 Mihaela Stefan
Mihaela Stefan Larissa C. Faustino
Larissa C. Faustino- Division of Endocrinology, Department of Medicine, Albert Einstein College of Medicine, Bronx, NY, USA
Production of thyroid-stimulating hormone receptor (TSHR) antibodies represents the hallmark of Graves’ disease (GD) pathogenesis. Thus, for more than two decades the TSHR gene has been at the center of studies intended to elucidate its contribution to disease pathology. The advent of genome-wide association technology allowed to establish a strong association of the TSHR gene with GD. Subsequent fine-mapping studies narrowed the disease-susceptibility region to a 40 kb sequence in intron 1, where at least five GD-associated SNPs in tight linkage disequilibrium were identified. The current challenge is to understand the functional mechanisms by which these polymorphisms modify physiological processes and trigger disease. The aim of this review is to summarize the current knowledge on the role of the TSHR gene in GD pathogenesis, which has been gained through linkage and association studies, as well as to discuss the emerging mechanisms underlying biological implications of TSHR variants in the development of GD.
Introduction
Autoimmune thyroid diseases (AITD), including Graves’ disease (GD) and Hashimoto’s thyroiditis (HT), affect 2–5% of the general population, representing the most frequent autoimmune conditions (1, 2). Similar to other complex autoimmune diseases, it is believed that AITD occur when interactions of genetic susceptibility factors with environmental triggers lead to loss of immune self-tolerance at peripheral and central levels (3). During the last three decades, several approaches from linkage and association studies to candidate genes analysis and whole-genome screening enabled significant progress in the identification of genes that confer susceptibility to AITD. These genes are broadly grouped as either immune-modulating genes (including the HLA family, CD40, CD25, FOXP3, PTPN22) or thyroid-specific genes [thyroid-stimulating hormone receptor (TSHR) and TG].
The TSHR is unique among these susceptibility genes because it encodes for a protein that is both responsible for the clinical manifestations of the disease and is the direct target of autoimmune response in GD. The TSHR gene, located on chromosome 14q31, consists of 10 exons and encodes for a G protein-coupled receptor that plays a central role in the regulation of thyroid development, growth, and function. Indeed, TSHR-stimulating antibodies (TSAbs) are present in nearly all cases of GD, and severity of the disease correlates with TSAbs levels (4). Moreover, compelling evidence has correlated TSHR gene variants exclusively with GD susceptibility and not with HT development (5, 6). The aim of this review is to summarize the current knowledge of the role of TSHR gene in GD pathology, gained through linkage analysis, association, and functional studies.
Linkage Studies
Whole-genome linkage is a powerful technique for screening the human genome for major susceptibility genes, without any previous assumption on the mechanisms of genetic susceptibility to the disease. It is based on the principle that the probability of recombination between two loci is directly related to the genetic distance between them. Thus, if two loci comprising a polymorphism and a disease-related gene are close to each other on a chromosome, the alleles of the polymorphic loci will cosegregate with the disease within affected families because the likelihood of recombination between the polymorphism and the disease-related gene is very low. Single nucleotide polymorphisms (SNPs) and microsatellites are the main genetic polymorphisms screened in the linkage studies (7, 8).
Several linkage studies intended to detect GD-specific loci had limited success in demonstrating significant linkage (as well as association) of the TSHR gene with GD (9–13). However, Tomer et al. identified a large region of linkage on chromosome 14q31 containing the TSHR locus in whole-genome linkage studies in multiplex, multigenerational AITD families (14–17). They identified a locus, designated Graves’ disease 1 (GD-1) that included TSHR gene, as well as other potential candidate genes—such as estrogen receptor 2, deiodinase type 2, and immunoglobulin heavy locus (14, 17). Later, fine-mapping analysis by Tomer et al. confirmed that the susceptibility gene in the GD-1 locus was indeed the TSHR gene, even though a second gene in this locus, NRXN3, was also identified as a major GD gene (18).
Association Studies
Reflecting the importance of TSHR for GD pathogenesis, TSHR was the first non-MHC gene to be tested for association with the disease. Three germline missense mutations were initially described in patients with GD and proposed to be associated with the disease (19, 20): a substitution of aspartic acid (D) for histidine (H) in position 36 (D36H); a substitution of a proline (P) for threonine (T) in position 52 (P52T), and a substitution of aspartic acid (D) for glutamic acid (E) in position 727 (D727E). Two of these three mutations, D36H and P52T, are located in the putative ligand binding region of the extracellular domain of the TSHR, while the third one, D727E lies within the intracellular domain of the receptor.
However, studies regarding association of these SNPs with GD were contradictory. In 1995, Bahn and colleagues were first to report the association of the P52T polymorphism with AITD in female population (21), but in a subsequent study Watson et al. found no differences in the distribution of this polymorphism in GD patients as compared with autoimmune hypothyroidism patients and control individuals (22). Several subsequent studies reached inconsistent results, thus a clear association of P52T or D36H with GD was not confirmed (23–25). In 1999, Gabriel et al. reported that the C to G transition in the TSHR 727 codon leading to D727E variant has an increased frequency in patients with non-autoimmune thyroiditis (26), and this association was also recently reported in a small Turkish population (27). However, the D727E association with non-autoimmune thyroiditis was not supported by studies in large series of European Caucasian patients (28). The association of D727E polymorphism with autoimmune thyroiditis was supported by data from a case–control study in Russian populations (29) but was not confirmed in US Caucasian patients (13, 26). Finally, Tomer group performed a case–control study and meta-analysis combining the data from three independent studies and showed a very week association of D727E polymorphism and GD (13).
To date, no compelling evidence exists to support a role of these three TSHR polymorphisms in GD pathogenesis. Given the frequency of these variants in general population, it is believed that they are common polymorphisms, not implicated in development of GD (20). The lack of consistency among completed studies could be the consequence of ethnic differences, selection bias, and population stratification.
In the last decade, the association of common genetic variants with complex diseases was significantly facilitated by the increased ability to measure genetic variability of hundreds of markers in large cohorts of individuals. The advent of genome-wide association technology applied to large case–control studies allowed identification of disease-associated variants and their contribution to disease susceptibility. Applying this technology to AITD resulted in identification of new disease-associated loci, including TSHR, and provided unique insights into their genetic contribution to disease pathology. Table 1 summarizes the main studies conducted over the years that established association of the TSHR gene with GD risk.
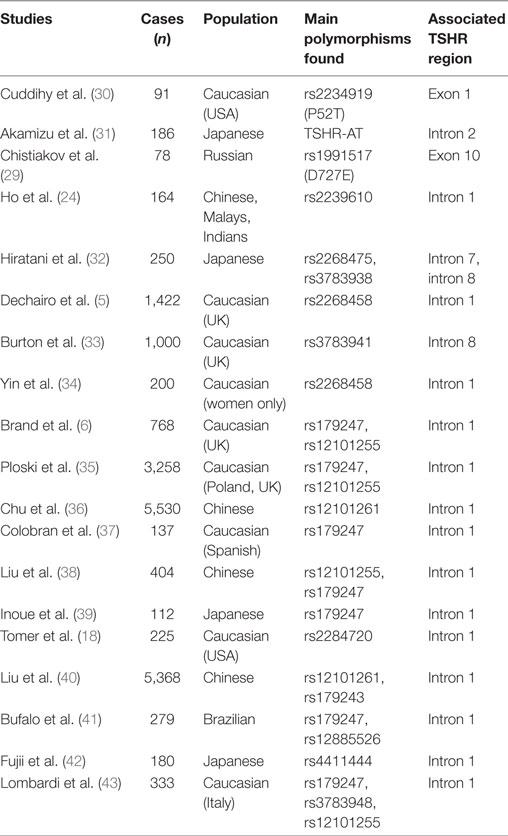
Table 1. Association studies of thyroid-stimulating hormone receptor (TSHR) gene with Graves’ disease risk.
In 2005, Dechairo et al. analyzed 40 SNPs mapping to a 600-kb sequence encompassing the TSHR gene in a Caucasian cohort of 1,056 AITD patients and 971 controls. They identified a haplotype associated with GD (OR = 1.7), but not with autoimmune hypothyroidism, and concluded that TSHR is a GD-specific susceptibility locus (5). Importantly, rs2268458, the SNP showing the strongest association (OR = 1.3) with GD mapped to TSHR intron 1; the association was confirmed in a large UK Caucasian cohort (5). The same year, an independent case–control study conducted in 400 patients with AITD and 238 controls of Japanese descent found several adjacent SNPs in TSHR intron 7 significantly associated with GD, but not with autoimmune hypothyroidism, suggesting that polymorphisms in the TSHR intron 7 could contribute to GD susceptibility (32). However, subsequent association studies conducted over several years in Caucasian populations could not replicate the association of TSHR intron 7 SNPs with GD (6, 35, 41).
In 2009, Brand et al. interrogated a panel of 98 SNPs spanning an 800 kb region of the TSHR gene in a cohort of 768 GD subjects and 768 controls (6). The SNPs showing the strongest association with GD, rs179247 (OR = 1.53), and rs12101255 (OR = 1.55) were located in TSHR intron 1 (6). The association of the TSHR intron 1 SNPs with GD was validated by further studies of several Caucasian (34, 35) and Brazilian (41) populations. In 2011, a large GWAS conducted by the China Consortium of the Genetics of Autoimmune Thyroid Disease in 1,536 individuals with GD and 1,516 controls confirmed TSHR as a primary susceptibility locus for GD by finding a robust association (OR = 1.35) of an intron 1 SNP (rs12101261) with the disease (36). Two years later, the same group conducted a fine-mapping study that established the association of two independent TSHR intron 1 susceptibility variants, rs1201261 and rs179243 in a large Chinese Han population (40). Noticeable, rs1201261 and rs179243 are in tight linkage disequilibrium (LD) with rs12101255 (r2 = 1.0) and rs2268458 (r2 = 0.91), respectively, which were found associated with GD in Caucasian populations of European descent (6).
Recently, three meta-analysis studies intended to refine the effects of rs179247 and rs12101255 SNPs on GD susceptibility concluded that there is a significant association between these SNPs in TSHR intron 1 and GD (44–46).
Collectively, the association studies conducted during the last decade provided compelling evidence and established TSHR as a GD-specific susceptibility locus. Furthermore, fine-mapping studies pointed to a unique susceptibility region located in TSHR intron 1, where at least five GD-associated SNPs were mapped: rs179247, rs2284720, rs12101255, rs12101261, and rs2268458 (Figure 1A).
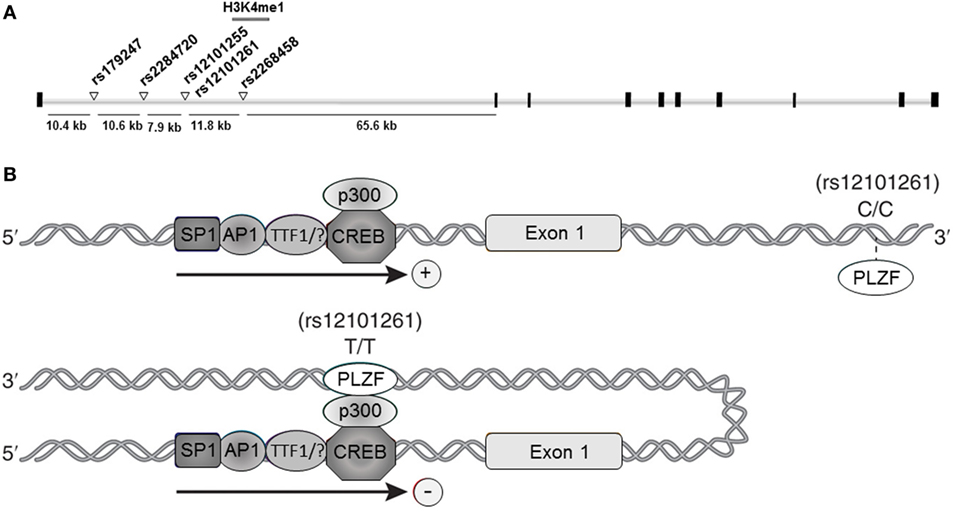
Figure 1. Thyroid-stimulating hormone receptor (TSHR) intron 1 Graves’ disease (GD)-associated DNA variants. (A) Schematic representation of the TSHR gene and the intron 1 GD-associated single nucleotide polymorphisms (SNPs). Black squares represent exons; white bars represent introns; white triangles in intron 1 represent the SNPs associated with GD susceptibility; gray bar overlapping the rs12101255 and rs12101261 represent the region characterized by H3K4me1 enrichment (47). (B) Proposed model for rs12101261 allele-dependent regulation of TSHR transcription. Upper panel: the presence of the disease-protective genotype (C/C) at the rs12101261 site prevents strong interactions with promyelocytic leukemia zinc finger protein (PLZF), allowing TSHR transcription; lower panel: the presence of the disease-associated genotype (T/T) facilitates binding of PLZF, triggering chromatin folding and interaction with TSHR promoter and transcriptional machinery.
Functional Relevance of TSHR Polymorphisms
The discovery of the TSHR GD-associated SNPs within a non-protein coding gene region raised questions about their potential effect on gene function, as well as their impact on disease pathology. Since DNA variants located in intronic or intergenic sites can impact different layers of gene regulation, identification of their functional role is often difficult. For example, by modifying the DNA sequence, non-coding SNPs can affect transcriptional factors’ (TFs) binding and thus modulate gene transcription; they can also impact RNA splicing and stability as well as posttranslation events (48). In addition, DNA variants can modulate, directly or indirectly, epigenetic marks such as DNA methylation, histone modifications, and microRNAs activity (48, 49). Recently, it has been shown that differential binding of TFs at sites harboring DNA variants triggers specific histone modifications, which can modify gene expression and determine the phenotype (50–52). In the case of disease-associated SNPs, such genetic–epigenetic interactions can increase the risk or even trigger disease in certain individuals.
To date, studies aimed to unveil the mechanistic role of TSHR intron 1 variants in gene function and thyroid autoimmunity pointed to two distinct mechanisms. The first proposed mechanism supports a role of the disease-associated SNPs in defective peripheral tolerance; the second mechanism supports the concept that the disease-associated intron 1 SNPs cause reduced intrathymic TSHR expression, leading to decreased central tolerance and increased risk of autoimmunity to TSHR.
Supportive of the first mechanism, Brand et al. proposed that the TSHR intron 1 GD-associated SNPs regulate mRNA spicing, resulting in increased levels of variants encoding a more auto-antigenic TSHR A-subunit (6). The authors measured the levels of full length TSHR (flTSHR) mRNA and of two TSHR truncated transcripts named ST4 and ST5 in thyroid tissues of 12 individuals and showed that the disease-risk alleles of 2 intron 1 SNPs (rs179247 and rs12101255) associate with increased ST4 and ST5 and with decreased flTSHR levels. They suggested that the truncated ST4 and ST5 variants could be translated into TSHR extracellular A-subunit, the main target of TSHR autoantibodies (6). However, the mechanisms by which the two SNPs interact with TSHR mRNA splicing were not addressed, and the authors did not exclude the possibility that other intron 1 SNPs in strong LD could also modulate TSHR transcription (53).
The second mechanism by which TSHR intron 1 variants could trigger thyroid autoimmunity through defective central tolerance was initially proposed by Pujol-Borrell group (37). By measuring TSHR mRNA levels in thymus and correlating them with the genotype of intron 1 SNPs, Colobran et al. found that individuals carrying the disease-protective genotype at the rs179247 site have higher levels of thymic TSHR mRNA than those with the disease-associated genotype (37). These findings, coupled with the fact that negative selection of autoreactive thymocytes is dose dependent (54), support the concept that, by modulating TSHR transcription, intron 1 disease-associated SNPs could modulate negative selection of TSHR-autoreactive T cells in the thymus. Thus, decreased TSHR thymic expression would facilitate the escape of TSHR-reactive T cells from central tolerance in genetically susceptible individuals, increasing the risk for AITD development. The same group showed that TSHR mRNA and protein are expressed in thymocytes from early stages of differentiation but are not detected in extra-thymic T cells. Moreover, thymic TSHR is functional, and TSAbs from GD patients can stimulate thymocytes through this receptor (55). Based on these findings, Pujol-Borrell group suggested that constant stimulation of thymic TSHR by TSAbs can be a potential mechanism explaining thymic hyperplasia, commonly observed in GD (55). Furthermore, the authors proposed that continuous TSAbs stimulation of thymocytes could lead to improved affinity and stimulating capability of TSHR cross-reactive low-affinity antibodies due to the interactions between egressing thymocytes and B-cells in the lymph nodes or the thyroid gland (55, 56). This would result in production of high-affinity TSAbs, the hallmark of GD pathogenesis.
Mechanistic insights into the contribution of TSHR intron 1 SNPs to AITD susceptibility through defective central tolerance were recently revealed by studies from Tomer group (47). Their work originated from the premise that the disease-associated variants can specifically interact, through epigenetic modifications, with environmental factors to trigger disease susceptibility. To reveal the functionality of the GD-associated SNPs, Stefan et al. (47) analyzed genome-wide modifications of histone 3 lysine 4 (K4)-monomethylated (H3K4me1), a chromatin mark often associated with distal enhancer elements (57), induced by interferon alpha (IFNα), a key cytokine secreted during viral infections, previously shown to trigger thyroid autoimmunity (58).
This approach led to identification of an open chromatin region marked by IFNα-induced H3K4me1 enrichment overlapping two adjacent TSHR intron 1 GD-associated SNPs: rs12101255 and rs12101261 (47) (Figure 1A). Functional studies revealed that the region overlapping the rs12101261 site harbors a regulatory element that functions through binding of the transcriptional repressor, promyelocytic leukemia zinc finger protein (PLZF). PLZF binding was shown to be restricted at the disease-associated variant of the rs12101261 site and was correlated with lower thymic TSHR mRNA levels in individuals carrying the disease-predisposing genotype, as compared with individuals carrying the disease-protective genotype. The authors proposed that loss of proper genetic–epigenetic interactions due to microenvironmental influences, such as sustained IFNα production during viral infections, would affect the regulation of TSHR susceptible variants resulting in impaired gene expression. In thymus, the lower TSHR expression triggered by the susceptible genotype would likely facilitate escape from central tolerance and increases the risk of autoimmunity to TSHR.
However, the underlying mechanisms by which the cis-regulatory element at the rs12101261 site modulates TSHR transcription have still to be experimentally addressed. A possible regulatory model involves long-range chromatin interactions determined by the rs12101261 genotype. In such model, the presence of the disease-protective genotype (C/C) would cause a weak interaction of PLZF with chromatin at the rs12101261 site, resulting in active TSHR transcription (Figure 1B, upper panel). The presence of the disease-associated genotype (T/T) would enable strong PLZF binding at the cis-element, triggering formation of chromatin loops and direct interactions between PLZF and TSHR promoter, resulting in inhibition of transcription (Figure 1B, lower panel). Thus, allele-dependent differences in chromatin folding would trigger allele-dependent differences in gene expression. These chromatin interactions, still to be experimentally addressed, are likely cell specific, and different factors (e.g., TFs) and SNPs may control TSHR expression in different tissues.
Concluding Remarks
Studies in the last 15 years established a robust association of TSHR gene with GD, and the disease-associated locus was recently fine-mapped within 40 kb region in intron 1. Moreover, it has become clear that whether TSHR SNPs interfere with gene expression in thymus leading to the escape of TSHR-reactive T cells from central tolerance or defects in peripheral tolerance are involved, these variants are unlikely to act alone, and interactions with epigenetic and environmental factors as well as combinatorial effects should be considered.
Although important advances have been made in our understanding of the role of TSHR polymorphisms in AITD, questions still persist. What are the mechanisms by which TSHR polymorphisms predispose to disease? Which are the environmental factors that unequivocally contribute to disease development and how they interact with susceptible variants? Can genetic variants be translated into markers predicting disease development? Does susceptibility of epigenetic markers to environmental triggers have a role in the functionality of the disease-associated variants? To answer these questions, more work and close collaborations of molecular biologists and clinical scientists as well as more integrated approaches are needed. It is hoped that such knowledge would open the road toward the development of new, targeted, and preventive therapies based on the individual’s particular susceptibility.
Author Contributions
All authors listed have made substantial, direct, and intellectual contribution to the work and approved it for publication.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
The authors thank Dr. Yaron Tomer for reviewing the manuscript, helpful comments, and recommendations.
References
1. McLachlan SM, Rapoport B. Breaking tolerance to thyroid antigens: changing concepts in thyroid autoimmunity. Endocr Rev (2014) 35:59–105. doi:10.1210/er.2013-1055
2. Lee HJ, Li CW, Hammerstad SS, Stefan M, Tomer Y. Immunogenetics of autoimmune thyroid diseases: a comprehensive review. J Autoimmun (2015) 64:82–90. doi:10.1016/j.jaut.2015.07.009
3. Tomer Y, Huber A. The etiology of autoimmune thyroid disease: a story of genes and environment. J Autoimmun (2009) 32:231–9. doi:10.1016/j.jaut.2009.02.007
4. Tomer Y. Mechanisms of autoimmune thyroid diseases: from genetics to epigenetics. Annu Rev Pathol (2014) 9:147–56. doi:10.1146/annurev-pathol-012513-104713
5. Dechairo BM, Zabaneh D, Collins J, Brand O, Dawson GJ, Green AP, et al. Association of the TSHR gene with Graves’ disease: the first disease specific locus. Eur J Hum Genet (2005) 13:1223–30. doi:10.1038/sj.ejhg.5201485
6. Brand OJ, Barrett JC, Simmonds MJ, Newby PR, McCabe CJ, Bruce CK, et al. Association of the thyroid stimulating hormone receptor gene (TSHR) with Graves’ disease. Hum Mol Genet (2009) 18:1704–13. doi:10.1093/hmg/ddp087
7. Lander E, Kruglyak L. Genetic dissection of complex traits: guidelines for interpreting and reporting linkage results. Nat Genet (1995) 11:241–7. doi:10.1038/ng1195-241
8. Allahabadia A, Gough SC. The different approaches to the genetic analysis of autoimmune thyroid disease. J Endocrinol (1999) 163:7–13. doi:10.1677/joe.0.1630007
9. de Roux N, Shields DC, Misrahi M, Ratanachaiyavong S, McGregor AM, Milgrom E. Analysis of the thyrotropin receptor as a candidate gene in familial Graves’ disease. J Clin Endocrinol Metab (1996) 81:3483–6. doi:10.1210/jcem.81.10.8855789
10. Kotsa KD, Watson PF, Weetman AP. No association between a thyrotropin receptor gene polymorphism and Graves’ disease in the female population. Thyroid (1997) 7:31–3. doi:10.1089/thy.1997.7.31
11. Allahabadia A, Heward JM, Mijovic C, Carr-Smith J, Daykin J, Cockram C, et al. Lack of association between polymorphism of the thyrotropin receptor gene and Graves’ disease in United Kingdom and Hong Kong Chinese patients: case control and family-based studies. Thyroid (1998) 8:777–80. doi:10.1089/thy.1998.8.777
12. Sakai K, Shirasawa S, Ishikawa N, Ito K, Tamai H, Kuma K, et al. Identification of susceptibility loci for autoimmune thyroid disease to 5q31-q33 and Hashimoto’s thyroiditis to 8q23-q24 by multipoint affected sib-pair linkage analysis in Japanese. Hum Mol Genet (2001) 10:1379–86. doi:10.1093/hmg/10.13.1379
13. Ban Y, Greenberg DA, Concepcion ES, Tomer Y. A germline single nucleotide polymorphism at the intracellular domain of the human thyrotropin receptor does not have a major effect on the development of Graves’ disease. Thyroid (2002) 12:1079–83. doi:10.1089/105072502321085171
14. Tomer Y, Barbesino G, Keddache M, Greenberg DA, Davies TF. Mapping of a major susceptibility locus for Graves’ disease (GD-1) to chromosome 14q31. J Clin Endocrinol Metab (1997) 82:1645–8. doi:10.1210/jc.82.5.1645
15. Tomer Y, Barbesino G, Greenberg DA, Concepcion E, Davies TF. Linkage analysis of candidate genes in autoimmune thyroid disease. III. Detailed analysis of chromosome 14 localizes Graves’ disease-1 (GD-1) close to multinodular goiter-1 (MNG-1). International Consortium for the Genetics of Autoimmune Thyroid Disease. J Clin Endocrinol Metab (1998) 83:4321–7. doi:10.1210/jc.83.12.4321
16. Tomer Y, Barbesino G, Greenberg DA, Concepcion E, Davies TF. Mapping the major susceptibility loci for familial Graves’ and Hashimoto’s diseases: evidence for genetic heterogeneity and gene interactions. J Clin Endocrinol Metab (1999) 84:4656–64. doi:10.1210/jc.84.12.4656
17. Tomer Y, Ban Y, Concepcion E, Barbesino G, Villanueva R, Greenberg DA, et al. Common and unique susceptibility loci in Graves and Hashimoto diseases: results of whole-genome screening in a data set of 102 multiplex families. Am J Hum Genet (2003) 73:736–47. doi:10.1086/378588
18. Tomer Y, Hasham A, Davies TF, Stefan M, Concepcion E, Keddache M, et al. Fine mapping of loci linked to autoimmune thyroid disease identifies novel susceptibility genes. J Clin Endocrinol Metab (2013) 98:E144–52. doi:10.1210/jc.2012-2408
19. Jacobson EM, Tomer Y. The CD40, CTLA-4, thyroglobulin, TSH receptor, and PTPN22 gene quintet and its contribution to thyroid autoimmunity: back to the future. J Autoimmun (2007) 28:85–98. doi:10.1016/j.jaut.2007.02.006
20. Tonacchera M, Pinchera A. Thyrotropin receptor polymorphisms and thyroid diseases. J Clin Endocrinol Metab (2000) 85:2637–9. doi:10.1210/jcem.85.8.6801
21. Cuddihy RM, Bryant WP, Bahn RS. Normal function in vivo of a homozygotic polymorphism in the human thyrotropin receptor. Thyroid (1995) 5:255–7. doi:10.1089/thy.1995.5.255
22. Watson PF, French A, Pickerill AP, McIntosh RS, Weetman AP. Lack of association between a polymorphism in the coding region of the thyrotropin receptor gene and Graves’ disease. J Clin Endocrinol Metab (1995) 80:1032–5. doi:10.1210/jc.80.3.1032
23. Simanainen J, Kinch A, Westermark K, Winsa B, Bengtsson M, Schuppert F, et al. Analysis of mutations in exon 1 of the human thyrotropin receptor gene: high frequency of the D36H and P52T polymorphic variants. Thyroid (1999) 9:7–11. doi:10.1089/thy.1999.9.7
24. Ho SC, Goh SS, Khoo DH. Association of Graves’ disease with intragenic polymorphism of the thyrotropin receptor gene in a cohort of Singapore patients of multi-ethnic origins. Thyroid (2003) 13:523–8. doi:10.1089/105072503322238773
25. Chistyakov DA, Savost’anov KV, Turakulov RI, Petunina NA, Trukhina LV, Kudinova AV, et al. Complex association analysis of graves disease using a set of polymorphic markers. Mol Genet Metab (2000) 70:214–8. doi:10.1006/mgme.2000.3007
26. Gabriel EM, Bergert ER, Grant CS, van Heerden JA, Thompson GB, Morris JC. Germline polymorphism of codon 727 of human thyroid-stimulating hormone receptor is associated with toxic multinodular goiter. J Clin Endocrinol Metab (1999) 84:3328–35. doi:10.1210/jcem.84.9.5966
27. Bayram B, Sonmez R, Bozari S, Onlu H, Turkoglu Z, Mutlu FS. The association between development and progression of multinodular goiter and thyroid-stimulating hormone receptor gene D727E and P52T polymorphisms. Genet Test Mol Biomarkers (2013) 17:109–14. doi:10.1089/gtmb.2012.0263
28. Muhlberg T, Herrmann K, Joba W, Kirchberger M, Heberling HJ, Heufelder AE. Lack of association of nonautoimmune hyperfunctioning thyroid disorders and a germline polymorphism of codon 727 of the human thyrotropin receptor in a European Caucasian population. J Clin Endocrinol Metab (2000) 85:2640–3. doi:10.1210/jcem.85.8.6704
29. Chistiakov DA, Savost’anov KV, Turakulov RI, Petunina N, Balabolkin MI, Nosikov VV. Further studies of genetic susceptibility to Graves’ disease in a Russian population. Med Sci Monit (2002) 8:CR180–4.
30. Cuddihy RM, Dutton CM, Bahn RS. A polymorphism in the extracellular domain of the thyrotropin receptor is highly associated with autoimmune thyroid disease in females. Thyroid (1995) 5:89–95. doi:10.1089/thy.1995.5.89
31. Akamizu T, Sale MM, Rich SS, Hiratani H, Noh JY, Kanamoto N, et al. Association of autoimmune thyroid disease with microsatellite markers for the thyrotropin receptor gene and CTLA-4 in Japanese patients. Thyroid (2000) 10:851–8. doi:10.1089/thy.2000.10.851
32. Hiratani H, Bowden DW, Ikegami S, Shirasawa S, Shimizu A, Iwatani Y, et al. Multiple SNPs in intron 7 of thyrotropin receptor are associated with Graves’ disease. J Clin Endocrinol Metab (2005) 90:2898–903. doi:10.1210/jc.2004-2148
33. C. Wellcome Trust Case Control, C. Australo-Anglo-American Spondylitis, Burton PR, Clayton DG, Cardon LR, Craddock N, et al. Association scan of 14,500 nonsynonymous SNPs in four diseases identifies autoimmunity variants. Nat Genet (2007) 39:1329–37. doi:10.1038/ng.2007.17
34. Yin X, Latif R, Bahn R, Tomer Y, Davies TF. Influence of the TSH receptor gene on susceptibility to Graves’ disease and Graves’ ophthalmopathy. Thyroid (2008) 18:1201–6. doi:10.1089/thy.2008.0098
35. Ploski R, Brand OJ, Jurecka-Lubieniecka B, Franaszczyk M, Kula D, Krajewski P, et al. Thyroid stimulating hormone receptor (TSHR) intron 1 variants are major risk factors for Graves’ disease in three European Caucasian cohorts. PLoS One (2010) 5:e15512. doi:10.1371/journal.pone.0015512
36. Chu X, Pan CM, Zhao SX, Liang J, Gao GQ, Zhang XM, et al. A genome-wide association study identifies two new risk loci for Graves’ disease. Nat Genet (2011) 43:897–901. doi:10.1038/ng.898
37. Colobran R, Armengol Mdel P, Faner R, Gartner M, Tykocinski LO, Lucas A, et al. Association of an SNP with intrathymic transcription of TSHR and Graves’ disease: a role for defective thymic tolerance. Hum Mol Genet (2011) 20:3415–23. doi:10.1093/hmg/ddr247
38. Liu L, Wu HQ, Wang Q, Zhu YF, Zhang W, Guan LJ, et al. Association between thyroid stimulating hormone receptor gene intron polymorphisms and autoimmune thyroid disease in a Chinese Han population. Endocr J (2012) 59:717–23. doi:10.1507/endocrj.EJ12-0024
39. Inoue N, Watanabe M, Katsumata Y, Hidaka Y, Iwatani Y. Different genotypes of a functional polymorphism of the TSHR gene are associated with the development and severity of Graves’ and Hashimoto’s diseases. Tissue Antigens (2013) 82:288–90. doi:10.1111/tan.12190
40. Liu BL, Yang SY, Liu W, Xue LQ, Chen X, Pan CM, et al. Refined association of TSH receptor susceptibility locus to Graves’ disease in the Chinese Han population. Eur J Endocrinol (2014) 170:109–19. doi:10.1530/EJE-13-0517
41. Bufalo NE, Dos Santos RB, Marcello MA, Piai RP, Secolin R, Romaldini JH, et al. TSHR intronic polymorphisms (rs179247 and rs12885526) and their role in the susceptibility of the Brazilian population to Graves’ disease and Graves’ ophthalmopathy. J Endocrinol Invest (2015) 38:555–61. doi:10.1007/s40618-014-0228-9
42. Fujii A, Inoue N, Watanabe M, Kawakami C, Hidaka Y, Hayashizaki Y, et al. TSHR gene polymorphisms in the enhancer regions are most strongly associated with the development of Graves’ disease, especially intractable disease, and of Hashimoto’s disease. Thyroid (2017) 27:111–9. doi:10.1089/thy.2016.0345
43. Lombardi A, Menconi F, Greenberg D, Concepcion E, Leo M, Rocchi R, et al. Dissecting the genetic susceptibility to Graves’ disease in a cohort of patients of Italian origin. Front Endocrinol (2016) 7:21. doi:10.3389/fendo.2016.00021
44. Qian X, Xu K, Jia W, Lan L, Zheng X, Yang X, et al. Association between TSHR gene polymorphism and the risk of Graves’ disease: a meta-analysis. J Biomed Res (2016) 30:466–75. doi:10.7555/JBR.30.20140144
45. Gong J, Jiang SJ, Wang DK, Dong H, Chen G, Fang K, et al. Association of polymorphisms of rs179247 and rs12101255 in thyroid stimulating hormone receptor intron 1 with an increased risk of Graves’ disease: a meta-analysis. J Huazhong Univ Sci Technolog Med Sci (2016) 36:473–9. doi:10.1007/s11596-016-1611-x
46. Xiong H, Wu M, Yi H, Wang X, Wang Q, Nadirshina S, et al. Genetic associations of the thyroid stimulating hormone receptor gene with Graves diseases and Graves ophthalmopathy: a meta-analysis. Sci Rep (2016) 6:30356. doi:10.1038/srep30356
47. Stefan M, Wei C, Lombardi A, Li CW, Concepcion ES, Inabnet WB III, et al. Genetic-epigenetic dysregulation of thymic TSH receptor gene expression triggers thyroid autoimmunity. Proc Natl Acad Sci U S A (2014) 111:12562–7. doi:10.1073/pnas.1408821111
48. Tak YG, Farnham PJ. Making sense of GWAS: using epigenomics and genome engineering to understand the functional relevance of SNPs in non-coding regions of the human genome. Epigenetics Chromatin (2015) 8:57. doi:10.1186/s13072-015-0050-4
49. Knight JC. Resolving the variable genome and epigenome in human disease. J Intern Med (2012) 271:379–91. doi:10.1111/j.1365-2796.2011.02508.x
50. Furey TS, Sethupathy P. Genetics. Genetics driving epigenetics. Science (2013) 342:705–6. doi:10.1126/science.1246755
51. McVicker G, van de Geijn B, Degner JF, Cain CE, Banovich NE, Raj A, et al. Identification of genetic variants that affect histone modifications in human cells. Science (2013) 342:747–9. doi:10.1126/science.1242429
52. Kilpinen H, Waszak SM, Gschwind AR, Raghav SK, Witwicki RM, Orioli A, et al. Coordinated effects of sequence variation on DNA binding, chromatin structure, and transcription. Science (2013) 342:744–7. doi:10.1126/science.1242463
53. Brand OJ, Gough SC. Genetics of thyroid autoimmunity and the role of the TSHR. Mol Cell Endocrinol (2010) 322:135–43. doi:10.1016/j.mce.2010.01.013
54. Eisenbarth GS. Type 1 diabetes: molecular, cellular and clinical immunology. Adv Exp Med Biol (2004) 552:306–10.
55. Gimenez-Barcons M, Colobran R, Gomez-Pau A, Marin-Sanchez A, Casteras A, Obiols G, et al. Graves’ disease TSHR-stimulating antibodies (TSAbs) induce the activation of immature thymocytes: a clue to the riddle of TSAbs generation? J Immunol (2015) 194:4199–206. doi:10.4049/jimmunol.1500183
56. Pujol-Borrell R, Gimenez-Barcons M, Marin-Sanchez A, Colobran R. Genetics of Graves’ disease: special focus on the role of TSHR gene. Horm Metab Res (2015) 47:753–66. doi:10.1055/s-0035-1559646
57. Harmston N, Lenhard B. Chromatin and epigenetic features of long-range gene regulation. Nucleic Acids Res (2013) 41:7185–99. doi:10.1093/nar/gkt499
Keywords: Graves’ disease, thyroid-stimulating hormone receptor, single nucleotide polymorphisms, GWAS, histone modifications
Citation: Stefan M and Faustino LC (2017) Genetics of Thyroid-Stimulating Hormone Receptor—Relevance for Autoimmune Thyroid Disease. Front. Endocrinol. 8:57. doi: 10.3389/fendo.2017.00057
Received: 25 January 2017; Accepted: 10 March 2017;
Published: 03 April 2017
Edited by:
Rauf Latif, Icahn School of Medicine at Mount Sinai, USAReviewed by:
Giuseppe Barbesino, Partners HealthCare, USATakao Ando, Nagasaki University Hospital, Japan
Copyright: © 2017 Stefan and Faustino. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Mihaela Stefan, bWloYWVsYS5zdGVmYW5AZWluc3RlaW4ueXUuZWR1