 Jacky K. Leung
Jacky K. Leung Marianne D. Sadar
Marianne D. Sadar- Department of Genome Sciences Centre, British Columbia Cancer Agency, Vancouver, BC, Canada
Androgen receptor (AR) is a validated drug target for prostate cancer based on its role in proliferation, survival, and metastases of prostate cancer cells. Unfortunately, despite recent improvements to androgen deprivation therapy and the advent of better antiandrogens with a superior affinity for the AR ligand-binding domain (LBD), most patients with recurrent disease will eventually develop lethal metastatic castration-resistant prostate cancer (CRPC). Expression of constitutively active AR splice variants that lack the LBD contribute toward therapeutic resistance by bypassing androgen blockade and antiandrogens. In the canonical pathway, binding of androgen to AR LBD triggers the release of AR from molecular chaperones which enable conformational changes and protein–protein interactions to facilitate its nuclear translocation where it regulates the expression of target genes. However, preceding AR function in the nucleus, initial binding of androgen to AR LBD in the cytoplasm may already initiate signal transduction pathways to modulate cellular proliferation and migration. In this article, we review the significance of signal transduction pathways activated by rapid, non-genomic signaling of the AR during the progression to metastatic CRPC and put into perspective the implications for current and novel therapies that target different domains of AR.
Castration-Resistant Prostate Cancer
Approximately 30% of patients will relapse after primary therapy (1). Androgen deprivation therapy, both surgical and biochemical castration, is the main treatment for relapsed patients and provides temporary relief to tumor burden; however, all patients will acquire lethal, castration-resistant prostate cancer (CRPC). Androgen receptor (AR)-targeted therapies for blocking the androgen signaling axis have improved, with more potent, second-generation antiandrogens such as enzalutamide, and 17-hydroxylase/17,20-lyase (CYP17) inhibitors, but these agents only increase median overall survival by approximately 4 months in chemotherapy-naïve patients (2, 3).
Most CRPC continues to depend on AR despite continued androgen deprivation therapy. This is apparent from a rising titer of serum prostate-specific antigen, which indicates biochemical failure and the transition of CRPC. Proposed mechanisms of continued AR activity throughout CRPC include upregulation of AR by amplification of the AR gene or overexpression of AR protein, synthesis of intratumoral androgens, stimulation of ligand-independent AR activity by epidermal growth factor (EGF) or interleukin-6 (IL-6) or by the mitogen-activated protein kinase (MAPK) cascade, phosphoinositide 3-kinase (PI3K)/Akt, and protein kinase A pathways, and the expression of constitutively active AR splice variants such as ARv567es and V7 (4–11).
Androgen receptor belongs to the nuclear receptor superfamily of proteins, along with the glucocorticoid receptor, progesterone receptor, mineralocorticoid, and estrogen receptors, which all share a modular structure composed of an unstructured N-terminal domain (NTD), a DNA-binding domain (DBD), and a C-terminal ligand-binding domain (LBD). These domains operate as individual folding units that contribute to the transformation of a transcriptionally active receptor. Unlike the LBD and DBD which are structured domains, the AR NTD is intrinsically disordered and mediates protein–protein interactions that are required for transactivation. During AR transactivation, the activation function 1 region (AF1) of the AR NTD acquires transient folding structures to bind transcriptional coactivators to bridge the AR to basal transcriptional machinery. Classic deletion analyses of the AR NTD identified two transcriptional activation units within AF1 (Tau1 and Tau5), which represent surfaces that mediate ligand-dependent and ligand-independent transactivation, respectively (12, 13).
In the canonical model, inactive AR is maintained in the cytosol by molecular chaperones that include heat shock proteins, co-chaperones, and cytoskeletal proteins (14). Binding of AR to its natural ligand dihydrotestosterone (DHT) or testosterone triggers dissociation of chaperones and induces conformational changes that enable AR dimerization and interactions with a cytoskeletal protein, Filamin A, to modulate nuclear translocation and target gene expression (15, 16). Even so, many of the cellular responses to androgen do not fit in the canonical model and do not require transcription mediated by AR. This is because ligand-transformed AR is able to associate with molecular substrates in the cytoplasm and inner leaflet of the cell membrane to activate intracellular kinase cascades. These actions are referred to as rapid, non-genomic signaling of AR and enhance cell proliferation and survival by rapid signal transduction (Figure 1). In contrast to altering gene expression and synthesizing new proteins which may take hours, non-genomic actions of AR precede transcriptional events and are generally observed within minutes after exposure to androgen. Herein, we review prominent intracellular signaling pathways activated by non-genomic AR signaling in prostate cells and offer perspective into their implications for therapies targeting the AR.
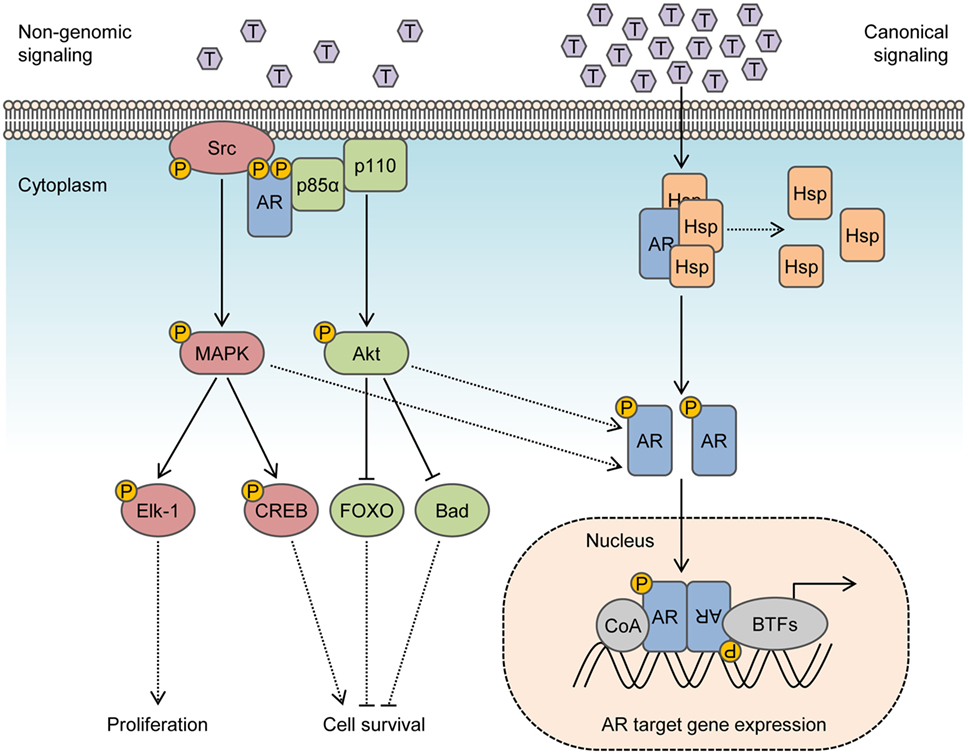
Figure 1. Integration of non-genomic signaling and canonical signaling of androgen receptor (AR). In the presence of low androgen levels (picomolar concentrations), AR interactions with Src kinase and p85α regulatory subunit of phosphoinositide 3-kinase activates mitogen-activated protein kinase (MAPK) and Akt pathways to enhance cell proliferation and survival in a non-genomic fashion. In the presence of high androgen levels (nanomolar concentrations), AR is activated in a canonical pathway to regulate the expression of target genes. Activation of MAPK and Akt by non-genomic signaling also enhances genomic AR signals by phosphorylating the AR or transcriptional coactivators.
Non-Genomic AR Signaling
Activation of Src Kinase
In response to androgen, a rapid association of AR with the non-receptor tyrosine kinase Src is responsible for enhancing cell proliferation through activation of the MAPK/ERK cascade. AR interacts with Src by binding of a polyproline sequence between residues 371 and 381 of the AR NTD to Src homology domain 3 (SH3). This association facilitates unfolding of Src and autophosphorylation to activate the Src kinase domain (17, 18). The importance of this interaction is emphasized in studies where deleting the polyproline sequence on AR or expressing peptides mimicking the polyproline region inhibits the activation of Src/ERK by AR and blocks the induction of human prostate or mammary cancer cell (LNCaP or MCF-7) growth by androgen (17, 19). Src/ERK induction is dependent on androgen concentration and is active in low to physiological androgen levels (0.1–10 nM) and inhibited by higher concentrations (100 nM) (20). This mirrors the biphasic effect of androgen, where low levels of androgens (0.01–0.1 nM) promote and high levels of androgens (1–100 nM) inhibit the growth of prostate cells (21–23). The biphasic effect of androgen to activate Src and stimulate proliferation is also observed in NIH3T3 fibroblast cells expressing low levels of AR, absent of AR nuclear translocation or transcription mediated by AR (24). These findings suggest that inhibitory effects on proliferation at high androgen levels come from a loss of transient AR protein–protein interactions with Src upon saturation of AR with ligand, rather than a diversion of AR to perform genomic functions. In NIH3T3 fibroblast cells, exposure to optimal concentration of androgen (10 nM) suppresses cell cycle progression, induces interactions between AR with the cytoskeletal protein Filamin A, and recruits integrin beta 1 to coordinate cell migration by activating focal adhesion kinase, paxillin, and Rac (25, 26). In malignant prostate tissue, cytoplasmic localization of Filamin A correlates with metastatic potential and an androgen-independent phenotype (27). Accordingly, forced nuclear localization of Filamin A may terminate non-genomic signals from AR supporting proliferation and restore bicalutamide sensitivity in C4-2 human prostate cancer cells, which exhibit androgen-independent growth (28).
Activation of the Ras-Raf-MAPK/ERK cascade is the primary mitogenic stimulus initiated by non-genomic AR effects observed in androgen-sensitive prostate cells. Levels of phospho-(p-) ERK1/2 peak within 5–30 min of exposure to DHT in LNCaP cells, PC-3 cells stably expressing wild-type AR, and primary prostatic stromal cells, but this is not detected in primary human genital skin fibroblasts until 16 h, implying cell-type specificity (20, 29). Activated ERK1/2 in response to androgen influences the activity of transcription factors in the nucleus that are independent of AR DNA binding, which in turn activates ETS domain-containing protein Elk-1 to regulate transcription of immediate early genes, including c-fos (29–31). Interestingly, antiandrogens (bicalutamide and flutamide) promoted the induction of p-ERK1/2 and induced transactivation of c-fos in reporter gene assays in PC3 cells only when wild-type AR was ectopically expressed (29). Moreover, p-ERK1/2 can also promote cell survival in a non-genomic manner by activating cAMP response element-binding protein (CREB) (20, 32). In a distinct manner, expression of a dominant negative CREB mutant in LNCaP cells abrogates DHT-induced protection against apoptosis, but does not prevent S-phase entry (20).
Aberrant Src activity is detected in malignant prostate cells and present in several AR-positive prostate cancer cell line models exhibiting androgen-independent growth (33–35). In low passage androgen-sensitive LNCaP cells, the ability for AR to activate Src and stimulate proliferation non-genomically is androgen dependent. However, in high passage LNCaP cells (more than 60 passages), AR interacts with Src constitutively and independently of androgen to promote growth under androgen-depleted conditions (20). In high passage LNCaP cells, the Src/MAP/ERK-1/2/CREB pathway is constitutively active, and only the MAPK inhibitor (PD98059) and not bicalutamide inhibits proliferation (20). Furthermore, while LNCaP cells do not typically form tumors in castrated hosts, high passage LNCaP cells do form tumors efficiently in castrated mice. C4-2 cells, a CRPC subline of LNCaP that can grow in castrated hosts, exhibit a threefold increase in protein expression of Src compared to androgen-sensitive LNCaP cells (36). Treatment of C4-2 cells with a Src inhibitor (PP2) inhibits growth, decreases invasive potential, and induces apoptosis, with synergy in combination with bicalutamide when bicalutamide alone has no inhibitory effects on this cell line (36). Supporting the role of Src in the progression of prostate cancer, immunohistochemistry of prostate tissue from the transgenic adenocarcinoma mouse prostate model shows a progressive increase (up to threefold) in positive staining of activated Src, as a function of age and cancer progression from 8 to 24 weeks (36). Since activating Src mutations are rare in human cancers, aberrant Src activity is presumably dependent on increased Src expression or stimulation by growth factors and interleukins abundant in the tumor microenvironment, including EGF, IGF, IL-6, IL-8 (37). Numerous studies provide evidence that Src inhibitors are effective in reducing proliferation and invasion of prostate cancer cell lines in vitro and demonstrate favorable antitumor activity in vivo using prostate cancer xenografts (38–40).
Cross Talk with the PI3K Pathway
Activation of the PI3K/Akt pathway is also triggered by non-genomic AR signaling. Direct interactions between ligand-activated AR and PI3K in the cytosol are mediated by binding of phosphotyrosine residues on the AR NTD to SH2 domain of p85α regulatory subunit of PI3K (41). Association of AR/p85α promotes activation of p110 catalytic subunit and generation of phosphatidylinositol-3,4,5-trisphosphate (PIP3) signaling lipids to induce activation of Akt kinase, which leads to regulation of transcription factors to inhibit apoptosis pathways and promote cell survival. Accumulation of Akt p-S473 is detected over 10–30 min upon androgen stimulation of non-tumoral VDEC cells or PC3 cells when stably expressing AR, supporting its role as a non-genomic AR stimulus (41). Activated Akt phosphorylates proapoptotic protein BAD and forkhead box FOXO proteins to maintain cell survival (41). Among Akt substrates, FOXO3a enhances AR expression by direct binding to the AR promoter (42), whereas FOXO1 may decrease AR transactivation by recruitment of histone deacetylase HDAC3 (43). Collectively, these emphasize the role of AR in maintaining cell survival independently of transcription, which is enhanced by PI3K/Akt signaling. Negative regulation of PI3K/Akt is facilitated by conversion of PIP3 to PIP2 by phosphatase and tensin homolog (PTEN), which is a commonly lost tumor suppressor in prostate cancer. Indeed, genomic alterations affecting the PI3K/Akt pathway are detected in 42% of primary prostate tumors and 100% of metastatic tumors (44). Furthermore, Akt may also bind to and phosphorylate residues S213 and S791 on AR to modulate transactivation (10). Interestingly, Akt represses AR activity in low passage LNCaP cells, but promotes AR transcription in high passage LNCaP cells (>60 passages) where the PI3K/Akt pathway is aberrantly active (8).
Phosphotryosine residues on the AR NTD mediate the interaction between AR and PI3K, which include Y267, Y363, and Y534 (45). These residues are phosphorylated in response to EGF by Cdc42-associated kinase Ack on Y267 and Y363, or Src on Y534 (46, 47). Src-mediated phosphorylation of AR Y534 enhances the stability of AR by preventing interactions with E3 ligase CHIP that promote proteasomal degradation (48). Mutation of AR Y534 to phenylalanine disrupts the ability for AR to localize in the nucleus in a ligand-independent manner and inhibits AR transactivation in reporter gene assays in response to EGF or low concentrations of androgen (47). Studies in fibroblast cells support a ternary complex of Src/AR/p85α as a non-genomic AR signal (24). It is likely that an AR/Src complex is formed first by rapid signaling, followed by phosphorylation of AR Y534 by Src to recruit the p85α regulatory subunit of PI3K. Thus, non-genomic activation of AR and interactions between Src/AR and p85α can trigger concurrent activation of the MAPK and PI3K/Akt pathways to enhance cell proliferation and cell survival under androgen-depleted conditions.
Convergence with Ligand-Independent Activation of AR
Our lab identified that the AR NTD can be activated independently of androgen, by intracellular kinase signals, cytokines, and osteoblast-derived factors (6, 7, 49). Ligand-independent activation of AR may be enhanced by the pathways activated in non-genomic AR signaling. Specifically, MAPK/ERK activation in response to IL-6 increases transactivation of the AR NTD through interactions with STAT3 (7). IL-8 stimulates androgen-independent growth and transactivation of AR in a Src and ERK-dependent manner (50). EGF signaling enhances ligand-independent AR transactivation and promotes activation of Src and the Ras-Raf-MAPK/ERK pathway (9, 51, 52). Notably, activated Src is also able to stimulate the STAT pathway and enhance signals by growth factor kinases, such as EGFR (53, 54). Moreover, activation of the MAPK/ERK and PI3K/Akt kinase pathways non-genomically can enhance ligand-independent transactivation of AR by modulating interactions with coactivators including steroid receptor coactivator-1 and androgen receptor-associated protein 70 (55, 56).
Implications for Prostate Cancer Therapies
Current AR Therapies
Non-genomic AR signaling pathways may likely contribute to resistance to androgen deprivation therapy and antiandrogens. Prostate cancer patients undergoing maximal androgen deprivation therapy with surgical orchiectomy or LHRH analog combined with an antiandrogen (bicalutamide or flutamide) still have low levels of testosterone in their serum (less than 0.1 nM), which is within the range of AR to mediate non-genomic responses (57, 58). Levels of androgen in CRPC tissue are at least 10-fold higher, in the 1–3 nM range (59). Bicalutamide and flutamide are not effective in blocking non-genomic activation of AR and show an inherent ability to induce p-ERK1/2 and c-fos by acting through AR in androgen-depleted conditions (29). More potent second-generation antiandrogens, such as enzalutamide, apalutamide (ARN-509), and darolutamide (ODM-201), have greater affinity for the AR LBD and may reduce nuclear translocation of full-length AR, resulting in accumulation AR in the cytosol (60–62). In the cytosol, the AR NTD has the potential to activate Src and PI3K, to drive tumor growth and survival. Concentrating AR in the cytoplasm may also effectively lower the androgen requirements for optimal growth, or work in concert with Src phosphorylation of AR Y534 to sensitize AR to low androgen concentrations. Furthermore, a consequence of AR inhibition is reduced expression of FKBP5, an AR target gene that is a chaperone for the phosphatase PHLPP targeting p-Akt. Accordingly, enhanced tumor survival by overactive Akt signaling, which is exacerbated by PTEN loss, is common in enzalutamide-resistant tumors (63).
Antiandrogens bind the AR LBD to inhibit full-length AR and thus have no effects on constitutively active AR splice variants, such as ARv567es and V7, which do not express a functional LBD. Expression of ARv567es and V7 is repressed by androgen and increased in the absence of androgen, thereby mediating transcription of target genes when the full-length AR is not transactivated by androgen under castrate conditions. ARv567es is predominantly nuclear (64), whereas the cellular localization of V7 is more variable. In malignant prostate tissue, V7 is predominantly localized in the cytoplasm but nuclear in CRPC (65). Due to the nuclear localization of ARv567es and V7 in CRPC, it has been proposed that they do not mediate non-genomic signaling. In support of this concept, expression of ARv567es results in decreased levels of IGF-1R mRNA and other genes (64) known to be upregulated by non-genomic signaling (66). Other splice variants have been discovered without any genomic function and are likely to carry out non-genomic actions, mainly AR8 and AR23. AR8 encodes solely the AR NTD and a unique 33-amino acid sequence at the C-terminus (67). AR8 has no transcriptional activity and localizes on the cell membrane, possibly by palmitoylation of one or two cysteines in the unique C-terminal sequence (67). Supporting a role in non-genomic signaling, AR8 interacts with full-length AR and EGFR and associates with Src upon EGF stimulation (67). AR8 is endogenously expressed in several CRPC cell lines (C4-2B, 22Rv1, and CWR-R1) and enhances androgen-independent growth (67). AR23 is a cytosolic AR variant carrying a 23-amino acid insertion between the zinc fingers of the AR DBD (68). Interestingly, AR23 is able to potentiate transcription mediated by full-length AR and influence transactivation of NF-kB in reporter gene assays, even though upon ligand binding it is unable to enter the nucleus and regulate gene expression (68).
Novel AR Therapies
Inhibitors targeting the AR NTD might be promising candidates for blocking non-genomic AR signals that enhance tumor growth or survival. To date, two families of compounds have been discovered that bind to the AF1 region of the AR NTD and inhibit transactivation. These are sintokamide A and EPI compounds (69, 70). Both of these agents demonstrate promising results for inducing the regression of enzalutamide-resistant CRPC xenografts in mice (70, 71). Sintokamide A and EPI have different mechanisms of action by binding unique sites on AF1. Nuclear magnetic resonance spectroscopy analyses demonstrate that EPI-001 binds to a specific pocket formed by Tau5 of AR NTD with contact to three regions, residues 353–364, 397–407, and 433–466 (72), that overlaps with the residues 371–381 containing the polyproline sequence required to interact with Src. EPI blocks protein–protein interactions on the AR NTD, between AF1 and CREB-binding protein and TFIIF of the transcription preinitiation complex, and between full-length AR and STAT3, which binds within residues 234–558 of the AR NTD (7, 69, 70). EPI-506 is the first AR NTD antagonist to be tested in clinical trials for metastatic CRPC (NCT02606123). A clinically important advantage to AR NTD inhibitors is that they do not depend on the presence of AR LBD for inhibiting the transcriptional activity of full-length AR as well as AR splice variants. Non-genomic signals from full-length AR, or possibly membrane-associated or cytosolic AR splice variants, such as AR8 or AR23, may be prevented by AR NTD inhibitors that block interaction with Src (Figure 2). Another potential strategy to prevent non-genomic action of AR is to degrade the AR protein. However, all AR degraders to date bind the AR LBD or other non-AR targets. AR degraders that have being tested in clinical trials for CRPC include galeterone (TOK-001) and niclosamide (73–75). The randomized phase III clinical study (NCT02438007) for galeterone was halted recently due to the unlikelihood of meeting its primary endpoint, whereas niclosamide currently entered phase I trials as a combination treatment with enzalutamide (NCT02532114).
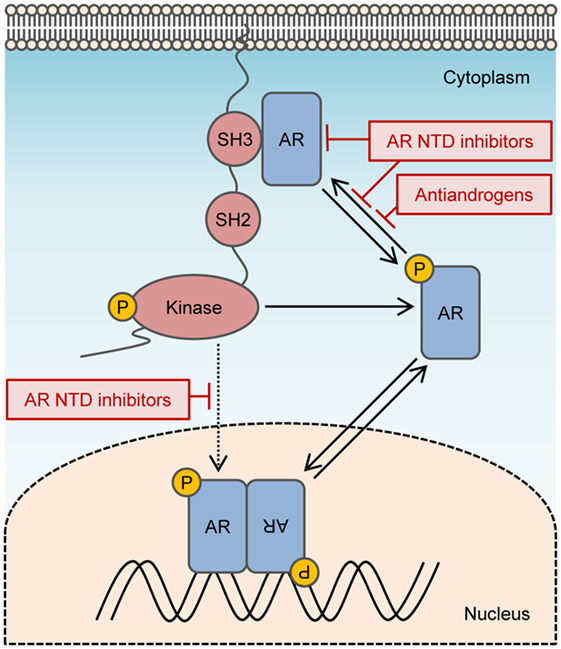
Figure 2. Targeting non-genomic actions of androgen receptor (AR). Non-genomic signaling of AR is activated by interactions between the polyproline domain of the AR N-terminal domain (NTD) and Src homology domain 3 of Src. Activated Src can enhance the transactivation of AR directly by phosphorylating AR Y534 or indirectly by stimulating alternate kinase pathways that modulate AR activity. AR NTD inhibitors may prevent non-genomic signals from AR by blocking Src interaction with the AR NTD and can block both ligand-dependent and ligand-independent transactivation of AR.
Combined Inhibition of AR and Non-Genomic Substrates
Along with androgen deprivation therapy, non-genomic AR signaling substrates may need pharmacological inhibition to provide maximum benefit to CRPC patients. Several inhibitors of Src family kinases have been tested in a clinical setting for prostate cancer, notably inhibitors that target kinase activity, dasatinib (BMSS-354825) and saracatinib (AZD0530); and KX2-391, a peptidomimetic that blocks the substrate binding site of Src. Clinical studies indicate that targeting Src or inhibiting activated downstream kinase pathways in isolation is ineffective for CRPC (37, 76–79). Likewise, inhibitors of PI3K, Akt, or mTOR have also demonstrated limited use in clinical practice as single agents (80, 81). This is probably because targeting non-genomic AR signals does not protect against ligand-dependent activation of AR and transcription of AR target genes. Inhibition of both non-genomic and genomic pathways of AR may be necessary to eradicate tumor dependency of AR. Concurrent inhibition of AR and non-genomic AR components may prove useful for prostate cancer patients with progression after primary therapy. Many of these strategies are currently under investigation and show promising results in preclinical models of CRPC (82–85).
Author Contributions
JL and MS both provided substantial contributions to the conception of the work and interpretation of literature. JL provided the first draft and MS revised the work for important intellectual content. JL and MS both approved the version submitted. JL and MS agreed to be accountable for all aspects of the work in ensuring that questions related to the accuracy or integrity of any part of the work are appropriately investigated and resolved.
Conflict of Interest Statement
MS is a director and officer of ESSA Pharma Inc. that has licensed EPI and sintokamide technologies. MS has equity and receives consulting fees from ESSA Pharma Inc. JL declares that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Funding
This research was supported by funding to MS from USA National Cancer Institutes, Grant 2R01CA105304.
References
1. Golabek T, Belsey J, Drewa T, Kolodziej A, Skoneczna I, Milecki P, et al. Evidence-based recommendations on androgen deprivation therapy for localized and advanced prostate cancer. Cent European J Urol (2016) 69(2):131–8. doi:10.5173/ceju.2016.812
2. Ryan CJ, Smith MR, Fizazi K, Saad F, Mulders PF, Sternberg CN, et al. Abiraterone acetate plus prednisone versus placebo plus prednisone in chemotherapy-naive men with metastatic castration-resistant prostate cancer (COU-AA-302): final overall survival analysis of a randomised, double-blind, placebo-controlled phase 3 study. Lancet Oncol (2015) 16(2):152–60. doi:10.1016/S1470-2045(14)71205-7
3. Beer TM, Armstrong AJ, Rathkopf D, Loriot Y, Sternberg CN, Higano CS, et al. Enzalutamide in men with chemotherapy-naive metastatic castration-resistant prostate cancer: extended analysis of the phase 3 PREVAIL study. Eur Urol (2016) 71(2):151–4. doi:10.1016/j.eururo.2016.07.032
4. Karantanos T, Evans CP, Tombal B, Thompson TC, Montironi R, Isaacs WB. Understanding the mechanisms of androgen deprivation resistance in prostate cancer at the molecular level. Eur Urol (2015) 67(3):470–9. doi:10.1016/j.eururo.2014.09.049
5. Imamura Y, Sadar MD. Androgen receptor targeted therapies in castration-resistant prostate cancer: bench to clinic. Int J Urol (2016) 23(8):654–65. doi:10.1111/iju.13137
6. Sadar MD. Androgen-independent induction of prostate-specific antigen gene expression via cross-talk between the androgen receptor and protein kinase A signal transduction pathways. J Biol Chem (1999) 274(12):7777–83. doi:10.1074/jbc.274.12.7777
7. Ueda T, Bruchovsky N, Sadar MD. Activation of the androgen receptor N-terminal domain by interleukin-6 via MAPK and STAT3 signal transduction pathways. J Biol Chem (2002) 277(9):7076–85. doi:10.1074/jbc.M108255200
8. Lin HK, Hu YC, Yang L, Altuwaijri S, Chen YT, Kang HY, et al. Suppression versus induction of androgen receptor functions by the phosphatidylinositol 3-kinase/Akt pathway in prostate cancer LNCaP cells with different passage numbers. J Biol Chem (2003) 278(51):50902–7. doi:10.1074/jbc.M300676200
9. Gregory CW, Fei X, Ponguta LA, He B, Bill HM, French FS, et al. Epidermal growth factor increases coactivation of the androgen receptor in recurrent prostate cancer. J Biol Chem (2004) 279(8):7119–30. doi:10.1074/jbc.M307649200
10. Wen Y, Hu MC, Makino K, Spohn B, Bartholomeusz G, Yan DH, et al. HER-2/neu promotes androgen-independent survival and growth of prostate cancer cells through the Akt pathway. Cancer Res (2000) 60(24):6841–5.
11. Nazareth LV, Weigel NL. Activation of the human androgen receptor through a protein kinase A signaling pathway. J Biol Chem (1996) 271(33):19900–7. doi:10.1074/jbc.271.33.19900
12. Jenster G, van der Korput JA, Trapman J, Brinkmann AO. Functional domains of the human androgen receptor. J Steroid Biochem Mol Biol (1992) 41(3–8):671–5. doi:10.1016/0960-0760(92)90402-5
13. Chamberlain NL, Whitacre DC, Miesfeld RL. Delineation of two distinct type 1 activation functions in the androgen receptor amino-terminal domain. J Biol Chem (1996) 271(43):26772–8. doi:10.1074/jbc.271.43.26772
14. Smith DF, Toft DO. Minireview: the intersection of steroid receptors with molecular chaperones: observations and questions. Mol Endocrinol (2008) 22(10):2229–40. doi:10.1210/me.2008-0089
15. Ozanne DM, Brady ME, Cook S, Gaughan L, Neal DE, Robson CN. Androgen receptor nuclear translocation is facilitated by the f-actin cross-linking protein filamin. Mol Endocrinol (2000) 14(10):1618–26. doi:10.1210/mend.14.10.0541
16. Loy CJ, Sim KS, Yong EL. Filamin-A fragment localizes to the nucleus to regulate androgen receptor and coactivator functions. Proc Natl Acad Sci U S A (2003) 100(8):4562–7. doi:10.1073/pnas.0736237100
17. Migliaccio A, Castoria G, Di Domenico M, de Falco A, Bilancio A, Lombardi M, et al. Steroid-induced androgen receptor-oestradiol receptor beta-Src complex triggers prostate cancer cell proliferation. EMBO J (2000) 19(20):5406–17. doi:10.1093/emboj/19.20.5406
18. Zarif JC, Lamb LE, Schulz VV, Nollet EA, Miranti CK. Androgen receptor non-nuclear regulation of prostate cancer cell invasion mediated by Src and matriptase. Oncotarget (2015) 6(9):6862–76. doi:10.18632/oncotarget.3119
19. Migliaccio A, Varricchio L, De Falco A, Castoria G, Arra C, Yamaguchi H, et al. Inhibition of the SH3 domain-mediated binding of Src to the androgen receptor and its effect on tumor growth. Oncogene (2007) 26(46):6619–29. doi:10.1038/sj.onc.1210487
20. Unni E, Sun S, Nan B, McPhaul MJ, Cheskis B, Mancini MA, et al. Changes in androgen receptor nongenotropic signaling correlate with transition of LNCaP cells to androgen independence. Cancer Res (2004) 64(19):7156–68. doi:10.1158/0008-5472.CAN-04-1121
21. Berns EM, de Boer W, Mulder E. Androgen-dependent growth regulation of and release of specific protein(s) by the androgen receptor containing human prostate tumor cell line LNCaP. Prostate (1986) 9(3):247–59. doi:10.1002/pros.2990090305
22. Olea N, Sakabe K, Soto AM, Sonnenschein C. The proliferative effect of “anti-androgens” on the androgen-sensitive human prostate tumor cell line LNCaP. Endocrinology (1990) 126(3):1457–63. doi:10.1210/endo-126-3-1457
23. de Launoit Y, Veilleux R, Dufour M, Simard J, Labrie F. Characteristics of the biphasic action of androgens and of the potent antiproliferative effects of the new pure antiestrogen EM-139 on cell cycle kinetic parameters in LNCaP human prostatic cancer cells. Cancer Res (1991) 51(19):5165–70.
24. Castoria G, Lombardi M, Barone MV, Bilancio A, Di Domenico M, Bottero D, et al. Androgen-stimulated DNA synthesis and cytoskeletal changes in fibroblasts by a nontranscriptional receptor action. J Cell Biol (2003) 161(3):547–56. doi:10.1083/jcb.200211099
25. Castoria G, D’Amato L, Ciociola A, Giovannelli P, Giraldi T, Sepe L, et al. Androgen-induced cell migration: role of androgen receptor/filamin A association. PLoS One (2011) 6(2):e17218. doi:10.1371/journal.pone.0017218
26. Castoria G, Giovannelli P, Di Donato M, Ciociola A, Hayashi R, Bernal F, et al. Role of non-genomic androgen signalling in suppressing proliferation of fibroblasts and fibrosarcoma cells. Cell Death Dis (2014) 5:e1548. doi:10.1038/cddis.2014.497
27. Bedolla RG, Wang Y, Asuncion A, Chamie K, Siddiqui S, Mudryj MM, et al. Nuclear versus cytoplasmic localization of filamin A in prostate cancer: immunohistochemical correlation with metastases. Clin Cancer Res (2009) 15(3):788–96. doi:10.1158/1078-0432.CCR-08-1402
28. Ghosh PM, Malik SN, Bedolla RG, Wang Y, Mikhailova M, Prihoda TJ, et al. Signal transduction pathways in androgen-dependent and -independent prostate cancer cell proliferation. Endocr Relat Cancer (2005) 12(1):119–34. doi:10.1677/erc.1.00835
29. Peterziel H, Mink S, Schonert A, Becker M, Klocker H, Cato AC. Rapid signalling by androgen receptor in prostate cancer cells. Oncogene (1999) 18(46):6322–9. doi:10.1038/sj.onc.1203032
30. Yang SH, Yates PR, Whitmarsh AJ, Davis RJ, Sharrocks AD. The Elk-1 ETS-domain transcription factor contains a mitogen-activated protein kinase targeting motif. Mol Cell Biol (1998) 18(2):710–20. doi:10.1128/MCB.18.2.710
31. Marais R, Wynne J, Treisman R. The SRF accessory protein Elk-1 contains a growth factor-regulated transcriptional activation domain. Cell (1993) 73(2):381–93. doi:10.1016/0092-8674(93)90237-K
32. Bonni A, Brunet A, West AE, Datta SR, Takasu MA, Greenberg ME. Cell survival promoted by the Ras-MAPK signaling pathway by transcription-dependent and -independent mechanisms. Science (1999) 286(5443):1358–62. doi:10.1126/science.286.5443.1358
33. Gelman IH. Androgen receptor activation in castration-recurrent prostate cancer: the role of Src-family and Ack1 tyrosine kinases. Int J Biol Sci (2014) 10(6):620–6. doi:10.7150/ijbs.8264
34. Posadas EM, Al-Ahmadie H, Robinson VL, Jagadeeswaran R, Otto K, Kasza KE, et al. FYN is overexpressed in human prostate cancer. BJU Int (2009) 103(2):171–7. doi:10.1111/j.1464-410X.2008.08009.x
35. Zardan A, Nip KM, Thaper D, Toren P, Vahid S, Beraldi E, et al. Lyn tyrosine kinase regulates androgen receptor expression and activity in castrate-resistant prostate cancer. Oncogenesis (2014) 3:e115. doi:10.1038/oncsis.2014.30
36. Asim M, Siddiqui IA, Hafeez BB, Baniahmad A, Mukhtar H. Src kinase potentiates androgen receptor transactivation function and invasion of androgen-independent prostate cancer C4-2 cells. Oncogene (2008) 27(25):3596–604. doi:10.1038/sj.onc.1211016
37. Varkaris A, Katsiampoura AD, Araujo JC, Gallick GE, Corn PG. Src signaling pathways in prostate cancer. Cancer Metastasis Rev (2014) 33(2–3):595–606. doi:10.1007/s10555-013-9481-1
38. Nam S, Kim D, Cheng JQ, Zhang S, Lee JH, Buettner R, et al. Action of the Src family kinase inhibitor, dasatinib (BMS-354825), on human prostate cancer cells. Cancer Res (2005) 65(20):9185–9. doi:10.1158/0008-5472.CAN-05-1731
39. Lombardo LJ, Lee FY, Chen P, Norris D, Barrish JC, Behnia K, et al. Discovery of N-(2-chloro-6-methyl-phenyl)-2-(6-(4-(2-hydroxyethyl)-piperazin-1-yl)-2-methylpyrimidin-4-ylamino)thiazole-5-carboxamide (BMS-354825), a dual Src/Abl kinase inhibitor with potent antitumor activity in preclinical assays. J Med Chem (2004) 47(27):6658–61. doi:10.1021/jm049486a
40. Recchia I, Rucci N, Festuccia C, Bologna M, MacKay AR, Migliaccio S, et al. Pyrrolopyrimidine c-Src inhibitors reduce growth, adhesion, motility and invasion of prostate cancer cells in vitro. Eur J Cancer (2003) 39(13):1927–35. doi:10.1016/S0959-8049(03)00394-0
41. Baron S, Manin M, Beaudoin C, Leotoing L, Communal Y, Veyssiere G, et al. Androgen receptor mediates non-genomic activation of phosphatidylinositol 3-OH kinase in androgen-sensitive epithelial cells. J Biol Chem (2004) 279(15):14579–86. doi:10.1074/jbc.M306143200
42. Yang L, Xie S, Jamaluddin MS, Altuwaijri S, Ni J, Kim E, et al. Induction of androgen receptor expression by phosphatidylinositol 3-kinase/Akt downstream substrate, FOXO3a, and their roles in apoptosis of LNCaP prostate cancer cells. J Biol Chem (2005) 280(39):33558–65. doi:10.1074/jbc.M504461200
43. Liu P, Li S, Gan L, Kao TP, Huang H. A transcription-independent function of FOXO1 in inhibition of androgen-independent activation of the androgen receptor in prostate cancer cells. Cancer Res (2008) 68(24):10290–9. doi:10.1158/0008-5472.CAN-08-2038
44. Taylor BS, Schultz N, Hieronymus H, Gopalan A, Xiao Y, Carver BS, et al. Integrative genomic profiling of human prostate cancer. Cancer Cell (2010) 18(1):11–22. doi:10.1016/j.ccr.2010.05.026
45. Gioeli D, Paschal BM. Post-translational modification of the androgen receptor. Mol Cell Endocrinol (2012) 352(1–2):70–8. doi:10.1016/j.mce.2011.07.004
46. Mahajan NP, Liu Y, Majumder S, Warren MR, Parker CE, Mohler JL, et al. Activated Cdc42-associated kinase Ack1 promotes prostate cancer progression via androgen receptor tyrosine phosphorylation. Proc Natl Acad Sci U S A (2007) 104(20):8438–43. doi:10.1073/pnas.0700420104
47. Guo Z, Dai B, Jiang T, Xu K, Xie Y, Kim O, et al. Regulation of androgen receptor activity by tyrosine phosphorylation. Cancer Cell (2006) 10(4):309–19. doi:10.1016/j.ccr.2006.08.021
48. DaSilva J, Gioeli D, Weber MJ, Parsons SJ. The neuroendocrine-derived peptide parathyroid hormone-related protein promotes prostate cancer cell growth by stabilizing the androgen receptor. Cancer Res (2009) 69(18):7402–11. doi:10.1158/0008-5472.CAN-08-4687
49. Blaszczyk N, Masri BA, Mawji NR, Ueda T, McAlinden G, Duncan CP, et al. Osteoblast-derived factors induce androgen-independent proliferation and expression of prostate-specific antigen in human prostate cancer cells. Clin Cancer Res (2004) 10(5):1860–9. doi:10.1158/1078-0432.CCR-0974-3
50. Lee LF, Louie MC, Desai SJ, Yang J, Chen HW, Evans CP, et al. Interleukin-8 confers androgen-independent growth and migration of LNCaP: differential effects of tyrosine kinases Src and FAK. Oncogene (2004) 23(12):2197–205. doi:10.1038/sj.onc.1207344
51. Roberts PJ, Der CJ. Targeting the Raf-MEK-ERK mitogen-activated protein kinase cascade for the treatment of cancer. Oncogene (2007) 26(22):3291–310. doi:10.1038/sj.onc.1210422
52. Ponguta LA, Gregory CW, French FS, Wilson EM. Site-specific androgen receptor serine phosphorylation linked to epidermal growth factor-dependent growth of castration-recurrent prostate cancer. J Biol Chem (2008) 283(30):20989–1001. doi:10.1074/jbc.M802392200
53. Turkson J, Bowman T, Garcia R, Caldenhoven E, De Groot RP, Jove R. Stat3 activation by Src induces specific gene regulation and is required for cell transformation. Mol Cell Biol (1998) 18(5):2545–52. doi:10.1128/MCB.18.5.2545
54. Garcia R, Bowman TL, Niu G, Yu H, Minton S, Muro-Cacho CA, et al. Constitutive activation of Stat3 by the Src and JAK tyrosine kinases participates in growth regulation of human breast carcinoma cells. Oncogene (2001) 20(20):2499–513. doi:10.1038/sj.onc.1204349
55. Ueda T, Mawji NR, Bruchovsky N, Sadar MD. Ligand-independent activation of the androgen receptor by interleukin-6 and the role of steroid receptor coactivator-1 in prostate cancer cells. J Biol Chem (2002) 277(41):38087–94. doi:10.1074/jbc.M203313200
56. Lin HK, Yeh S, Kang HY, Chang C. Akt suppresses androgen-induced apoptosis by phosphorylating and inhibiting androgen receptor. Proc Natl Acad Sci U S A (2001) 98(13):7200–5. doi:10.1073/pnas.121173298
57. Gomella LG. Effective testosterone suppression for prostate cancer: is there a best castration therapy? Rev Urol (2009) 11(2):52–60.
58. Heyns CF, Simonin MP, Grosgurin P, Schall R, Porchet HC; South African Triptorelin Study G. Comparative efficacy of triptorelin pamoate and leuprolide acetate in men with advanced prostate cancer. BJU Int (2003) 92(3):226–31. doi:10.1046/j.1464-410X.2003.04308.x
59. Mohler JL, Gregory CW, Ford OH III, Kim D, Weaver CM, Petrusz P, et al. The androgen axis in recurrent prostate cancer. Clin Cancer Res (2004) 10(2):440–8. doi:10.1158/1078-0432.CCR-1146-03
60. Efstathiou E, Titus M, Wen S, Hoang A, Karlou M, Ashe R, et al. Molecular characterization of enzalutamide-treated bone metastatic castration-resistant prostate cancer. Eur Urol (2015) 67(1):53–60. doi:10.1016/j.eururo.2014.05.005
61. Clegg NJ, Wongvipat J, Joseph JD, Tran C, Ouk S, Dilhas A, et al. ARN-509: a novel antiandrogen for prostate cancer treatment. Cancer Res (2012) 72(6):1494–503. doi:10.1158/0008-5472.CAN-11-3948
62. Fizazi K, Massard C, Bono P, Jones R, Kataja V, James N, et al. Activity and safety of ODM-201 in patients with progressive metastatic castration-resistant prostate cancer (ARADES): an open-label phase 1 dose-escalation and randomised phase 2 dose expansion trial. Lancet Oncol (2014) 15(9):975–85. doi:10.1016/S1470-2045(14)70240-2
63. Carver BS, Chapinski C, Wongvipat J, Hieronymus H, Chen Y, Chandarlapaty S, et al. Reciprocal feedback regulation of PI3K and androgen receptor signaling in PTEN-deficient prostate cancer. Cancer Cell (2011) 19(5):575–86. doi:10.1016/j.ccr.2011.04.008
64. Sun S, Sprenger CC, Vessella RL, Haugk K, Soriano K, Mostaghel EA, et al. Castration resistance in human prostate cancer is conferred by a frequently occurring androgen receptor splice variant. J Clin Invest (2010) 120(8):2715–30. doi:10.1172/JCI41824
65. Guo Z, Yang X, Sun F, Jiang R, Linn DE, Chen H, et al. A novel androgen receptor splice variant is up-regulated during prostate cancer progression and promotes androgen depletion-resistant growth. Cancer Res (2009) 69(6):2305–13. doi:10.1158/0008-5472.CAN-08-3795
66. Pandini G, Mineo R, Frasca F, Roberts CT Jr, Marcelli M, Vigneri R, et al. Androgens up-regulate the insulin-like growth factor-I receptor in prostate cancer cells. Cancer Res (2005) 65(5):1849–57. doi:10.1158/0008-5472.CAN-04-1837
67. Yang X, Guo Z, Sun F, Li W, Alfano A, Shimelis H, et al. Novel membrane-associated androgen receptor splice variant potentiates proliferative and survival responses in prostate cancer cells. J Biol Chem (2011) 286(41):36152–60. doi:10.1074/jbc.M111.265124
68. Jagla M, Feve M, Kessler P, Lapouge G, Erdmann E, Serra S, et al. A splicing variant of the androgen receptor detected in a metastatic prostate cancer exhibits exclusively cytoplasmic actions. Endocrinology (2007) 148(9):4334–43. doi:10.1210/en.2007-0446
69. Andersen RJ, Mawji NR, Wang J, Wang G, Haile S, Myung JK, et al. Regression of castrate-recurrent prostate cancer by a small-molecule inhibitor of the amino-terminus domain of the androgen receptor. Cancer Cell (2010) 17(6):535–46. doi:10.1016/j.ccr.2010.04.027
70. Banuelos CA, Tavakoli I, Tien AH, Caley DP, Mawji NR, Li Z, et al. Sintokamide A is a novel antagonist of androgen receptor that uniquely binds activation function-1 in its amino-terminal domain. J Biol Chem (2016) 291(42):22231–43. doi:10.1074/jbc.M116.734475
71. Yang YC, Banuelos CA, Mawji NR, Wang J, Kato M, Haile S, et al. Targeting androgen receptor activation function-1 with EPI to overcome resistance mechanisms in castration-resistant prostate cancer. Clin Cancer Res (2016) 22(17):4466–77. doi:10.1158/1078-0432.CCR-15-2901
72. De Mol E, Fenwick RB, Phang CT, Buzon V, Szulc E, de la Fuente A, et al. EPI-001, A compound active against castration-resistant prostate cancer, targets transactivation unit 5 of the androgen receptor. ACS Chem Biol (2016) 11(9):2499–505. doi:10.1021/acschembio.6b00182
73. Yu Z, Cai C, Gao S, Simon NI, Shen HC, Balk SP. Galeterone prevents androgen receptor binding to chromatin and enhances degradation of mutant androgen receptor. Clin Cancer Res (2014) 20(15):4075–85. doi:10.1158/1078-0432.CCR-14-0292
74. Purushottamachar P, Godbole AM, Gediya LK, Martin MS, Vasaitis TS, Kwegyir-Afful AK, et al. Systematic structure modifications of multitarget prostate cancer drug candidate galeterone to produce novel androgen receptor down-regulating agents as an approach to treatment of advanced prostate cancer. J Med Chem (2013) 56(12):4880–98. doi:10.1021/jm400048v
75. Liu C, Lou W, Zhu Y, Nadiminty N, Schwartz CT, Evans CP, et al. Niclosamide inhibits androgen receptor variants expression and overcomes enzalutamide resistance in castration-resistant prostate cancer. Clin Cancer Res (2014) 20(12):3198–210. doi:10.1158/1078-0432.CCR-13-3296
76. Yu EY, Duan F, Muzi M, Deng X, Chin BB, Alumkal JJ, et al. Castration-resistant prostate cancer bone metastasis response measured by 18F-fluoride PET after treatment with dasatinib and correlation with progression-free survival: results from American College of Radiology Imaging Network 6687. J Nucl Med (2015) 56(3):354–60. doi:10.2967/jnumed.114.146936
77. Araujo JC, Trudel GC, Saad F, Armstrong AJ, Yu EY, Bellmunt J, et al. Docetaxel and dasatinib or placebo in men with metastatic castration-resistant prostate cancer (READY): a randomised, double-blind phase 3 trial. Lancet Oncol (2013) 14(13):1307–16. doi:10.1016/S1470-2045(13)70479-0
78. Posadas EM, Ahmed RS, Karrison T, Szmulewitz RZ, O’Donnell PH, Wade JL III, et al. Saracatinib as a metastasis inhibitor in metastatic castration-resistant prostate cancer: a University of Chicago phase 2 consortium and DOD/PCF prostate cancer clinical trials consortium study. Prostate (2016) 76(3):286–93. doi:10.1002/pros.23119
79. Antonarakis ES, Heath EI, Posadas EM, Yu EY, Harrison MR, Bruce JY, et al. A phase 2 study of KX2-391, an oral inhibitor of Src kinase and tubulin polymerization, in men with bone-metastatic castration-resistant prostate cancer. Cancer Chemother Pharmacol (2013) 71(4):883–92. doi:10.1007/s00280-013-2079-z
80. Bitting RL, Armstrong AJ. Targeting the PI3K/Akt/mTOR pathway in castration-resistant prostate cancer. Endocr Relat Cancer (2013) 20(3):R83–99. doi:10.1530/ERC-12-0394
81. Statz CM, Patterson SE, Mockus SM. mTOR inhibitors in castration-resistant prostate cancer: a systematic review. Target Oncol (2016). doi:10.1007/s11523-016-0453-6
82. Toren P, Kim S, Cordonnier T, Crafter C, Davies BR, Fazli L, et al. Combination AZD5363 with enzalutamide significantly delays enzalutamide-resistant prostate cancer in preclinical models. Eur Urol (2015) 67(6):986–90. doi:10.1016/j.eururo.2014.08.006
83. Ramamurthy VP, Ramalingam S, Gediya L, Kwegyir-Afful AK, Njar VC. Simultaneous targeting of androgen receptor (AR) and MAPK-interacting kinases (MNKs) by novel retinamides inhibits growth of human prostate cancer cell lines. Oncotarget (2015) 6(5):3195–210. doi:10.18632/oncotarget.3084
84. Sarker D, Reid AH, Yap TA, de Bono JS. Targeting the PI3K/AKT pathway for the treatment of prostate cancer. Clin Cancer Res (2009) 15(15):4799–805. doi:10.1158/1078-0432.CCR-08-0125
Keywords: androgen receptor, non-genomic signaling, prostate cancer, AR antagonists, Src kinase, MAPK/ERK signaling, PI3K/Akt signaling
Citation: Leung JK and Sadar MD (2017) Non-Genomic Actions of the Androgen Receptor in Prostate Cancer. Front. Endocrinol. 8:2. doi: 10.3389/fendo.2017.00002
Received: 29 November 2016; Accepted: 05 January 2017;
Published: 17 January 2017
Edited by:
Shyama Majumdar, University of Illinois at Chicago, USAReviewed by:
Antimo Migliaccio, Università degli Studi della Campania “L. Vanvitelli”, ItalyGabriella Castoria, Seconda Università degli Studi di Napoli, Italy
Copyright: © 2017 Leung and Sadar. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Marianne D. Sadar, bXNhZGFyQGJjZ3NjLmNh