (2011) 79(4):639–48. doi:10.1124/mol.110.069104
40. Hayashi M, Shimada Y, Nishimura Y, Hama T, Tanaka T. Genomic organization, chromosomal localization, and alternative splicing of the human phosphodiesterase 8B gene. Biochem Biophys Res Commun (2002) 297(5):1253–8. doi:10.1016/S0006-291X(02)02371-9
41. Gamanuma M, Yuasa K, Sasaki T, Sakurai N, Kotera J, Omori K. Comparison of enzymatic characterization and gene organization of cyclic nucleotide phosphodiesterase 8 family in humans. Cell Signal (2003) 15(6):565–74. doi:10.1016/S0898-6568(02)00146-8
42. Horvath A, Giatzakis C, Robinson-White A, Boikos S, Levine E, Griffin K, et al. Adrenal hyperplasia and adenomas are associated with inhibition of phosphodiesterase 11A in carriers of PDE11A sequence variants that are frequent in the population. Cancer Res (2006) 66(24):11571–5. doi:10.1158/0008-5472.CAN-06-2914
43. Rothenbuhler A, Horvath A, Libé R, Faucz FR, Fratticci A, Raffin Sanson ML, et al. Identification of novel genetic variants in phosphodiesterase 8B (PDE8B), a cAMP-specific phosphodiesterase highly expressed in the adrenal cortex, in a cohort of patients with adrenal tumours. Clin Endocrinol (Oxf) (2012) 77(2):195–9. doi:10.1111/j.1365-2265.2012.04366.x
44. Wilmot Roussel H, Vezzosi D, Rizk-Rabin M, Barreau O, Ragazzon B, René-Corail F, et al. Identification of gene expression profiles associated with cortisol secretion in adrenocortical adenomas. J Clin Endocrinol Metab (2013) 98(6):E1109–21. doi:10.1210/jc.2012-4237
45. Wayman C, Phillips S, Lunny C, Webb T, Fawcett L, Baxendale R, et al. Phosphodiesterase 11 (PDE11) regulation of spermatozoa physiology. Int J Impot Res (2005) 17(3):216–23. doi:10.1038/sj.ijir.3901307
46. Yuasa K, Kotera J, Fujishige K, Michibata H, Sasaki T, Omori K. Isolation and characterization of two novel phosphodiesterase PDE11A variants showing unique structure and tissue-specific expression. J Biol Chem (2000) 275(40):31469–79. doi:10.1074/jbc.M003041200
47. Jäger R, Russwurm C, Schwede F, Genieser HG, Koesling D, Russwurm M. Activation of PDE10 and PDE11 phosphodiesterases. J Biol Chem (2012) 287(2):1210–9. doi:10.1074/jbc.M111.263806
48. Carney JA, Gaillard RC, Bertherat J, Stratakis CA. Familial micronodular adrenocortical disease, Cushing syndrome, and mutations of the gene encoding phosphodiesterase 11A4 (PDE11A). Am J Surg Pathol (2010) 34(4):547–55. doi:10.1097/PAS.0b013e3181d31f49
49. Hsiao HP, Kirschner LS, Bourdeau I, Keil MF, Boikos SA, Verma S, et al. Clinical and genetic heterogeneity, overlap with other tumor syndromes, and atypical glucocorticoid hormone secretion in adrenocorticotropin-independent macronodular adrenal hyperplasia compared with other adrenocortical tumors. J Clin Endocrinol Metab (2009) 94(8):2930–7. doi:10.1210/jc.2009-0516
50. Beuschlein F, Fassnacht M, Assié G, Calebiro D, Stratakis CA, Osswald A, et al. Constitutive activation of PKA catalytic subunit in adrenal Cushing’s syndrome. N Engl J Med (2014) 370(11):1019–28. doi:10.1056/NEJMoa1310359
51. Cao Y, He M, Gao Z, Peng Y, Li Y, Li L, et al. Activating hotspot L205R mutation in PRKACA and adrenal Cushing’s syndrome. Science (2014) 344(6186):913–7. doi:10.1126/science.1249480
52. Goh G, Scholl UI, Healy JM, Choi M, Prasad ML, Nelson-Williams C, et al. Recurrent activating mutation in PRKACA in cortisol-producing adrenal tumors. Nat Genet (2014) 46(6):613–7. doi:10.1038/ng.2956
53. Fragoso MC, Domenice S, Latronico AC, Martin RM, Pereira MA, Zerbini MC, et al. Cushing’s syndrome secondary to adrenocorticotropin-independent macronodular adrenocortical hyperplasia due to activating mutations of GNAS1 gene. J Clin Endocrinol Metab (2003) 88(5):2147–51. doi:10.1210/jc.2002-021362
54. Bertherat J, Groussin L, Sandrini F, Matyakhina L, Bei T, Stergiopoulos S, et al. Molecular and functional analysis of PRKAR1A and its locus (17q22-24) in sporadic adrenocortical tumors: 17q losses, somatic mutations, and protein kinase A expression and activity. Cancer Res (2003) 63(17):5308–19.
55. Louiset E, Stratakis CA, Perraudin V, Griffin KJ, Libé R, Cabrol S, et al. The paradoxical increase in cortisol secretion induced by dexamethasone in primary pigmented nodular adrenocortical disease involves a glucocorticoid receptor-mediated effect of dexamethasone on protein kinase A catalytic subunits. J Clin Endocrinol Metab (2009) 94(7):2406–13. doi:10.1210/jc.2009-0031
56. Thiel A, Reis AC, Haase M, Goh G, Schott M, Willenberg HS, et al. PRKACA mutations in cortisol-producing adenomas and adrenal hyperplasia: a single-center study of 60 cases. Eur J Endocrinol (2015) 172(6):677–85. doi:10.1530/EJE-14-1113
57. Ceyhan O, Birsoy K, Hoffman CS. Identification of biologically active PDE11-selective inhibitors using a yeast-based high-throughput screen. Chem Biol (2012) 19(1):155–63. doi:10.1016/j.chembiol.2011.12.010
Keywords: phosphodiesterases, adrenocortical tumors, adrenal hyperplasia, Cushing syndrome, genetics, Carney complex, cAMP
Citation: Hannah-Shmouni F, Faucz FR and Stratakis CA (2016) Alterations of Phosphodiesterases in Adrenocortical Tumors. Front. Endocrinol. 7:111. doi: 10.3389/fendo.2016.00111
Received: 10 July 2016; Accepted: 02 August 2016;
Published: 30 August 2016
Edited by:
Andre Lacroix, Université de Montréal, CanadaReviewed by:
Cristina L. Ronchi, University Hospital of Wuerzburg, GermanyDelphine Vezzosi, Centre Hospitalier Universitaire, France
Copyright: © 2016 Hannah-Shmouni, Faucz and Stratakis. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Constantine A. Stratakis, c3RyYXRha2NAbWFpbC5uaWguZ292
†Submitting author: Fady Hannah-Shmouni, ZmFkeS5oYW5uYWgtc2htb3VuaUBuaWguZ292