 Isabel Marian Hartmann Quadros*
Isabel Marian Hartmann Quadros* Giovana Camila Macedo
Giovana Camila Macedo Liz Paola Domingues
Liz Paola Domingues Cristiane Aparecida Favoretto
Cristiane Aparecida Favoretto
- Department of Psychobiology, Escola Paulista de Medicina, Universidade Federal de São Paulo, São Paulo, São Paulo, Brazil
Alcohol is the most commonly used and abused substance worldwide. The emergence of alcohol use disorders, and alcohol dependence in particular, is accompanied by functional changes in brain reward and stress systems, which contribute to escalated alcohol drinking and seeking. Corticotropin-releasing factor (CRF) systems have been critically implied in the transition toward problematic alcohol drinking and alcohol dependence. This review will discuss how dysregulation of CRF function contributes to the vulnerability for escalated alcohol drinking and other consequences of alcohol consumption, based on preclinical evidence. CRF signaling, mostly via CRF1 receptors, seems to be particularly important in conditions of excessive alcohol taking and seeking, including during early and protracted withdrawal, relapse, as well as during withdrawal-induced anxiety and escalated aggression promoted by alcohol. Modulation of CRF1 function seems to exert a less prominent role over low to moderate alcohol intake, or to species-typical behaviors. While CRF mechanisms in the hypothalamic–pituitary–adrenal axis have some contribution to the neurobiology of alcohol abuse and dependence, a pivotal role for extra-hypothalamic CRF pathways, particularly in the extended amygdala, is well characterized. More recent studies further suggest a direct modulation of brain reward function by CRF signaling in the ventral tegmental area, nucleus accumbens, and the prefrontal cortex, among other structures. This review will further discuss a putative role for other components of the CRF system that contribute for the overall balance of CRF function in reward and stress pathways, including CRF2 receptors, CRF-binding protein, and urocortins, a family of CRF-related peptides.
Overview
For a few decades now, a critical role for stress in the induction, maintenance, and relapse to drug and alcohol dependence has been increasingly investigated. Indeed, drug dependence has been hypothesized as a “stress surfeit disorder” (1). Dysregulation of brain stress systems would contribute to the transition from drug-taking, which is primarily motivated by reward-seeking, toward escalated drug-taking that becomes mainly instigated by dysphoria and negative reinforcement. As the very primary player mediating brain stress responses, the corticotropin-releasing factor [CRF; also known as corticotropin-releasing hormone (CRH)] has received great attention in drug dependence. This review will discuss evidence provided by preclinical studies focusing on the critical participation of CRF and its closely related peptides urocortins, in escalated alcohol drinking, and other important alcohol-related behaviors. Also, apart from its role in more traditional brain stress circuits, this review will discuss increasing evidence for the CRF system as a direct modulator of brain reward pathways, suggesting that CRF/urocortin signaling may also modulate alcohol’s effects at earlier stages of alcohol dependence. The impact of alcohol exposure on CRF/urocortin signaling and plasticity will be discussed, as potential neuroadaptive mechanisms recruited during the establishment of alcohol dependence. Pharmacological manipulations targeting CRF receptors are presented as promising tools for reducing excessive alcohol drinking, withdrawal-related anxiety as well as alcohol-induced aggression and behavioral sensitization induced by alcohol. The participation of the hypothalamic–pituitary–adrenal (HPA) stress axis is addressed at specific points, but specifically as a downstream target engaged by brain CRF [for reviews on HPA axis and alcohol, refer to Stephens and Wand (2) and Edwards et al. (3)]. Finally, considerations are made on a few human alcohol studies evaluating polymorphisms in CRF-related genes. This review does not attempt to discuss alcohol studies using genetically modified animals for CRF-related genes, a subject vastly covered by a recent review on CRF systems and alcohol (4).
We refer to several review articles published on CRF and drug/alcohol dependence [e.g., Ref. (5–7)] as well as a recent review specifically focusing on CRF mechanisms involved in alcohol use disorders (AUDs) and alcohol-induced neuroplasticity (4).
Alcohol and Alcohol Use Disorders
Alcohol is the most widely consumed psychotropic substance worldwide. Likewise, the harmful use of alcohol ranks within the five main risk factors for disease, disability, and death in the globe (8). AUDs are responsible for ~6% of all deaths throughout the world, even considering potential beneficial effects of low-risk alcohol drinking (8). Consequences of alcohol misuse can be both acute (e.g., acute intoxication, heavy episodic drinking, driving under the influence of alcohol, alcohol-related violence) and chronic (e.g., AUDs, gastrointestinal diseases including liver cirrhosis, cancers), and cause harm to the drinker, to other individuals, and to society in general. Thus, alcohol consumption is responsible for a large burden in public health, with serious economic and social impact.
Alcohol-related health harm is proportional to the amount of alcohol consumed, with a dose–response relationship. About 13% of the alcohol-drinking population will show a pattern of heavy episodic drinking, or “binge” drinking, which is more commonly associated with alcohol-related injury, including driving accidents and violent behavior (9). AUDs are the main neuropsychiatric condition promoted by alcohol consumption and encompass both the harmful use of alcohol and alcohol dependence. Alcohol dependence affects ~5% of the adult (15 years and older) population worldwide (8) and consists of a variety of behavioral, cognitive, and physiological symptoms that follow repeated alcohol use. According to the International Classification of Diseases, main features of alcohol dependence include a strong desire to consume alcohol; difficulties in controlling the frequency and amount of alcohol consumption; persistent use despite perceived harmful consequences; a higher priority given to obtaining and using alcohol relative to other activities and obligations; increased tolerance; and sometimes a physiological withdrawal state (10).
Despite the complexity of biological, environmental, and social factors that influence drug and AUDs, animal models have been instrumental in providing evidence for behavioral, pharmacological, and neurobiological mechanisms underlying acute and repeated alcohol exposure [for a review, see Ref. (11)]. This review will focus primarily on findings stemming from preclinical studies and how they contribute to our understanding of CRF/urocortin mechanisms in alcohol dependence. Of particular interest are models of voluntary alcohol consumption, including home cage drinking (e.g., two-bottle choice, with intermittent, limited, or continuous access conditions; the “drinking in the dark” model, etc.) and operant self-administration (animals respond by pressing a lever or nose poking in order to obtain an alcohol reward). Using these experimental models, it is also possible to promote escalated, excessive alcohol intake by using protocols of repeated and prolonged voluntary access to the substance, usually followed by one or multiple cycles of withdrawal. Examples are procedures such as the alcohol-deprivation effect (12) and dependent-like alcohol consumption in subsets of chronically drinking rats and mice [e.g., Ref. (13, 14)]. Other widely used procedures for escalated drinking rely on forced alcohol exposure in order to obtain reliable and consistent exposure to high alcohol concentrations, such as the alcohol vapor chamber (15, 16), as well as alcohol-containing liquid diet [see also Ref. (17) for a review]. Such excessive alcohol exposure is usually associated with withdrawal symptoms upon short-term withdrawal, and dysphoria and anxiety during protracted abstinence. These negative features trigger and motivate increased levels of voluntary alcohol self-administration [e.g., Ref. (18); see review by Heilig and Koob (5)].
Different from other drugs of abuse, such as cocaine, amphetamines, and morphine, pharmacological mechanisms for alcohol include a variety of targets in the central nervous system. Synaptic actions of alcohol have been described with several neurotransmitter-gated ionotropic receptors (e.g., GABAA, glutamate NMDA and AMPA receptors, serotonin 5-HT3 receptors, etc.), ion channels (e.g., different subtypes of calcium and potassium channels), as well as intracellular downstream signaling proteins also involved in metabotropic receptors cascades (e.g., diacylglycerol, protein kinase A, protein kinase C, etc.), as extensively reviewed by Lovinger and Roberto (19). Additionally, presynaptic effects of alcohol may be observed, with a facilitation or potentiation of presynaptic GABA release, but not glutamate, as revealed by electrophysiological studies using slice preparations or isolated neurons [reviewed by Lovinger and Roberto (19) and Siggins et al. (20)]. Chronic alcohol exposure produces consistent neuroadaptive changes in the function of both ionotropic and metabotropic glutamate receptors, for example, with the upregulation of NMDA receptors [e.g., Ref. (21–25)]. Likewise, GABAA receptors are markedly affected in their subunit composition, sensitivity, and function by chronic alcohol treatment [see reviews by Grobin et al. (26) and Kumar et al. (27)]. These and many other changes in synaptic function, which vary in different brain regions, are considered critical for the development of tolerance and dependence to alcohol (19).
The effects of alcohol are importantly modulated by neuropeptides, including opioid peptides, neuropeptide Y, orphanin/nociceptin, and orexin. This review will focus on the family of neuropeptides comprised of CRF and its closely related peptides, the urocortins. The next section will briefly describe the physiology of CRF/urocortin signaling in the brain, and subsequently we will discuss the critical role of CRF signaling in AUDs.
A Glance at CRF/Urocortin Systems in the Brain
The 41-amino acid neuropeptide CRF was identified in 1981 by Vale and colleagues and has long been associated with neural, endocrine, autonomic, and immune responses to stress (28). Acting as a neuromodulator, the availability of CRF is critically determined by the CRF-binding protein (CRF-BP), a glycoprotein that regulates the extracellular availability of CRF to bind to its receptors (29, 30). CRF exerts its effects via interaction with two G-protein coupled receptors, namely CRFR1 and CRFR2 (CRF receptor 1 and 2, respectively), which are found in several variants (31). However, CRF has a 10-fold higher affinity for CRFR1 relative to CRFR2 (32). Urocortins, another set of peptides in the CRF family, are the main endogenous ligands for CRFR2, showing similar affinity for both receptor subtypes (urocortin 1), or binding almost exclusively to CRFR2 [urocortins 2 and 3; (33)]. Activation of both CRFR1 and CRFR2 preferentially lead to the activation of cyclic-AMP second messenger pathways (34). The two receptor subtypes are differentially distributed in the brain, with overlapping regions (35). CRFR1 are more ubiquitously found. In rodents, high densities of CRFR1 are found in the anterior hypophysis, cerebral cortex, cerebellum, amygdala, hippocampus, and striatum (36), whereas CRFR2 are more limited to the mesencephalon, raphé nuclei, lateral septum, amygdala, and hypothalamus (37), as recently reviewed by Phillips et al. (4). Although not the focus of the current review, CRF and urocortin peptides, as well as receptors and binding protein, are widely distributed in peripheric organs and tissues, including the gastrointestinal tract, cardiovascular, and immune systems, where they integrate systemic stress responses and participate in other functions [see reviews by Fekete and Zorrilla (33), Pan and Kastin (38), Stengel and Taché (39)].
As a major modulator of systemic neuroendocrine stress responses, hypothalamic CRF drives the HPA axis, with secondary modulation by the neuropeptide arginine vasopressin [e.g., Ref. (28, 40, 41)]. Produced by parvocellular and magnocellular neurons of the paraventricular nucleus of the hypothalamus (PVN), AVP has a limited capacity to activate the HPA axis by itself (4, 42). However, the neuropeptide AVP seems to increase the effects of CRF on HPA axis by synergistic activation of its V1b receptor in the anterior pituitary (4, 42, 43). Produced by parvocellular neurons of the PVN, CRF is released in the median eminence to reach the anterior hypophysis (or pituitary), where it binds to densely expressed receptors, CRFR1 (44). Adrenocorticotropic hormone (ACTH) is then secreted into the blood stream by the adenohypophysis (45). Once secreted, ACTH stimulates the production and release of glucocorticoids from the cortex of adrenal glands (cortisol in humans, corticosterone in rodents). Glucocorticoids will then act upon high- and low-affinity receptors (mineralocorticoid and glucocorticoid receptors, respectively), widely distributed in the periphery and the brain. Cortisol and corticosterone promote adaptive responses to environmental challenges and stressors, including changes in energy metabolism, physiological, and behavioral responses. HPA axis function is importantly regulated via inhibitory feedback by glucocorticoids, ultimately reducing activity of PVN neurons and CRF release. For a more complete discussion on HPA axis signaling and functions, we refer to reviews by Herman et al. (45), McEwen (46), Myers et al. (47), Sapolsky et al. (48), and Ulrich-Lai and Herman (49).
Wide distribution of cell bodies and fibers with immunoreactivity for CRF is found in stress-related pathways involving amygdalar nuclei [especially the central nucleus of the amygdala (CeA)], the bed nucleus of the stria terminalis (BNST), and the PVN, with ascending projections to forebrain structures as well as descending innervations to the brain stem (31). High levels of CRF are also detected in the hippocampus, thalamus, locus coeruleus, raphé nuclei, and other mesencephalic structures (50, 51). Detection of neurons and fibers containing urocortin peptides are limited to fewer brain structures and projection sites in rodents, although a wider distribution of brain urocortin can be found in human and non-human primates (38, 52, 53). As extensively reviewed by Fekete and Zorrilla (33), urocortin 1 (Ucn1) is primarily synthesized in the centrally projecting Edinger–Westphal nucleus, a midbrain structure, and other secondary sites. Descending fibers of Ucn1 are found in many regions, including the substantia nigra, the dorsal raphe nucleus, and periaqueductal gray, while ascending fibers of Ucn1 are found in the lateral septum, BNST, hypothalamus, and other structures. Urocortin 2 (Ucn2) is synthesized in the PVN and other hypothalamic nuclei, as well as in the locus coeruleus, but its projection targets are unknown (33). Urocortin 3 (Ucn3) is produced in hypothalamic and amygdala regions. Ucn3 fibers project from the amygdala to the hypothalamic ventral premammillary nucleus, while fibers from undertermined origins are also found in other hypothalamic regions, in lateral septum, BNST, medial amygdala, and ventral hippocampus [for reviews, see Ref. (33, 54)].
Briefly, biological functions mediated by CRF and urocortin peptides include not only stress responses but also regulation of appetitive responses, such as feeding and exploratory behaviors, and a variety of social behaviors (38, 55, 56). In several of these functions, CRF receptor subtypes often play different roles (57, 58). For example, stress and anxiety-like responses are thought to be primarily initiated by CRF/urocortin 1 activation of CRFR1 signaling, while CRFR2 activation has been associated with anxiolytic effects and/or termination of stress responses (38, 59). As another example, CRF and urocortins inhibit feeding behavior likely via CRFR2 signaling, although both CRFR1 and CRFR2 are involved in stress-induced anorexigenic effects (54). Thus, biological responses involving CRF and urocortins are wide and complex, and recruitment of specific CRF receptors will vary according to each particular behavior and/or pathology, in a brain region-dependent manner.
Through actions on CRFR1 and/or CRFR2, CRF and urocortin peptides set the pace for brain monoaminergic function in regions, such as the locus coeruleus (primary site for noradrenergic neurons in the brain), the dorsal and median raphé (serotonin neurons), and the ventral tegmental area (VTA), where reward-related dopamine neurons are located [e.g., Ref. (57, 60–62)]. Of particular interest to this review, dopamine neurons in the VTA receive CRF inputs from fibers that originate in the BNST, the CeA and, to a lesser extent, the hypothalamic PVN (63). In the VTA, CRF stimulates firing of dopamine neurons likely via CRFR1 signaling, modulating dopamine output to the nucleus accumbens (64). On the other hand, when CRF is directly applied in the nucleus accumbens, it can increase dopamine release and promote appetitive behavior (65). Exposure to acute stress is well known to induce activation of this dopamine pathway arising from the VTA and projecting to the nucleus accumbens and the prefrontal cortex, typically promoting increased drug-taking and/or drug-seeking behaviors [see review by Holly and Miczek (66)]. Interestingly, the ability of stress to trigger activation of dopamine brain reward pathways may be a downstream event from stress-induced increased CRF signaling within the VTA. For example, acute exposure to footshock stress promotes marked increases in CRF levels in the VTA and is associated with dopamine increases in the VTA and with reinstatement of cocaine-seeking behavior in cocaine-experienced rats (67). Remarkably, these CRF effects seem to be primarily mediated by CRFR2 in the VTA (68). More recently, acute or repeated social defeat stress were also shown to phasically increase CRF levels in the VTA, but with regional heterogeneity (anterior vs. posterior subregions of VTA), while repeated stress promotes long-term increases in CRF tonus in both subregions (69). In this case, both CRFR1 and CRFR2 modulated cocaine-seeking behaviors in rats with a history of social stress, according to the subregion of the VTA (69). Evidence for the involvement of CRF/urocortin signaling in alcohol drinking and other alcohol-related behaviors will be specifically addressed in the following sections.
Modulation of Alcohol Consumption by Pharmacological Manipulations of CRF/Urocortin Signaling
Extensive evidence points to a critical role for CRF and CRFR1 receptors in escalated alcohol consumption [(70–74); see review by Phillips et al. (4)]. As summarized in Table 1, antalarmin – a non-peptidergic CRFR1 antagonist – reduces free-choice alcohol drinking in rats given intermittent access to alcohol (72). Mice chronically exposed to alcohol vapor showed reduced escalated alcohol self-administration when treated with antalarmin prior to the withdrawal drinking session (75). In this study, control mice showed no changes in alcohol self-administration when treated with antalarmin, suggesting that only excessive drinking, in this case withdrawal-induced drinking, was sensitive to CRFR1 treatment (75). Binge drinking in C57BL6 mice was attenuated by another CRFR1 antagonist, CP-154,526, using the drinking-in-the-dark protocol (76). However, alcohol consumption was only reduced in high drinking conditions (blood alcohol ~80 mg/dl), not in moderate drinking conditions (blood alcohol ~40 mg/dl or lower). Other systemically administered CRFR1 antagonists were also effective in attenuating drinking-in-the-dark consumption, such as CP 376,395 and NBI-27914, despite non-selective effects reducing water and calory intake as well (77). The possibility that reduced alcohol drinking due to CRFR1 blockade could be secondary to broader effects on fluid and calory intake needs to be further investigated (77). A couple of studies failed to observe an attenuation of alcohol intake by CRFR1 antagonist when animals had continuous access to the drug [e.g., Ref. (78, 79)].
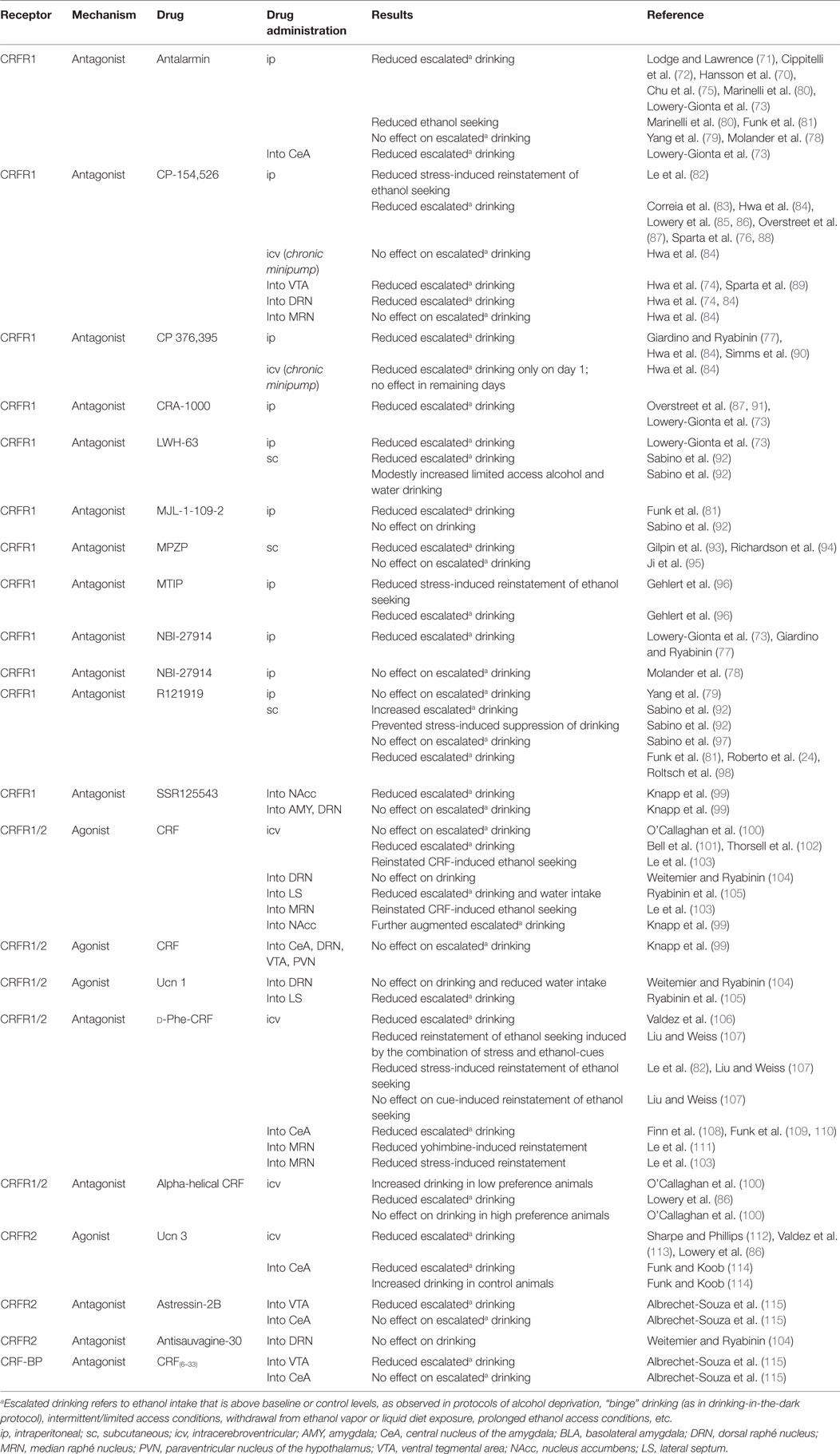
Table 1. Summarized effects of pharmacological manipulations of CRF/urocortin targets on alcohol-drinking studies.
Overall, these studies suggest that systemic CRFR1 antagonists present selective actions in reducing escalated alcohol drinking, but not moderate drinking, as summarized in Table 1 [e.g., Ref. (75, 76, 88)]. One particular study suggests that these effects are likely extra-hypothalamic and independent from HPA axis activation, since adrenalectomized mice show similar binge alcohol intake as controls, and CRFR1 blockade remains effective in attenuating drinking in adrenalectomized animals (86). Non-selective peptidergic CRF receptor antagonists, such as alpha-helical CRF or d-Phe-CRF, usually present a similar profile of effects as those of selective CRFR1 antagonists. In particular, icv infusion of d-Phe-CRF has been shown to reduce escalated alcohol drinking (106) and prevent reinstatement of stress-induced alcohol seeking (82, 107), just like more selective CRFR1 antagonists (see Table 1). When a selective CRFR1 antagonist was given chronically via icv minipumps, however, reduced drinking was only observed on the first drinking day and not on the following test sessions, suggesting that the chronic, continuous blockade of CRFR1 may not be as effective (84).
Important brain regions for the anti-drinking effects of selective CRFR1 and non-selective CRFR1/2 antagonists include brain reward and stress-related regions. For example, infusion of CP-154,526 into the VTA attenuated binge drinking in C57BL/6J mice (89). A different study also showed a reduction in two-bottle choice drinking in both rats and mice after intra-VTA infusion of CP-154,526 (74). CRFR1 signaling in the dorsal raphé nucleus (DRN) is also recruited for escalated alcohol drinking in rats and mice, likely due to a CRFR1 modulation of serotonergic output to the prefrontal cortex (74, 84). On the other hand, while CRFR1 in the median raphé nucleus may not modulate drinking (84), blockade of CRFR1/2 in this region prevents stress-induced reinstatement of alcohol seeking (103, 111). Infusion of either the non-selective antagonist d-Phe-CRF or antalarmin into the CeA also attenuates binge drinking (73) and withdrawal-induced alcohol self-administration (108–110). Notably, the preferential, but non-selective, activation of CRFR1 by CRF itself, whether icv or in different brain regions, produces inconsistent outcomes in alcohol drinking [e.g., Ref. (100, 101, 104, 105)]. Nonetheless, infusion of CRF into the ventricles or into the median raphé promotes reinstatement of alcohol-seeking behavior (103).
Using a different procedure, high alcohol-preferring P rats were exposed to three cycles of voluntary alcohol drinking (5 days), with 2-day withdrawal periods in between cycles (99). During withdrawal periods, rats received the infusion of a CRFR1 antagonist, SSR 125543, into the nucleus accumbens prior to exposure to restraint stress. Escalated drinking was promoted by the multiple cycles of withdrawal and stress exposure, and was attenuated by the blockade of CRFR1 in the accumbens during withdrawal periods (99). Using the same protocol, CRFR1 blockade in the DRN or the amygdala failed to affect escalated alcohol drinking (99). Interestingly, intra-accumbens infusion of CRF itself during the withdrawal periods (as a substitute for restraint stress) augmented alcohol consumption but had no effects on drinking when microinjected into the VTA, amygdala, DRN, or the paraventricular nucleus of the hypothalamus (99). Thus, plastic changes in accumbal CRFR1 seem to be recruited during cycles of withdrawal and stress, which underlie escalated drinking.
A few studies also suggest the involvement of urocortin peptides and CRFR2 in the modulation of escalated alcohol drinking. In mice, infusion of Ucn 1 (a non-selective agonist at CRFR1/2) into the lateral septum, but not into the dorsal raphé, significantly blunts binge alcohol drinking, likely due to a preferential action on CRFR2 (105). The selective activation of CRFR2 with icv administration of Ucn 3 reduced alcohol intake in mice (86, 112) and rats (113). Interestingly, this attenuation of escalated alcohol consumption can also be observed after infusion of Ucn 3 into the CeA of rats withdrawn from chronic alcohol vapor (114). On the other hand, blockade of CRFR2 in the CeA with astressin-2B failed to affect binge alcohol drinking in mice (115). Thus, these studies suggest that it is the activation, not the blockade of CRFR2, which prevents or attenuates excessive alcohol drinking, icv, or in regions such as the lateral septum or the CeA. However, one recent study reported decreased alcohol intake after infusion of a selective CRFR2 antagonist into the VTA (115), suggesting that different roles for CRFR2 may emerge from different brain pathways.
In summary, both subtypes of CRF receptors seem to be important during escalated alcohol drinking. While consistent and extensive evidence weighs toward CRFR1 modulation of excessive alcohol intake, increasing studies are currently suggesting that a balance between CRFR1 and CRFR2 activation/blockade may be critical in determining the final outcome. In particular brain regions such as the CeA, it seems that the inhibition of CRFR1 signaling and/or facilitation of CRFR2 function will blunt excessive alcohol drinking. On the other hand, blockade of either CRFR1 or CRFR2 in the VTA is effective in attenuating drinking (74, 89, 115). Thus, both CRFR1 and CRFR2 are important pharmacological targets that can contribute to treatment of AUDs. Furthermore, targets, such as the CRF-BP, have just started to emerge as interesting modulators of alcohol drinking, as recently suggested by Albrechet-Souza et al. (115).
CRF/Urocortin Modulation of Other Alcohol-Related Behaviors
Conditioned Place Preference
Conditioned place preference is a common model for indirectly assessing drug reward and drug seeking, by associating drug-induced reinforcing effects to a particular environment [for reviews, see Ref. (116–118)]. In this model, alcohol has reliably produced conditioned reward as evidenced by increased time spent in the alcohol-paired environment, relative to a different saline-paired environment, when the animal is tested in the absence of the drug (i.e., conditioned preference).
Genetic manipulations of the CRF system seem to affect the conditioned reinforcing effects of alcohol. Olive et al. (119) showed that transgenic CRF knockout mice fail to present preference to the alcohol-paired environment after conditioning sessions with a moderate dose of alcohol (2 g/kg), in a protocol that promotes significant preference in wild-type controls. Nonetheless, with a higher conditioning dose of alcohol (3 g/kg), both wild-type and knockout mice presented conditioned preference for the alcohol-associated context (119). These results suggest that CRF may have a facilitatory role in alcohol-conditioned reward, but other mechanisms contribute as well.
Urocortin1 knockout mice failed to present preference to the alcohol-paired environment (2 g/kg conditioning dose), and this strain also presents decreased alcohol intake and preference in a two-bottle choice protocol (120). Nonetheless, alcohol-conditioned aversive effects were preserved in this strain, suggesting that different mechanisms underlie conditioned reward and conditioned aversion (120). Furthermore, CRFR2 knockout mice do not display alcohol-induced conditioned place preference (120). Since Ucn1 acts on both CRFR1 and CRFR2, authors suggest that Ucn1-mediated activation of CRFR2 would be critical for the conditioned effects of alcohol. These were the only two studies assessing conditioned place preference for alcohol and CRF/urocortin systems. Determination of specific mechanisms requires assessment of the role of CRFR1 in this model, and further pharmacological studies.
Behavioral Sensitization
Similar to other drugs of abuse, repeated injections of alcohol promotes neuroadaptation in brain reward pathways, rendering the brain more vulnerable and sensitive to drug-induced reward and stimulation [see reviews by Robinson and Berridge (121, 122), Steketee and Kalivas (123), Vanderschuren and Pierce (124)]. Behaviorally, this neurochemical sensitization may be accompanied by progressively augmented motor stimulation responses or “behavioral sensitization” [e.g., Ref. (125–129)]. Different from other drugs, however, alcohol-induced locomotor sensitization is not easily demonstrated in rats1 (130, 131) but is well described in some inbred and outbred strains of mice [e.g., Ref. (132–135); see also Ref. (136) for a report on adolescent macaques]. Also, a putative role for sensitized behavioral responses to alcohol has been suggested as a vulnerability factor for alcohol dependence in humans (137, 138).
Components of the CRF system play a critical role in alcohol-induced behavioral sensitization, as also reviewed by Phillips et al. (4). Studies with transgenic mice show that CRF and CRFR1 are required for alcohol sensitization (139, 140). Mice homozygous for CRF gene depletion do not show a sensitized locomotor response to alcohol after repeated injections, while heterozygous animals develop normal levels of sensitization (140). CRFR1 knockout mice failed to develop alcohol sensitization and also show a reduction in the acute response to alcohol (119, 139). On the other hand, knockout mouse lines for CRFR2 or urocortin 1 both develop normal levels of sensitization (139), suggesting no particular role for CRFR2 in alcohol sensitization.
As shown in Table 2, pharmacological blockade of CRFR1 receptors with CP-154,526 (in doses of 10 mg/kg and higher; ip) consistently abolishes the expression of a sensitized locomotor stimulation after a 10-day alcohol treatment regimen, without affecting the acute response to alcohol in DBA/2J mice (139, 141). During the induction phase, the effects of the CRFR1 antagonist are less clear, with significant attenuation of alcohol sensitization only at a high 30 mg/kg dose of CP-154,526 [no effects at lower doses; (139, 141)]. No studies were found assessing the pharmacological manipulation of CRFR1 in different inbred or outbred lines of mice other than DBA/2J, nor were these CRFR1 pharmacological manipulations tested in particular brain sites, as shown in Table 2. Future studies should broaden our understanding of the critical CRF/CRFR1 mechanisms underlying alcohol sensitization. Furthermore, while glucocorticoids seem to play a role in alcohol sensitization (139, 140, 142–144), it remains to be determined whether glucocorticoid involvement in this process is directly mediated by upstream CRF signaling.
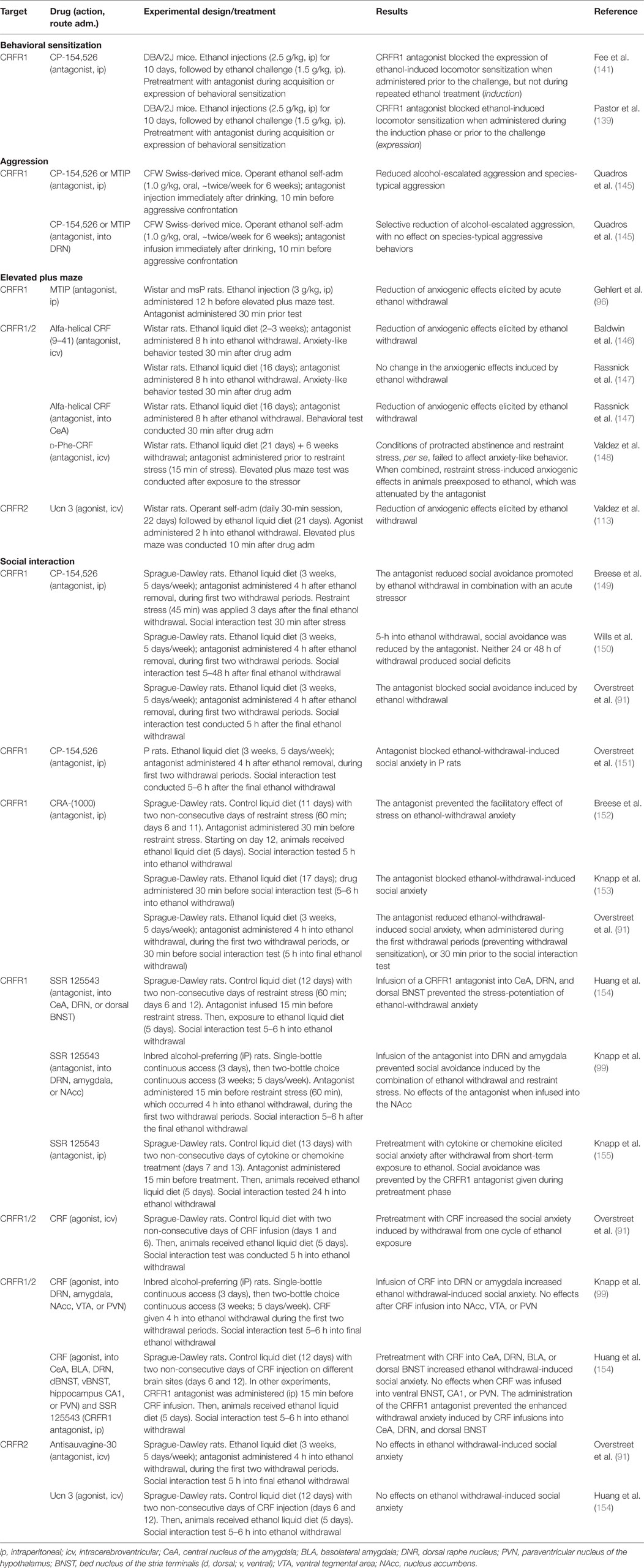
Table 2. Pharmacological manipulations of CRF/urocortin targets on alcohol-related behaviors.
Alcohol-Escalated Aggression
Moderate doses of alcohol may promote escalated levels of aggressive behaviors in mice, rats, monkeys, and humans [see reviews from Miczek et al. (156, 157)]. In mice, alcohol-heightened aggression seems to involve serotonin transmission arising from the DRN and involving different serotonin receptors and terminal regions [e.g., Ref. (158–161)]. Interestingly, CRF receptors are well established critical modulators of serotonin function in the dorsal raphé [e.g., Ref. (57, 58, 162), for reviews], and we have observed a critical role for raphé CRFR1 in the modulation of alcohol-related aggression in male mice (145). Systemic administration of two different CRFR1 antagonists (CP-154,526 and MTIP) was shown to dose-dependently reduce aggression not only in mice that were consuming a low dose of alcohol (1.0 g/kg) but also in mice under control conditions (after water drinking). However, when the CRFR1 antagonists were administered intra-dorsal raphé, a selective reduction in alcohol-escalated aggression was reported, with no other effects on species-typical aggressive behavior (145). Furthermore, the anti-aggressive effects of CP-154,526 seem to rely upon serotonin transmission, with increased serotonin output to the prefrontal cortex as a result of CRFR1 antagonism in alcohol-drinking animals (145).
Alcohol Withdrawal-Induced Effects on Anxiety-Like Behaviors
In models with extensive and prolonged exposure to alcohol (e.g., alcohol vapor chamber or feeding from an alcohol-containing liquid diet), animals may present alcohol withdrawal effects within a few hours or days post termination of alcohol treatment, as well as protracted abstinence signs. Increases in anxiety-like behavior are a well characterized consequence of alcohol withdrawal, particularly within the first hours of withdrawal. Using the elevated plus maze, withdrawal-induced anxiety-like behavior was attenuated by icv administration of the non-selective CRFR1/R2 antagonist, alpha-helical CRF (146). However, a subsequent study only observed anti-anxiety effects when alpha-helical CRF was injected into the CeA, but not icv (147). In protracted abstinence from a 3-week alcohol exposure, anxiogenic effects in the plus maze emerged upon exposure to a stressor, selectively in alcohol “dependent” subjects. This effect was prevented by centrally blocking CRF receptors prior to the stressor (148). Specific roles for CRFR1 or CRFR2 mechanisms in this particular model are still inconclusive. On the one hand, the activation of CRFR2 with urocortin 3 reduced anxiety behavior after withdrawal from chronic alcohol (113). On the other hand, selectively blocking CRFR1 receptors reduced anxiety-like behavior after withdrawal from a single alcohol injection (96).
A greater number of studies addressed social anxiety as an index of alcohol withdrawal, as assessed in social interaction tests, as shown in Table 2. Most commonly, these studies exposed rats to an alcohol liquid diet, with a single or repeated cycles of withdrawal (e.g., one cycle consists of 5 days of alcohol diet, followed by 2 days of alcohol withdrawal). This protocol promotes reduced social interaction when animals are tested a few hours into withdrawal, usually after the third cycle of alcohol exposure (see experimental designs on Table 2). Pharmacological antagonism of CRFR1 consistently attenuates repeated alcohol-withdrawal-induced social anxiety. Different CRFR1 antagonists are effective in reducing social avoidance when administered shortly prior to the social interaction test [e.g., Ref. (91, 149, 153)]. Interestingly, blocking CRFR1 signaling during the initial withdrawal days of the cycles of alcohol exposure/withdrawal also prevents the typical social avoidance response, even though animals are tested in the absence of the antagonist [e.g., Ref. (91, 149–151)]. This suggests that cumulative neuroadaptations involving CRF occur over the repeated cycles of exposure and withdrawal, and by blocking CRFR1 receptors attenuates the deleterious effects of withdrawal on social anxiety. A few brain regions have been shown to participate in the CRFR1 modulation of withdrawal-related anxiety, including the DRN and the CeA, but not the nucleus accumbens (99).
Furthermore, exposure to stressors or to an infusion of CRF may additionally aggravate alcohol withdrawal-related social anxiety with a single cycle of alcohol diet exposure (91, 154), or with a two-bottle choice protocol (99). Infusion of CRF directly into the DRN, CeA, BNST, and the basolateral amygdala (BLA), either prior to alcohol exposure or during withdrawal periods of drinking cycles, also potentiates social avoidance associated with alcohol withdrawal (99). These effects of CRF are likely mediated by CRFR1, since urocortin 3 infusion does not replicate the CRF potentiation of alcohol withdrawal (154), neither does a CRFR2 antagonist block withdrawal-induced social anxiety (91).
Consequences of Alcohol Exposure on the CRF/Urocortin System
Extensive and consistent evidence supports alcohol’s actions on the CRF systems, modulating both HPA axis function as well as extra-hypothalamic CRF signaling in different brain regions. However, as shown in Table 3, consequences of alcohol exposure on CRF/urocortin system and the HPA axis may vary according to several variables, such as the route of alcohol administration, duration of alcohol exposure, alcohol withdrawal period, age, sex, animal strain, and CRF-related targets. Because the primary interest of this review is focused on alcohol and CRF-related peptides and signaling, data on ACTH and corticosterone were only included from studies that also directly assessed CRF-related targets. For reviews analyzing alcohol’s effects on HPA axis, with focus on glucocorticoid systems, please refer to Rose et al. (163), Stephens and Wand (2), and Edwards et al. (3).
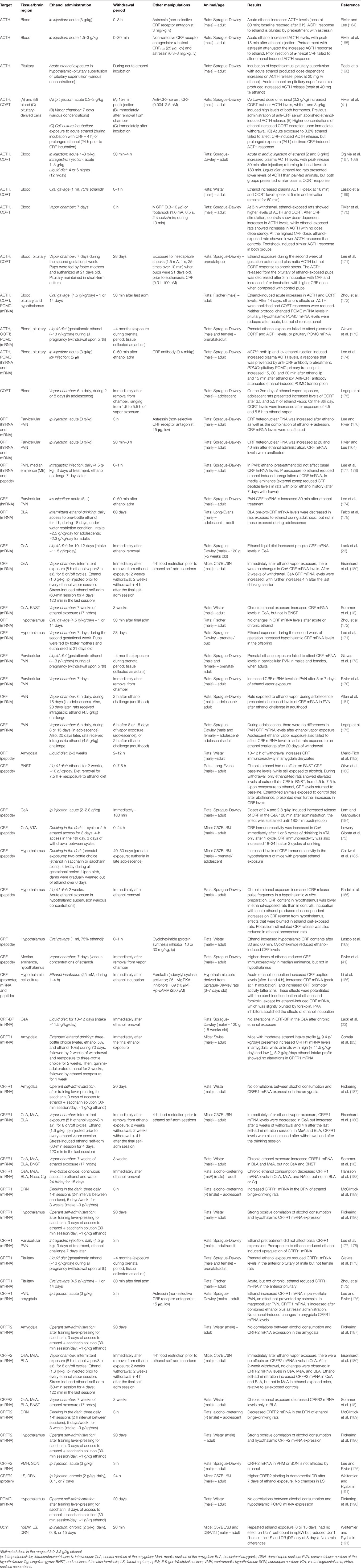
Table 3. Consequences of ethanol exposure on CRF/urocortin systems.
As shown in Table 3, acute alcohol exposure, whether administered ip or by gavage, promotes activation of the HPA axis, as evidenced by dose-dependent increases on plasma ACTH and corticosterone with doses of 1 g/kg and higher [e.g., Ref. (41, 164, 169, 172)]. Increases in ACTH can also be elicited with icv administration of alcohol (174) and can be shown in vitro in a hypothalamus–pituitary preparation (166). Importantly, alcohol’s actions on ACTH and corticosterone release seem to be primarily mediated by alcohol-induced CRF activation in the hypothalamus [see review by Rivier (192)]. Pretreatment with antagonists of CRF receptors as well as with anti-CRF antibody, both prevent or attenuate alcohol-induced increases in ACTH and corticosterone [e.g., Ref. (164, 165, 174)]. Consistent with this hypothesis, heteronuclear RNA (precursor of mRNA) for CRF and CRF peptide levels are reliably increased in the hypothalamus after acute alcohol administration (164, 169, 174, 176). Hypothalamic CRF mRNA levels are not as reliably affected by acute alcohol, perhaps due to the kinetics of transcription processes (172, 176). In hypothalamic cell culture, alcohol incubation produced increases in CRF mRNA, CRF promoter activity, as well as CRF peptide, in a process that requires adenylate cyclase-PKA signaling (186). Extra-hypothalamic sites, such as the central amygdala, also show augmented release of CRF peptide as a result of acute alcohol administration (184). Concerning CRF receptors, mRNA levels may be affected by acute alcohol exposure, but results are scarce and inconsistent (172, 176), as shown in Table 3.
Chronic or repeated alcohol exposure promote less consistent changes on CRF/urocortin function, when components of the CRF system are analyzed under alcohol influence or immediately (within 60 min) after removal from alcohol exposure. Some reports suggest the development of tolerance in HPA axis responses after repeated alcohol treatment, with lower ACTH and corticosterone responses to alcohol [e.g., Ref. (172, 175); but see Ref. (41, 167, 168)]. Concerning CRF contents in the hypothalamus and/or pituitary, there are reports of no changes (41, 172), decreases (41, 166, 177, 178), or increases (170) in CRF peptide, mRNA, and/or hnRNA after chronic alcohol. For example, in the PVN, alcohol-induced upregulation of CRF hnRNA and alcohol-induced CRF release were reduced in rats with a history of alcohol exposure (166, 177, 178). In the CeA, repeated alcohol was reported to increase detection of CRF mRNA or immunoreactivity [(73, 193); but see Ref. (188)], although a lack of alteration was also found (180). In amygdalar nuclei, a reduction in CRFR1 gene expression, but not CRFR2, was detected after alcohol vapor exposure (180) or after 15 days of voluntary of alcohol drinking in an alcohol-preferring strain (188). After extended access to alcohol for over 90 days, only mice with a profile of moderate alcohol intake showed increased CRFR1 mRNA in the amygdala, which was not the case for mice with high or low intake profiles (83). In the BNST, no changes in CRF immunoreactivity were seen while animals still had access to alcohol in a liquid diet, but increases in CRF emerged after 4 h into withdrawal from alcohol (183). Interestingly, repeated alcohol failed to change the number of urocortin1 cells in the Edinger–Westphal nucleus, but it decreased urocortin1 fibers to the lateral septum and the DRN (191).
Short-term withdrawal (here defined as an arbitrary window of 1–24 h after the final alcohol exposure) from repeated/chronic alcohol administration is usually associated with increased anxiety-like behavior and escalated alcohol self-administration, as discussed in previous sections. Acute withdrawal (i.e., 3 h) from a 7-day alcohol vapor exposure was characterized by increased plasma ACTH and corticosterone levels, and also associated with increased CRF mRNA levels in the PVN (170). However, maximal CRF-mediated ACTH response was reduced in alcohol-exposed rats (170). A different study showed that after 1-day withdrawal from chronic alcohol liquid diet, CRF immunoreactivity was not changed in the hypothalamus but was reduced in the amygdala, hippocampus, and frontal cortex (194). These changes were also accompanied by lower plasma corticosterone concentrations at acute withdrawal. In a classic study on acute alcohol withdrawal, dialyzate CRF levels in the amygdala are shown to increase in great proportions throughout the first 12 h of withdrawal from chronic alcohol vapor exposure (182). Increases in CRF immunoreactivity in CeA are also observed after repeated cycles of binge alcohol drinking in mice, persisting for 18–24 h into alcohol withdrawal (73). In another stress-related region, the BNST, CRF peptide levels increase after 4.5 h into withdrawal from an alcohol liquid diet. Remarkably, CRF levels in the BNST return back to baseline levels when rats regain access to the alcohol-containing liquid diet (183). However, these effects of acute alcohol withdrawal may be age-dependent. Exposure to alcohol vapor or binge drinking for 8–15 days in adolescent rats produces no changes in CRF transcripts or peptides in the PVN (175, 195), although a reduced CRF immunoreactivity in the CeA is observed (195). As shown in Table 3, a few studies also reported changes in CRF receptors’ transcripts after short-term alcohol withdrawal in the PVN (177, 178) and the DRN (189), as well as in CRFR2 binding levels in the DRN (191).
Finally, critical adaptations in the CRF/urocortin are recruited during protracted abstinence from chronic alcohol exposure (i.e., at least 7 days of withdrawal, but usually longer than 2 weeks). For example, rat’s exposure to repeated alcohol administrations followed by a 7-day withdrawal, show reduced CRF peptide levels in the median eminence of the hypothalamus, despite no changes in CRF or CRFR1 transcripts in the PVN (177, 178). However, animals preexposed to the drug show a blunted upregulation of CRF and CRFR1 transcripts in the PVN in response to an alcohol challenge (177, 178). These data, and others, support a dampened function of HPA axis function after chronic alcohol followed by prolonged withdrawal, which is also supported by evidence of low baseline corticosterone levels after 3-weeks withdrawal from chronic alcohol [e.g., Ref. (194)]. As shown in Table 3, prenatal exposure to alcohol also dysregulates elements of the HPA axis in adolescent and adults animals, although differences in neuroadaptations arise according to the duration and methodology of in utero alcohol exposure [e.g., Ref. (171, 173, 185)]. Chronic exposure to alcohol during adolescence may lead to reduced HPA axis response to alcohol in early adulthood, as shown by reduced CRF mRNA in the PVN (181). However, such outcome was not observed when adolescents were exposed to alcohol as binge drinking (195). Thus, recruitment of neuroadaptive changes in CRF/urocortin elements in the HPA axis depend on age of alcohol exposure, the duration and protocol for alcohol exposure (vapor, liquid diet, oral intake), as well as age at time of assessment of CRF/urocortin targets.
In the amygdala, and particularly within the CeA, there is consistent evidence of upregulated CRF signaling after protracted withdrawal from chronic alcohol exposure. For example, there are increased levels of CRF transcript in the CeA (18, 180) or immunoreactivity for CRF in the amygdala (194) after 3 or more weeks of withdrawal from alcohol. These changes are accompanied by increased CRFR1 gene expression within the CeA and/or in other amygdalar nuclei (BLA, MeA). Changes in CRFR2 transcripts in the amygdala were less consistent within different studies (18, 180). One study also reported a decrease in CRF mRNA in the BLA in adult, but not adolescent rats after 18 days of restricted access to alcohol drinking followed by a 60-day withdrawal (179). Although BNST levels of CRF seem to be upregulated during acute withdrawal (183), BNST may not be particularly engaged in protracted abstinence, as no changes in transcripts for CRF or CRFR1 were seen in this region (18).
In summary, alcohol exposure importantly impacts CRF/urocortin components and function, both within the HPA axis and in extra-hypothalamic sites. In general, acute alcohol reliably upregulates CRF transcripts and/or peptide within the hypothalamus, promoting activation of the HPA axis with increases in ACTH and corticosterone. When CRF/urocortin signaling is assessed shortly after chronic alcohol exposure, relevant but quite variable and inconsistent effects may be detected, depending on brain region, alcohol exposure protocol, and the age of animals. After protracted abstinence from alcohol, a more reliable profile emerges, characterized by hyperactivity of brain CRF signaling, particularly within the amygdala. Although the studies are limited, existing research suggests the involvement of urocortins as targets for alcohol’s effects, as well as changes in CRFR2 mRNA/protein levels, and therefore these molecules should be further investigated. Also, target brain regions were highly limited to the hypothalamus, amygdala, and BNST, with a few exceptions. A wider characterization of alcohol modulation of CRF/urocortin signaling should include the VTA, accumbens, DRN, prefrontal cortex, and other reward-related structures, which may contribute to our understanding of alcohol-induced neuroadaptations that influence alcohol reward and withdrawal.
Involvement of CRF Systems in Human Alcoholism
This review will discuss a few human studies regarding HPA axis function in AUDs. For a vast discussion on this literature, we also recommend reviews by Lovallo (196); Rose et al. (163); Stephens and Wand (2). As described in animal studies, human studies confirm an activation of HPA axis by alcohol as demonstrated by increases in plasma cortisol after an acute alcohol administration (197). This cortisol response seems to require ACTH release from the hypophysis, as alcohol failed to increase cortisol levels in patients with lesions in the anterior hypophysis (197). In heavy drinkers and dependent subjects, there is an attenuation of alcohol-induced cortisol response (198), as well as reduced cortisol and ACTH levels upon CRF stimulation (199–201). Dependent individuals also show an attenuated cortisol response to moderate mental and physical stressors (202–205). Upon alcohol withdrawal, plasma cortisol is elevated during the first week, but with protracted abstinence, cortisol levels drop below normal range (206–208). Together, these studies suggest a blunted HPA function in response to drugs and stressors in alcoholics, supporting a contribution of the HPA axis in the transition to dependence (209).
Studies targeting the CRF system and alcohol in humans mostly rely on genetic approaches, with evidence of associations between AUDs and polymorphisms in genes encoding for CRFR1 and CRF-BP. Chen et al. (210) showed that variations in the CRFR1 gene are associated with alcohol dependence. In another study, patterns of alcohol consumption were assessed in a teenage sample, as well as in a sample of dependent adults. In teenagers with little to moderate alcohol exposure, polymorphisms in CRFR1 gene were associated with the amount of alcohol consumed, but not with the frequency of drinking episodes (211). In alcohol-dependent adults, an association was also detected between CRFR1 polymorphism and the amount of alcohol consumed (211). Different studies show gene–environment interactions, in which variants of the CRFR1 gene interact with a history of exposure to stressful events and predict heavy alcohol drinking (212, 213) and age of drinking initiation (213). On the other hand, protective effects for alcohol dependence were shown for a gene–environment interaction involving a CRFR1 haplotype and childhood sexual abuse in an Australian population (214).
Electroencephalogram phenotypes have been identified for anxiety-prone alcoholics (215, 216). A “linkage scan” indicated CRF-BP as a candidate gene for electroencephalogram profiles in two distinct populations. Further, genetic variations in CRF-BP were associated with anxiety disorders and AUDs (217). There is also evidence that polymorphisms in CRF-BP modulate stress-induced craving in heavy drinkers (218) and are involved in the relationship between negative reactivity to stress and negative consequences to alcohol consumption (219). Thus, CRF-BP gene variations may play particular a role in stress-related alcohol dependence. Furthermore, an interaction of polymorphisms in CRFR1 and CRF-BP genes predicts increased risk for AUDs in schizophrenic patients (220). Thus, genetic studies, so far, point to some polymorphisms in CRFR1 and CRF-BP genes, which can contribute to the susceptibility for AUDs.
Summary and Perspectives
Increasing evidence suggests that a recruitment of CRF/urocortin mechanisms is more prominent in individuals with increased sensitivity to and/or vulnerability for alcohol-induced effects (5). After repeated and chronic exposure to alcohol, rats present hyperactive extra-hypothalamic CRF activity, as indicated by increases in CRF immunoreactivity and/or increases in mRNA for CRF and its receptors in amygdala nuclei and the BNST (18, 182, 183). Consistently, increased alcohol seeking and intake can be attenuated after the administration of antagonists to CRF type 1 receptors (systemically, icv or intra-amygdala), particularly in animals with previous alcohol history (5, 18, 81, 106, 109, 110) or in high alcohol-consuming mice (76). While blocking CRFR1 attenuates alcohol drinking, this effect can also be achieved with the activation of CRFR2 signaling, suggesting opposite roles for CRFR1 and CRFR2 in the modulation of excessive alcohol intake [e.g., Ref. (86, 105, 113)]. However, manipulation of CRF receptor signaling in different brain regions may reveal differential effects and interactions between CRFR1 and CRFR2 [e.g., Ref. (73, 99, 105, 115)]. Pharmacological studies start to unveil a role for both CRF receptors within brain reward pathways, as well as CRF-BP, as critical modulators of escalated alcohol drinking [e.g., Ref. (74, 84, 89, 115)]. Furthermore, the finding that different CRF mechanisms and pathways may be engaged during escalated drinking or alcohol withdrawal-related anxiety (99) gives rise to an intriguing path for further investigation. Moreover, CRF/urocortin signaling is also recruited during other alcohol-related effects, including alcohol-induced behavioral sensitization, alcohol-escalated aggression, and alcohol withdrawal-related anxiety. Determination of specific mechanisms for the engagement of CRF, urocortins, CRF-BP, and CRF receptors, after acute and chronic alcohol exposure still remain to be clarified. While effects of alcohol on the HPA axis, the amygdala, and other stress-related structures have been more widely characterized, promising contributions of CRF/urocortin signaling in brain reward pathways and other structures are starting to emerge. Finally, there is hope that new molecules targeting components of the CRF/urocortin system will be made available for testing in preclinical experimental settings and advancing to clinical trials, providing helpful therapeutic tools for the treatment of AUDs.
Author Contributions
IQ and GM proposed and outlined the contents of the review. IQ, GM, LD, and CF wrote different sections of the text, prepared the corresponding tables, and organized the bibliography. IQ revised the manuscript text and tables. All authors carefully read, revised, and approved the entire text.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Funding
This work was supported by Associação Fundo de Incentivo à Pesquisa (AFIP) and Coordenação de Aperfeiçoamento de Pessoal de Nível Superior (CAPES). GM is the recipient of a fellowship from Fundação de Amparo à Pesquisa do Estado de São Paulo (Fapesp #2013/01562-2), LD had a fellowship from Conselho Nacional Científico e Tecnológico (CNPq #131585/2014-2), CF had a fellowship from Fapesp (#2014/14623-2), and currently receives a fellowship from CNPq (#141532/2016-5).
Footnote
- ^This affirmation refers exclusively to locomotor sensitization induced by alcohol. Neurochemical sensitization of the dopamine reward system and sensitization to other behaviors and drugs (including cross-sensitization between alcohol and other drugs; withdrawal-induced sensitization of anxiety-like behaviors, etc.) have been extensively demonstrated in rats.
References
1. Koob GF, Buck CL, Cohen A, Edwards S, Park PE, Schlosburg JE, et al. Addiction as a stress surfeit disorder. Neuropharmacology (2014) 76:370–82. doi:10.1016/j.neuropharm.2013.05.024
2. Stephens MA, Wand G. Stress and the HPA axis: role of glucocorticoids in alcohol dependence. Alcohol Res (2012) 34(4):468–83.
3. Edwards S, Little HJ, Richardson HN, Vendruscolo LF. Divergent regulation of distinct glucocorticoid systems in alcohol dependence. Alcohol (2015) 49(8):811–6. doi:10.1016/j.alcohol.2015.04.004
4. Phillips TJ, Reed C, Pastor R. Preclinical evidence implicating corticotropin-releasing factor signaling in ethanol consumption and neuroadaptation. Genes Brain Behav (2015) 14(1):98–135. doi:10.1111/gbb.12189
5. Heilig M, Koob GF. A key role for corticotropin-releasing factor in alcohol dependence. Trends Neurosci (2007) 30(8):399–406. doi:10.1016/j.tins.2007.06.006.A
6. Sarnyai Z, Shaham Y, Heinrichs SC. The role of corticotropin-releasing factor in drug addiction. Pharmacol Rev (2001) 53:209–43.
7. Zorrilla EP, Logrip ML, Koob GF. Corticotropin releasing factor: a key role in the neurobiology of addiction. Front Neuroendocrinol (2014) 35:234–44. doi:10.1016/j.yfrne.2014.01.001
8. WHO. Global Information System on Alcohol and Health. World Health Statistics (2014). Available from: http://www.who.int/gho/publications/world_health_statistics/EN_WHS2014_Part3.pdf
9. Gowing LR, Ali RL, Allsop S, Marsden J, Turf EE, West R, et al. Global statistics on addictive behaviours: 2014 status report. Addiction (2015) 110(6):904–19. doi:10.1111/add.12899
10. WHO. International Classification of Diseases and Related Health Problems, 10th Revision. Geneva: World Health Organization (1992).
11. Sanchis-Segura C, Spanagel R. Behavioural assessment of drug reinforcement and addictive features in rodents: an overview. Addict Biol (2006) 11(1):2–38. doi:10.1111/j.1369-1600.2006.00012.x
12. Spanagel R, Hölter SM. Pharmacological validation of a new animal model of alcoholism. J Neural Transm (2000) 107(6):669–80. doi:10.1007/s007020070068
13. Fachin-Scheit DJ, Ribeiro AF, Pigatto G, Goeldner FO, de Lacerda RB. Development of a mouse model of ethanol addiction: naltrexone efficacy in reducing consumption but not craving. J Neural Transm (2006) 113(9):1305–21. doi:10.1007/s00702-005-0416-z
14. Wolffgramm J. An ethopharmacological approach to the development of drug addiction. Neurosci Biobehav Rev (1991) 15(4):515–9. doi:10.1016/S0149-7634(05)80142-3
15. Goldstein DB, Pal N. Alcohol dependence produced in mice by inhalation of ethanol: grading the withdrawal reaction. Science (1971) 172(3980):288–90. doi:10.1126/science.172.3980.288
16. Rogers J, Wiener SG, Bloom FE. Long-term ethanol administration methods for rats: advantages of inhalation over intubation or liquid diets. Behav Neural Biol (1979) 27(4):466–86. doi:10.1016/S0163-1047(79)92061-2
17. Becker HC. Behavioral neurobiology of alcohol addiction. In: Sommer WH, Spanagel R, editors. Current Topics in Behavioral Neurosciences. Vol. 13. Berlin, Heidelberg: Springer Berlin Heidelberg (2013).
18. Sommer WH, Rimondini R, Hansson AC, Hipskind PA, Gehlert DR, Barr CS, et al. Upregulation of voluntary alcohol intake, behavioral sensitivity to stress, and amygdala crhr1 expression following a history of dependence. Biol Psychiatry (2008) 63(2):139–45. doi:10.1016/j.biopsych.2007.01.010
19. Lovinger DM, Roberto M. Synaptic effects induced by alcohol. Curr Top Behav Neurosci (2013) 13:31–86. doi:10.1007/7854_2011_143
20. Siggins GR, Roberto M, Nie Z. The tipsy terminal: presynaptic effects of ethanol. Pharmacol Ther (2005) 107(1):80–98. doi:10.1016/j.pharmthera.2005.01.006
21. Chandler LJ, Sutton G, Norwood D, Sumners C, Crews FT. Chronic ethanol increases N-methyl-d-aspartate-stimulated nitric oxide formation but not receptor density in cultured cortical neurons. Mol Pharmacol (1997) 51(5):733–40.
22. Chandler LJ, Norwood D, Sutton G. Chronic ethanol upregulates NMDA and AMPA, but not kainate receptor subunit proteins in rat primary cortical cultures. Alcohol Clin Exp Res (1999) 23(2):363–70. doi:10.1111/j.1530-0277.1999.tb04123.x
23. Lack AK, Diaz MR, Chappell A, DuBois DW, McCool BA. Chronic ethanol and withdrawal differentially modulate pre- and postsynaptic function at glutamatergic synapses in rat basolateral amygdala. J Neurophysiol (2007) 98(6):3185–96. doi:10.1152/jn.00189.2007
24. Roberto M, Cruz MT, Gilpin NW, Sabino V, Schweitzer P, Bajo M, et al. Corticotropin releasing factor-induced amygdala gamma-aminobutyric acid release plays a key role in alcohol dependence. Biol Psychiatry (2010) 67(9):831–9. doi:10.1016/j.biopsych.2009.11.007
25. Cozzoli DK, Goulding SP, Zhang PW, Xiao B, Hu JH, Ary AW, et al. Binge drinking upregulates accumbens mGluR5-Homer2-PI3K signaling: functional implications for alcoholism. J Neurosci (2009) 29(27):8655–68. doi:10.1523/JNEUROSCI.5900-08.2009
26. Grobin AC, Matthews DB, Devaud LL, Morrow AL. The role of GABA(A) receptors in the acute and chronic effects of ethanol. Psychopharmacology (1998) 139(1–2):2–19. doi:10.1007/s002130050685
27. Kumar S, Porcu P, Werner DF, Matthews DB, Diaz-Granados JL, Helfand RS, et al. The role of GABA(A) receptors in the acute and chronic effects of ethanol: a decade of progress. Psychopharmacology (2009) 205(4):529–64. doi:10.1007/s00213-009-1562-z
28. Vale W, Spiess J, Rivier C, Rivier J. Characterization of a 41-residue ovine hypothalamic peptide that stimulates secretion of corticotropin and beta-endorphin. Science (1981) 213(4514):1394–7. doi:10.1126/science.6267699
29. Behan DP, De Souza EB, Lowry PJ, Potter E, Sawchenko P, Vale WW. Corticotropin releasing factor (CRF) binding protein: a novel regulator of CRF and related peptides. Front Neuroendocrinol (1995) 16(4):362–82. doi:10.1006/frne.1995.1013
30. Seasholtz AF, Valverde RA, Denver RJ. Corticotropin-releasing hormone-binding protein: biochemistry and function from fishes to mammals. J Endocrinol (2002) 175(1):89–97. doi:10.1677/joe.0.1750089
31. Bale TL, Vale WW. CRF and CRF receptors: role in stress responsivity and other behaviors. Annu Rev Pharmacol Toxicol (2004) 44(January):525–57. doi:10.1146/annurev.pharmtox.44.101802.121410
32. Perrin M, Donaldson C, Chen R, Blount A, Berggren T, Bilezikjian L, et al. Identification of a second corticotropin-releasing factor receptor gene and characterization of a cDNA expressed in heart. Proc Natl Acad Sci U S A (1995) 92(7):2969–73. doi:10.1073/pnas.92.7.2969
33. Fekete EM, Zorrilla EP. Physiology, pharmacology, and therapeutic relevance of urocortins in mammals: ancient CRF paralogs. Front Neuroendocrinol (2007) 28(1):1–27. doi:10.1016/j.yfrne.2006.09.002
34. Perrin MH, Vale WW. Corticotropin releasing factor receptors and their ligand family. Ann N Y Acad Sci (1999) 885:312–28. doi:10.1111/j.1749-6632.1999.tb08687.x
35. Van Pett K, Viau V, Bittencourt JC, Chan RK, Li HY, Arias C, et al. Distribution of mRNAs encoding CRF receptors in brain and pituitary of rat and mouse. J Comp Neurol (2000) 428(2):191–212. doi:10.1002/1096-9861(20001211)428:2<191::AID-CNE1>3.0.CO;2-U
36. Chalmers DT, Lovenberg TW, De Souza EB. Localization of novel corticotropin-releasing factor receptor (CRF2) mRNA expression to specific subcortical nuclei in rat brain: comparison with CRF1 receptor mRNA expression. J Neurosci (1995) 15(10):6340–50.
37. Lovenberg TW, Chalmers DT, Liu C, De Souza EB. CRF2 alpha and CRF2 beta receptor mRNAs are differentially distributed between the rat central nervous system and peripheral tissues. Endocrinology (1995) 136(9):4139–42. doi:10.1210/endo.136.9.7544278
38. Pan W, Kastin AJ. Urocortin and the brain. Prog Neurobiol (2008) 84(2):148–56. doi:10.1016/j.pneurobio.2007.10.008
39. Stengel A, Taché Y. Corticotropin-releasing factor signaling and visceral response to stress. Exp Biol Med (2010) 235(10):1168–78. doi:10.1258/ebm.2010.009347
40. Rivier C, Rivier J, Vale W. Inhibition of adrenocorticotropic hormone secretion in the rat by immunoneutralization of corticotropin-releasing factor. Science (1982) 218(4570):377–9. doi:10.1126/science.6289439
41. Rivier C, Bruhn T, Vale W. Effect of ethanol on the hypothalamic-pituitary-adrenal axis in the rat: role of corticotropin-releasing factor (CRF). J Pharmacol Exp Ther (1984) 229(1):127–31. doi:10.1111/j.1530-0277.2001.tb02133.x
42. Papadimitriou A, Priftis KN. Regulation of the hypothalamic-pituitary-adrenal axis. Neuroimmunomodulation (2009) 16(5):265–71. doi:10.1159/000216184
43. Scott LV, Dinan TG. Vasopressin and the regulation of hypothalamic-pituitary-adrenal axis function: implications for the pathophysiology of depression. Life Sci (1998) 62(22):1985–98. doi:10.1016/S0024-3205(98)00027-7
44. Steckler T, Holsboer F. Corticotropin-releasing hormone receptor subtypes and emotion. Biol Psychiatry (1999) 46(11):1480–508. doi:10.1016/S0006-3223(99)00170-5
45. Herman JP, Figueiredo H, Mueller NK, Ulrich-Lai Y, Ostrander MM, Choi DC, et al. Central mechanisms of stress integration: hierarchical circuitry controlling hypothalamo-pituitary-adrenocortical responsiveness. Front Neuroendocrinol (2003) 24(3):151–80. doi:10.1016/j.yfrne.2003.07.001
46. McEwen BS. Allostasis, allostatic load, and the aging nervous system: role of excitatory amino acids and excitotoxicity. Neurochem Res (2000) 25(9–10):1219–31. doi:10.1023/A:1007687911139
47. Myers B, McKlveen JM, Herman JP. Glucocorticoid actions on synapses, circuits, and behavior: implications for the energetics of stress. Front Neuroendocrinol (2014) 35(2):180–96. doi:10.1016/j.yfrne.2013.12.003
48. Sapolsky RM. Stress hormones: good and bad. Neurobiol Dis (2000) 7(5):540–2. doi:10.1006/nbdi.2000.0350
49. Ulrich-Lai YM, Herman JP. Neural regulation of endocrine and autonomic stress responses. Nat Rev Neurosci (2009) 10(6):397–409. doi:10.1038/nrn2647
50. Morin SM, Ling N, Liu XJ, Kahl SD, Gehlert DR. Differential distribution of urocortin- and corticotropin-releasing factor-like immunoreactivities in the rat brain. Neuroscience (1999) 92:281–91. doi:10.1016/S0306-4522(98)00732-5
51. Swanson LW, Sawchenko PE, Rivier J, Vale WW. Organization of ovine corticotropin-releasing factor immunoreactive cells and fibers in the rat brain: an immunohistochemical study. Neuroendocrinology (1983) 36:165–86. doi:10.1159/000123454
52. Takahashi K, Totsune K, Sone M, Murakami O, Satoh F, Arihara Z, et al. Regional distribution of urocortin-like immunoreactivity and expression of urocortin mRNA in the human brain. Peptides (1998) 19(4):643–7. doi:10.1016/S0196-9781(98)00012-6
53. Vasconcelos LA, Donaldson C, Sita LV, Casatti CA, Lotfi CF, Wang L, et al. Urocortin in the central nervous system of a primate (Cebus apella): sequencing, immunohistochemical, and hybridization histochemical characterization. J Comp Neurol (2003) 463(2):157–75. doi:10.1002/cne.10742
54. Stengel A, Taché Y. CRF and urocortin peptides as modulators of energy balance and feeding behavior during stress. Front Neurosci (2014) 8:52. doi:10.3389/fnins.2014.00052
55. Hostetler CM, Ryabinin AE. The CRF system and social behavior: a review. Front Neurosci (2013) 7:92. doi:10.3389/fnins.2013.00092
56. Koob GF, Heinrichs SC. A role for corticotropin releasing factor and urocortin in behavioral responses to stressors. Brain Res (1999) 848(1–2):141–52. doi:10.1016/S0006-8993(99)01991-5
57. Valentino RJ, Commons KG. Peptides that fine-tune the serotonin system. Neuropeptides (2005) 39(1):1–8. doi:10.1016/j.npep.2004.09.005
58. Valentino RJ, Lucki I, Van Bockstaele E. Corticotropin-releasing factor in the dorsal raphe nucleus: linking stress coping and addiction. Brain Res (2010) 1314:29–37. doi:10.1016/j.brainres.2009.09.100
59. Ryabinin AE, Tsoory MM, Kozicz T, Thiele TE, Neufeld-Cohen A, Chen A, et al. Urocortins: CRF’s siblings and their potential role in anxiety, depression and alcohol drinking behavior. Alcohol (2012) 46(4):349–57. doi:10.1016/j.alcohol.2011.10.007
60. Borgland SJ, Ungless MA, Bonci A. Convergent actions of orexin/hypocretin and CRF on dopamine neurons: emerging players in addiction. Brain Res (2010) 1314:139–44. doi:10.1016/j.brainres.2009.10.068
61. George O, Le Moal M, Koob GF. Allostasis and addiction: role of the dopamine and corticotropin-releasing factor systems. Physiol Behav (2011) 106:58–64. doi:10.1016/j.physbeh.2011.11.004
62. Van Bockstaele EJ, Valentino RJ. Neuropeptide regulation of the locus coeruleus and opiate-induced plasticity of stress responses. Adv Pharmacol (2013) 68:405–20. doi:10.1016/B978-0-12-411512-5.00019-1
63. Rodaros D, Caruana DA, Amir S, Stewart J. Corticotropin-releasing factor projections from limbic forebrain and paraventricular nucleus of the hypothalamus to the region of the ventral tegmental area. Neuroscience (2007) 150(1):8–13. doi:10.1016/j.neuroscience.2007.09.043
64. Wanat MJ, Hopf FW, Stuber GD, Phillips PE, Bonci A. Corticotropin-releasing factor increases mouse ventral tegmental area dopamine neuron firing through a protein kinase C-dependent enhancement of Ih. J Physiol (2008) 586(8):2157–70. doi:10.1113/jphysiol.2007.150078
65. Lemos JC, Wanat MJ, Smith JS, Reyes BA, Hollon NG, Van Bockstaele EJ, et al. Severe stress switches CRF action in the nucleus accumbens from appetitive to aversive. Nature (2012) 490(7420):402–6. doi:10.1038/nature11436
66. Holly EN, Miczek KA. Ventral tegmental area dopamine revisited: effects of acute and repeated stress. Psychopharmacology (2016) 233(2):163–86. doi:10.1007/s00213-015-4151-3
67. Wang B, Shaham Y, Zitzman D, Azari S, Wise RA, You ZB. Cocaine experience establishes control of midbrain glutamate and dopamine by corticotropin-releasing factor: a role in stress-induced relapse to drug seeking. J Neurosci (2005) 25(22):5389–96. doi:10.1523/JNEUROSCI.0955-05.2005
68. Wang B, You ZB, Rice KC, Wise RA. Stress-induced relapse to cocaine seeking: roles for the CRF(2) receptor and CRF-binding protein in the ventral tegmental area of the rat. Psychopharmacology (2007) 193(2):283–94. doi:10.1007/s00213-007-0782-3
69. Holly EN, Boyson CO, Montagud-Romero S, Stein DJ, Gobrogge KL, DeBold JF, et al. Episodic social stress-escalated cocaine self-administration: role of phasic and tonic corticotropin releasing factor in the anterior and posterior ventral tegmental area. J Neurosci (2016) 36(14):4093–105. doi:10.1523/JNEUROSCI.2232-15.2016
70. Hansson AC, Cippitelli A, Sommer WH, Fedeli A, Björk K, Soverchia L, et al. Variation at the rat Crhr1 locus and sensitivity to relapse into alcohol seeking induced by environmental stress. Proc Natl Acad Sci U S A (2006) 103(41):15236–41. doi:10.1073/pnas.0604419103
71. Lodge DJ, Lawrence AJ. The CRF1 receptor antagonist antalarmin reduces volitional ethanol consumption in isolation-reared fawn-hooded rats. Neuroscience (2003) 117(2):243–7. doi:10.1016/S0306-4522(02)00793-5
72. Cippitelli A, Damadzic R, Singley E, Thorsell A, Ciccocioppo R, Eskay RL, et al. Pharmacological blockade of corticotropin-releasing hormone receptor 1 (CRH1R) reduces voluntary consumption of high alcohol concentrations in non-dependent Wistar rats. Pharmacol Biochem Behav (2012) 100(3):522–9. doi:10.1016/j.pbb.2011.10.016
73. Lowery-Gionta EG, Navarro M, Li C, Pleil KE, Rinker JA, Cox BR, et al. Corticotropin releasing factor signaling in the central amygdala is recruited during binge-like ethanol consumption in C57BL/6J mice. J Neurosci (2012) 32(10):3405–13. doi:10.1523/jneurosci.6256-11.2012
74. Hwa LS, Debold JF, Miczek KA. Alcohol in excess: CRF1 receptors in the rat and mouse VTA and DRN. Psychopharmacology (2013) 225(2):313–27. doi:10.1007/s00213-012-2820-z
75. Chu K, Koob GF, Cole M, Zorrilla EP, Roberts AJ. Dependence-induced increases in ethanol self-administration in mice are blocked by the CRF1 receptor antagonist antalarmin and by CRF1 receptor knockout. Pharmacol Biochem Behav (2007) 86(4):813–21. doi:10.1016/j.pbb.2007.03.009
76. Sparta DR, Sparrow AM, Lowery EG, Fee JR, Knapp DJ, Thiele TE. Blockade of the corticotropin releasing factor type 1 receptor attenuates elevated ethanol drinking associated with drinking in the dark procedures. Alcohol Clin Exp Res (2008) 32(2):259–65. doi:10.1111/j.1530-0277.2007.00575.x
77. Giardino WJ, Ryabinin AE. CRF1 receptor signaling regulates food and fluid intake in the drinking-in-the-dark model of binge alcohol consumption. Alcohol Clin Exp Res (2013) 37(7):1161–70. doi:10.1111/acer.12076
78. Molander A, Vengeliene V, Heilig M, Wurst W, Deussing JM, Spanagel R. Brain-specific inactivation of the Crhr1 gene inhibits post-dependent and stress-induced alcohol intake, but does not affect relapse-like drinking. Neuropsychopharmacology (2012) 37(4):1047–56. doi:10.1038/npp.2011.297
79. Yang X, Wang S, Rice KC, Munro CA, Wand GS. Restraint stress and ethanol consumption in two mouse strains. Alcohol Clin Exp Res (2008) 32(5):840–52. doi:10.1111/j.1530-0277.2008.00632.x
80. Marinelli PW, Funk D, Juzytsch W, Harding S, Rice KC, Shaham Y, et al. The CRF1 receptor antagonist antalarmin attenuates yohimbine-induced increases in operant alcohol self-administration and reinstatement of alcohol seeking in rats. Psychopharmacology (2007) 195(3):345–55. doi:10.1007/s00213-007-0905-x
81. Funk CK, Zorrilla EP, Lee MJ, Rice KC, Koob GF. Corticotropin-releasing factor 1 antagonists selectively reduce ethanol self-administration in ethanol-dependent rats. Biol Psychiatry (2007) 61(1):78–86. doi:10.1016/j.biopsych.2006.03.063
82. Le AD, Harding S, Juzytsch W, Watchus J, Shalev U, Shaham Y. The role of corticotrophin-releasing factor in stress-induced relapse to alcohol-seeking behavior in rats. Psychopharmacology (2000) 150(3):317–24. doi:10.1007/s002130000411
83. Correia D, Martynhak BJ, Pereira M, Siba IP, Ribeiro AF, Camarini R, et al. Reduction of ethanol intake by corticotropin-releasing factor receptor-1 antagonist in ‘heavy-drinking’ mice in a free-choice paradigm. Psychopharmacology (2015) 232(15):2731–9. doi:10.1007/s00213-015-3909-y
84. Hwa LS, Shimamoto A, Kayyali T, Norman KJ, Valentino RJ, DeBold JF, et al. Dissociation of μ-opioid receptor and CRF-R1 antagonist effects on escalated ethanol consumption and mPFC serotonin in C57BL/6J mice. Addict Biol (2016) 21:111–24. doi:10.1111/adb.12189
85. Lowery EG, Sparrow AM, Breese GR, Knapp DJ, Thiele TE. The CRF-1 receptor antagonist, CP-154,526, attenuates stress-induced increases in ethanol consumption by BALB/cJ mice. Alcohol Clin Exp Res (2008) 32(2):240–8. doi:10.1111/j.1530-0277.2007.00573.x
86. Lowery EG, Spanos M, Navarro M, Lyons AM, Hodge CW, Thiele TE. CRF-1 antagonist and CRF-2 agonist decrease binge-like ethanol drinking in C57BL/6J mice independent of the HPA axis. Neuropsychopharmacology (2010) 35(6):1241–52. doi:10.1038/npp.2009.209
87. Overstreet DH, Knapp DJ, Breese GE. Drug challenges reveal differences in mediation of stress facilitation of voluntary alcohol drinking and withdrawal-induced anxiety in alcohol-preferring P rats. Alcohol Clin Exp Res (2007) 31(9):1473–81. doi:10.1111/j.1530-0277.2007.00445.x
88. Sparta DR, Ferraro FM, Fee JR, Knapp DJ, Breese GR, Thiele TE. The alcohol deprivation effect in C57BL/6J mice is observed using operant self-administration procedures and is modulated by CRF-1 receptor signaling. Alcohol Clin Exp Res (2009) 33(1):31–42. doi:10.1111/j.1530-0277.2008.00808.x
89. Sparta DR, Hopf FW, Gibb SL, Cho SL, Stuber GD, Messing RO, et al. Binge ethanol-drinking potentiates corticotropin releasing factor R1 receptor activity in the ventral tegmental area. Alcohol Clin Exp Res (2013) 37(10):1680–7. doi:10.1111/acer.12153
90. Simms JA, Nielsen CK, Li R, Bartlett SE. Intermittent access ethanol consumption dysregulates CRF function in the hypothalamus and is attenuated by the CRF-R1 antagonist, CP-376395. Addict Biol (2014) 19(4):606–11. doi:10.1111/adb.12024
91. Overstreet DH, Knapp DJ, Breese GR. Modulation of multiple ethanol withdrawal-induced anxiety-like behavior by CRF and CRF1 receptors. Pharmacol Biochem Behav (2004) 77(2):405–13. doi:10.1016/j.pbb.2003.11.010
92. Sabino V, Cottone P, Koob GF, Steardo L, Lee MJ, Rice KC, et al. Dissociation between opioid and CRF1 antagonist sensitive drinking in Sardinian alcohol-preferring rats. Psychopharmacology (2006) 189(2):175–86. doi:10.1007/s00213-006-0546-5
93. Gilpin NW, Richardson HN, Koob GF. Effects of CRF 1-receptor and opioid-receptor antagonists on dependence-induced increases in alcohol drinking by alcohol-preferring (P) rats. Alcohol Clin Exp Res (2008) 32(9):1535–42. doi:10.1111/j.1530-0277.2008.00745.x
94. Richardson HN, Zhao Y, Fekete EM, Funk CK, Wirsching P, Janda KD, et al. MPZP: a novel small molecule corticotropin-releasing factor type 1 receptor (CRF1) antagonist. Pharmacol Biochem Behav (2008) 88(4):497–510. doi:10.1016/j.pbb.2007.10.008
95. Ji D, Gilpin NW, Richardson HN, Rivier CL, Koob GF. Effects of naltrexone, duloxetine, and a corticotropin-releasing factor type 1 receptor antagonist on binge-like alcohol drinking in rats. Behav Pharmacol (2008) 19(1):1–12. doi:10.1097/FBP.0b013e3282f3cf70
96. Gehlert DR, Cippitelli A, Thorsell A, Lê AD, Hipskind PA, Hamdouchi C, et al. 3-(4-chloro-2-morpholin-4-Yl-thiazol-5-Yl)-8-(1-ethylpropyl)-2,6-dimethyl-imidazo[1,2-B]pyridazine: a novel brain-penetrant, orally available corticotropin-releasing factor receptor 1 antagonist with efficacy in animal models of alcoholism. J Neurosci (2007) 27(10):2718–26. doi:10.1523/jneurosci.4985-06.2007
97. Sabino V, Kwak J, Rice KC, Cottone P. Pharmacological characterization of the 20% alcohol intermittent access model in Sardinian alcohol-preferring rats: a model of binge-like drinking. Alcohol Clin Exp Res (2013) 37(4):635–43. doi:10.1111/acer.12008
98. Roltsch EA, Baynes BB, Mayeux JP, Whitaker AM, Baiamonte BA, Gilpin NW. Predator odor stress alters corticotropin-releasing factor-1 receptor (CRF1R)-dependent behaviors in rats. Neuropharmacology (2014) 79(April):83–9. doi:10.1016/j.neuropharm.2013.11.005
99. Knapp DJ, Overstreet DH, Huang M, Wills TA, Whitman BA, Angel RA, et al. Effects of a stressor and corticotrophin releasing factor on ethanol deprivation-induced ethanol intake and anxiety-like behavior in alcohol-preferring P rats. Psychopharmacology (2011) 218(1):179–89. doi:10.1007/s00213-011-2366-5
100. O’Callaghan MJ, Croft AP, Jacquot C, Little HJ. The hypothalamopituitary-adrenal axis and alcohol preference. Brain Res Bull (2005) 68(3):171–8. doi:10.1016/j.brainresbull.2005.08.006
101. Bell SM, Reynolds JG, Thiele TE, Gan J, Figlewicz DP, Woods SC. Effects of third intracerebroventricular injections of corticotropin-releasing factor (CRF) on ethanol drinking and food intake. Psychopharmacology (1998) 139(1–2):128–35. doi:10.1007/s002130050697
102. Thorsell A, Slawecki CJ, Ehlers CL. Effects of neuropeptide Y and corticotropin-releasing factor on ethanol intake in Wistar rats: interaction with chronic ethanol exposure. Behav Brain Res (2005) 161(1):133–40. doi:10.1016/j.bbr.2005.01.016
103. Le AD, Harding S, Juzytsch W, Fletcher PJ, Shaham Y. The role of corticotropin-releasing factor in the median raphe nucleus in relapse to alcohol. J Neurosci (2002) 22(18):7844–9.
104. Weitemier AZ, Ryabinin AE. Urocortin 1 in the dorsal raphe regulates food and fluid consumption, but not ethanol preference in C57BL/6J mice. Neuroscience (2006) 137(4):1439–45. doi:10.1016/j.neuroscience.2005.10.021
105. Ryabinin AE, Yoneyama N, Tanchuck MA, Mark GP, Finn DA. Urocortin 1 microinjection into the mouse lateral septum regulates the acquisition and expression of alcohol consumption. Neuroscience (2008) 151(3):780–90. doi:10.1016/j.neuroscience.2007.11.014
106. Valdez GR, Roberts AJ, Chan K, Davis H, Brennan M, Zorrilla EP, et al. Increased ethanol self-administration and anxiety-like behavior during acute ethanol withdrawal and protracted abstinence: regulation by corticotropin-releasing factor. Alcohol Clin Exp Res (2002) 26(10):1494–501. doi:10.1111/j.1530-0277.2002.tb02448.x
107. Liu X, Weiss F. Additive effect of stress and drug cues on reinstatement of ethanol seeking: exacerbation by history of dependence and role of concurrent activation of corticotropin-releasing factor and opioid mechanisms. J Neurosci (2002) 22(18):7856–61.
108. Finn DA, Snelling C, Fretwell AM, Tanchuck MA, Underwood L, Cole M, et al. Increased drinking during withdrawal from intermittent ethanol exposure is blocked by the CRF receptor antagonist D-Phe-CRF(12-41). Alcohol Clin Exp Res (2007) 31(6):939–49. doi:10.1111/j.1530-0277.2007.00379.x
109. Funk CK, O’Dell LE, Crawford EF, Koob GF. Corticotropin-releasing factor within the central nucleus of the amygdala mediates enhanced ethanol self-administration in withdrawn, ethanol-dependent rats. J Neurosci (2006) 26(44):11324–32. doi:10.1523/JNEUROSCI.3096-06.2006
110. Funk D, Li Z, Lê AD. Effects of environmental and pharmacological stressors on c-Fos and corticotropin-releasing factor mRNA in rat brain: relationship to the reinstatement of alcohol seeking. Neuroscience (2006) 138(1):235–43. doi:10.1016/j.neuroscience.2005.10.062
111. Le AD, Funk D, Coen K, Li Z, Shaham Y. Role of corticotropin-releasing factor in the median raphe nucleus in yohimbine-induced reinstatement of alcohol seeking in rats. Addict Biol (2013) 18(3):448–51. doi:10.1111/j.1369-1600.2011.00374.x
112. Sharpe AL, Phillips TJ. Central urocortin 3 administration decreases limited-access ethanol intake in nondependent mice. Behav Pharmacol (2009) 20(4):346–51. doi:10.1097/FBP.0b013e32832f01ba
113. Valdez GR, Sabino V, Koob GF. Increased anxiety-like behavior and ethanol self-administration in dependent rats: reversal via corticotropin-releasing factor-2 receptor activation. Alcohol Clin Exp Res (2004) 28(6):865–72. doi:10.1097/01.ALC.0000128222.29875.40
114. Funk CK, Koob GF. A CRF(2) agonist administered into the central nucleus of the amygdala decreases ethanol self-administration in ethanol-dependent rats. Brain Res (2007) 1155(June):172–8. doi:10.1016/j.brainres.2007.04.009
115. Albrechet-Souza L, Hwa LS, Han X, Zhang WY, DeBold JF, Miczek KA. Corticotropin releasing factor binding protein and CRF2 receptors in the ventral tegmental area: modulation of ethanol binge drinking in C57BL/6J mice. Alcohol Clin Exp Res (2015) 39(9):1609–18. doi:10.1111/acer.12825
116. Cunningham CL, Gremel CM, Groblewski PA. Drug-induced conditioned place preference and aversion in mice. Nat Protoc (2006) 1(4):1662–70. doi:10.1038/nprot.2006.279
117. Huston JP, Silva MAS, Topic B, Müller CP. What’s conditioned in conditioned place preference? Trends Pharmacol Sci (2013) 34(3):162–6. doi:10.1016/j.tips.2013.01.004
118. Tzschentke TM. Measuring reward with the conditioned place preference (CPP) paradigm: update of the last decade. Addict Biol (2007) 12(3–4):227–462. doi:10.1111/j.1369-1600.2007.00070.x
119. Olive MF, Mehmert KK, Koenig HN, Camarini R, Kim JA, Nannini MA, et al. A role for corticotropin releasing factor (CRF) in ethanol consumption, sensitivity, and reward as revealed by CRF-deficient mice. Psychopharmacology (2003) 165(2):181–7. doi:10.1007/s00213-002-1248-2
120. Giardino WJ, Cocking DL, Kaur S, Cunningham CL, Ryabinin AE. Urocortin-1 within the centrally-projecting Edinger-Westphal nucleus is critical for ethanol preference. PLoS One (2011) 6(10):e26997. doi:10.1371/journal.pone.0026997
121. Robinson TE, Berridge KC. The neural basis of drug craving: an incentive-sensitization theory of addiction. Brain Res Rev (1993) 18(3):247–91. doi:10.1016/0165-0173(93)90013-P
122. Robinson TE, Berridge KC. Incentive-sensitization and addiction. Addiction (2001) 96(1):103–14. doi:10.1080/09652140020016996
123. Steketee JD, Kalivas PW. Drug wanting: behavioral sensitization and relapse to drug-seeking behavior. Pharmacol Rev (2011) 63(2):348–65. doi:10.1124/pr.109.001933
124. Vanderschuren LJMJ, Pierce RC. Sensitization processes in drug addiction. Curr Top Behav Neurosci (2010) 3:179–95. doi:10.1007/7854_2009_21
125. Abrahao KP, Souza-Formigoni MLO. Behavioral sensitization to ethanol results in cross-sensitization to MK-801 but not to NMDA administered intra-accumbens. Behav Brain Res (2012) 235(2):218–24. doi:10.1016/j.bbr.2012.07.034
126. Masur J, Boerngen R. The excitatory component of ethanol in mice: a chronic study. Pharmacol Biochem Behav (1980) 13(6):777–80. doi:10.1016/0091-3057(80)90206-3
127. Post RM, Rose H. Increasing effects of repetitive cocaine administration in the rat. Nature (1976) 260(5553):731–2. doi:10.1038/260731a0
128. Quadros IMH, Nobrega JN, Hipólide DC, de Lucca EM, Souza-Formigoni MLO. Differential propensity to ethanol sensitization is not associated with altered binding to D1 receptors or dopamine transporters in mouse brain. Addict Biol (2002) 7(3):291–9. doi:10.1080/13556210220139505
129. Segal DS, Mandell AJ. Long-term administration of d-amphetamine: progressive augmentation of motor activity and stereotypy. Pharmacol Biochem Behav (1974) 2(2):249–55. doi:10.1016/0091-3057(74)90060-4
130. Correa M, Arizzi MN, Betz A, Mingote S, Salamone JD. Locomotor stimulant effects of intraventricular injections of low doses of ethanol in rats: acute and repeated administration. Psychopharmacology (2003) 170(4):368–75. doi:10.1007/s00213-003-1557-0
131. Hoshaw BA, Lewis MJ. Behavioral sensitization to ethanol in rats: evidence from the Sprague-Dawley strain. Pharmacol Biochem Behav (2001) 68(4):685–90. doi:10.1016/S0091-3057(01)00489-0
132. Camarini R, Hodge CW. Ethanol preexposure increases ethanol self-administration in C57BL/6J and DBA/2J mice. Pharmacol Biochem Behav (2004) 79(4):623–32. doi:10.1016/j.pbb.2004.09.012
133. Masur J, Oliveira de Souza ML, Zwicker AP. The excitatory effect of ethanol: absence in rats, no tolerance and increased sensitivity in mice. Pharmacol Biochem Behav (1986) 24(5):1225–8. doi:10.1016/0091-3057(86)90175-9
134. Phillips TJ, Dickinson S, Burkhart-Kasch S. Behavioral sensitization to drug stimulant effects in C57BL/6J and DBA/2J inbred mice. Behav Neurosci (1994) 108(4):789–803. doi:10.1037/0735-7044.108.4.789
135. Phillips TJ, Huson M, Gwiazdon C, Burkhart-Kasch S, Shen EH. Effects of acute and repeated ethanol exposures on the locomotor activity of BXD recombinant inbred mice. Alcohol Clin Exp Res (1995) 19(2):269–78. doi:10.1111/j.1530-0277.1995.tb01502.x
136. Schwandt ML, Higley JD, Suomi SJ, Heilig M, Barr CS. Rapid tolerance and locomotor sensitization in ethanol-naïve adolescent rhesus macaques. Alcohol Clin Exp Res (2008) 32(7):1217–28. doi:10.1111/j.1530-0277.2008.00676.x
137. Newlin DB, Thomson JB. Chronic tolerance and sensitization to alcohol in sons of alcoholics. Alcohol Clin Exp Res (1991) 15(3):399–405. doi:10.1111/j.1530-0277.1991.tb00537.x
138. Newlin DB, Thomson JB. Chronic tolerance and sensitization to alcohol in sons of alcoholics: II. Replication and reanalysis. Exp Clin Psychopharmacol (1999) 7(3):234–43. doi:10.1037/1064-1297.7.3.234
139. Pastor R, McKinnon CS, Scibelli AC, Burkhart-Kasch S, Reed C, Ryabinin AE, et al. Corticotropin-releasing factor-1 receptor involvement in behavioral neuroadaptation to ethanol: a urocortin1-independent mechanism. Proc Natl Acad Sci U S A (2008) 105(26):9070–5. doi:10.1073/pnas.0710181105
140. Pastor R, Reed C, Meyer PJ, McKinnon C, Ryabinin AE, Phillips TJ. Role of corticotropin-releasing factor and corticosterone in behavioral sensitization to ethanol. J Pharmacol Exp Ther (2012) 341(2):455–63. doi:10.1124/jpet.111.190595
141. Fee JR, Sparta DR, Picker MJ, Thiele TE. Corticotropin releasing factor-1 receptor antagonist, CP-154,526, blocks the expression of ethanol-induced behavioral sensitization in DBA/2J mice. Neuroscience (2007) 150(1):14–21. doi:10.1016/j.neuroscience.2007.08.027
142. Kawakami SE, Quadros IMH, Takahashi S, Suchecki D. Long maternal separation accelerates behavioural sensitization to ethanol in female, but not in male mice. Behav Brain Res (2007) 184(2):109–16. doi:10.1016/j.bbr.2007.06.023
143. Kawakami SE, Quadros IMH, Machado RB, Suchecki D. Sex-dependent effects of maternal separation on plasma corticosterone and brain monoamines in response to chronic ethanol administration. Neuroscience (2013) 253:55–66. doi:10.1016/j.neuroscience.2013.08.031
144. Roberts AJ, Lessov CN, Phillips TJ. Critical role for glucocorticoid receptors in stress- and ethanol-induced locomotor sensitization. J Pharmacol Exp Ther (1995) 275(2):790–7.
145. Quadros IM, Hwa LS, Shimamoto A, Carlson J, DeBold JF, Miczek KA. Prevention of alcohol-heightened aggression by CRF-R1 antagonists in mice: critical role for DRN-PFC serotonin pathway. Neuropsychopharmacology (2014) 39(12):2874–83. doi:10.1038/npp.2014.139
146. Baldwin HA, Rassnick S, Rivier J, Koob GF, Britton KT. CRF antagonist reverses the ‘anxiogenic’ response to ethanol withdrawal in the rat. Psychopharmacology (1991) 103(2):227–32. doi:10.1007/BF02244208
147. Rassnick S, Heinrichs SC, Britton KT, Koob GF. Microinjection of a corticotropin-releasing factor antagonist into the central nucleus of the amygdala reverses anxiogenic-like effects of ethanol withdrawal. Brain Res (1993) 605(1):25–32. doi:10.1016/0006-8993(93)91352-S
148. Valdez GR, Zorrilla EP, Roberts AJ, Koob GF. Antagonism of corticotropin-releasing factor attenuates the enhanced responsiveness to stress observed during protracted ethanol abstinence. Alcohol (2003) 29(2):55–60. doi:10.1016/S0741-8329(03)00020-X
149. Breese GR, Overstreet DH, Knapp DJ, Navarro M. Prior multiple ethanol withdrawals enhance stress-induced anxiety-like behavior: inhibition by CRF1- and benzodiazepine-receptor antagonists and a 5-HT1a-receptor agonist. Neuropsychopharmacology (2005) 30(9):1662–9. doi:10.1038/sj.npp.1300706
150. Wills TA, Knapp DJ, Overstreet DH, Breese GR. Sensitization, duration, and pharmacological blockade of anxiety-like behavior following repeated ethanol withdrawal in adolescent and adult rats. Alcohol Clin Exp Res (2009) 33(3):455–63. doi:10.1111/j.1530-0277.2008.00856.x
151. Overstreet DH, Knapp DJ, Breese GR. Pharmacological modulation of repeated ethanol withdrawal-induced anxiety-like behavior differs in alcohol-preferring P and Sprague-Dawley rats. Pharmacol Biochem Behav (2005) 81(1):122–30. doi:10.1016/j.pbb.2005.03.006
152. Breese GR, Knapp DJ, Overstreet DH. Stress sensitization of ethanol withdrawal-induced reduction in social interaction: inhibition by CRF-1 and benzodiazepine receptor antagonists and a 5-HT1A-receptor agonist. Neuropsychopharmacology (2004) 29(3):470–82. doi:10.1038/sj.npp.1300282
153. Knapp DJ, Overstreet DH, Moy SS, Breese GR. SB242084, flumazenil, and CRA1000 block ethanol withdrawal-induced anxiety in rats. Alcohol (2004) 32(2):101–11. doi:10.1016/j.alcohol.2003.08.007
154. Huang MM, Overstreet DH, Knapp DJ, Angel R, Wills TA, Navarro M, et al. Corticotropin-releasing factor (CRF) sensitization of ethanol withdrawal-induced anxiety-like behavior is brain site specific and mediated by CRF-1 receptors: relation to stress-induced sensitization. J Pharmacol Exp Ther (2010) 332(1):298–307. doi:10.1124/jpet.109.159186
155. Knapp DJ, Whitman BA, Wills TA, Angel RA, Overstreet DH, Criswell HE, et al. Cytokine involvement in stress may depend on corticotrophin releasing factor to sensitize ethanol withdrawal anxiety. Brain Behav Immun (2011) 25(Suppl1):S146–54. doi:10.1016/j.bbi.2011.02.018
156. Miczek KA, Faccidomo S, De Almeida RMM, Bannai M, Fish EW, Debold JF. Escalated aggressive behavior: new pharmacotherapeutic approaches and opportunities. Ann N Y Acad Sci (2004) 1036:336–55. doi:10.1196/annals.1330.021
157. Miczek KA, Takahashi A, Gobrogge KL, Hwa LS, de Almeida RMM. Escalated aggression in animal models: shedding new light on mesocorticolimbic circuits. Curr Opin Behav Sci (2015) 3:90–5. doi:10.1016/j.cobeha.2015.02.007
158. Chiavegatto S, Quadros IMH, Ambar G, Miczek KA. Individual vulnerability to escalated aggressive behavior by a low dose of alcohol: decreased serotonin receptor mRNA in the prefrontal cortex of male mice. Genes Brain Behav (2010) 9(1):110–9. doi:10.1111/j.1601-183X.2009.00544.x
159. Faccidomo S, Bannai M, Miczek KA. Escalated aggression after alcohol drinking in male mice: dorsal raphé and prefrontal cortex serotonin and 5-HT(1B) receptors. Neuropsychopharmacology (2008) 33(12):2888–99. doi:10.1038/npp.2008.7
160. Faccidomo S, Quadros IM, Takahashi A, Fish EW, Miczek KA. Infralimbic and dorsal raphé microinjection of the 5-HT(1B) receptor agonist CP-93,129: attenuation of aggressive behavior in CFW male mice. Psychopharmacology (2012) 222(1):117–28. doi:10.1007/s00213-011-2629-1
161. Miczek KA, de Almeida RM. Oral drug self-administration in the home cage of mice: alcohol-heightened aggression and inhibition by the 5-HT1B agonist anpirtoline. Psychopharmacology (2001) 157(4):421–9. doi:10.1007/s002130100831
162. Fox JH, Lowry CA. Corticotropin-releasing factor-related peptides, serotonergic systems, and emotional behavior. Front Neurosci (2013) 7:169. doi:10.3389/fnins.2013.00169
163. Rose AK, Shaw SG, Prendergast MA, Little HJ. The importance of glucocorticoids in alcohol dependence and neurotoxicity. Alcohol Clin Exp Res (2010) 34(12):2011–8. doi:10.1111/j.1530-0277.2010.01298.x
164. Rivier C, Lee S. Acute alcohol administration stimulates the activity of hypothalamic neurons that express corticotropin-releasing factor and vasopressin. Brain Res (1996) 726(1–2):1–10. doi:10.1016/0006-8993(96)00301-0
165. Rivier C, Rivier J, Lee S. Importance of pituitary and brain receptors for corticotrophin-releasing factor in modulating alcohol-induced ACTH secretion in the rat. Brain Res (1996) 721(1–2):83–90. doi:10.1016/0006-8993(96)00164-3
166. Redei E, Rittenhouse PA, Revskoy S, McGivern RF, Aird F. A novel endogenous corticotropin release inhibiting factor. Ann N Y Acad Sci (1998) 840:456–69. doi:10.1111/j.1749-6632.1998.tb09584.x
167. Ogilvie K, Lee S, Rivier C. Effect of three different modes of alcohol administration on the activity of the rat hypothalamic-pituitary-adrenal axis. Alcohol Clin Exp Res (1997) 21(3):467–76. doi:10.1111/j.1530-0277.1997.tb03792.x
168. Ogilvie KM, Lee S, Rivier C. Role of arginine vasopressin and corticotropin-releasing factor in mediating alcohol-induced adrenocorticotropin and vasopressin secretion in male rats bearing lesions of the paraventricular nuclei. Brain Res (1997) 744(1):83–95. doi:10.1016/S0006-8993(96)01082-7
169. Laszlo FA, Varga C, Pávó I, Gardi J, Vecsernyés M, Gálfi M, et al. Vasopressin pressor receptor-mediated activation of HPA axis by acute ethanol stress in rats. Am J Physiol Regul Integr Comp Physiol (2001) 280(2):R458–65.
170. Rivier C, Imaki T, Vale W. Prolonged exposure to alcohol: effect on CRF mRNA levels, and CRF- and stress-induced ACTH secretion in the rat. Brain Res (1990) 520:1–5. doi:10.1016/0006-8993(90)91685-A
171. Lee S, Imaki T, Vale W, Rivier C. Effect of prenatal exposure to ethanol on the activity of the hypothalamic-pituitary-adrenal axis of the offspring: importance of the time of exposure to ethanol and possible modulating mechanisms. Mol Cell Neurosci (1990) 1(2):168–77. doi:10.1016/1044-7431(90)90022-V
172. Zhou Y, Franck J, Spangler R, Maggos CE, Ho A, Kreek MJ. Reduced hypothalamic POMC and anterior pituitary CRF1 receptor mRNA levels after acute, but not chronic, daily ‘binge’ intragastric alcohol administration. Alcohol Clin Exp Res (2000) 24(10):1575–82. doi:10.1111/j.1530-0277.2000.tb04577.x
173. Glavas MM, Ellis L, Yu WK, Weinberg J. Effects of prenatal ethanol exposure on basal limbic-hypothalamic-pituitary-adrenal regulation: role of corticosterone. Alcohol Clin Exp Res (2007) 31(9):1598–610. doi:10.1111/j.1530-0277.2007.00460.x
174. Lee S, Selvage D, Hansen K, Rivier C. Site of action of acute alcohol administration in stimulating the rat hypothalamic-pituitary-adrenal axis: comparison between the effect of systemic and intracerebroventricular injection of this drug on pituitary and hypothalamic responses. Endocrinology (2004) 145(10):4470–9. doi:10.1210/en.2004-0110
175. Logrip ML, Rivier C, Lau C, Im S, Vaughan J, Lee S. Adolescent alcohol exposure alters the rat adult hypothalamic-pituitary-adrenal axis responsiveness in a sex-specific manner. Neuroscience (2013) 235:174–86. doi:10.1016/j.neuroscience.2012.12.069
176. Lee S, Rivier C. Alcohol increases the expression of type 1, but not type 2 alpha corticotropin-releasing factor (CRF) receptor messenger ribonucleic acid in the rat hypothalamus. Brain Res Mol Brain Res (1997) 52(1):78–89. doi:10.1016/S0169-328X(97)00226-X
177. Lee S, Schmidt ED, Tilders FJ, Rivier C. Effect of repeated exposure to alcohol on the response of the hypothalamic-pituitary-adrenal axis of the rat: I. role of changes in hypothalamic neuronal activity. Alcohol Clin Exp Res (2001) 25(1):98–105. doi:10.1111/j.1530-0277.2001.tb02132.x
178. Lee S, Smith GW, Vale W, Lee KF, Rivier C. Mice that lack corticotropin-releasing factor (CRF) receptors type 1 show a blunted ACTH response to acute alcohol despite up-regulated constitutive hypothalamic CRF gene expression. Alcohol Clin Exp Res (2001) 25(3):427–33. doi:10.1111/j.1530-0277.2001.tb02231.x
179. Falco AM, Bergstrom HC, Bachus SE, Smith RF. Persisting changes in basolateral amygdala mRNAs after chronic ethanol consumption. Physiol Behav (2009) 96(1):169–73. doi:10.1016/j.physbeh.2008.09.019
180. Eisenhardt M, Hansson AC, Spanagel R, Bilbao A. Chronic intermittent ethanol exposure in mice leads to an up-regulation of CRH/CRHR1 signaling. Alcohol Clin Exp Res (2015) 39(4):752–62. doi:10.1111/acer.12686
181. Allen CD, Rivier CL, Lee SY. Adolescent alcohol exposure alters the central brain circuits known to regulate the stress response. Neuroscience (2011) 182:162–8. doi:10.1016/j.neuroscience.2011.03.003
182. Merlo-Pich E, Lorang M, Yeganeh M, Rodriguez de Fonseca F, Raber J, et al. Increase of extracellular corticotropin-releasing factor-like immunoreactivity levels in the amygdala of awake rats during restraint stress and ethanol withdrawal as measured by microdialysis. J Neurosci (1995) 15(8):5439–47.
183. Olive MF, Koenig HN, Nannini MA, Hodge CW. Elevated extracellular CRF levels in the bed nucleus of the stria terminalis during ethanol withdrawal and reduction by subsequent ethanol intake. Pharmacol Biochem Behav (2002) 72(1–2):213–20. doi:10.1016/S0091-3057(01)00748-1
184. Lam MP, Gianoulakis C. Effects of acute ethanol on corticotropin-releasing hormone and β-endorphin systems at the level of the rat central amygdala. Psychopharmacology (2011) 218(1):229–39. doi:10.1007/s00213-011-2337-x
185. Caldwell KK, Goggin SL, Tyler CR, Allan AM. Prenatal alcohol exposure is associated with altered subcellular distribution of glucocorticoid and mineralocorticoid receptors in the adolescent mouse hippocampal formation. Alcohol Clin Exp Res (2014) 38(2):392–400. doi:10.1111/acer.12236
186. Li Z, Kang SS, Lee S, Rivier C. Effect of ethanol on the regulation of corticotropin-releasing factor (CRF) gene expression. Mol Cell Neurosci (2005) 29(3):345–54. doi:10.1016/j.mcn.2005.04.002
187. Pickering C, Avesson L, Lindblom J, Liljequist S, Schiöth HB. Identification of neurotransmitter receptor genes involved in alcohol self-administration in the rat prefrontal cortex, hippocampus and amygdala. Prog Neuropsychopharmacol Biol Psychiatry (2007) 31(1):53–64. doi:10.1016/j.pnpbp.2006.06.010
188. Hansson AC, Cippitelli A, Sommer WH, Ciccocioppo R, Heilig M. Region-specific down-regulation of Crhr1 gene expression in alcohol-preferring msP rats following ad lib access to alcohol. Addict Biol (2007) 12(1):30–4. doi:10.1111/j.1369-1600.2007.00050.x
189. McClintick JN, McBride WJ, Bell RL, Ding Z, Liu Y, Xuei X, et al. Gene expression changes in serotonin, GABA-A receptors, neuropeptides and ion channels in the dorsal raphe nucleus of adolescent alcohol-preferring (P) rats following binge-like alcohol drinking. Pharmacol Biochem Behav (2015) 129:87–96. doi:10.1016/j.pbb.2014.12.007
190. Pickering C, Avesson L, Liljequist S, Lindblom J, Schiöth HB. the Role of hypothalamic peptide gene expression in alcohol self-administration behavior. Peptides (2007) 28(12):2361–71. doi:10.1016/j.peptides.2007.09.011
191. Weitemier AZ, Ryabinin AE. Brain region-specific regulation of urocortin 1 innervation and corticotropin-releasing factor receptor type 2 binding by ethanol exposure. Alcohol Clin Exp Res (2005) 29(9):1610–20. doi:10.1097/01.alc.0000179363.44542.05
192. Rivier C. Role of hypothalamic corticotropin-releasing factor in mediating alcohol-induced activation of the rat hypothalamic-pituitary-adrenal axis. Front Neuroendocrinol (2014) 35(2):221–33. doi:10.1016/j.yfrne.2013.10.005
193. Läck AK, Floyd DW, McCool BA. Chronic ethanol ingestion modulates proanxiety factors expressed in rat central amygdala. Alcohol (2005) 36(2):83–90. doi:10.1016/j.alcohol.2005.07.004
194. Zorrilla EP, Valdez GR, Weiss F. Changes in levels of regional CRF-like-immunoreactivity and plasma corticosterone during protracted drug withdrawal in dependent rats. Psychopharmacology (2001) 158(4):374–81. doi:10.1007/s002130100773
195. Allen CD, Lee S, Koob GF, Rivier C. Immediate and prolonged effects of alcohol exposure on the activity of the hypothalamic-pituitary-adrenal axis in adult and adolescent rats. Brain Behav Immun (2011) 25(Suppl 1):S50–60. doi:10.1016/j.bbi.2011.01.016
196. Lovallo WR. The hypothalamic-pituitary-adrenocortical axis in addiction. Int J Psychophysiol (2006) 59(3):193–4. doi:10.1016/j.ijpsycho.2005.10.006
197. Jenkins JS, Connolly J. Adrenocortical response to ethanol in man. Br Med J (1968) 2(5608):804–5. doi:10.1136/bmj.2.5608.804
198. King A, Munisamy G, Wit H, Lin S. Attenuated cortisol response to alcohol in heavy social drinkers. Int J Psychophysiol (2006) 59(3):203–9. doi:10.1016/j.ijpsycho.2005.10.008
199. Adinoff B, Krebaum SR, Chandler PA, Ye W, Morton BB, Williams MJ. Dissection of hypothalamic-pituitary-adrenal axis pathology in 1-month-abstinent alcohol-dependent men, part 1: adrenocortical and pituitary glucocorticoid responsiveness. Alcohol Clin Exp Res (2005) 29(4):517–27. doi:10.1097/01.ALC.0000158940.05529.0A
200. Adinoff B, Krebaum SR, Chandler PA, Ye W, Morton BB, Williams MJ. Dissection of hypothalamic-pituitary-adrenal axis pathology in 1-month-abstinent alcohol-dependent men, part 2: response to ovine corticotropin-releasing factor and naloxone. Alcohol Clin Exp Res (2005) 29(4):528–37. doi:10.1097/01.ALC.0000158939.25531.EE
201. Wand GS, Dobs AS. Alterations in the hypothalamic-pituitary-adrenal axis in actively drinking alcoholics. J Clin Endocrinol Metab (1991) 72(6):1290–5. doi:10.1210/jcem-72-6-1290
202. Errico AL, Parsons OA, King AC, Lovallo WL. Attenuated cortisol response to biobehavioral stressors in sober alcoholics. J Stud Alcohol (1993) 54(4):393–8. doi:10.15288/jsa.1993.54.393
203. Bernardy NC, King AC, Parsons OA, Lovallo WE. Altered cortisol response in sober alcoholics: an examination of contributing factors. Alcohol (1996) 13(5):493–8. doi:10.1016/0741-8329(96)00043-2
204. Lovallo WR, Dickensheets SL, Myers DA, Thomas TL, Nixon SJ. Blunted stress cortisol response in abstinent alcoholic and polysubstance-abusing men. Alcohol Clin Exp Res (2000) 24(5):651–8. doi:10.1111/j.1530-0277.2000.tb02036.x
205. Margraf HW, Moyer CA, Ashford LE, Lavalle LW. Adrenocortical function in alcoholics. J Surg Res (1967) 7(2):55–62. doi:10.1016/0022-4804(67)90035-2
206. Esel E, Sofuoglu S, Aslan SS, Kula M, Yabanoglu I, Turan MT. Plasma levels of beta-endorphin, adrenocorticotropic hormone and cortisol during early and late alcohol withdrawal. Alcohol Alcohol (2001) 36(6):572–6. doi:10.1093/alcalc/36.6.572
207. Keedwell PA, Poon L, Papadopoulos AS, Marshall EJ, Checkley SA. Salivary cortisol measurements during a medically assisted alcohol withdrawal. Addict Biol (2001) 6(3):247–56. doi:10.1080/13556210120056580
208. Majumdar SK, Shaw GK, Bridges PK. Relationship between plasma adrenocorticotrophic hormone and cortisol concentrations in chronic alcoholic patients with depression. Drug Alcohol Depend (1989) 23(2):111–6. doi:10.1016/0376-8716(89)90015-X
209. Koob GF, Le Moal M. Drug addiction, dysregulation of reward, and allostasis. Neuropsychopharmacology (2001) 24(2):97–129. doi:10.1016/S0893-133X(00)00195-0
210. Chen ACH, Manz N, Tang Y, Rangaswamy M, Almasy L, Kuperman S, et al. Single-nucleotide polymorphisms in corticotropin releasing hormone receptor 1 gene (CRHR1) are associated with quantitative trait of event-related potential and alcohol dependence. Alcohol Clin Exp Res (2010) 34(6):988–96. doi:10.1111/j.1530-0277.2010.01173.x
211. Treutlein J, Kissling C, Frank J, Wiemann S, Dong L, Depner M, et al. Genetic association of the human corticotropin releasing hormone receptor 1 (CRHR1) with binge drinking and alcohol intake patterns in two independent samples. Mol Psychiatry (2006) 11(6):594–602. doi:10.1038/sj.mp.4001813
212. Blomeyer D, Treutlein J, Esser G, Schmidt MH, Schumann G, Laucht M. Interaction between CRHR1 gene and stressful life events predicts adolescent heavy alcohol use. Biol Psychiatry (2008) 63(2):146–51. doi:10.1016/j.biopsych.2007.04.026
213. Schmid B, Blomeyer D, Treutlein J, Zimmermann US, Buchmann AF, Schmidt MH, et al. Interacting effects of CRHR1 gene and stressful life events on drinking initiation and progression among 19-year-olds. Int J Neuropsychopharmacol (2010) 13(6):703–14. doi:10.1017/S1461145709990290
214. Nelson EC, Agrawal A, Pergadia ML, Wang JC, Whitfield JB, Saccone FS, et al. H2 haplotype at chromosome 17q21.31 protects against childhood sexual abuse-associated risk for alcohol consumption and dependence. Addict Biol (2010) 15(1):1–11. doi:10.1111/j.1369-1600.2009.00181.x
215. Vogel F. The genetic basis of the normal human electroencephalogram (EEG). Humangenetik (1970) 10(2):91–114. doi:10.1007/BF00295509
216. Enoch MA, White KV, Harris CR, Robin RW, Ross J, Rohrbaugh JW, et al. Association of low-voltage alpha EEG with a subtype of alcohol use disorders. Alcohol Clin Exp Res (1999) 23(8):1312–9. doi:10.1111/j.1530-0277.1999.tb04352.x
217. Enoch MA, Shen PH, Ducci F, Yuan Q, Liu J, White KV, et al. Common genetic origins for EEG, alcoholism and anxiety: the role of CRH-BP. PLoS One (2008) 3(10):e3620. doi:10.1371/journal.pone.0003620
218. Ray LA. Stress-induced and cue-induced craving for alcohol in heavy drinkers: preliminary evidence of genetic moderation by the OPRM1 and CRH-BP genes. Alcohol Clin Exp Res (2011) 35(1):166–74. doi:10.1111/j.1530-0277.2010.01333.x
219. Tartter M, Ray L. A prospective study of stress and alcohol craving in heavy drinkers. Pharmacol Biochem Behav (2012) 101(4):625–31. doi:10.1016/j.pbb.2012.03.007
220. Ribbe K, Ackermann V, Schwitulla J, Begemann M, Papiol S, Grube S, et al. Prediction of the risk of comorbid alcoholism in schizophrenia by interaction of common genetic variants in the corticotropin-releasing factor system. Arch Gen Psychiatry (2011) 68(12):1247–56. doi:10.1001/archgenpsychiatry.2011.100
Keywords: alcohol, addiction, alcohol self-administration, neuropeptides, animal models, mesocorticolimbic system, sensitization, alcohol-related aggression
Citation: Quadros IMH, Macedo GC, Domingues LP and Favoretto CA (2016) An Update on CRF Mechanisms Underlying Alcohol Use Disorders and Dependence. Front. Endocrinol. 7:134. doi: 10.3389/fendo.2016.00134
Received: 06 July 2016; Accepted: 09 September 2016;
Published: 21 October 2016
Edited by:
Carol F. Elias, University of Michigan, USAReviewed by:
Andrey E. Ryabinin, Oregon Health & Science University, USAKazuhiro Takahashi, Tohoku University, Japan
Copyright: © 2016 Quadros, Macedo, Domingues and Favoretto. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Isabel Marian Hartmann Quadros, aXNhYmVsLnF1YWRyb3NAdW5pZmVzcC5icg==