 Helge L. Waldum
Helge L. Waldum Liv Sagatun
Liv Sagatun Patricia Mjønes
Patricia Mjønes- 1Department of Gastroenterology and Hepatology, St Olav’s Hospital, Trondheim, Norway
- 2Department of Cancer Research and Molecular Medicine, Faculty of Medicine, Norwegian University of Science and Technology, Trondheim, Norway
- 3Department of Pathology, St Olav’s Hospital, Trondheim, Norway
- 4Department of Laboratory Medicine, Children and Women’s Health, Norwegian University of Science and Technology, Trondheim, Norway
Gastric cancer although occurring in reduced frequency is still an important disease, partly because of the bad prognosis when occurring in western countries. This decline in occurrence may mainly be due to the reduced prevalence of Helicobacter pylori (Hp) infection, which is the most important cause of gastric cancer. There exist many different pathological classifications of gastric carcinomas, but the most useful seems to be the one by Lauren into intestinal and diffuse types since these types seldom transform into the other and also have different epidemiology. During the nearly 30 years that have passed since the groundbreaking description of Hp as the cause of gastritis and gastric cancer, a continuous search for the mechanism by which Hp infection causes gastric cancer has been done. Interestingly, it is mainly atrophic gastritis of the oxyntic mucosa that predisposes to gastric cancer possibly by inducing hypoacidity and hypergastrinemia. There are many arguments in favor of an important role of gastrin and its target cell, the enterochromaffin-like cell, in gastric carcinogenesis. The role of gastrin in gastric carcinogenesis implies caution in the long-term treatment with inhibitors of gastric acid secretion inducing secondary hypergastrinemia, in a common disease like gastroesophageal reflux disease.
Gastric cancer has been one of the most prevalent cancers and combined with a high mortality, gastric cancer has been among the leading causes of cancer death. However, there has been a continuous decline in the prevalence of gastric cancers during the last decades. Unfortunately, in spite of widespread use of upper gastrointestinal endoscopy, the prognosis in patients affected in the western world has not been much improved. This is in contrast to Japan where gastric cancer often is diagnosed at an early phase allowing removal and a better prognosis (1, 2). This is partly due to special screening programs in Japan having a high prevalence of gastric cancer (1, 2), resulting in diagnoses at a curable stage. In most western countries, the prevalence is much lower (3), and therefore, no such program has been implemented. The cause of the geographical differences in the prevalence of gastric cancer most probably is increased prevalence of Helicobacter pylori (Hp) in East Asia as well as a higher frequency of atrophic oxyntic gastritis (4). The prognosis of gastric cancer is better in patients from East Asia even when living in the west possibly due to less aggressive biology (5). In this review, we will focus on the role of gastrin in the etiology of gastric cancer and at the same time give an explanation of the decline in frequency. We will only cover cancers originating from epithelial cells (carcinomas) and will not discuss the importance of Epstein–Barr virus that plays a role neither in gastric carcinogenesis (6) nor in human papilloma virus, which has a less established impact (7).
The Gastric Mucosa
The mucosa of the stomach has traditionally been divided into three parts: the cardiac, the oxyntic, and the antral mucosa. During the last decades, it has, however, been discussed whether the cardiac mucosa occurs normally or represents metaplastic mucosa (8, 9). In the oxyntic mucosa, the highly specialized glands contain the acid-producing parietal cell, the pepsinogen-producing chief cell, and the regulatory, histamine-producing [enterochromaffin-like (ECL)] cell, which are specific for the oxyntic glands. These cells are not found in the antral glands where instead the gastrin-producing G-cell is localized. Previously, a sharp border between the oxyntic and antral mucosa was presumed, but recent work has shown that there is overlap with oxyntic glandular elements occurring in the antral mucosa (10). Nevertheless, taking into consideration the differences between the oxyntic and the antral mucosa, it should be obvious that gastric carcinomas should be classified anatomically according to mucosa of origin and not as presently only into cardiac and distal carcinomas with the latter comprising both oxyntic and antral starting point.
Embryology of the Gastric Mucosa
The gastrointestinal tract is derived from the endoderm. Stem cells located at the neck of the glands divide and differentiate into specialized cells while moving into the crypts of the glands (parietal and chief cells) or to the surface becoming specialized cells producing mucus and bicarbonate, which make the gastric mucosa like the mucosa of the duodenal bulb, able to resist the highly acidic and proteolytic gastric juice. There are many regulatory neuroendocrine (NE) cells in the gastric mucosa. The NE cells in man were previously claimed not to divide (11) in contrast to similar cells in rodents (12, 13). Now it is, however, established that NE cells also in man do divide as shown for the β-cell (14) and indirectly for the gastric ECL cell by the selective and concentration-dependent trophic effect by gastrin (15). In the gastric mucosa, the ability to self-replicate is unique to the ECL cell and probably the other NE cells and in contrast to other mucosal cells that are formed by differentiation of cells originating from stem cells. Nevertheless, studies have indicated that also the NE cell originate from a common stem cell (16, 17), and thus not coming from the neural crest as proposed by Pearse and Polak (18) based on the similarities between NE cells at different locations and neural cells. Although there seems to be rather firm evidence for stem cell origin of NE cells in the intestine and the antrum (16, 19), this has not been convincingly shown for NE cells in the oxyntic mucosa.
Properties of NE Cells
Whatever the embryology, the NE cells have a unique position among the mucosal cells in their ability to divide. Moreover, they produce signal substances that affect the function of neighboring cells. The signal substances are delivered via a paracrine route or via synaptic-like transmission from neuron-resembling projections (20, 21) or reaching cells via the blood working as hormones. The NE cells have secretory granules that may be detected by the general markers belonging to the chromogranins (22), and by cell-specific signal substances such as gastrin, ghrelin, somatostatin, enteroglucagon, and many others. Interestingly from an embryological point of view, the synaptic vesicle marker synaptophysin (23) is found in NE cells in common with neurons. In the gastrointestinal tract, there are about 30–40 different NE cells producing separate signal substances. Although the different types of NE cells look quite similar, most of the NE tumors originate from only a few of them, typically the ECL cell in the stomach and the enterochromaffin (EC) cell in the gut. The reason for this discrepancy in tumourigenecity is not known, but could be related to differences in growth regulation as well as to different signal substances with different effect on surrounding tissue. Thus, not only the function (24) but also the proliferation (25) of the ECL cell in the stomach is regulated by the hormone gastrin. Gastrin is often elevated in gastric hypoacidity either due to atrophic gastritis (26) or drug treatment (27). Moreover, the signal substance of the ECL cell is histamine having profound vascular effects, which presumably may favor invasion and spread. In the small intestine, the serotonin-producing EC cell is the principal cell of origin of NE tumors. The regulation of the EC cell function and proliferation is mainly unknown, but it is possible that the serotonin production via its vascular effects is the cause of invasion and spread at an early phase, which is in many ways a hallmark of this tumor. Although the NE cells do proliferate, they do so very slowly which was the background for the dispute whether they divide or not (11, 28). The slow proliferation of normal NE cells is reflected in the relatively benign course of NE tumors where the patient may live asymptomatically for more than a decade with such a tumor or the first metastasis may manifest itself after more than two decades.
Predisposing Conditions for Gastric Cancer
Gastritis with metaplasia has for long been known to predispose to gastric cancer (29). In fact, it was shown that gastric cancer virtually only occurred in stomachs with gastritis (29). When Hp was shown to be the cause of most types of gastritis (30), it was natural to examine the role of Hp in gastric cancer, and it was soon shown to be the most important etiological factor of this malignancy (31). Very recently, a meta-analysis showed that Hp eradication reduced the risk of gastric cancer (32). However, also the other type of gastritis, the so-called autoimmune gastritis affecting only the oxyntic mucosa predisposes to gastric cancer (33). Thus, it seems that the gastritis and not Hp itself is the carcinogenic mechanism. Nevertheless, throughout the 25 years since the description of Hp as the cause of gastritis (30) and gastric carcinoma (31), there have been numerous studies on the mechanism by which Hp induces gastric carcinoma, all of them without success. When Hp gastritis is confined to the antral mucosa, it predisposes to duodenal ulcer (34), but not to gastric carcinoma. On the contrary, it is well known that duodenal ulcer in a way protects against development of gastric carcinoma (35). The risk of gastric carcinoma increases only when the gastritis affects the oxyntic mucosa (36) and mainly when oxyntic atrophy has developed (36). The presence of oxyntic gastric atrophy may indirectly be assessed by low serum pepsinogen I (37) and even better when combined with determination of gastrin (38). In atrophic gastritis, intestinal metaplasia may with time develop (39). Intestinal metaplasia has been associated with an additional cancer risk but could also just reflect long-standing oxyntic atrophy as patients with intestinal metaplasia are older than those with oxyntic atrophy without intestinal metaplasia (39, 40). Anyhow, the sequence in gastric carcinogenesis postulated by Correa where the mucosa goes through phases of gastritis, atrophic gastritis, intestinal metaplasia, dysplasia of different degrees, and finally neoplasia has had great impact for a long time (41). However, in a recent Swedish study, there was little difference in risk of gastric cancer between patients with atrophic gastritis and those with intestinal metaplasia (42). As in other parts of the gastrointestinal tract, polyps in the stomach may predispose to gastric carcinomas, although the polyp carcinoma sequence in the stomach is much less typical than in the colon. Typically, the adenomatous polyps, which are rather seldom, are the polyps with the highest malignant potential. To be complete, it has to be added that ECL cell-derived neuroendocrine tumors (NETs) are associated with both Hp infectious gastritis (43) and autoimmune gastritis (44). Mutation in the gene CDH1 coding for E-cadherin predisposes to gastric carcinoma of diffuse type (45), and quite recently missense mutation of one of the genes of the proton pump, resulting in anacidity from birth was reported to give ECL cell-derived NETs of different degree of malignancy at an early age (46, 47).
Classification of Gastric Carcinomas
Quite recently, a molecular classification of gastric carcinomas based on occurrence of mutations was described (48). Although such a system may have impact on the choice of treatment at the time of classification, it may not tell much about the cell of origin, which is mandatory for understanding of carcinogenesis and prophylaxis of tumors. In that respect, older histological classifications may be more useful.
There are many classification systems for gastric carcinomas, but the system proposed by Lauren seems to be the most relevant (49). Thus, his classification into intestinal and diffuse types where the intestinal type shows glandular structures, whereas the diffuse type lacks such differentiation is increasingly used most likely because the carcinomas do not transform into each other over time (49). In fact, the stable phenotype of the carcinomas during the course of the disease may suggest important biological differences that are supported by different epidemiology for the types of cancer (50). The main weakness with Lauren’s classification is that about 15% of the carcinomas are difficult to classify due to overlapping traits (49). Although gastric carcinomas of diffuse type lack a hallmark of adenocarcinomas (glandular growth pattern), they have nevertheless been classified as adenocarcinomas due to presumed mucin content of the cancer cells (49). The presence of mucin has been based on PAS positivity. However, PAS positivity is an unspecific histochemical method reacting with glycoproteins/peptides in general (51), and we have shown that the diffuse gastric carcinomas with the highest content of PAS positive material, the signet ring cells, are positive for NE markers both by immunohistochemistry (52) and in situ hybridization (53). In general, gastric carcinomas of diffuse type often show NE differentiation (54, 55), and it might be that this type actually is a NE carcinoma (Figure 1). In this context, it should be recalled that it has for long been known that NETs (formerly called carcinoids) can show glandular growth pattern (56, 57) and mucin positivity (58). Since the gastric carcinomas with endocrine differentiation (which mainly are of diffuse type), more specifically seem to be of ECL cell origin (59), the principal regulator of ECL cell function as well as growth, gastrin, naturally becomes of central interest in gastric carcinogenesis.
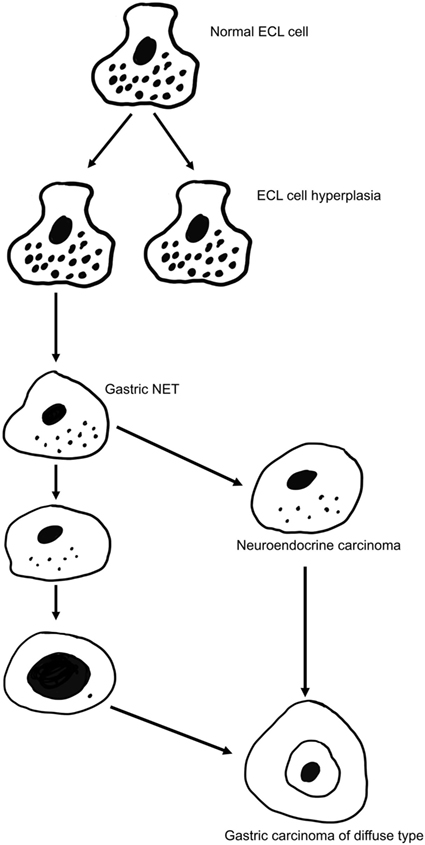
Figure 1. The gradual transition of an enterochromaffin-like (ECL) cell into a highly malignant carcinoma cell of gastric carcinoma of diffuse type [from Waldum et al. (57) with permission].
Gastrin and Gastric NETs
More than 30 years ago, gastrin was realized to induce gastric NETs in man whether due to gastrinoma with increased gastric acid secretion (60) or being secondary to hypoacidity (61) causing Bordi to raise the question whether this was a hormone induced tumor (62). These tumors were regarded as rather benign and also very rare. Gastric NETs were therefore not seen as very important from a clinical point of view. With the description of NETs in the oxyntic mucosa of rodents dosed long term with inhibitors of gastric acid secretion belonging to the histamine receptor-2 blockers (H-2 blockers) like loxtidine (63) or the proton pump inhibitors (PPIs) omeprazole (64), the interest increased dramatically since these drug were so commonly used. It was soon realized that hypergastrinemia was the common pathogenic factor and that the degree and duration of hypoacidity and secondary hypergastrinemia determine the risk of tumor development. Thus, although very high doses of the commonly used H-2 blockers like ranitidine can induce such tumors in rodents (65), the doses used clinically carry little risk since the potency of H-2 blockers in clinical use is too low. Moreover, the tolerance ordinary H-2 blockers induce (66) also protects against long-term marked hypoacidity/hypergastrinemia. Loxtidine was the so-called unsurmountable H-2 blocker (63), and this compound was not developed into a drug due to the NETs seen in rodents. The PPIs are very efficient drugs in inhibiting gastric acid secretion (67), but at the same time risky due to hypergastrinemia. Rather early, it was shown that PPI treatment induced ECL cell hyperplasia (68), which caused rebound acid hypersecretion (69). A decade later, it was reported that a period of PPI dosed to healthy individuals induced dyspepsia (70) probably due to rebound acid hypersecretion. Studies based on treatment of PPIs for up to 10 years have not reported occurrence of gastric NETs (71, 72), and thus such treatment has therefore been claimed to be safe with regard to neoplasia. However, it has to be remembered that neoplasia and particularly with respect to tumors originating from NE cells, which are so slow proliferators, may take decades to develop. Moreover, already more than 15 years ago, the producer reported on the occurrence of reversible ECL cell NETs during omeprazole treatment (73), and we (74) and others (75, 76) have reported similar tumors during PPI treatment. Finally, and very importantly, a large kindred from Spain was reported to develop ECL cell tumors of different degree of malignancy at a young age (20s and 30s) due to missense mutation causing lack of function of the proton pump. Thus, the homozygote individuals who all developed tumors had been anacidic and hypergastrinemic from birth (46). Most of the tumors were classified as NETs although one tumor was diagnosed as an adenocarcinoma (46). Later, we have examined these tumors and have concluded that the adenocarcinoma more correctly should have been classified as a NE neoplasm (47). We have also described a gastric NE carcinoma that developed after about 12 years of PPI treatment (77). Oxyntic gastric neoplasia has been reported in all conditions with hypergastrinemia in animals as well as man. It is therefore no doubt that hypergastrinemia may lead to gastric NETs. The dominating role of gastrin in the pathogenesis of gastric NETs is demonstrated by their eradication by the treatment with the gastrin antagonist netazepide (78). We have also demonstrated that treatment with a long-acting somatostatin analog makes gastric NETs disappear or shrink (79). This effect was most probably due to a direct effect on the tumor cells since the reduction in blood gastrin was only marginal (79). On the other hand, patients operated with antrectomi due to peptic ulcer either as Billroth-I or particularly Billroth-II do develop after long latency carcinomas in the remnant stomach even though they are hypogastrinemic (80). However, not only the ECL cell but also other NE cells in the gastric mucosa could possibly give rise to tumors. Thus, we have described a case where a cancer of the remnant stomach originating from the D cell, developed decades after the Billroth-II procedure (81). It has not yet been examined, but it is conceivable that gastrin has a negative trophic effect on the D cell in the oxyntic mucosa.
Gastrin and Gastric Carcinomas
The main cause of gastric carcinomas is Hp infection (31) and mainly when inducing atrophy of the oxyntic mucosa (36). Atrophic oxyntic gastritis leads to reduced acid secretion, gastric hypoacidity, and secondary hypergastrinemia (26). We have repeatedly shown that gastric carcinomas, especially of diffuse type, express NE markers (54, 55) and more specifically ECL cell markers (59), which incriminate gastrin in the pathogenesis. We have described a patient with pernicious anemia who developed a gastric NET that was removed endoscopically and after 1 year had recurrence of the tumor, resulting in gastrectomy and who died some years later of general metastasis (82). During the 5-year period after the first diagnosis of the gastric NET, the tumor showed progressive loss of NE markers and a dramatic increase in proliferation demonstrating the transition from a gastric NET into a highly malignant carcinoma (82). The close relationship between gastric NETs and gastric carcinomas is also reflected by the co-occurrence of NETs and carcinomas in autoimmune gastritis (83) as well as Hp gastritis predisposing both to gastric cancer (31) and gastric NETs (43). A few studies have reported hypergastrinemia in patients with gastric carcinoma (84–86). The gastric carcinomas of diffuse type quite possibly originate from the ECL cell (54, 55, 59) by dedifferentiation. Carcinomas of intestinal type are also associated to atrophic oxyntic gastritis (87), but this type of gastric carcinoma more seldom express NE markers and therefore more likely develop from the stem cells. However, gastrin may nevertheless be important in the carcinogenic process leading to carcinoma of intestinal type by stimulating the proliferation of the stem cell, either by a possible but not proven gastrin receptor on this cell or indirectly by a positive trophic effect of mediators from the ECL cell, among them Reg protein is the most likely candidate (88) (Figure 2).
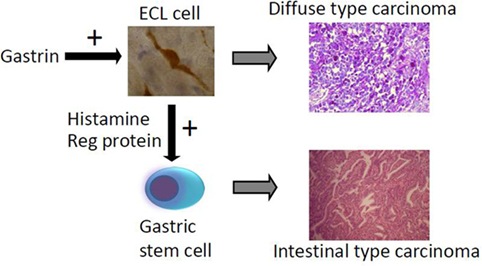
Figure 2. The way hypergastrinemia may induce gastric carcinoma of both diffuse and intestinal types [from Waldum et al. (89) with permission].
Since the acknowledgment of the central role of Hp infection in gastric carcinogenesis (31), there has been an unsuccessful search for the mechanism. However, it seems logical to focus on the gastritis since also the so-called autoimmune gastritis predisposes to gastric cancer (33). Moreover, oxyntic gastric atrophy is a common feature in conditions causing gastric cancer (36), and therefore, we recently proposed that hypergastrinemia could be the factor incriminated in Hp gastric carcinogenesis (89). The risk of long-term profound inhibition of gastric acid secretion particularly by the very efficient PPIs is increasingly recognized (90, 91). To conclude this part, gastrin seems to be the most important factor in gastric carcinogenesis.
Consequences of the Dominating Role of Gastrin in Gastric Carcinogenesis
The understanding of the fundamental role of gastrin in gastric carcinogenesis is of clinical importance. Thus, we should avoid long-term treatment particularly in children and young adults due to their long life expectancy, with inhibitors of gastric secretion leading to hypergastrinemia. Presently, long-term treatment with PPI is mainly indicated in patients with gastroesophageal reflux disease (GERD). In young GERD patients, H-2 blocker treatment should be the first choice in order to reach the goals (acceptable symptomatic relief and healing of lesions) at the lowest level of acid inhibition as possible. The alternative of anti-reflux surgery must also be carefully evaluated in these patients. When PPI treatment is applied, gastrin and chromogranin A, which in this setting are good markers of the ECL cell mass (22, 69, 92), should be monitored. It may in the future be possible to treat young patients with oxyntic atrophic gastritis and hypergastrinemia with a gastrin antagonist as prophylaxis against cancer. Furthermore, at which stage of tumor development gastrin loses its effect must be elucidated, in order to determine whether a gastrin antagonist could be used in the treatment. Indirectly, this may be assessed by examination of the tumor cells for the presence of the gastrin receptor either by immunohistochemistry or by in situ hybridization.
Conclusion
Gastrin and its target cell, the ECL cell, probably play a central role in gastric carcinogenesis.
Author Contributions
HW has been the leader of this research throughout many decades. LS has participated in the work during the last years making a thesis on the subject. She has participated active in making the manuscript. PM is a pathologist presently making her doctoral degree on the classification of gastric and renal carcinomas. She has done the evaluation of tumor tissue during the last years and has also participated in the writing of the manuscript.
Conflict of Interest Statement
The research behind this publication has been conducted in the absence of any commercial or financial relationship that could be construed as a potential conflict of interest.
Funding
The funding behind this research has been provided by the hospital and university.
References
1. Verdecchia A, Mariotto A, Gatta G, Bustamante-Teixeira MT, Ajiki W. Comparison of stomach cancer incidence and survival in four continents. Eur J Cancer (2003) 39:1603–9. doi: 10.1016/S0959-8049(03)00360-5
2. Suh YS, Yang HK. Screening and early detection of gastric cancer: East versus West. Surg Clin North Am (2015) 95:1053–66. doi:10.1016/j.suc.2015.05.012
3. Crew KD, Neugut AI. Epidemiology of gastric cancer. World J Gastroenterol (2006) 12:354–62. doi:10.3748/wjg.v12.i3.354
4. Naylor GM, Gotoda T, Dixon M, Shimoda T, Gatta L, Owen R, et al. Why does Japan have a high incidence of gastric cancer? Comparison of gastritis between UK and Japanese patients. Gut (2006) 55:1545–52. doi:10.1136/gut.2005.080358
5. Theuer CP, Kurosaki T, Ziogas A, Butler J, Anton-Culver H. Asian patients with gastric carcinoma in the United States exhibit unique clinical features and superior overall and cancer specific survival rates. Cancer (2000) 89:1883–92. doi:10.1002/1097-0142(20001101)89:9<1883::AID-CNCR3>3.3.CO;2-8
6. Shinozaki-Ushiku A, Kunita A, Fukayama M. Update on Epstein-Barr virus and gastric cancer (review). Int J Oncol (2015) 46:1421–34. doi:10.3892/ijo.2015.2856
7. Snietura M, Waniczek D, Piglowski W, Kopec A, Nowakowska-Zajdel E, Lorenc Z, et al. Potential role of human papilloma virus in the pathogenesis of gastric cancer. World J Gastroenterol (2014) 20:6632–7. doi:10.3748/wjg.v20.i21.6632
8. Chandrasoma PT, Der R, Ma Y, Dalton P, Taira M. Histology of the gastroesophageal junction: an autopsy study. Am J Surg Pathol (2000) 24:402–9. doi:10.1097/00000478-200003000-00009
9. Kilgore SP, Ormsby AH, Gramlich TL, Rice TW, Richter JE, Falk GW, et al. The gastric cardia: fact or fiction? Am J Gastroenterol (2000) 95:921–4. doi:10.1111/j.1572-0241.2000.01930.x
10. Choi E, Roland JT, Barlow BJ, O’Neal R, Rich AE, Nam KT, et al. Cell lineage distribution atlas of the human stomach reveals heterogeneous gland populations in the gastric antrum. Gut (2014) 63:1711–20. doi:10.1136/gutjnl-2013-305964
11. Barrett P, Hobbs RC, Coates PJ, Risdon RA, Wright NA, Hall PA. Endocrine cells of the human gastrointestinal tract have no proliferative capacity. Histochem J (1995) 27:482–6. doi:10.1007/BF02388805
12. Ryberg B, Tielemans Y, Axelson J, Carlsson E, Hakanson R, Mattson H, et al. Gastrin stimulates the self-replication rate of enterochromaffinlike cells in the rat stomach. Effects of omeprazole, ranitidine, and gastrin-17 in intact and antrectomized rats. Gastroenterology (1990) 99:935–42. doi:10.1016/0016-5085(90)90610-D
13. Tielemans Y, Willems G, Sundler F, Hakanson R. Self-replication of enterochromaffin-like cells in the mouse stomach. Digestion (1990) 45:138–46. doi:10.1159/000200235
14. Dor Y, Brown J, Martinez OI, Melton DA. Adult pancreatic beta-cells are formed by self-duplication rather than stem-cell differentiation. Nature (2004) 429:41–6. doi:10.1038/nature02520
15. Delle Fave G, Marignani M, Moretti A, D’Ambra G, Martino G, Annibale B. Hypergastrinemia and enterochromaffin-like cell hyperplasia. Yale J Biol Med (1998) 71:291–301.
16. Thompson M, Fleming KA, Evans DJ, Fundele R, Surani MA, Wright NA. Gastric endocrine cells share a clonal origin with other gut cell lineages. Development (1990) 110:477–81.
17. Thompson EM, Price YE, Wright NA. Kinetics of enteroendocrine cells with implications for their origin: a study of the cholecystokinin and gastrin subpopulations combining tritiated thymidine labelling with immunocytochemistry in the mouse. Gut (1990) 31:406–11. doi:10.1136/gut.31.4.406
18. Pearse AG, Polak JM. Neural crest origin of the endocrine polypeptide (APUD) cells of the gastrointestinal tract and pancreas. Gut (1971) 12:783–8. doi:10.1136/gut.12.10.783
19. Andrew A, Kramer B, Rawdon BB. The origin of gut and pancreatic neuroendocrine (APUD) cells – the last word? J Pathol (1998) 186:117–8. doi:10.1002/(SICI)1096-9896(1998100)186:2<117::AID-PATH152>3.0.CO;2-J
20. Larsson LI, Goltermann N, de Magistris L, Rehfeld JF, Schwartz TW. Somatostatin cell processes as pathways for paracrine secretion. Science (1979) 205:1393–5. doi:10.1126/science.382360
21. Gustafsson BI, Bakke I, Hauso O, Kidd M, Modlin IM, Fossmark R, et al. Parietal cell activation by arborization of ECL cell cytoplasmic projections is likely the mechanism for histamine induced secretion of hydrochloric acid. Scand J Gastroenterol (2011) 46:531–7. doi:10.3109/00365521.2011.558113
22. Facer P, Bishop AE, Lloyd RV, Wilson BS, Hennessy RJ, Polak JM. Chromogranin: a newly recognized marker for endocrine cells of the human gastrointestinal tract. Gastroenterology (1985) 89:1366–73. doi:10.1016/0016-5085(85)90657-2
23. Wiedenmann B, Waldherr R, Buhr H, Hille A, Rosa P, Huttner WB. Identification of gastroenteropancreatic neuroendocrine cells in normal and neoplastic human tissue with antibodies against synaptophysin, chromogranin A, secretogranin I (chromogranin B), and secretogranin II. Gastroenterology (1988) 95:1364–74. doi:10.1016/0016-5085(88)90374-5
24. Sandvik AK, Waldum HL, Kleveland PM, Schulze Sognen B. Gastrin produces an immediate and dose-dependent histamine release preceding acid secretion in the totally isolated, vascularly perfused rat stomach. Scand J Gastroenterol (1987) 22:803–8. doi:10.3109/00365528708991918
25. Brenna E, Waldum HL. Trophic effect of gastrin on the enterochromaffin like cells of the rat stomach: establishment of a dose response relationship. Gut (1992) 33:1303–6. doi:10.1136/gut.33.10.1303
26. Korman MG, Strickland RG, Hansky J. Serum gastrin in chronic gastritis. Br Med J (1971) 2:16–8. doi:10.1136/bmj.2.5752.16
27. Pounder R. Changes of plasma gastrin concentration associated with drugs for peptic ulceration. In: Walsh JH, editor. Gastrin. New York, NY: Raven Press (1993). p. 319–34.
28. Waldum HL, Brenna E. Non-proliferative capacity of endocrine cells of the human gastro-intestinal tract. Histochem J (1996) 28:397–8. doi:10.1007/BF02331403
29. Morson BC. Intestinal metaplasia of the gastric mucosa. Br J Cancer (1955) 9:365–76. doi:10.1038/bjc.1955.35
30. Marshall BJ, Warren JR. Unidentified curved bacilli in the stomach of patients with gastritis and peptic ulceration. Lancet (1984) 1:1311–5. doi:10.1016/S0140-6736(84)91816-6
31. Parsonnet J, Friedman GD, Vandersteen DP, Chang Y, Vogelman JH, Orentreich N, et al. Helicobacter pylori infection and the risk of gastric carcinoma. N Engl J Med (1991) 325:1127–31. doi:10.1056/NEJM199110173251603
32. Lee YC, Chiang TH, Chou CK, Tu YK, Liao WC, Wu MS, et al. Association between Helicobacter pylori eradication and gastric cancer incidence: a systematic review and meta-analysis. Gastroenterology (2016) 150:1113–24.e5. doi:10.1053/j.gastro.2016.01.028
33. Walker IR, Strickland RG, Ungar B, Mackay IR. Simple atrophic gastritis and gastric carcinoma. Gut (1971) 12:906–11. doi:10.1136/gut.12.11.906
34. Levi S, Beardshall K, Haddad G, Playford R, Ghosh P, Calam J. Campylobacter pylori and duodenal ulcers: the gastrin link. Lancet (1989) 1:1167–8. doi:10.1016/S0140-6736(89)92752-9
35. Hansson LE, Nyren O, Hsing AW, Bergstrom R, Josefsson S, Chow WH, et al. The risk of stomach cancer in patients with gastric or duodenal ulcer disease. N Engl J Med (1996) 335:242–9. doi:10.1056/NEJM199607253350404
36. Uemura N, Okamoto S, Yamamoto S, Matsumura N, Yamaguchi S, Yamakido M, et al. Helicobacter pylori infection and the development of gastric cancer. N Engl J Med (2001) 345:784–9. doi:10.1056/NEJMoa001999
37. Waldum HL, Burhol PG. Serum group I pepsinogens. Scand J Gastroenterol (1981) 16:449–51. doi:10.3109/00365528109181996
38. Sipponen P. Biomarkers in clinical practice: a tool to find subjects at high risk for stomach cancer. A personal view. Adv Med Sci (2006) 51:51–3.
39. Chen XY, van Der Hulst RW, Shi Y, Xiao SD, Tytgat GN, Ten Kate FJ. Comparison of precancerous conditions: atrophy and intestinal metaplasia in Helicobacter pylori gastritis among Chinese and Dutch patients. J Clin Pathol (2001) 54:367–70. doi:10.1136/jcp.54.5.367
40. de Vries AC, van Grieken NC, Looman CW, Casparie MK, de Vries E, Meijer GA, et al. Gastric cancer risk in patients with premalignant gastric lesions: a nationwide cohort study in the Netherlands. Gastroenterology (2008) 134:945–52. doi:10.1053/j.gastro.2008.01.071
41. Correa P. Human gastric carcinogenesis: a multistep and multifactorial process – First American Cancer Society Award Lecture on Cancer Epidemiology and Prevention. Cancer Res (1992) 52:6735–40.
42. Song H, Ekheden IG, Zheng Z, Ericsson J, Nyren O, Ye W. Incidence of gastric cancer among patients with gastric precancerous lesions: observational cohort study in a low risk Western population. BMJ (2015) 351:h3867. doi:10.1136/bmj.h3867
43. Sato Y, Iwafuchi M, Ueki J, Yoshimura A, Mochizuki T, Motoyama H, et al. Gastric carcinoid tumors without autoimmune gastritis in Japan: a relationship with Helicobacter pylori infection. Dig Dis Sci (2002) 47:579–85. doi:10.1023/A:1017972204219
44. Hsing AW, Hansson LE, McLaughlin JK, Nyren O, Blot WJ, Ekbom A, et al. Pernicious anemia and subsequent cancer. A population-based cohort study. Cancer (1993) 71:745–50. doi:10.1002/1097-0142(19930201)71:3<745::AID-CNCR2820710316>3.0.CO;2-1
45. Guilford P, Hopkins J, Harraway J, McLeod M, McLeod N, Harawira P, et al. E-cadherin germline mutations in familial gastric cancer. Nature (1998) 392:402–5. doi:10.1038/32918
46. Calvete O, Reyes J, Zuniga S, Paumard-Hernandez B, Fernandez V, Bujanda L, et al. Exome sequencing identifies ATP4A gene as responsible of an atypical familial type I gastric neuroendocrine tumour. Hum Mol Genet (2015) 24:2914–22. doi:10.1093/hmg/ddv054
47. Fossmark R, Calvete O, Mjones P, Benitez J, Waldum HL. ECL-cell carcinoids and carcinoma in patients homozygous for an inactivating mutation in the gastric H(+) K(+) ATPase alpha subunit. APMIS (2016) 124:561–6. doi:10.1111/apm.12546
48. Network TCGAR. Comprehensive molecular characterization of gastric adenocarcinoma. Nature (2014) 513:202–9. doi:10.1038/nature13480
49. Lauren P. The two histological main types of gastric carcinoma: diffuse and so-called intestinal-type carcinoma. An attempt at histo-clinical classification. Acta Pathol Microbiol Scand (1965) 64:31–49.
50. Fuchs CS, Mayer RJ. Gastric carcinoma. N Engl J Med (1995) 333:32–41. doi:10.1056/NEJM199507063330107
51. Karsten FH. The chemistry of Schiff’s reagent. In: Bourne GH, editor. International Review of Cytology. New York: Academic Press (1960). p. 1–100.
52. Bakkelund K, Fossmark R, Nordrum I, Waldum H. Signet ring cells in gastric carcinomas are derived from neuroendocrine cells. J Histochem Cytochem (2006) 54:615–21. doi:10.1369/jhc.5A6806.2005
53. Sordal O, Qvigstad G, Nordrum IS, Sandvik AK, Gustafsson BI, Waldum H. The PAS positive material in gastric cancer cells of signet ring type is not mucin. Exp Mol Pathol (2014) 96:274–8. doi:10.1016/j.yexmp.2014.02.008
54. Waldum HL, Aase S, Kvetnoi I, Brenna E, Sandvik AK, Syversen U, et al. Neuroendocrine differentiation in human gastric carcinoma. Cancer (1998) 83:435–44. doi:10.1002/(SICI)1097-0142(19980801)83:3<435::AID-CNCR11>3.0.CO;2-X
55. Waldum H, Haugen O, Isaksen C, Mecsei R, Sandvik A. Are diffuse gastric carcinomas neuroendocrine tumours (ECL-omas)? Eur J Gastroenterol Hepatol (1991) 3:245–9.
56. Grossi C, Lattes R. Carcinoid tumors of the stomach. Cancer (1956) 9:698–711. doi:10.1002/1097-0142(195607/08)9:4<698::AID-CNCR2820090412>3.0.CO;2-I
57. Waldum HL, Brenna E, Sandvik AK. Relationship of ECL cells and gastric neoplasia. Yale J Biol Med (1998) 71:325–35.
58. Whitehead R, Cosgrove C. Mucins and carcinoid tumours. Pathology (1979) 11:473–8. doi:10.3109/00313027909059024
59. Qvigstad G, Qvigstad T, Westre B, Sandvik AK, Brenna E, Waldum HL. Neuroendocrine differentiation in gastric adenocarcinomas associated with severe hypergastrinemia and/or pernicious anemia. APMIS (2002) 110:132–9. doi:10.1034/j.1600-0463.2002.100302.x
60. Solcia E, Capella C, Fiocca R, Rindi G, Rosai J. Gastric argyrophil carcinoidosis in patients with Zollinger-Ellison syndrome due to type 1 multiple endocrine neoplasia. A newly recognized association. Am J Surg Pathol (1990) 14:503–13. doi:10.1097/00000478-199006000-00001
61. Borch K, Renvall H, Liedberg G. Gastric endocrine cell hyperplasia and carcinoid tumors in pernicious anemia. Gastroenterology (1985) 88:638–48. doi:10.1016/0016-5085(85)90131-3
62. Bordi C, D’Adda T, Pilato FP, Ferrari C. Carcinoid (ECL cell) tumor of the oxyntic mucosa of the stomach: a hormone-dependent neoplasm? In: Fenoglio-Preiser C, Wolf M, Rilke R, editors. Progress in Surgical Pathology. (Vol. 8), Philadelphia, PA: Field & WOOD (1988). p. 177–95.
63. Poynter D, Pick CR, Harcourt RA, Selway SA, Ainge G, Harman IW, et al. Association of long lasting unsurmountable histamine H2 blockade and gastric carcinoid tumours in the rat. Gut (1985) 26:1284–95. doi:10.1136/gut.26.12.1284
64. Havu N. Enterochromaffin-like cell carcinoids of gastric mucosa in rats after life-long inhibition of gastric secretion. Digestion (1986) 35(Suppl 1):42–55. doi:10.1159/000199381
65. Havu N, Mattsson H, Ekman L, Carlsson E. Enterochromaffin-like cell carcinoids in the rat gastric mucosa following long-term administration of ranitidine. Digestion (1990) 45:189–95. doi:10.1159/000200245
66. Nwokolo CU, Smith JT, Gavey C, Sawyerr A, Pounder RE. Tolerance during 29 days of conventional dosing with cimetidine, nizatidine, famotidine or ranitidine. Aliment Pharmacol Ther (1990) 4(Suppl 1):29–45.
67. Sharma BK, Walt RP, Pounder RE, Gomes MD, Wood EC, Logan LH. Optimal dose of oral omeprazole for maximal 24 hour decrease of intragastric acidity. Gut (1984) 25:957–64. doi:10.1136/gut.25.9.957
68. Lamberts R, Creutzfeldt W, Stockmann F, Jacubaschke U, Maas S, Brunner G. Long-term omeprazole treatment in man: effects on gastric endocrine cell populations. Digestion (1988) 39:126–35. doi:10.1159/000199615
69. Waldum HL, Arnestad JS, Brenna E, Eide I, Syversen U, Sandvik AK. Marked increase in gastric acid secretory capacity after omeprazole treatment. Gut (1996) 39:649–53. doi:10.1136/gut.39.5.649
70. Reimer C, Sondergaard B, Hilsted L, Bytzer P. Proton-pump inhibitor therapy induces acid-related symptoms in healthy volunteers after withdrawal of therapy. Gastroenterology (2009) 137:80–7, 7.e1. doi:10.1053/j.gastro.2009.03.058
71. Laine L, Ahnen D, McClain C, Solcia E, Walsh JH. Review article: potential gastrointestinal effects of long-term acid suppression with proton pump inhibitors. Aliment Pharmacol Ther (2000) 14:651–68. doi:10.1046/j.1365-2036.2000.00768.x
72. Lundell L, Vieth M, Gibson F, Nagy P, Kahrilas PJ. Systematic review: the effects of long-term proton pump inhibitor use on serum gastrin levels and gastric histology. Aliment Pharmacol Ther (2015) 42:649–63. doi:10.1111/apt.13324
73. Advisory Commitee. Omeprazole Magnesium Tablets. Wilmington, DE: Astra Zeneca (2000). p. 117–41.
74. Jianu CS, Fossmark R, Viset T, Qvigstad G, Sordal O, Marvik R, et al. Gastric carcinoids after long-term use of a proton pump inhibitor. Aliment Pharmacol Ther (2012) 36:644–9. doi:10.1111/apt.12012
75. Cavalcoli F, Zilli A, Conte D, Ciafardini C, Massironi S. Gastric neuroendocrine neoplasms and proton pump inhibitors: fact or coincidence? Scand J Gastroenterol (2015) 50:1397–403. doi:10.3109/00365521.2015.1054426
76. Nandy N, Hanson JA, Strickland RG, McCarthy DM. Solitary gastric carcinoid tumor associated with long-term use of omeprazole: a case report and review of the literature. Dig Dis Sci (2016) 61:708–12. doi:10.1007/s10620-015-4014-0
77. Jianu CS, Lange OJ, Viset T, Qvigstad G, Martinsen TC, Fougner R, et al. Gastric neuroendocrine carcinoma after long-term use of proton pump inhibitor. Scand J Gastroenterol (2012) 47:64–7. doi:10.3109/00365521.2011.627444
78. Fossmark R, Sordal O, Jianu CS, Qvigstad G, Nordrum IS, Boyce M, et al. Treatment of gastric carcinoids type 1 with the gastrin receptor antagonist netazepide (YF476) results in regression of tumours and normalisation of serum chromogranin A. Aliment Pharmacol Ther (2012) 36:1067–75. doi:10.1111/apt.12090
79. Fykse V, Sandvik AK, Qvigstad G, Falkmer SE, Syversen U, Waldum HL. Treatment of ECL cell carcinoids with octreotide LAR. Scand J Gastroenterol (2004) 39:621–8. doi:10.1080/00365520410005225
80. Northfield TC, Hall CN. Carcinoma of the gastric stump: risks and pathogenesis. Gut (1990) 31:1217–9. doi:10.1136/gut.31.11.1217
81. Waldum HL, Haugen OA, Brenna E. Do neuroendocrine cells, particularly the D-cell, play a role in the development of gastric stump cancer? Cancer Detect Prev (1994) 18:431–6.
82. Qvigstad G, Falkmer S, Westre B, Waldum HL. Clinical and histopathological tumour progression in ECL cell carcinoids (“ECLomas”). APMIS (1999) 107:1085–92. doi:10.1111/j.1699-0463.1999.tb01513.x
83. Kokkola A, Sjoblom SM, Haapiainen R, Sipponen P, Puolakkainen P, Jarvinen H. The risk of gastric carcinoma and carcinoid tumours in patients with pernicious anaemia. A prospective follow-up study. Scand J Gastroenterol (1998) 33:88–92. doi:10.1080/00365529850166266
84. Rakic S, Milicevic MN. Serum gastrin levels in patients with intestinal and diffuse type of gastric cancer. Br J Cancer (1991) 64:1189. doi:10.1038/bjc.1991.489
85. Fossmark R, Sagatun L, Nordrum IS, Sandvik AK, Waldum HL. Hypergastrinemia is associated with adenocarcinomas in the gastric corpus and shorter patient survival. APMIS (2015) 123:509–14. doi:10.1111/apm.12380
86. McGuigan JE, Trudeau WL. Serum and tissue gastrin concentrations in patients with carcinoma of the stomach. Gastroenterology (1973) 64:22–5.
87. El-Zimaity HM, Ota H, Graham DY, Akamatsu T, Katsuyama T. Patterns of gastric atrophy in intestinal type gastric carcinoma. Cancer (2002) 94:1428–36. doi:10.1002/cncr.10375
88. Fukui H, Kinoshita Y, Maekawa T, Okada A, Waki S, Hassan S, et al. Regenerating gene protein may mediate gastric mucosal proliferation induced by hypergastrinemia in rats. Gastroenterology (1998) 115:1483–93. doi:10.1016/S0016-5085(98)70027-7
89. Waldum HL, Hauso O, Sordal OF, Fossmark R. Gastrin may mediate the carcinogenic effect of Helicobacter pylori infection of the stomach. Dig Dis Sci (2015) 60:1522–7. doi:10.1007/s10620-014-3468-9
90. Waldum HL, Hauso O, Brenna E, Qvigstad G, Fossmark R. Does long-term profound inhibition of gastric acid secretion increase the risk of ECL cell-derived tumors in man? Scand J Gastroenterol (2016) 51:1–7. doi:10.3109/00365521.2016.1143527
91. Aronson JK. Inhibiting the proton pump: mechanisms, benefits, harms, and questions. BMC Med (2016) 14:172. doi:10.1186/s12916-016-0724-1
Keywords: carcinogenesis, classification of cancer, gastric cancer, gastrin, hormones, neuroendocrine neoplasia
Citation: Waldum HL, Sagatun L and Mjønes P (2017) Gastrin and Gastric Cancer. Front. Endocrinol. 8:1. doi: 10.3389/fendo.2017.00001
Received: 16 November 2016; Accepted: 03 January 2017;
Published: 17 January 2017
Edited by:
Hubert Vaudry, University of Rouen, FranceReviewed by:
Tullio Florio, University of Genova, ItalyJoseph Pisegna, University of California Los Angeles, USA
Terry Moody, National Cancer Institute, USA
Copyright: © 2017 Waldum, Sagatun and Mjønes. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Helge L. Waldum, aGVsZ2Uud2FsZHVtQG50bnUubm8=