 Xavier Bertagna
Xavier Bertagna- Service des Maladies Endocriniennes et Métaboliques, Centre de Référence des Maladies Rares de la Surrénale, Faculté de Médecine Paris Descartes, Université Paris 5, Hôpital Cochin, Paris, France
Chronic ACTH excess leads to chronic cortisol excess, without escape phenomenon, resulting in Cushing’s syndrome. Excess adrenal androgens also occur: in females, they will overcompensate the gonadotrophic loss, inducing high testosterone; in males, they will not compensate it, inducing low testosterone. Chronic ACTH excess leads to chronic adrenal mineralocorticoid excess and low aldosterone levels: after an acute rise, aldosterone plasma levels resume low values after a few days when ACTH is prolonged. Two other mineralocorticoids in man, cortisol and 11 deoxycorticosterone (DOC), at the zona fasciculata, will not escape the long-term effect of chronic ACTH excess and their secretion rates will remain elevated in parallel. Over all, the concomitant rise in cortisol and 11 DOC will more than compensate the loss of aldosterone, and eventually create a state of chronic mineralocorticoid excess, best evidenced by the accompanying suppression of the renin plasma levels, a further contribution to the suppression of aldosterone secretion. Prolonged in vivo stimulation with ACTH leads to an increase in total adrenal protein and RNA synthesis. Cell proliferation is indicated by an increase in total DNA the resulting adrenocortical hyperplasia participates in the amplified response of the chronically stimulated gland, and the weight of each gland can be greatly increased. The growth-stimulatory effect of ACTH in vivo most likely proceeds through the activation of a local and complex network of autocrine growth factors and their own receptors; a number of compounds, including non-ACTH proopiomelanocortin peptides such as γ3-MSH, have been shown to exert some adrenocortical growth effect.
Introduction
The pituitary–adrenal axis is a central actor in Endocrinology. Through it, the fine tuning of corticosteroids secretion is maintained, from fetal to adult life, under basal and stressful conditions, with immediate and/or long-term consequences. Altered ACTH secretion induces catastrophic clinical situations: adrenal insufficiency on the one hand, Cushing’s syndrome on the other hand. Both are debilitating conditions that severely alter the quality of life, create multiple complications, and, ultimately may lead to premature death. Besides ACTH-dependent Cushing’s syndrome, there are many situations where ACTH is chronically oversecreted. This review will examine the effects of chronic ACTH excess on adrenal cortex in man, and concentrate on steroid secretion and adrenal cortex growth.
Rare and particular situations will also be addressed.
The Various Situations in Man with Chronic ACTH Excess
The many situations that are associated with chronic ACTH excess in man are presented in Table 1. Depending on the «quality» of the original adrenal cortex they can be artificially distinguished in three different groups.
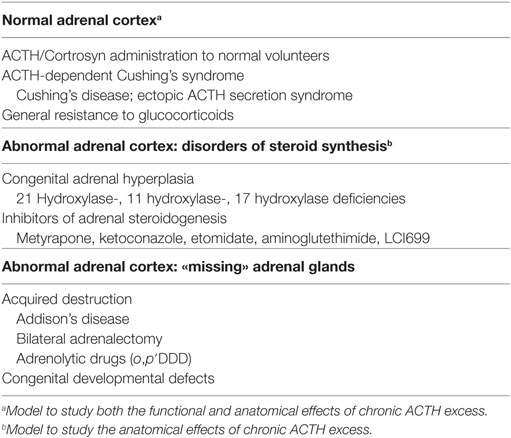
Table 1. Conditions with chronic ACTH excess in man.
There Are Three Situations Where the Adrenal Cortex Is Originally, Functionally, and Anatomically Normal
– Healthy volunteers administered with exogenous ACTH or its synthetic analog Cortrosyn (ACTH1–24).
– Patients with ACTH-dependent Cushing’s syndrome, either Cushing’s disease or the ectopic ACTH secretion syndrome. Excess ACTH is chronically produced by a pituitary or a non-pituitary tumor and acts on a basically normal adrenal cortex.
– Patients with the syndrome of general resistance to glucocorticoids.
These three situations allow measuring the effects of chronic ACTH excess on both corticosteroid secretions and adrenal cortex growth.
There Are Two Situations Where the Adrenal Cortex Has, Congenital or Acquired, Intrinsic Steroidogenic Defects
– The various types of congenital adrenal hyperplasias (CAHs) associated with altered cortisol synthesis.
– Treatment of ACTH-dependent Cushing’s syndrome with anticortisolic drugs that inhibit steroidogenesis (metyrapone, ketoconazole, etomidate, aminoglutethimide, LCI699).
In both situations, chronic ACTH excess is an adapted response to chronic cortisol deprivation. They preclude studying the effects of chronic ACTH excess on corticosteroid secretions. Still the effects on adrenocortical growth can be evaluated in a pertinent fashion.
In the Last Situations, the Adrenal Cortex Is Simply—And Anatomically—Missing or Compromised
– Bilateral adrenalectomy, usually for ACTH-dependent Cushing’s syndrome.
– Adrenolytic treatments (o,p′DDD) of ACTH-dependent Cushing’s syndrome.
– Acquired adrenal destructions (infections, hemorrhagies, autoimmune, bilateral metastases…).
– Congenital developmental defects (genetic syndromes).
Evidently, none of these situations help to study the «adrenal» effects of chronic ACTH excess. They would be, however, adapted to study the non-adrenal effects of chronic ACTH—and proopiomelanocortin (POMC)-related peptides excess. Indeed, by opposition with the sole situation where ACTH itself—or its synthetic analog Cortrosyn—are exogenously administered to healthy volunteers, all other situations with chronic excess of endogenous ACTH are accompanied by parallel excess of other non-ACTH POMC-derived peptides.
Chronic ACTH Excess in Man and Corticosteroid Secretion
Cortisol
Chronic ACTH Excess Leads to Chronic Cortisol Excess
Experiments in healthy volunteers, more than half a century ago, have shown the effects of repeated ACTH administrations (1, 2).
– When the same dose of ACTH is exogenously administered daily in man, a stepwise increase in daily cortisol secretion is observed over the days [see Figure 8 in Ref. (1)].
– This «amplifying» phenomenon has now a molecular explanation: adrenocortical cells exposed to ACTH in vitro acquire an increased number of ACTH receptors (MC2R) and an increased rate of protein Gs expression (3–6).
– Through the cAMP pathway the binding of ACTH and the transducing apparatus are both amplified, explaining the higher sensitivity and the greater response potential of chronically stimulated cells.
– No escape phenomenon is observed, although the cortisol oversecretion tends to plateau after several days.
– Over a wide range, the cortisol response is proportional to the dose of administered ACTH.
In patients with ACTH-dependent Cushing’s syndrome, chronic excess of endogenous ACTH also leads to chronic cortisol excess. Interestingly in many patients with Cushing’s disease, cortisol excess is associated with «normal» ACTH plasma values compared with those seen in the morning in normal subjects (7, 8). These values are considered «abnormally» normal or «inappropriate» in face of the hypercortisolism that should normally totally suppress ACTH secretion. Furthermore, these levels remain constant over the day, with no circadian variation, and ACTH acts on hyperresponsive adrenocortical cells; indeed, acute ACTH stimulation of the hyperplastic adrenals in Cushing’s disease patients triggers a much higher and more lasting response than would be observed in normal subjects given the same dose [see Figure 2 in Ref. (2)].
Chronic ACTH excess in case of non-pituitary tumors (the ectopic ACTH secretion syndrome) has the exact same consequences on cortisol secretion. Because ACTH plasma levels are often higher in these patients, they also have—in general—higher cortisol oversecretion (8).
In the syndrome of general resistance to cortisol, the glucocorticoid receptor type 2 is mutated with a loss of function. It is an autosomal dominant familial disease (9–11). All cells and tissues have lost their normal sensitivity to cortisol. At the hypothalamic–pituitary level, it is felt as an apparent lack of cortisol, which—naturally—induces an adapted response with chronic increase in ACTH secretion (12). This natural human situation offers a privileged model to observe the response of a perfectly normal adrenal cortex to chronic ACTH excess: unsurprisingly chronic cortisol excess is observed, but without the clinical features of Cushing’s syndrome. Together with cortisol, other ACTH-dependent corticosteroids, androgens dehydroepiandrosterone (DHEA), and mineralocorticoids deoxycorticosterone (DOC) are oversecreted by the zona fasciculata (see further).
A state of chronic and acquired general resistance to cortisol can be artificially created in Cushing’s disease patients chronically treated with RU486 (Mifepristone). The drug is an antagonist to the glucocorticoid receptor type 2, the acute administration of which triggers an immediate pituitary ACTH retort in normal subjects (13, 14). As expected, under long-term RU486 administration some clinical peripheral features of the Cushing’s syndrome are ameliorated such as hyperglycemia (15); as expected also, in the patients with Cushing’s disease, the hypothalamic–pituitary–adrenal axis is stimulated (by the “apparent” cortisol deprivation) and ACTH is acutely and/or chronically increased further with plasma levels raising up ca. three times above their baseline initial values (16, 17). In the SEISMIC study, several patients developed high blood pressure, edema, and hypokelmia suggestive of a state of chronic hypermineralocorticism (17). All ACTH-dependent corticosteroids presumably increase in parallel: cortisol, the clinical impact of which remains blunted at the glucocorticoid receptor by the drug; but also adrenal androgens and DOC, the actions of which are not opposed at their respective androgen and mineralocorticoid receptors (for both DOC and cortisol) and which may thus induce serious clinical features: possibly hyperandogenism in women; hypertension and hypokalemia in both sexes (18).
Adrenocortical Androgens
Chronic ACTH Excess Leads to Chronic Adrenal Androgens Excess with Contrasted Impacts in Females and Males
Adrenal androgens are produced by the zona fasciculata/reticularis, and—as cortisol—are under the same—and dominant if not exclusive—control of ACTH and the cAMP pathway. Acute administration to healthy volunteers induces abrupt increase in circulating adrenal androgens (19). The situation has been particularly studied in Cushing’s disease patients. The specific adrenal androgen DHEA sulfate (DHEAS) is chronically increased both in males and females (20). Thus DHEA, DHEAS, and Δ-4-androstenedione are elevated in Cushing’s disease patients (21–23). Yet, the impact of chronic ACTH excess upon the overall androgenic status is contrasted between females and males:
– In both females and males, chronic excess of androgens, and cortisol, both lead to suppressed gonadotroph function directly at the hypothalamic–pituitary levels.
– In females, because the pituitary gonadotrophic function normally accounts for only half of the total circulating androgens, the excess adrenal androgens will eventually overcompensate the gonadotrophic loss and induce an overall excess of circulating androgens: their peripheral transformation to testosterone and dihydrotestosterone may lead to a moderate state of androgen excess in females with its clinical impact: hirsutism, infertility [see Figure 3 in Ref. (22)].
– In males, because the pituitary gonadotrophic/gonadal function normally accounts for the vast majority of circulating androgens (ca. 90%), the excess of adrenal androgens will not compensate that which has been lost due to the cortisol-induced suppression of the gonadotrophic/gonadal function. The overall circulating androgens will eventually be abnormally low (testosterone), with a clinical impact: decreased sexual activity, infertility (24).
In a way similar to that observed with cortisol, chronic increase in adrenal androgens will be observed in other situations with chronic ACTH excess such as the ectopic ACTH secretion syndrome, and the general resistance to glucocorticoids. In the latter situation, as evoked earlier, excess androgens in females may be the dominant symptom that should alert on the diagnosis in the absence of «Cushing’s» features; it may provoke precocious puberty in children (12).
In patients with Cushing’s disease treated with the antiglucocorticoid RU486, the inescapable rise in ACTH secretion will—theoretically—increase further the state of hyperandrogenism in females and this should be a major drawback for the use of this compound …. Surprisingly, there is no report on the clinical or hormonal androgen status of women in the SEISMIC study (17).
Dissociation between cortisol and adrenal androgens is observed, however, in the particular situation of patients resuming normal corticotroph function after long-term ACTH excess. After successful pituitary surgery in Cushing’s disease patients, DHEAS remains suppressed for months or years after plasma cortisol has normalized (21).
Mineralocorticoids
Chronic ACTH Excess Leads to Chronic Adrenal Mineralocorticoid Excess and Low Aldosterone Levels
It has long been known that acute ACTH administration to man leads to immediate aldosterone secretion; yet this effect is only transient and is not maintained when ACTH is prolonged over periods of time, and aldosterone plasma levels resume low values after a few days (25, 26).
Yet this “escape” phenomenon on aldosterone will not prevent the establishment of a state of chronic mineralocorticoid excess, for two reasons:
– This “escape” will only concern aldosterone, at the zona glomerulosa. Its exact mechanism is not entirely understood: it is suggested that increased concentrations of cortisol in the adrenal cortex directly inactivates the last steps of aldosterone synthesis in the ZG (27); there is also evidence that initial ZG cells undergo a differentiation process toward cortisol producing cells (28).
– Two other mineralocorticoids in man, cortisol and 11 DOC, at the zona fasciculata, will not escape the long-term effect of chronic ACTH excess and their secretion rates will remain elevated in parallel (29).
Over all, the concomitant rise in cortisol and DOC, even though each of these molecules is intrinsically less potent than aldosterone at the mineralocorticoid receptor, will more than compensate the loss of aldosterone, and eventually create a state of chronic mineralocorticoid excess, best evidenced by the accompanying suppression of the renin plasma levels, a further—if not exclusive—contribution to the suppression of aldosterone secretion (30). This general mineralocorticoid effect is in correlation with the level of ACTH excess: its clinical consequences (high blood pressure, hypokalemic alkalosis) are more frequent in patients with the ectopic ACTH secretion syndrome than in those with Cushing’s disease (31).
As expected, patients with the syndrome of general resistance to glucocorticoids have parallel increases in cortisol and DOC, and hypokalemic hypertension may be another—sometime predominant—clinical presentation in these patients, as reported in the first published cases (9).
These complications may also occur in patients with Cushing’s disease treated by RU486 (Mifepristone). The drug induces a further increase in cortisol and probably DOC (this latter steroid rarely if ever measured in these patients!), which both can interact, without any opposition, at the mineralocorticoid receptor (18), and see Figure 1.
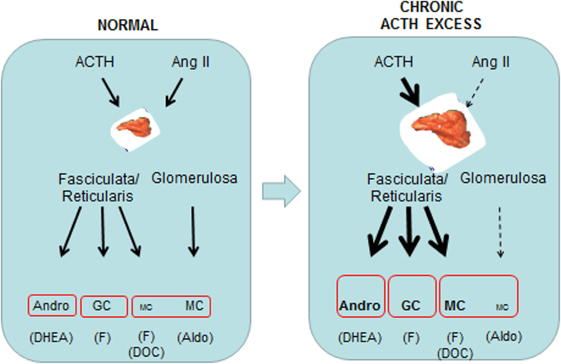
Figure 1. Schematic overview of the comparative effects of normal and prolonged ACTH excess in man on adrenal cortical steroids at the zona glomerulosa and the zona fasciculata/reticularis.
Chronic ACTH Excess in Man and Adrenal Cortex Growth
The central role of ACTH on the adrenal cortex trophicity has long been known. Hypophysectomy results in adrenal cortex atrophy that is restored by the sole administration of ACTH (32). Thus, in vivo, ACTH is the predominant if not the exclusive trophic factor for the adrenals. More recently, various animal models, which eliminate ACTH or its receptor (MC2R) in transgenic mice, have confirmed the central role of ACTH to maintain normal adrenal cortex growth (33–35). Prolonged in vivo stimulation with chronic ACTH administration or oversecretion eventually leads to an increase in total adrenal protein and RNA synthesis. Cell proliferation is indicated by an increase in total DNA (32) the resulting adrenocortical hyperplasia participates in the amplified response of the chronically stimulated gland, and the weight of each gland can be greatly increased.
The exact mechanism whereby ACTH promotes adrenocortical growth still is complex and remains partially understood, since in vitro studies show a paradoxical negative effect of ACTH on adrenocortical cell proliferation (36) The growth-stimulatory effect of ACTH in vivo most likely proceeds through the activation of a local and complex network of autocrine growth factors and their own receptors; a number substances, including non-ACTH POMC peptides such as γ3-MSH, have been shown to exert some adrenocortical growth effect (28).
In ACTH-dependent Cushing’s syndrome, chronic ACTH excess leads to bilateral adrenal hyperplasia: both adrenals are enlarged, their weight is increased in comparison with normal glands, and the histological appearance shows diffuse widening of the fasciculata/reticularis zona (Figure 2). This hyperplasia is typically homogeneous and rather symmetrical. In some cases however, it may be asymmetrical, and/or one or the two glands may bear nodular zones or authentic nodules embedded within the diffuse hyperplasia. Today, CT-Scan allows to see the adrenal hyperplasia in vivo in patients with ACTH-dependent Cushing’s syndrome, and to observe its fate after the suppression of ACTH, that is after successful pituitary surgery: the two enlarged glands and the nodules progressively shrink and can even become atrophic until normal ACTH secretion is spontaneously restored, which may take months or years (37, 38). In parallel to these anatomical changes, baseline cortisol and its response to the acute stimulation by ACTH is suppressed, and progressively resume over months or years. As already mentioned, cortisol response is restored more rapidly than androgen response.
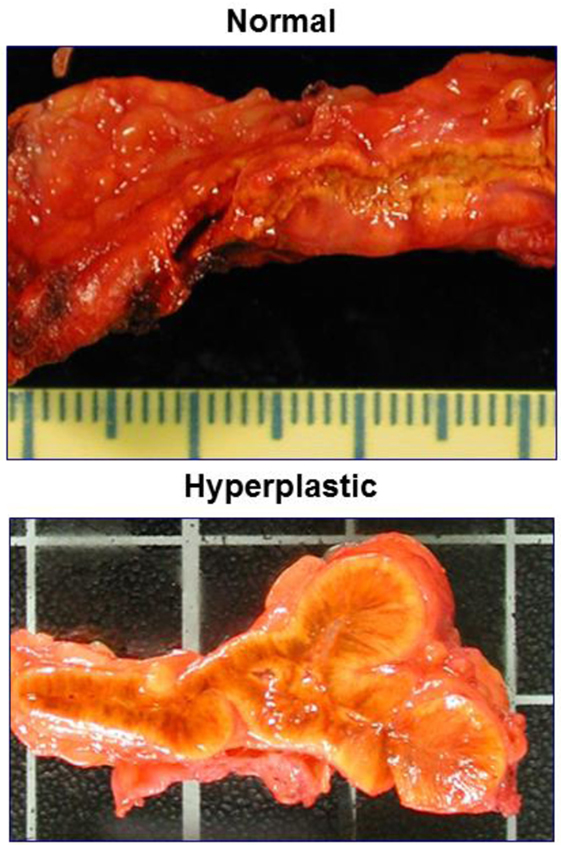
Figure 2. Comparing the macroscopic aspect of normal adrenals and those of a Cushing’s disease patient.
A rather similar presentation is observed in patients with CAH: both glands are enlarged, and become smaller under glucocorticoid administration (Figure 3).
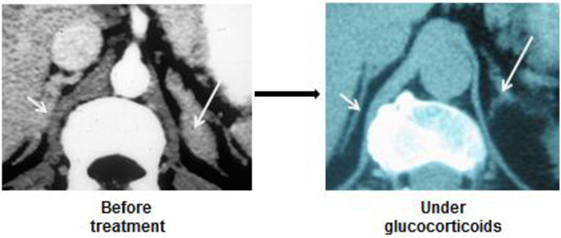
Figure 3. CT-scan images of enlarged and nodular adrenal glands in a patient with 21 hydroxylase deficiency. Regression under ACTH suppression by large doses of glucocorticoids.
Some studies seem to demonstrate that chronic ACTH excess also favors the appearance of adrenocortical nodules, and it has been suggested that some of them ultimately become autonomous (i.e., ACTH-independent). Yet, it is common observation that suppression of chronic ACTH excess, in Cushing’s or CAH patients, reduces both the hyperplastic and nodular parts of the enlarged glands (Figure 3).
Interestingly, the growth promoting effect of chronic ACTH excess can be exerted also at distance, on adrenal rests:
– Testicular adrenal rests may develop as local tumors which impinge the normal spermatogenesis, and may become cause of infertility in poorly controlled male patients with CAH (39). There is some evidence that lowering ACTH plasma levels may shrink these tumors which, in contrast with the normal testicular tissue, are loaded with the MC2R (40). The close phenotype shared by TART and fetal adrenals, including classical markers of adrenal steroidogenesis, highly favors the hypothesis that TART develops from an original adrenal cortical cell type.
– In Cushing’s disease patients, particularly those treated by “total” bilateral adrenalectomy, a large increase in ACTH secretion can be triggered. It may play a role in the growth of adrenal rests, locally (adrenal extrusions that have escaped the surgeon tools!), but also at distance, in the testis, and many other places (41, 42).
Rare and Particular Situations
Mimicking Chronic ACTH Excess
There are several pathological conditions where pituitary ACTH is actually suppressed and cortisol is oversecreted in response to molecular phenomenon which occur directly at the adrenal level, and, somehow, mimick chronic ACTH excess:
– ACTH may be produced locally by tumoral adrenal cells, and its autocrine action may participate in cortisol oversecretions in cases of bilateral macronodular adrenal hyperplasia (43).
– In the absence of ACTH, its signaling pathway may be nevertheless overactivated and generate excess of corticosteroid secretion: adrenocortical tumors which express illegitimate G-protein coupled receptors (44, 45), activated mutated MC2R (46), mutated PRKAR1A in the Carney complex (47, 48), and mutated PKACa in adrenocortical adenomas (49).
– It has also been suggested auto antibodies acting at the ACTH receptor might be responsible for excess cortisol secretion in some cases of Wulffraat (50). Yet little confirmation has been obtained since the original paper.
It is interesting to observe that all these situations eventually result in the overactivation of the cAMP signaling pathway, and always occur in benign tumors. They concur with the idea that chronic ACTH excess, or chronic activation of its signaling pathway at the same time may have some growth effect but also a differentiation action.
When “Normal” ACTH Is Too Much!
In the syndrome of apparent mineralocorticoid excess, the loss of function of 11 hydroxysteroid dehydrogenase type II, particularly at the kidney, locally enhances the mineralocorticoid action of cortisol (51).
In the glucocorticoid-remediable aldosteronism, gene rearrangement induces the “ectopic” expression of the Aldo synthase gene within the ZF cells and thus provokes the oversecretion of aldosterone under the action of ACTH (52).
In these two situations, the HPA axis and cortisol secretion are normal. Yet, it drives a state of “apparent” or “real” mineralocorticoid excess, and the treatment option is indeed to suppress ACTH secretion.
Non-ACTH POMC Peptides
As mentioned earlier, ACTH is part of a larger polypeptide precursor, POMC the enzymatic processing of which liberates ACTH itself and a number of other “non-ACTH” POMC-derived peptides. Among these peptides, the lipotropins (beta- and gamma-lipotropins) exert a definitive action on the melanocytes in man, and are responsible, together with ACTH, for skin hyperpigmentation that is observed in all situations of excess endogenous ACTH secretion. Beta-endorphin is an opioid peptide, which is a processing product of POMC. It circulates in blood, in parallel with the ACTH that is secreted by normal or tumoral pituitary or non-pituitary corticotroph cells. Yet, even at extremely high plasma values, circulating beta-endorphin has no known actions in man, and does not exert any analgesic action (53). The analgesic effect of beta-endorphin is entirely due to that which is produced locally in the brain by a set of POMC-expressing neurons: the circulating beta-endorphin does not cross the blood–brain barrier.
Experimental evidence suggest that non-ACTH N-terminal POMC peptides may exert a growth effect on the adrenal cortex (54, 55). The small peptide N-POMC1–28, which bears two intramolecular disulfide bridges, is responsible for this action which has also been described in the human adreno cortical tumors cells NCI-H295 (56). Yet the receptor for the N-POMC1–28 peptide remains elusive (55).
Chronic Stress and Pseudo-Cushing
Pseudo-Cushing corresponds to these situations in man where an authentic hypercortisolism is biologically present (increased urinary cortisol, abnormal response to dexamethasone suppression test) and may be confused with “endogenous” Cushing’s disease. Yet, in Pseudo-Cushing there is no pituitary adenoma; the ACTH excess is thought to be “functional,” driven by the overactivity of the hypothalamus and oversecretion of corticotrophin-releasing hormone under chronic stress, severe depression, intense physical activity, and anorexia nervosa (57).
Author Contributions
The author confirms being the sole contributor of this work and approved it for publication.
Conflict of Interest Statement
The author declares that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1. Liddle GW, Island D, Rinfret AP, Forsham PH. Factors enhancing the response of the human adrenal to corticotropin: is there an adrenal growth factor? J Clin Endocrinol Metab (1954) 14:839–58. doi: 10.1210/jcem-14-8-839
2. Liddle GW. Regulation of adrenocortical function in man. In: Christy NP, editor. The Human Adrenal Cortex. New York: Harper and Row Publisher (1971). p. 41–68.
3. Penhoat A, Jailiard C, Saez JM. Corticotropin positively regulates its own receptors and cAMP response in cultured bovine adrenal cells. Proc Natl Acad Sci U S A (1989) 86:4978–81. doi:10.1073/pnas.86.13.4978
4. Begeot M, Langlois D, Penhoat A, Saez JM. Variations in guanine-binding proteins (Gs, Gi) in cultured bovine adrenal cells. Consequences on the effects of phorbol and cholera-toxin-induced cAMP production. Eur J Biochem (1988) 174:317–21. doi:10.1111/j.1432-1033.1988.tb14100.x
5. Mountjoy KG, Robbins LS, Mortrud MT, Cone RD. The cloning of a family of genes that encode the melanocortin receptors. Science (1992) 257:1248–51. doi:10.1126/science.1325670
6. Penhoat A, Lebrethon MC, Bégeot M, Saez JM. Regulation of ACTH receptor mRNA and binding sites by ACTH and angiotensin II in cultured human and bovine adrenal fasciculata cells. Endocr Res (1995) 21(1–2):157–68. doi:10.3109/07435809509030431
7. Kuhn JM, Proeschel MF, Seurin DJ, Bertagna XY, Luton JP, Girard FL. Comparative assessment of ACTH and lipotropin plasma levels in the diagnosis and follow-up of patients with Cushing’s syndrome: a study of 210 cases. Am J Med (1989) 86(6 Pt 1):678–84. doi:10.1016/0002-9343(89)90443-9
8. Bertagna X, Guignat L, Raux-Demay MC, Guilhaume B, Girard F. Cushing’s disease. 3rd ed. In: Melmed S, editor. The Pituitary. London, UK: Academic Press (2011). p. 533–617.
9. Vingerhoeds AC, Thijssen JH, Schwarz F. Spontaneous hypercortisolism without Cushing’s syndrome. J Clin Endocrinol Metab (1976) 43(5):1128–33. doi:10.1210/jcem-43-5-1128
10. Chrousos GP, Vingerhoeds A, Brandon D, Eil C, Pugeat M, DeVroede M, et al. Primary cortisol resistance in man. A glucocorticoid receptor-mediated disease. J Clin Invest (1982) 69(6):1261–9. doi:10.1172/JCI110565
11. Malchoff DM, Brufsky A, Reardon G, McDermott P, Javier EC, Bergh CH, et al. A mutation of the glucocorticoid receptor in primary cortisol resistance. J Clin Invest (1993) 91(5):1918–25. doi:10.1172/JCI116410
12. Charmandari E, Kino T, Ichijo T, Chrousos GP. Generalized glucocorticoid resistance: clinical aspects, molecular mechanisms, and implications of a rare genetic disorder. J Clin Endocrinol Metab (2008) 93(5):1563–72. doi:10.1210/jc.2008-0040
13. Bertagna X, Bertagna C, Luton JP, Husson JM, Girard F. The new steroid analog RU 486 inhibits glucocorticoid action in man. J Clin Endocrinol Metab (1984) 59(1):25–8. doi:10.1210/jcem-59-1-25
14. Gaillard RC, Riondel A, Muller AF, Herrmann W, Baulieu EE. RU 486: a steroid with antiglucocorticosteroid activity that only disinhibits the human pituitary-adrenal system at a specific time of day. Proc Natl Acad Sci U S A (1984) 81(12):3879–82. doi:10.1073/pnas.81.12.3879
15. Fleseriu M, Biller BM, Findling JW, Molitch ME, Schteingart DE, Gross C, et al. Mifepristone, a glucocorticoid receptor antagonist, produces clinical and metabolic benefits in patients with Cushing’s syndrome. J Clin Endocrinol Metab (2012) 97(6):2039–49. doi:10.1210/jc.2011-3350
16. Bertagna X, Bertagna C, Laudat MH, Husson JM, Girard F, Luton JP. Pituitary-adrenal response to the antiglucocorticoid action of RU 486 in Cushing’s syndrome. J Clin Endocrinol Metab (1986) 63(3):639–43. doi:10.1210/jcem-63-3-639
17. Fleseriu M, Findling JW, Koch CA, Schlaffer SM, Buchfelder M, Gross C. Changes in plasma ACTH levels and corticotroph tumor size in patients with Cushing’s disease during long-term treatment with the glucocorticoid receptor antagonist mifepristone. J Clin Endocrinol Metab (2014) 99(10):3718–27. doi:10.1210/jc.2014-1843
18. Castinetti F, Fassnacht M, Johanssen S, Terzolo M, Bouchard P, Chanson P, et al. Merits and pitfalls of mifepristone in Cushing’s syndrome. Eur J Endocrinol (2009) 160(6):1003–10. doi:10.1530/EJE-09-0098
19. Reisch N, Slawik M, Zwermann O, Beuschlein F, Reincke M. Genetic influence of an ACTH receptor promoter polymorphism on adrenal androgen secretion. Eur J Endocrinol (2005) 153(5):711–5. doi:10.1530/eje.1.02015
20. Barbetta L, Dall’Asta C, Re T, Colombo P, Travaglini P, Ambrosi B. Androgen secretion in ectopic ACTH syndrome and in Cushing’s disease: modifications before and after surgery. Horm Metab Res (2001) 33(10):596–601. doi:10.1055/s-2001-17906
21. Yamaji T, Ishibashik M, Sekihara H, Itabashi A, Yanaihara T. Serum dehydroepiandrosterone sulfate in Cushing’s syndrome. J Clin Endocrinol Metab (1984) 1984:1164–8. doi:10.1210/jcem-59-6-1164
22. Halpin DM, Burrin JM, Joplin GF. Serum testosterone levels in women with Cushing’s disease. Acta Endocrinol (Copenh) (1990) 122(1):71–5.
23. Smals AG, Kloppenborg PW, Benraad TJ. Plasma testosterone profiles in Cushing’s syndrome. J Clin Endocrinol Metab (1977) 45(2):240–5. doi:10.1210/jcem-45-2-240
24. Luton JP, Thieblot P, Valcke JC, Mahoudeau JA, Bricaire H. Reversible gonadotropin deficiency in male Cushing’s disease. J Clin Endocrinol Metab (1977) 45(3):488–95.
25. Gaillard RC, Riondel AM, Favrod-Coune CA, Vallotton MB, Muller AF. Aldosterone escape to chronic ACTH administration in man. Acta Endocrinol (Copenh) (1983) 103(1):116–24.
26. Rauh W, Levine LS, Gottesdiener K, New MI. Mineralocorticoids, salt balance and blood pressure after prolonged ACTH administration in juvenile hypertension. Klin Wochenschr (1978) 56(Suppl 1):161–7. doi:10.1007/BF01477468
27. Bird IM, Pasquarette MM, Rainey WE, Mason JI. Differential control of 17 alpha hydroxylase and 3-beta hydroxyl dehydrogenase expression in human adrenocortical H295R cells. J Clin Endocrinol Metab (1996) 81:2171–8. doi:10.1210/jcem.81.6.8964847
28. Gallo-Payet N. Adrenal and extra-adrenal functions of ACTH. J Mol Endocrinol (2016) 56(4):T135–56. doi:10.1530/JME-15-0257
29. Kater CE, Biglieri EG, Brust N, Chang B, Hirai J, Irony I. Stimulation and suppression of the mineralocorticoid hormones in normal subjects and adrenocortical disorders. Endocr Rev (1989) 10(2):149–64. doi:10.1210/edrv-10-2-149
30. Oelkers W. Prolonged ACTH infusion suppresses aldosterone secretion in spite of high renin activity. Acta Endocrinol (Copenh) (1985) 108:91–7.
31. Schambelan M, Slaton PE Jr, Biglieri EG. Mineralocorticoid production in hyperadrenocorticism. Role in pathogenesis of hypokalemic alkalosis. Am J Med (1971) 51(3):299–303. doi:10.1016/0002-9343(71)90264-6
32. Gill GN. ACTH regulation of the adrenal cortex. In: Gill GN, editor. Pharmacology of Adrenal Cortical Hormones. New York: Pergamon (1979). p. 35–66.
33. Chida D, Nakagawa S, Nagai S, Sagara H, Katsumata H, Imaki T, et al. Melanocortin 2 receptor is required for adrenal gland development, steroidogenesis, and neonatal gluconeogenesis. Proc Natl Acad Sci U S A (2007) 104:18205–10. doi:10.1073/pnas.0706953104
34. Coll AP, Challis BG, Yeo GS, Snell K, Piper SJ, Halsall D, et al. The effects of proopiomelanocortin deficiency on murine adrenal development and responsiveness to adrenocorticotropin. Endocrinology (2004) 145:4721–7. doi:10.1210/en.2004-0491
35. Karpac J, Ostwald D, Bui S, Hunnewell P, Shankar M, Hochgeschwender U. Development, maintenance, and function of the adrenal gland in early postnatal proopiomelanocortin-null mutant mice. Endocrinology (2005) 146:2555–62. doi:10.1210/en.2004-1290
36. Hornsby PJ. The mechanism of action of ACTH in the adrenal cortex. In: Cooke BA, King RJB, Van Der Molen HJ, editors. Hormones and Their Actions, Part II. Amsterdam: Elsevier Science Publishers BV (Biomedical Division) (1988). p. 193–210.
37. Imaki T, Naruse M, Takano K. Adrenocortical hyperplasia associated with ACTH-dependent Cushing’s syndrome: comparison of the size of adrenal glands with clinical and endocrinological data. Endocr J (2004) 51(1):89–95. doi:10.1507/endocrj.51.89
38. Albiger NM, Occhi G, Sanguin F, Iacobone M, Casarrubea G, Ferasin S, et al. Adrenal nodules in patients with Cushing’s disease: prevalence, clinical significance and follow-up. J Endocrinol Invest (2011) 34:204–9. doi:10.3275/7349
39. Ahmad IC, Yilmaz TF, Kocakoç E. Doppler ultrasonography and magnetic resonance imaging findings of testicular adrenal rest tissue in a patient with 11 β hydroxilase deficiency. Case report. Med Ultrason (2014) 16(4):383–5.
40. Lottrup G, Nielsen JE, Skakkebaek NE, Juul A, Rajpert-De Meyts E. Abundance of DLK1, differential expression of CYP11B1, CYP21A2 and MC2R, and lack of INSL3 distinguish testicular adrenal rest tumours from Leydig cell tumours. Eur J Endocrinol (2015) 172(4):491–9. doi:10.1530/EJE-14-0810
41. Baranetsky NG, Zipser RD, Goebelsmann U, Kurman RJ, March CM, Morimoto I, et al. Adrenocorticotropin-dependent virilizing paraovarian tumors in Nelson’s syndrome. J Clin Endocrinol Metab (1979) 49(3):381–6. doi:10.1210/jcem-49-3-381
42. Carpenter PC, Wahner HW, Salassa RM, Duick DS. Demonstration of steroid-producing gonadal tumors by external scanning with the use of NP-59. Mayo Clin Proc (1979) 54(5):332–4.
43. Louiset E, Duparc C, Young J, Renouf S, Tetsi Nomigni M, Boutelet I, et al. Intraadrenal corticotropin in bilateral macronodular adrenal hyperplasia. N Engl J Med (2013) 369(22):2115–25. doi:10.1056/NEJMoa1215245
44. Lacroix A, Bolté E, Tremblay J, Dupré J, Poitras P, Fournier H, et al. Gastric inhibitory polypeptide-dependent cortisol hypersecretion – a new cause of Cushing’s syndrome. N Engl J Med (1992) 327(14):974–80. doi:10.1056/NEJM199210013271402
45. Reznik Y, Allali-Zerah V, Chayvialle JA, Leroyer R, Leymarie P, Travert G, et al. Food-dependent Cushing’s syndrome mediated by aberrant adrenal sensitivity to gastric inhibitory polypeptide. N Engl J Med (1992) 327(14):981–6. doi:10.1056/NEJM199210013271403
46. Swords FM, Baig A, Malchoff DM, Malchoff CD, Thorner MO, King PJ, et al. Impaired desensitization of a mutant adrenocorticotropin receptor associated with apparent constitutive activity. Mol Endocrinol (2002) 16(12):2746–53. doi:10.1210/me.2002-0099
47. Kirschner LS, Carney JA, Pack SD, Taymans SE, Giatzakis C, Cho YS, et al. Mutations of the gene encoding the protein kinase A type I-alpha regulatory subunit in patients with the Carney complex. Nat Genet (2000) 26(1):89–92. doi:10.1038/79238
48. Groussin L, Jullian E, Perlemoine K, Louvel A, Leheup B, Luton JP, et al. Mutations of the PRKAR1A gene in Cushing’s syndrome due to sporadic primary pigmented nodular adrenocortical disease. J Clin Endocrinol Metab (2002) 87(9):4324–9. doi:10.1210/jc.2002-020592
49. Beuschlein F, Fassnacht M, Assié G, Calebiro D, Stratakis CA, Osswald A, et al. Constitutive activation of PKA catalytic subunit in adrenal Cushing’s syndrome. N Engl J Med (2014) 370(11):1019–28. doi:10.1056/NEJMoa1310359
50. Wulffraat NM, Drexhage HA, Wiersinga WM, van der Gaag RD, Jeucken P, Mol JA. Immunoglobulins of patients with Cushing’s syndrome due to pigmented adrenocortical micronodular dysplasia stimulate in vitro steroidogenesis. J Clin Endocrinol Metab (1988) 66(2):301–7. doi:10.1210/jcem-66-2-301
51. Stewart PM, Corrie JE, Shackleton CH, Edwards CR. Syndrome of apparent mineralocorticoid excess. A defect in the cortisol-cortisone shuttle. J Clin Invest (1988) 82(1):340–9.
52. Lifton RP, Dluhy RG, Powers M, Rich GM, Cook S, Ulick S, et al. A chimaeric 11 beta-hydroxylase/aldosterone synthase gene causes glucocorticoid-remediable aldosteronism and human hypertension. Nature (1992) 355(6357):262–5.
53. Willer JC, Sheng-Shu L, Bertagna X, Girard F. Pituitary beta-endorphin not involved in pain control in some pathophysiological conditions. Lancet (1984) 2(8397):295–6.
54. Estivariz FE, Iturriza F, McLean C, Hope J, Lowry PJ. Stimulation of adrenal mitogenesis by N-terminal proopiocortin peptides. Nature (1982) 297(5865):419–22.
55. Bicknell AB. N-terminal POMC peptides and adrenal growth. J Mol Endocrinol (2016) 56(4):T39–48. doi:10.1530/JME-15-0269
56. Fassnacht M, Hahner S, Hansen IA, Kreutzberger T, Zink M, Adermann K, et al. N-terminal proopiomelanocortin acts as a mitogen in adrenocortical tumor cells and decreases adrenal steroidogenesis. J Clin Endocrinol Metab (2003) 88(5):2171–9. doi:10.1210/jc.2002-021318
Keywords: adrenal, ACTH excess, Cushing’s syndrome, cortisol, androgens, mineralocorticoids, adrenal cortex growth
Citation: Bertagna X (2017) Effects of Chronic ACTH Excess on Human Adrenal Cortex. Front. Endocrinol. 8:43. doi: 10.3389/fendo.2017.00043
Received: 02 December 2016; Accepted: 20 February 2017;
Published: 08 March 2017
Edited by:
Nicole Gallo-Payet, Université de Sherbrooke, CanadaReviewed by:
Maria Candida Barisson Villares Fragoso, University of São Paulo, BrazilDuarte L. Pignatelli, IPATIMUP, Portugal
Copyright: © 2017 Bertagna. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Xavier Bertagna, eGF2aWVyLmJlcnRhZ25hQGFwaHAuZnI=