 Leonid E. Fridlyand
Leonid E. Fridlyand Natalia A. Tamarina1
Natalia A. Tamarina1 Andrew V. Schally
Andrew V. Schally Louis H. Philipson
Louis H. Philipson- 1Department of Medicine, Kovler Diabetes Center, The University of Chicago, Chicago, IL, USA
- 2VA Medical Center, Miami, FL, USA
- 3Department of Pathology and Medicine, Division of Endocrinology and Hematology-Oncology, Sylvester Comprehensive Cancer Center, University of Miami Miller School of Medicine, Miami, FL, USA
- 4Department of Pediatrics, The University of Chicago, Chicago, IL, USA
Growth hormone-releasing hormone (GHRH) is produced by the hypothalamus and stimulates growth hormone synthesis and release in the anterior pituitary gland. In addition, GHRH is an important regulator of cellular functions in many cells and organs. Expression of GHRH G-Protein Coupled Receptor (GHRHR) has been demonstrated in different peripheral tissues and cell types, including pancreatic islets. Among the peripheral activities, recent studies demonstrate a novel ability of GHRH analogs to increase and preserve insulin secretion by beta-cells in isolated pancreatic islets, which makes them potentially useful for diabetes treatment. This review considers the role of GHRHR in the beta-cell and addresses the unique engineered GHRH agonists and antagonists for treatment of type 2 diabetes mellitus. We discuss the similarity of signaling pathways activated by GHRHR in pituitary somatotrophs and in pancreatic beta-cells and possible ways as to how the GHRHR pathway can interact with glucose and other secretagogues to stimulate insulin secretion. We also consider the hypothesis that novel GHRHR agonists can improve glucose metabolism in Type 2 diabetes by preserving the function and survival of pancreatic beta-cells. Wound healing and cardioprotective action with new GHRH agonists suggest that they may prove useful in ameliorating certain diabetic complications. These findings highlight the future potential therapeutic effectiveness of modulators of GHRHR activity for the development of new therapeutic approaches in diabetes and its complications.
Introduction
Type 2 diabetes mellitus (T2DM) is an important metabolic disease affecting almost 30 million Americans with an estimated $250 billion lost yearly, due to effects of morbidity and mortality on total medical costs and lost wages. T2DM is associated with a progressive decline in insulin secretion by pancreatic beta-cells in the face of insulin resistance (1). Despite its importance, we do not fully understand the complex interplay of molecular signals and signal transduction events that control beta-cell functionality and survival. This limits our ability to develop novel approaches for prevention and treatment of diabetes.
The beta-cell membrane contains a profusion of G-protein coupled receptors (GPCRs) that are critical for the regulation of insulin secretion by hormones and neurotransmitters (2–4). Growth hormone-releasing hormone (GHRH) is an important regulator not only of growth hormone secretion but also of a variety of cellular functions in many cells and organs. Expression of GHRH G-protein coupled receptor (GHRHR) has been demonstrated in different peripheral tissues and cell types, including pancreatic islets (5, 6).
Recent studies demonstrate a novel ability of GHRH analogs to increase and preserve insulin secretion by beta-cells in islets and diabetic mice (7, 8) that makes them potentially useful for treatment of T2DM. Remarkable results from the study of new GHRH agonists in wound healing and cardiovascular performance could also provide novel treatments in patients with diabetes (5, 7, 9). This review addresses the possible role of GHRHR and its unique engineered agonists and antagonists for treating diabetes and its complications.
GHRH and Its Analogs
Hypothalamic growth hormone-releasing hormone is one of the “humoral factors” that is critical for growth hormone secretion. The discovery of hypothalamic hormones, such as thyrotropin-releasing hormone, luteinizing hormone-releasing hormone (also known as gonadotropin-releasing hormone), which regulate the secretion of anterior pituitary hormones led to the awarding of the Nobel Prize (1977) to one of us (Andrew V. Schally) (10). GHRH, expressed in the arcuate nucleus of the hypothalamus and released into portal vasculature, directly stimulates growth hormone synthesis and secretion from the pituitary somatotropes by activating the corresponding GHRH receptors (5, 11). GHRHR is present in several other tissues, such as myocardium, lymphocytes, testes, ovaries, skin, and pancreas and is involved in a variety of biological processes (5). The roles of GHRHR in other cells and tissues continue to be explored. In addition, GHRHR have been detected in various tumor cells and in some stem cells (5, 6).
It should be noted that GHRH undergoes rapid enzymatic degradation in blood. Dipeptidylpeptidase IV inactivates the active form of GHRH in blood to its more stable inactive metabolite GHRH(3-44)-NH2 (12, 13). For this reason, concentration of active GHRH (that is produced in the hypothalamus) in blood may be insignificant, and so without significant influence on organs beyond the pituitary somatotropes. Interestingly, inhibitors to dipeptidylpeptidase IV are in widespread use now for type 2 diabetes treatment to increase GLP-1 concentration in blood (14). These agents should also lead to increased GHRH blood concentration. However, this interesting possibility and the effect of GHRH on various target tissues where the GHRHR is expressed have not been investigated.
Accumulating evidence also suggests that, in addition to the neuroendocrine action of GHRH, extrahypothalamic GHRH has been implicated in many peripheral actions via autocrine/paracrine mechanisms. Exogenous GHRH can regulate proliferation, survival, apoptosis, and differentiation in several tissues and cell types (5, 15).
The GHRHR is a member of the class II B GPCR family, which couples predominantly to the Gs-adenylate cyclase-cAMP signaling pathway. Peptide hormones that activate class II GPCRs include GHRH, secretin, glucagon-like peptides, gastric-inhibitory peptide (GIP), pituitary adenylate cyclase-activating peptide, corticotropin-releasing hormone, vasoactive intestinal peptide, parathyroid hormone, and calcitonin-related peptides (16, 17).
The mechanism of the acute action of GHRH on the pituitary somatotrope to increase growth hormone synthesis and secretion has been studied (Figure 1). Binding of GHRH to its receptor activates a stimulatory G protein, which activates adenylyl cyclase to produce cAMP, leading to activation of protein kinase A (PKA). This stimulates an influx of calcium, most likely through plasma membrane depolarization, and activation of voltage-sensitive Ca2+ channels. Increased Ca2+ and cAMP stimulates the growth hormone exocytosis process (18–21). For example, forskolin (adenylate cyclase activator) increases Ca2+ influx in somatotrophs, and inhibition of phosphodiesterase increases the electrical activity of somatotrophs confirming the relevance of cAMP in GHRH action (22). Regulated secretion of growth hormone involves movement of secretory vesicles along microtubules, transient “docking” in the cell membrane, and subsequent release of vesicles (21).
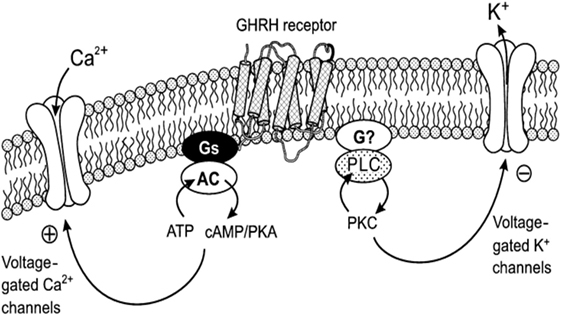
Figure 1. Mechanism of the action of GHRH on Ca2+ and K+ channels: coupling with protein kinase A (PKA) and protein kinase C (PKC) systems. This diagram illustrates the coupling of the Ca2+ and K+ channels with GHRH receptors. cAMP–PKA system mediates the action of GHRH on voltage-gated Ca2+ currents, and the PKC system is essential for the action of GHRH on voltage-gated K+ currents in somatotropes. AC, adenylyl cyclase; PLC, phospholipase C. Reprinted by permission from Macmillian Publishers Ltd., from Ref (23), Figure 11.
In pituitary somatotrophs, upon binding of the ligand GHRH to the GHRHR, the activated second messengers include not only the adenylate cyclase–cAMP–PKA and Ca2+-calmodulin but also inositol phosphate–diacylglycerol–protein kinase C (PKC), L-type calcium channels, and arachidonic acid–eicosanoic pathways as well, these ultimately result in the stimulation of growth hormone production and secretion (23–25). Increased cAMP also stimulates PKA to activate the cAMP response element-binding protein (CREB), which stimulates GHRHR gene transcription.
It is also likely that GHRH function relates to the ability to stimulate somatotroph cell proliferation. The activation of MAP kinase and ERK phosphorylation has been observed in the pituitary in a cAMP/PKA/PKC-dependent manner (26, 27). Alternatively, GHRH can stimulate the Ras/MAPK via βγ-subunits, to promote cell growth (26). In the myocardium, GHRHR-mediated inhibition of apoptosis involves modulation of ERK1 and ERK2 and PI3K−Akt signaling because ERK1/2- and PI3K/Akt-specific inhibitors abolished these effects (28).
Numerous high affinity and high specificity agonists and antagonists of GHRHR have been developed (9, 29–31). There are remarkable results from studies of GHRH agonists for wound healing, cardioprotective action, and protection from pneumolysin-induced pulmonary permeability edema (5, 7, 9, 32, 33). On other hand, GHRHR antagonists show prominent effects in augmenting apoptosis and decreasing proliferation of multiple types of cancer cells (30, 34, 35).
Effects of GHRH and Relevant GHRHR Agonists in Pancreatic Beta-Cell and Islets
Insulin is produced by pancreatic beta-cells in the islets of Langerhans. GHRH receptors have been described in primary as well as clonal pancreatic beta-cells (insulinoma cells) and isolated islets (7, 8, 36, 37). Human GHRH can acutely stimulate insulin secretion from isolated rodent islets and dispersed beta-cells (38, 39) and from perfused dog pancreas (40). Intravenous injection of human GHRH to rats increased plasma concentration of insulin being released into the hepatic portal vein (39). In another functional assay, pretreatment with synthetic GHRH analogs improved the engraftment and the metabolic function of islets, following transplantation to streptozotocin (STZ)-induced diabetic mice (36). Pretreatment of rat islets with the GHRH agonist, JI-36, significantly enhanced graft function by improving glucose tolerance and increasing beta-cell insulin reserve in rats (41). Novel high affinity and high specificity agonists of GHRHR improve insulin secretion and preserve beta-cells and islets in lethality assays (7, 8). Based on these findings, GHRH and its corresponding receptor hold promising therapeutic potential for improving beta-cell function and possibly treating T2DM.
Interestingly, the discovery of GHRH was due in part to the recognition of ectopic GHRH secretion from human pancreatic islet tumors causing ectopic acromegaly (42–45). GHRH was, thus, found in human pancreatic tumor tissue extracts, leading to its structural elucidation (44). These data suggest that perhaps GHRH is expressed at low levels by pancreatic islet cells and possibly during development also at low levels. This suggests that GHRH may be part of a paracrine system in islets, but this possibility has not yet been investigated. It is also possible that the GHRHR exerts an influence on cell function even without receptor activation through some tonic receptor function.
Despite these advances, the details of GHRHR expression, signaling pathways, and function in pancreatic islet cells have not been fully elucidated. We will consider the possible mechanisms of regulation of insulin secretion as well as mechanisms relating to beta-cell proliferation to evaluate the possible roles of GHRHR activation.
The primary role of pancreatic beta-cells is to regulate metabolism by sensing changes in blood glucose concentration and responding by secreting precisely regulated amounts of insulin. The action of hormones and neurotransmitters contribute to such signaling and amplify the glucose-stimulated insulin secretion (GSIS) (46) (Figure 2). GSIS is Ca2+-dependent and is regulated by metabolic signals generated by glucose catabolism. Glucose-dependent signal transduction begins with uptake of glucose into beta-cells via the GLUT2 transporter. Cytoplasmic glucose molecules are rapidly phosphorylated by glucokinase and converted to pyruvate in the cytosol via the glycolytic pathway, then oxidized within the mitochondria via the tricarboxylic acid cycle and oxidative phosphorylation pathways, respectively. Glucose catabolism generates ATP. The membrane potential of beta-cells is controlled by KATP channels. Under basal conditions, sufficient KATP channels are open so that the plasma membrane is hyperpolarized. Blocking KATP channels by an ATP-dependent mechanism initiates plasma membrane depolarization that opens voltage-gated Ca2+ channels; Ca2+ enters the beta-cell from the extracellular milieu through of these channels and increases cytoplasmic Ca2+. Glucose-induced increases in cytoplasmic Ca2+ and insulin secretion are directly correlated (46, 47).
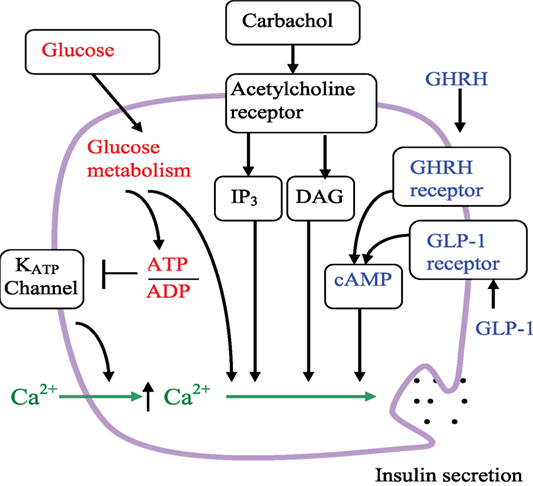
Figure 2. Schematic of the beta-cell signaling pathways and hypothetical role of GHRHR. Arrows indicate activation or an increase in concentration. The line ending with a bar indicates inhibition or closure. Glucose metabolism increases the ATP/ADP ratio, leading to the closure of KATP channels, reduction of K+ efflux, membrane depolarization, increase of intracellular Ca2+, and insulin secretion. Glucose also leads to insulin secretion through amplifying pathways that are independent of KATP channels. Carbachol stimulation enhanced insulin secretion via the acetylcholine (muscarinic) receptor and phospholipase C pathways. IP3 is inositol 1,4,5-trisphosphate, DAG is diacylglycerol. As depicted, GLP-1 and GHRH both enhance insulin secretion via the cAMP pathway.
The beta-cell membrane contains a profusion of GPCRs that are implicated in regulation of insulin secretion by hormones and neurotransmitters. GPCRs may have complimentary or antagonistic actions on insulin secretion (2–4). For example, a stimulation of insulin secretion by food begins with a “cephalic” phase due to sensory stimulation by sight and taste of food. This is largely mediated by the release of acetylcholine from nerves innervating pancreatic islets. The subsequent cholinergic stimulation via the muscarinic (acetylcholine) GPCR leads to an activation of the phospholipase C (PLC) pathway (48, 49).
Incretin hormones also play a critical role in insulin secretory response following meal ingestion. These hormones have significant influence on GSIS primarily through activation of the cAMP pathway that also leads to plasma membrane depolarization and an increase in cytoplasmic Ca2+ (50, 51). For example, glucagon-like peptide I (GLP-1) is one such potent incretin hormone activating the GPCR alpha(s) that can increase cAMP and activate the PKA pathway in beta-cells. GLP-1 agonists (and DPP4 inhibitors to prolong the half-life of endogenous incretins) have been successfully adopted for T2DM treatment (3, 50).
Growth hormone-releasing hormone and the incretin hormones such as GLP-I and GIP belong to the same class of the structurally related hormones that activate class B GPCRs. As discussed above, these incretin hormones activate the Gs-adenylate cyclase-cAMP signaling pathway, that is, this mechanism can be identical to that of GHRHR in somatotroph cells (see Figures 1 and 2). Interestingly, GHRHR agonists significantly increased the levels of cellular cAMP in rat beta-cell line (INS-1) (7). For this reason, it is reasonable to conclude that GHRHR agonists also employ a cAMP-based signaling mechanism and, therefore, they would have beneficial effects on insulin secretion and beta-cell survival (7). There is also a possibility that activation of cAMP pathway can lead to an increase in cytoplasmic Ca2+ concentration that can activate PLC and correspondingly activate this pathway [see, for example, Ref. (4)].
Interestingly, activation of the cAMP pathway in beta-cells by the incretin hormones leads to increased insulin secretion in part due to plasma membrane depolarization and an increase in cytoplasmic Ca2+ [for review see Ref. (4, 52)]. Mechanisms by which GHRH increases growth hormone-release also include plasma membrane depolarization and an increase in cytoplasmic Ca2+ (see above) that also point to significant similarities between the mechanisms of GLP-1 in beta-cells and the mechanisms of GHRH in the pituitary somatotropes.
Activation of GHRH receptors may also lead to activation of gene transcription, proliferation, and survival in beta-cells. For example, the mechanisms of beta-cell proliferation and survival include ERK and Akt signaling pathways (53). GHRHR agonists activate these pathways in various cell types (see above). Experiments in rat insulinoma cells (INS-1) showed that the GHRH agonist MR-409 significantly increased cell proliferation and induced activation of ERK and Akt pathways (7).
GHRH G-protein coupled receptor agonists significantly increased the levels of the phosphorylation of CREB in rat beta-cell line (INS-1) (7) as well as in somatotrophs, where GHRHR agonists can stimulate growth hormone gene transcription (see above). This may also be similar to one of the mechanisms of GLP-1. GLP-1 has also been shown to promote beta-cell proliferation and survival in rodents by activating ERK and Akt pathways (50). cAMP induced by GLP-1 caused elevated phosphorylation of CREB/activating-transcription-factor-1 in insulin-secreting beta-cells (54). However, the exact mode(s) of GHRHR signaling in the pancreatic islets and the most important mechanisms of stimulation of insulin secretion and/or beta-cell survival are unknown.
Causal interrelationships between GHRHR agonists and the variety of GPCRs in beta-cells and the role of such networks in insulin secretion are unknown. We have recently employed our general beta-cell mathematical modeling approach for beta-cell GPCRs for a comparison of action of GPCRs for GLP-1 and GIP (4). Both of them couple predominantly to the Gs-adenylate cyclase-cAMP signaling pathway. Based on those models, we suggest that GHRHR agonists can have a similar role as GIP in its interaction with GLP-1. In this case, GHRHR agonists can act in a competitive manner with GLP-1 in their mechanisms of stimulating insulin secretion. This testable hypothesis remains to be directly demonstrated.
There are several possible ways as to how pancreatic beta-cell GHRH signaling can have implications in T2DM treatment. One of the root causes of T2DM is the altered signaling system in beta-cells, which leads to decreased insulin production and exocytosis. Our previous published data and that of several other groups suggest that some signaling systems in insulin-secreting cells are damaged or attenuated in diabetic states. “Diabetic conditions” – such as hyperglycemia and hyperlipidemia – can lead to the loss of the GLP-1 receptor (GLP-1R) from the cell surface and, thereby, impair GLP-1 signaling, which may underlie the reduced clinical efficacy of GLP-1R activators (50, 51). GHRHR agonists can have beneficial effects under these conditions since these agonists activate the same cAMP pathway as GLP-1R, assuming the GHRHR is not also downregulated. Therefore, we can hypothesize that glycemic insensitivity to GLP-1R agonists in T2DM can be improved by simultaneous or sequential application of GHRHR agonists, thus replacing a possible deficit of GLP-1R.
Ex vivo treatment of isolated islets with GHRH agonists may also improve results of islet transplantation in animal models. Preconditioning of encapsulated pancreatic islets with GHRHR agonists significantly enhanced graft function by improving glucose tolerance and increasing beta-cell insulin reserve in diabetic rats (41). This effect is of sufficient interest to further examine it in human islet transplantation.
Interestingly, GHRH stimulated and GHRH antagonist inhibited the expression of the major antioxidant enzymes in the LNCaP human prostate cancer line (55). Additional expression of the major antioxidant enzymes may have additional benefits in T2DM (56) as well as T1D (57), and this may be another potentially beneficial effect of GHRHR agonists in both major types of diabetes.
Based on these studies, we suggest that GHRHR analogs have the potential to enhance beta-cell function, proliferation, and survival in vivo. Further studies with human islets and beta-cells will help determine if the GHRHR expression levels and signaling systems are similar in human and rodent models.
Diabetes and Activity of GHRH Beyond Beta-Cells
Remarkable results from the study of new GHRH agonists in wound healing and cardiovascular performance could also suggest novel treatments in patients with diabetes or perhaps help understand the pathways involved (5, 7, 9). GHRHR antagonists may target certain complications of diabetes, especially in Type 1 diabetes and insulin-dependent T2DM, where insulin production by the beta-cell is at least clinically insignificant. For example, GHRH antagonism may improve some of the lipid, renal, and vascular complications of low insulin-associated diabetes (58). Another potential target for GHRH antagonists could be diabetic retinopathy, which is the main cause of blindness in patients with diabetes and diabetic nephropathy (glomerulosclerosis) (30). Despite remarkable advances in treatment and prevention of these complications, they are still dramatic components of the long-term costs of diabetes.
Gastrointestinal effects are also complications of diabetes. There was upregulation of GHRHR expression in intestinal cells in a mouse model of type 2 diabetes (58). Treatment with the GHRHR antagonist, MIA-602, interfered with GLP-1-dependent diabetes-related dyslipidemia in mice. It also decreased the plasma levels of GLP-1, glucagon, and TRL in these mice (58), which might lead to worsening of diabetes rather than improvement. Cross-talk between the GHRHR antagonist and acetylcholine signaling (M3 receptor) was observed in the aorta, where MIA-602 prevented the diabetes-related block of carbachol-mediated vasodilation (58).
Interestingly, human GHRH can reduce glucagon release from isolated mouse islets (39). We can explain this by the increased insulin secretion that suppresses glucagon secretion in this case [see, for example, Ref. (59)]. However, enhanced glucagon secretion by islet cells in diabetes was also lowered by application of antagonist MIA-602 (58). The decreased glucagon secretion in this case can be explained by cAMP decrease in alpha cells [see, for example, Ref. (60)] through blocking the corresponding GHRH receptor when insulin secretion is insignificant in islets from diabetic animals or humans. Decreased glucagon release by GHRHR antagonists could have a beneficial effect in diabetes through decreasing hepatic glucose production and perhaps decreasing ketogenesis (59, 61).
Conclusion
This review of recent data with GHRHR agonists shows them to be capable of acutely increasing insulin secretion and enhancing rodent beta-cell proliferation and survival, when administered systemically. On the other hand, the modulators of GHRHR activity may be useful in ameliorating certain complications of diabetes. Studies are currently ongoing to determine the dose and treatment regimes of GHRHR modulators for the treatment of other diseases. The results demonstrate a clear connection of GHRH and its receptor with glucose metabolism and pancreatic beta-cell function. We believe that there is a sound basis for further studies evaluating GHRHR agonists and/or antagonists as promising therapeutic agents for diabetes and its complications.
Author Contributions
LF, NT, AS, and LP reviewed the literature, designed the work, and, together, wrote and edited the paper.
Conflict of Interest Statement
The authors have no commercial or financial relationship to disclose that could have influenced the redaction of the present review.
Funding
This work was supported by grants from the National Institutes of Health to LP (DK063493 and DK092616), and by University of Chicago Diabetes Research and Training Center funding from the NIH (DK020595, PI G.I. Bell). Various studies on GHRH analogs by the group of AS, cited in this manuscript were supported by the Medical Research Service of the Department of Veterans Affairs.
Abbreviations
GHRH, growth hormone-releasing hormone; GHRHR, GHRH G-protein coupled receptor; GLP-1, glucagon-like peptide 1; GLP-1R, receptor for GLP-1; GSIS, glucose-stimulated insulin secretion; GPCR, G-protein coupled receptor; KATP channels, ATP-sensitive K+ channels; PKA, protein kinase A; PLC, phospholipase C, T2DM, type 2 diabetes mellitus.
References
1. Kahn SE, Cooper ME, Del Prato S. Pathophysiology and treatment of type 2 diabetes: perspectives on the past, present, and future. Lancet (2014) 383(9922):1068–83. doi:10.1016/S0140-6736(13)62154-6
2. Winzell MS, Ahren B. G-protein-coupled receptors and islet function-implications for treatment of type 2 diabetes. Pharmacol Ther (2007) 116(3):437–48. doi:10.1016/j.pharmthera.2007.08.002
3. Amisten S, Salehi A, Rorsman P, Jones PM, Persaud SJ. An atlas and functional analysis of G-protein coupled receptors in human islets of Langerhans. Pharmacol Ther (2013) 139(3):359–91. doi:10.1016/j.pharmthera.2013.05.004
4. Fridlyand LE, Philipson LH. Pancreatic beta cell G-protein coupled receptors and second messenger interactions: a systems biology computational analysis. PLoS One (2016) 11(5):e0152869. doi:10.1371/journal.pone.0152869
5. Kiaris H, Chatzistamou I, Papavassiliou AG, Schally AV. Growth hormone-releasing hormone: not only a neurohormone. Trends Endocrinol Metab (2011) 22(8):311–7. doi:10.1016/j.tem.2011.03.006
6. Ziegler CG, Brown JW, Schally AV, Erler A, Gebauer L, Treszl A, et al. Expression of neuropeptide hormone receptors in human adrenal tumors and cell lines: antiproliferative effects of peptide analogues. Proc Natl Acad Sci U S A (2009) 106(37):15879–84. doi:10.1073/pnas.0907843106
7. Zhang X, Cui T, He J, Wang H, Cai R, Popovics P, et al. Beneficial effects of growth hormone-releasing hormone agonists on rat INS-1 cells and on streptozotocin-induced NOD/SCID mice. Proc Natl Acad Sci U S A (2015) 112(44):13651–6. doi:10.1073/pnas.1518540112
8. Schubert U, Schmid J, Lehmann S, Zhang XY, Morawietz H, Block NL, et al. Transplantation of pancreatic islets to adrenal gland is promoted by agonists of growth-hormone-releasing hormone. Proc Natl Acad Sci U S A (2013) 110(6):2288–93. doi:10.1073/pnas.1221505110
9. Cai R, Schally AV, Cui T, Szalontay L, Halmos G, Sha W, et al. Synthesis of new potent agonistic analogs of growth hormone-releasing hormone (GHRH) and evaluation of their endocrine and cardiac activities. Peptides (2014) 52:104–12. doi:10.1016/j.peptides.2013.12.010
10. Schally AV. Aspects of hypothalamus regulation of the pituitary gland with major emphasis on its implications for the control of reproductive processes. Nobel lecture, 8 December 1977. In: Lindsten J, editor. Nobel Lectures, Physiology or Medicine 1071-1980. Singapore: Word Scientific Publishing Co (1992). p. 405–38.
11. Mayo KE, Godfrey PA, Suhr ST, Kulik DJ, Rahal JO. Growth hormone-releasing hormone: synthesis and signaling. Recent Prog Horm Res (1995) 50:35–73.
12. Frohman LA, Downs TR, Heimer EP, Felix AM. Dipeptidylpeptidase IV and trypsin-like enzymatic degradation of human growth hormone-releasing hormone in plasma. J Clin Invest (1989) 83(5):1533–40. doi:10.1172/JCI114049
13. Okimura Y, Chihara K, Abe H, Kaji H, Kodama H, Kita T, et al. Plasma disappearance half-time and metabolic clearance rate of exogenous human growth hormone-releasing hormone-(1-44)-NH2 in normal subjects. Endocrinol Jpn (1986) 33(6):875–81. doi:10.1507/endocrj1954.33.875
14. Nauck M. Incretin therapies: highlighting common features and differences in the modes of action of glucagon-like peptide-1 receptor agonists and dipeptidyl peptidase-4 inhibitors. Diabetes Obes Metab (2016) 18(3):203–16. doi:10.1111/dom.12591
15. Granata R. Peripheral activities of growth hormone-releasing hormone. J Endocrinol Invest (2016) 39(7):721–7. doi:10.1007/s40618-016-0440-x
16. Frohman LA, Kineman RD. Growth hormone-releasing hormone and pituitary development, hyperplasia and tumorigenesis. Trends Endocrinol Metab (2002) 13(7):299–303. doi:10.1016/S1043-2760(02)00613-6
17. Martin B, Lopez de Maturana R, Brenneman R, Walent T, Mattson MP, Maudsley S. Class II G protein-coupled receptors and their ligands in neuronal function and protection. Neuromolecular Med (2005) 7(1–2):3–36. doi:10.1385/NMM:7:1-2:003
18. Holl RW, Thorner MO, Leong DA. Intracellular calcium concentration and growth hormone secretion in individual somatotropes: effects of growth hormone-releasing factor and somatostatin. Endocrinology (1988) 122(6):2927–32. doi:10.1210/endo-122-6-2927
19. Tsaneva-Atanasova K, Sherman A, van Goor F, Stojilkovic SS. Mechanism of spontaneous and receptor-controlled electrical activity in pituitary somatotrophs: experiments and theory. J Neurophysiol (2007) 98(1):131–44. doi:10.1152/jn.00872.2006
20. Lin-Su K, Wajnrajch MP. Growth hormone releasing hormone (GHRH) and the GHRH receptor. Rev Endocr Metab Disord (2002) 3(4):313–23. doi:10.1023/A:1020949507265
21. Anderson LL, Scanes CG. Nanobiology and physiology of growth hormone secretion. Exp Biol Med (Maywood) (2012) 237(2):126–42. doi:10.1258/ebm.2011.011306
22. Stojilkovic SS, Tabak J, Bertram R. Ion channels and signaling in the pituitary gland. Endocr Rev (2010) 31(6):845–915. doi:10.1210/er.2010-0005
23. Chen C, Xu R, Clarke IJ, Ruan M, Loneragan K, Roh SG. Diverse intracellular signalling systems used by growth hormone-releasing hormone in regulating voltage-gated Ca2+ or K channels in pituitary somatotropes. Immunol Cell Biol (2000) 78(4):356–68. doi:10.1046/j.1440-1711.2000.00917.x
24. Muller EE, Locatelli V, Cocchi D. Neuroendocrine control of growth hormone secretion. Physiol Rev (1999) 79(2):511–607.
25. Yang SK, Wang K, Parkington H, Chen C. Involvement of tetrodotoxin-resistant Na+ current and protein kinase C in the action of growth hormone (GH)-releasing hormone on primary cultured somatotropes from GH-green fluorescent protein transgenic mice. Endocrinology (2008) 149(9):4726–35. doi:10.1210/en.2008-0405
26. Pombo CM, Zalvide J, Gaylinn BD, Dieguez C. Growth hormone-releasing hormone stimulates mitogen-activated protein kinase. Endocrinology (2000) 141(6):2113–9. doi:10.1210/endo.141.6.7513
27. Zeitler P, Siriwardana G. Stimulation of mitogen-activated protein kinase pathway in rat somatotrophs by growth hormone-releasing hormone. Endocrine (2000) 12(3):257–64. doi:10.1385/ENDO:12:3:257
28. Granata R, Trovato L, Gallo MP, Destefanis S, Settanni F, Scarlatti F, et al. Growth hormone-releasing hormone promotes survival of cardiac myocytes in vitro and protects against ischaemia-reperfusion injury in rat heart. Cardiovasc Res (2009) 83(2):303–12. doi:10.1093/cvr/cvp090
29. Izdebski J, Pinski J, Horvath JE, Halmos G, Groot K, Schally AV. Synthesis and biological evaluation of superactive agonists of growth hormone-releasing hormone. Proc Natl Acad Sci U S A (1995) 92(11):4872–6. doi:10.1073/pnas.92.11.4872
30. Schally AV, Varga JL, Engel JB. Antagonists of growth-hormone-releasing hormone: an emerging new therapy for cancer. Nat Clin Pract Endocrinol Metab (2008) 4(1):33–43. doi:10.1038/ncpendmet0677
31. Zarandi M, Varga JL, Schally AV, Horvath JE, Toller GL, Kovacs M, et al. Lipopeptide antagonists of growth hormone-releasing hormone with improved antitumor activities. Proc Natl Acad Sci U S A (2006) 103(12):4610–5. doi:10.1073/pnas.0511348103
32. Kanashiro-Takeuchi RM, Szalontay L, Schally AV, Takeuchi LM, Popovics P, Jaszberenyi M, et al. New therapeutic approach to heart failure due to myocardial infarction based on targeting growth hormone-releasing hormone receptor. Oncotarget (2015) 6(12):9728–39. doi:10.18632/oncotarget.3303
33. Lucas R, Sridhar S, Rick FG, Gorshkov B, Umapathy NS, Yang G, et al. Agonist of growth hormone-releasing hormone reduces pneumolysin-induced pulmonary permeability edema. Proc Natl Acad Sci U S A (2012) 109(6):2084–9. doi:10.1073/pnas.1121075109
34. Letsch M, Schally AV, Busto R, Bajo AM, Varga JL. Growth hormone-releasing hormone (GHRH) antagonists inhibit the proliferation of androgen-dependent and -independent prostate cancers. Proc Natl Acad Sci U S A (2003) 100(3):1250–5. doi:10.1073/pnas.0337496100
35. Szalontay L, Schally AV, Popovics P, Vidaurre I, Krishan A, Zarandi M, et al. Novel GHRH antagonists suppress the growth of human malignant melanoma by restoring nuclear p27 function. Cell Cycle (2014) 13(17):2790–7. doi:10.4161/15384101.2015.945879
36. Ludwig B, Ziegler CG, Schally AV, Richter C, Steffen A, Jabs N, et al. Agonist of growth hormone-releasing hormone as a potential effector for survival and proliferation of pancreatic islets. Proc Natl Acad Sci U S A (2010) 107(28):12623–8. doi:10.1073/pnas.1005098107
37. Schmid J, Ludwig B, Schally AV, Steffen A, Ziegler CG, Block NL, et al. Modulation of pancreatic islets-stress axis by hypothalamic releasing hormones and 11beta-hydroxysteroid dehydrogenase. Proc Natl Acad Sci U S A (2011) 108(33):13722–7. doi:10.1073/pnas.1110965108
38. Green IC, Southern C, Ray K. Mechanism of action of growth-hormone-releasing hormone in stimulating insulin secretion in vitro from isolated rat islets and dispersed islet cells. Horm Res (1990) 33(5):199–204. doi:10.1159/000181509
39. Bailey CJ, Wilkes LC, Flatt PR, Conlon JM, Buchanan KD. Effects of growth hormone-releasing hormone on the secretion of islet hormones and on glucose homeostasis in lean and genetically obese-diabetic (ob/ob) mice and normal rats. J Endocrinol (1989) 123(1):19–24. doi:10.1677/joe.0.1230019
40. Hermansen K, Kappelgaard AM. Characterization of growth hormone-releasing hormone stimulation of the endocrine pancreas: studies with alpha- and beta-adrenergic and cholinergic antagonists. Acta Endocrinol (Copenh) (1987) 114(4):589–94.
41. Ludwig B, Rotem A, Schmid J, Weir GC, Colton CK, Brendel MD, et al. Improvement of islet function in a bioartificial pancreas by enhanced oxygen supply and growth hormone releasing hormone agonist. Proc Natl Acad Sci U S A (2012) 109(13):5022–7. doi:10.1073/pnas.1201868109
42. Frohman LA. Ectopic hormone production. Am J Med (1981) 70(5):995–7. doi:10.1016/0002-9343(81)90847-0
43. Ghazi AA, Amirbaigloo A, Dezfooli AA, Saadat N, Ghazi S, Pourafkari M, et al. Ectopic acromegaly due to growth hormone releasing hormone. Endocrine (2013) 43(2):293–302. doi:10.1007/s12020-012-9790-0
44. Rivier J, Spiess J, Thorner M, Vale W. Characterization of a growth hormone-releasing factor from a human pancreatic islet tumour. Nature (1982) 300(5889):276–8. doi:10.1038/300276a0
45. Shibasaki T, Shizume K, Masuda A, Nakahara M, Hizuka N, Miyakawa M, et al. Plasma growth hormone response to growth hormone-releasing factor in acromegalic patients. J Clin Endocrinol Metab (1984) 58(1):215–7. doi:10.1210/jcem-58-1-212
46. Rorsman P, Braun M. Regulation of insulin secretion in human pancreatic islets. Annu Rev Physiol (2013) 75:155–79. doi:10.1146/annurev-physiol-030212-183754
47. Gilon P, Chae HY, Rutter GA, Ravier MA. Calcium signaling in pancreatic beta-cells in health and in type 2 diabetes. Cell Calcium (2014) 56(5):340–61. doi:10.1016/j.ceca.2014.09.001
48. Ahren B, Holst JJ. The cephalic insulin response to meal ingestion in humans is dependent on both cholinergic and noncholinergic mechanisms and is important for postprandial glycemia. Diabetes (2001) 50(5):1030–8. doi:10.2337/diabetes.50.5.1030
49. Seino Y, Miki T, Fujimoto W, Young Lee E, Takahashi Y, Minami K, et al. Cephalic phase insulin secretion is KATP channel independent. J Endocrinol (2013) 218(1):25–33. doi:10.1530/JOE-12-0579
50. Campbell JE, Drucker DJ. Pharmacology, physiology, and mechanisms of incretin hormone action. Cell Metab (2013) 17(6):819–37. doi:10.1016/j.cmet.2013.04.008
51. Rajan S, Dickson LM, Mathew E, Orr CM, Ellenbroek JH, Philipson LH, et al. Chronic hyperglycemia downregulates GLP-1 receptor signaling in pancreatic beta-cells via protein kinase A. Mol Metab (2015) 4(4):265–76. doi:10.1016/j.molmet.2015.01.010
52. Nadkarni P, Chepurny OG, Holz GG. Regulation of glucose homeostasis by GLP-1. Prog Mol Biol Transl Sci (2014) 121:23–65. doi:10.1016/B978-0-12-800101-1.00002-8
53. Wijesekara N, Krishnamurthy M, Bhattacharjee A, Suhail A, Sweeney G, Wheeler MB. Adiponectin-induced ERK and Akt phosphorylation protects against pancreatic beta cell apoptosis and increases insulin gene expression and secretion. J Biol Chem (2010) 285(44):33623–31. doi:10.1074/jbc.M109.085084
54. Malm HA, Mollet IG, Berggreen C, Orho-Melander M, Esguerra JL, Goransson O, et al. Transcriptional regulation of the miR-212/miR-132 cluster in insulin-secreting beta-cells by cAMP-regulated transcriptional co-activator 1 and salt-inducible kinases. Mol Cell Endocrinol (2016) 424:23–33. doi:10.1016/j.mce.2016.01.010
55. Barabutis N, Schally AV. Antioxidant activity of growth hormone-releasing hormone antagonists in LNCaP human prostate cancer line. Proc Natl Acad Sci U S A (2008) 105(51):20470–5. doi:10.1073/pnas.0811209106
56. Fridlyand LE, Philipson LH. Does the glucose-dependent insulin secretion mechanism itself cause oxidative stress in pancreatic beta-cells? Diabetes (2004) 53(8):1942–8. doi:10.2337/diabetes.53.8.1942
57. Padgett LE, Broniowska KA, Hansen PA, Corbett JA, Tse HM. The role of reactive oxygen species and proinflammatory cytokines in type 1 diabetes pathogenesis. Ann N Y Acad Sci (2013) 1281:16–35. doi:10.1111/j.1749-6632.2012.06826.x
58. Romero MJ, Lucas R, Dou H, Sridhar S, Czikora I, Mosieri EM, et al. Role of growth hormone-releasing hormone in dyslipidemia associated with experimental type 1 diabetes. Proc Natl Acad Sci U S A (2016) 113(7):1895–900. doi:10.1073/pnas.1525520113
59. Fridlyand LE, Philipson LH. A computational systems analysis of factors regulating alpha cell glucagon secretion. Islets (2012) 4(4):262–83. doi:10.4161/isl.22193
60. Elliott AD, Ustione A, Piston DW. Somatostatin and insulin mediate glucose-inhibited glucagon secretion in the pancreatic alpha-cell by lowering cAMP. Am J Physiol Endocrinol Metab (2015) 308(2):E130–43. doi:10.1152/ajpendo.00344.2014
Keywords: diabetic complications, GLP-1, islet, insulin, pancreatic beta-cell
Citation: Fridlyand LE, Tamarina NA, Schally AV and Philipson LH (2016) Growth Hormone-Releasing Hormone in Diabetes. Front. Endocrinol. 7:129. doi: 10.3389/fendo.2016.00129
Received: 17 May 2016; Accepted: 06 September 2016;
Published: 10 October 2016
Edited by:
Romesh Khardori, Eastern Virginia Medical School, USAReviewed by:
Riccarda Granata, University of Turin, ItalyBenjamin Glaser, Hadassah-Hebrew University Medical Center, Israel
Copyright: © 2016 Fridlyand, Tamarina, Schally and Philipson. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Leonid E. Fridlyand, bGZyaWRsaWFAbWVkaWNpbmUuYnNkLnVjaGljYWdvLmVkdQ==