 Lewan Parker
Lewan Parker Christopher S. Shaw
Christopher S. Shaw Nigel K. Stepto
Nigel K. Stepto Itamar Levinger
Itamar Levinger- 1Institute of Sport, Exercise and Active Living (ISEAL), College of Sport and Exercise Science, Victoria University, Melbourne, VIC, Australia
- 2Institute for Physical Activity and Nutrition, School of Exercise and Nutrition Sciences, Deakin University, Geelong, VIC, Australia
- 3Monash Centre for Health Research and Implementation, School of Public Health and Preventative Medicine, Monash University, Clayton, VIC, Australia
- 4Australian Institute for Musculoskeletal Science (AIMSS), Victoria University and Western Health, St. Albans, VIC, Australia
Physical inactivity, excess energy consumption, and obesity are associated with elevated systemic oxidative stress and the sustained activation of redox-sensitive stress-activated protein kinase (SAPK) and mitogen-activated protein kinase signaling pathways. Sustained SAPK activation leads to aberrant insulin signaling, impaired glycemic control, and the development and progression of cardiometabolic disease. Paradoxically, acute exercise transiently increases oxidative stress and SAPK signaling, yet postexercise glycemic control and skeletal muscle function are enhanced. Furthermore, regular exercise leads to the upregulation of antioxidant defense, which likely assists in the mitigation of chronic oxidative stress-associated disease. In this review, we explore the complex spatiotemporal interplay between exercise, oxidative stress, and glycemic control, and highlight exercise-induced reactive oxygen species and redox-sensitive protein signaling as important regulators of glucose homeostasis.
Introduction
Physical inactivity and excess adipose tissue are associated with the development of insulin resistance and type 2 diabetes mellitus (T2DM), which has reached epidemic proportions (1). Regular exercise can assist in the prevention and management of metabolic disease (2). Even a single session of exercise can improve glycemic control for up to 48 h postexercise (3–5). Improved glycemic control following acute and regular exercise occurs in part through improved insulin action and substrate metabolism in skeletal muscle (6, 7) by mechanisms that remain largely unknown. One potential mechanism may involve reactive oxygen species (ROS) and their paradoxical dual role in the pathophysiology of glucose homeostasis (8, 9). Considering that acute and chronic exercise training lead to alterations in oxidation–reduction (redox) homeostasis (10, 11), it is not surprising that redox biology has been proposed as a possible modulator of glycemic control and skeletal muscle adaptation to exercise (12–14). This review explores current evidence supporting exercise-induced ROS and skeletal muscle redox-sensitive protein signaling as important regulators of glucose homeostasis.
Exercise and Glycemic Control
Insulin-Stimulated Glucose Uptake
Glucose homeostasis is vital for organism survival and involves the complex interaction between intestinal glucose absorption, liver gluconeogenesis and glycogenolysis, and tissue glucose uptake (15). During conditions of elevated substrate availability, for example, a glucose load from a meal, elevated blood glucose is sensed by pancreatic β-cells resulting in the secretion of insulin to maintain glucose homeostasis (15). Under normal physiological conditions, insulin binds to the extracellular α-subunit of the insulin receptor promoting autophosphorylation of the transmembrane β-subunit on tyrosine residues 1158, 1162, and 1163 (16). Scaffolding proteins including Shc adapter protein isoforms, signal-regulatory protein family members, Gab-1, Cbl, adapter protein with a PH and SH2 domain, and insulin receptor substrates (IRS) are bound, and tyrosine residues phosphorylated to promote subsequent binding to phosphatidylinositol-3 kinase (PI3K) (17, 18). Activation of PI3K generates phosphatidylinositol (3,4,5)-trisphosphate (PIP3) that docks to and subsequently induces membrane translocation of the serine/threonine kinase Akt. PIP3 activation of phosphoinositide-dependent kinase-1 (PDK1) and the Rictor/mTOR complex 2 lead to dual phosphorylation of Akt on serine 473 and threonine residue 308 promoting subsequent activation of Akt kinase (19, 20). Increased Akt activity elicits phosphorylation of Akt substrate of 160 kDa (AS160; also known as TBC1D4) and TBC1D1 (21), promoting GTP loading and activation of Rab proteins releasing glucose transporter 4 (GLUT4) vesicles from intracellular compartments and promoting GLUT4 plasma membrane docking to facilitate glucose uptake (22–24).
Akt phosphorylation not only promotes GLUT4 translocation but also facilitates glycogen synthesis via inhibitory phosphorylation of glycogen synthase kinase 3 (GSK3) on Ser23 (GSK3α) and Ser9 (GSK3β) (25–27). PIP3 and PDK1 also activate atypical protein kinase C (PKC) isoforms ζ and λ, which are reported to facilitate GLUT4 vesicle trafficking and glucose uptake (28, 29). A summary of the canonical insulin signaling pathway is presented in Figure 1.
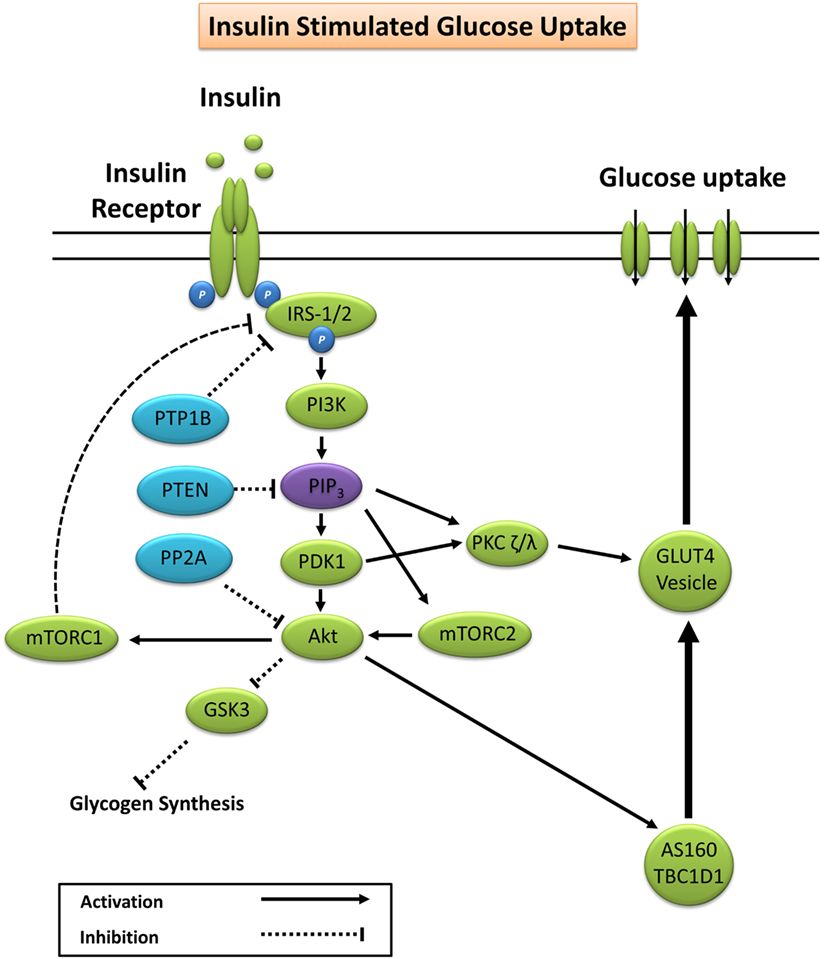
Figure 1. Primary signaling pathways involved in insulin-stimulated glucose uptake. Akt, protein kinase B; AS160, Akt substrate of 160 kDa; GLUT4, glucose transporter 4; GSK3, glycogen synthase kinase 3; IRS-1/2, insulin receptor substrates 1 and 2; mTORC1/2, mechanistic target of rapamycin complex 1/2; PDK1, phosphoinositide-dependent kinase-1; PI3K, phosphatidylinositol-3 kinase; PIP3, phosphatidylinositol (3,4,5)-trisphosphate; PKC, protein kinase C; PP2A, protein phosphatase 2; PTEN, phosphatase and tensin homolog; PTP1B, protein tyrosine phosphatase 1B.
Glucose Uptake during Exercise
Glucose uptake during exercise occurs in an exercise intensity- and exercise duration-dependent manner, which depends largely on a combination of increased glucose delivery, glucose transport, and glucose metabolism (7). Increased trafficking of GLUT4 to the plasma membrane during exercise occurs largely through mechanisms independent of insulin (7). These include the cellular detection of changes in Ca2+ concentration (30, 31), changes in the energy status (ATP) of the cell (32–35), remodeling of the actin cytoskeleton via GTPase Rac1 (36), and fiber type-specific mediation of nitric oxide (NO) synthase (37). The primary protein signaling pathways include contraction-induced activation of calcium (Ca2+)/calmodulin-dependent kinase, atypical PKC, calcineurin, 5′ adenosine monophosphate-activated protein kinase (AMPK), Akt, and mitogen-activated protein kinases (12, 38). Exercise-induced AMPK, and to a lesser extent Ca2+ signaling pathways (30, 31), elicits GLUT4 translocation and subsequent glucose uptake through phosphorylation and inactivation of the convergent glucose uptake signaling proteins AS160 and TBC1D1 (21, 24, 39–42) (Figure 2).
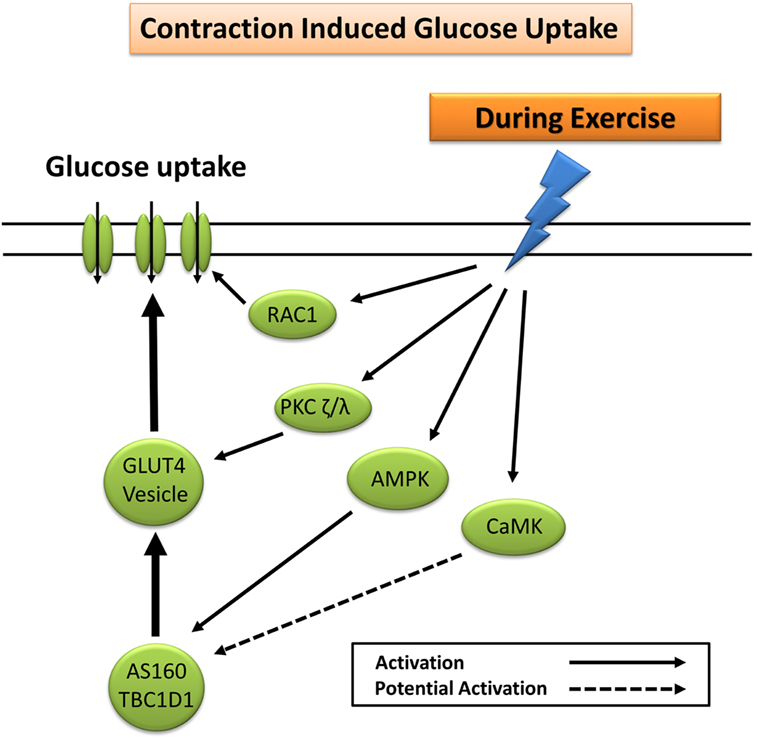
Figure 2. Primary signaling pathways involved in contraction-induced glucose uptake. AMPK, 5′ adenosine monophosphate-activated protein kinase; AS160, Akt substrate of 160 kDa; CaMK, Ca2+/calmodulin-dependent protein kinase; GLUT4, glucose transporter 4; PKC, protein kinase C; RAC1, ras-related C3 botulinum toxin substrate 1.
Postexercise Enhancement of Insulin Sensitivity
Glucose uptake during exercise is maintained in populations who are insulin resistant and/or have been diagnosed with type 2 diabetes (43). In contrast, basal and postexercise insulin-stimulated glucose uptake appears to be impaired and contribute to the development of chronic disease (8, 44, 45). Regular exercise in both healthy and clinical populations improves indices of glycemic control including glycated hemoglobin (HbA1c) and insulin sensitivity in a “dose”-dependent manner (duration and intensity) (2, 46). It is generally conceded that training-induced improvements in glycemic control lead to improved insulin action in part through the upregulation of key skeletal muscle glucose homeostasis regulatory proteins such as Akt1/2, AS160, AMPK, hexokinase 2, and importantly GLUT4 (6, 7). Improved insulin action may also occur through exercise-induced mitochondrial biogenesis and improved mitochondrial function in addition to the upregulation of antioxidant defenses that lead to improved redox homeostasis (6, 13).
In contrast to regular exercise, the transient enhancement of insulin sensitivity in the hours after acute exercise appear to occur independent of modifications to the insulin receptor, IRS1/2, PI3K, Akt, and/or GSK3 α/β proteins (3, 14, 47, 48). It has been reported that AS160 and TBC1D1, which converge downstream of insulin- and contraction-mediated glucose uptake signaling pathways, are associated with the postexercise enhancement of insulin sensitivity (14, 42, 49–53). Although decades of research have contributed to a greater understanding of exercise and glycemic control, the specific exercise-induced signaling mechanisms leading to the acute and long-term adaptations that favor enhanced glycemic control are less clear (3, 7). One potential mechanism may be through exercise-induced ROS and their capacity to act as second messengers for skeletal muscle cell signaling (13, 14, 54, 55).
Redox Homeostasis
Biological organisms are constantly undergoing oxidation–reduction (redox) reactions to maintain a redox environment that is optimal for cellular signaling (56). Under certain circumstances, excess ROS production can lead to oxidative damage and/or modification of lipids, proteins, RNA, and DNA, leading to a redox state that is often referred to as oxidative stress (57). ROS production in a biological system occurs through numerous sources including radiation, environmental pollutants, chemotherapeutics, psychological stress (58), normal and abnormal cellular substrate metabolism (9, 59), and mechanical and physiological stress induced through exercise (9, 11). ROS considered to be of biological importance, which includes hydroxyl radical (OH), superoxide anion , NO, peroxyl radical, peroxynitrite, hypochlorous acid, hydrogen peroxide (H2O2), singlet oxygen, and ozone (57, 60). It should be noted that reactive nitrogen species and reactive sulfur species also constitute separate radical groups with independent biological functions (61, 62); however, their discussion lies beyond the scope of this review.
Reactive oxygen species are capable of direct and/or indirect oxidative modification to proteins (63). Sustained oxidation of proteins can result in disruptions in the normal functioning of the proteome including protein inactivation (64), modification of the protein side chains, fragmentation of peptide bonds (65), and structural unfolding and conformational changes (66). Likewise, ROS are implicated in oxidative damage to DNA, a process that ultimately results in strand breakage, DNA–protein crosslinks and base alterations, and defective DNA transcription and translation leading to the synthesis of less protein and/or defective protein (67–69). In addition to DNA, both messenger and ribosomal RNA are vulnerable to oxidative damage, which can lead to the disturbance of translational process and impairment of protein synthesis (69). ROS-induced damage to mRNA occurs primarily through the formation of highly reactive free radicals such as the OH (70) and appears to be selective and independent of the abundance of the mRNA species (69). Although RNA is highly susceptible to oxidative damage, considerably more so than DNA, protein, and lipids (69), to the authors knowledge, research has yet to investigate the effect of exercise-induced ROS production on RNA damage and the subsequent effects on protein synthesis and exercise adaption.
Lipids, especially polyunsaturated fatty acids, are susceptible to oxidative degradation, a process referred to as lipid peroxidation, which can result in a chain reaction leading to subsequent formation of peroxyl radicals and hydroperoxides (71). In addition to the direct cellular damage caused by ROS-induced lipid peroxidation, secondary products from lipid peroxidation such as malondialdehyde, propanal, hexanal, and the highly toxic 4-hydroxynonenal (4-HNE) can elicit signaling events that contribute to the development of cardiometabolic disease (72–75).
Disturbances in redox homeostasis can lead to perturbed redox signaling and aberrant cellular functioning (56). Therefore, organisms have evolved to encompass a complex and interconnected antioxidant defense system, which helps maintain redox homeostasis through the reduction of ROS and/or ROS intermediates, subsequent termination of ROS-mediated chain reactions, and/or through ROS-induced damage repair mechanisms (60, 76). These defenses include a number of redox-buffering enzymes, proteins, and scavengers, such as superoxide dismutase (SOD), catalase (CAT), glutathione peroxidase (GPx)/reductase, thioredoxin, peroxiredoxin, inducible nitric oxide synthase (iNOS), gamma-glutamylcysteine synthetase, redox effector factor 1, nuclear factor erythroid 2-related factor 2, antioxidant response element, Kelch-like ECH-associated protein 1, uric acid, lipoic acid, bilirubin, coenzyme Q10, vitamin C, vitamin E, and carotenoids (57, 60, 77–82).
Oxidative Stress and Metabolic Health
Chronically elevated systemic oxidative stress is associated with over 100 pathological conditions including accelerated aging, cardiovascular disorders, insulin resistance, and T2DM (9, 57, 83). Considerable research has reported attenuated antioxidant defense and elevated basal oxidative stress in populations with chronic disease, often correlating with classical cardiometabolic risk factors such as increased circulating high-sensitivity C-reactive protein, greater waist-to-hip ratio, total cholesterol, triglycerides, and fasting blood glucose (84–90). As such, the measurement of basal systemic oxidative stress has been proposed as a marker for predicting the onset of a disease, assessing the progression of a disease, and evaluating the effect of pharmacological (e.g., antioxidant supplementation) and non-pharmacological (e.g., diet and exercise) therapies targeting oxidative stress-associated disease (81, 87, 91).
Exercise-Induced Oxidative Stress
Acute exercise elicits a transient state of elevated ROS, which depending on the type of exercise, duration and intensity, and antioxidant capacity of the individual, can result in oxidative stress (11, 87, 92). In contrast to chronic oxidative stress, the transient increase in ROS and oxidative stress elicited by most types of exercise (i.e., non-extreme muscle damaging exercise) are reported to be beneficial and a necessary requirement for optimal cellular functioning and adaptation to physiological stress (79).
Mechanisms for Exercise-Induced Oxidative Stress
The mechanisms of intracellular and extracellular ROS generation in skeletal muscle during exercise are reviewed in detail elsewhere (93–95). In brief, the primary mechanisms are suggested to include NADPH oxidase (96, 97), xanthine oxidase (98), NO synthase (99), and arachidonic acid release from cell membranes by phospholipase A2 (100), whereas mitochondrial electron leak is suggested to contribute only marginally during muscular contraction (101) (Figure 3). Other mechanisms that may contribute to elevated skeletal muscle and/or plasma oxidative stress include the oxidation of catecholamine (102), lactate accumulation (103, 104), elevated core body temperature (105), hemoglobin and myoglobin-mediated autooxidation (106–108), and postexercise inflammatory and phagocytic responses including ischemic reperfusion, cytokine secretion, and respiratory burst (109–111).
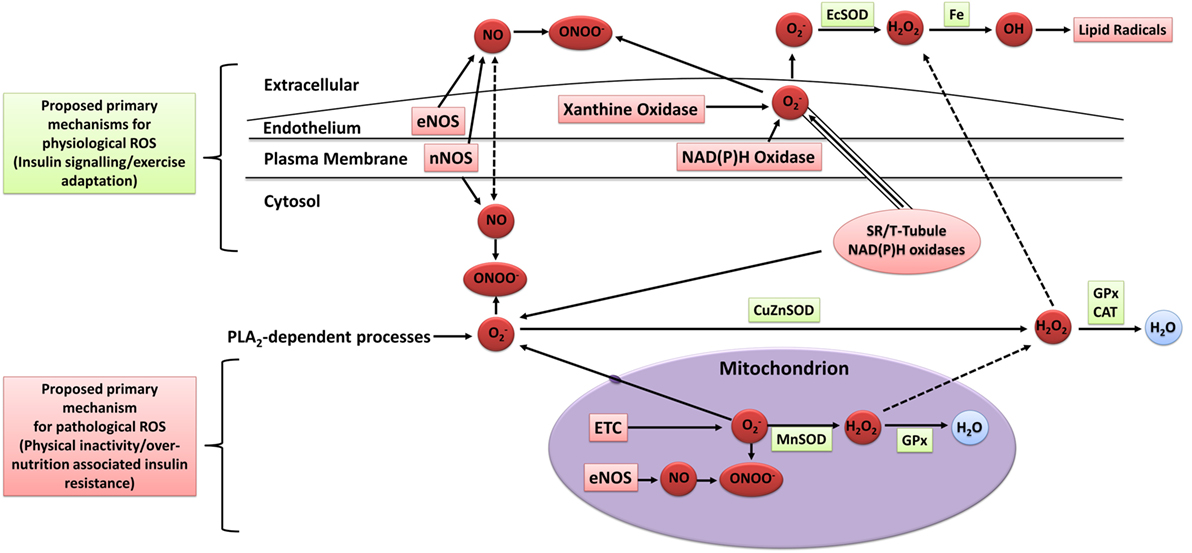
Figure 3. Sources of ROS in skeletal muscle. ETC, electron transport chain; eNOS, endothelial nitric oxide synthase; nNOS, neuronal nitric oxide synthase; NO, nitric oxide; ONOO−, peroxynitrite; OH, hydroxyl radical; , superoxide; H2O2, hydrogen peroxide; H2O, water; EcSOD, extracellular superoxide dismutase; MnSOD, manganese superoxide dismutase; CuZuSOD, copper–zinc superoxide dismutase; GPx, glutathione peroxidase; CAT, catalase; PLA2, phospholipase A2; Fe, iron; ROS, reactive oxygen species. Adapted from the study by Powers and Jackson (93) with permission.
Although plasma oxidative stress is commonly measured as an indicator of exercise-induced oxidative stress, the exact sources of systemic oxidative stress following skeletal muscle contraction are not well understood. Nevertheless, due to the large proportion of body mass that is constituted by skeletal muscle, it is proposed that skeletal muscle fibers, vascular cells, endothelial cells, and/or blood cells residing within skeletal tissue are the main contributors of both the exercise-induced local and systemic oxidative stress (95). Ex vivo skeletal muscle contraction studies have established the potential for skeletal muscle to elicit systemic oxidative stress (95, 112, 113). The specific cell types that contribute to skeletal muscle ROS production likely include vascular smooth muscle cells, endothelial cells, fibroblasts, erythrocytes, and white blood cells, with skeletal muscle fibers suggested to play the biggest role in the generation of extracellular ROS during and after exercise (95, 114, 115). Other tissues such as the heart, liver, and lungs may also contribute to the systemic increase in oxidative stress following acute exercise, but likely to a lesser degree (95).
Exercise-Induced Oxidative Stress and Metabolic Health
To date, the literature is equivocal in regards to the effect of acute exercise on biomarkers of oxidative stress and antioxidant activity (11). Inconsistencies in the literature likely result from variations in dietary intake, training status, exercise intensity (5, 11, 92, 116, 117), exercise duration (11, 118, 119), exercise mode (11, 119), tissues sampled (119), sampling time points (119, 120), as well as the variety and volatility of the biochemical assays used (121). Nevertheless, the general consensus is that acute exercise elicits a transient increase in systemic and localized oxidative stress and antioxidant defense, which, depending on the intensity and mode of exercise, can be detected for up to 4 days after exercise (11, 116, 122).
Excessive ROS production and/or oxidative stress induced through severe or extreme exercise regimes (e.g., ultraendurance events) in humans is associated with cellular disturbances promoting muscular fatigue (94, 123), aberrant upregulation of endogenous antioxidant defenses (124, 125), and impaired cognitive function (126). Similarly, impaired exercise tolerance and physiological responses have been documented in murine animal models (127). For example, Aoi et al. (128) reported that muscle damaging exercise in mice induced through downhill running increased skeletal muscle oxidative stress [thiobarbituric acid reactive substances (TBARS)] and resulted in 4-HNE-mediated impairment of the canonical insulin protein signaling pathway and decreased insulin-stimulated glucose uptake 24 h after exercise. Thus, under certain conditions, exercise-induced oxidative stress has the potential to elicit a deleterious redox environment conducive to impaired exercise capacity and health (Figure 4).
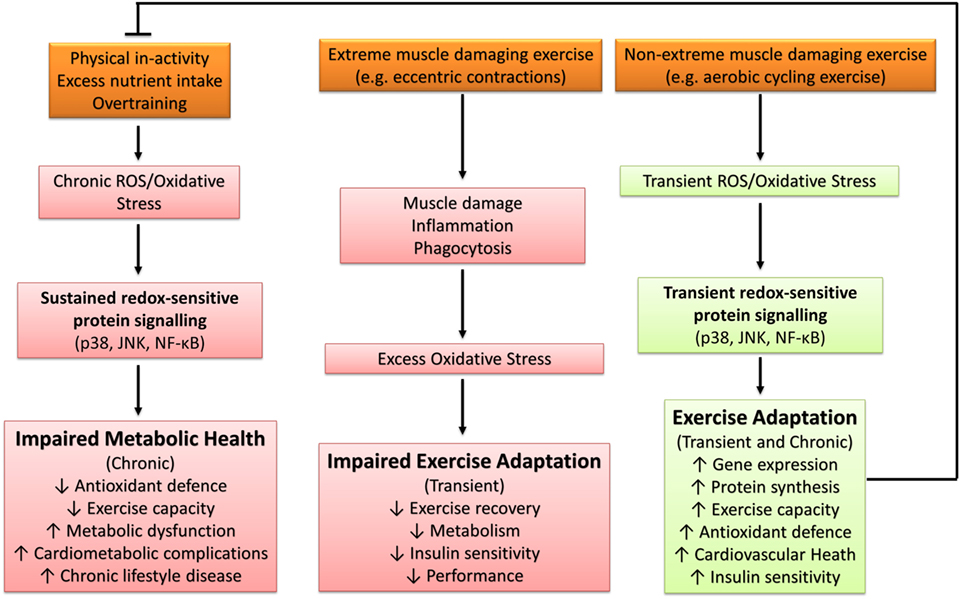
Figure 4. The influence of oxidative stress in health and disease. p38, p38 mitogen-activated protein kinases; JNK, c-Jun N-terminal kinases; NF-κB, nuclear factor kappa-light-chain-enhancer of activated B cells.
The pathological effects of exercise-induced oxidative stress likely stem from secondary muscle damage leading to phagocytic infiltration into skeletal muscle (129) and subsequent generation of ROS (130, 131). In support, Nikolaidis et al. (122) reported that muscle damaging exercise (75 lengthening knee flexions) significantly increased serum oxidative stress (TBARS, oxidized GSH, and protein carbonyls) and serum antioxidant defense (CAT activity, uric acid, bilirubin, and total antioxidant capacity), which lasted for up to 4 days after exercise. When a second identical bout of exercise was performed 3 weeks later, indices of muscle damage were lower, including improved isometric torque, which coincided with attenuation of the postexercise systemic redox response (122). Thus, the acute exercise-induced oxidative stress impairment of exercise performance, recovery, and metabolic health appears to occur independently from the transient and immediate increase in oxidative stress measured during and after exercise and is likely attenuated with subsequent exercise-induced oxidative stress insults (e.g., exercise training).
The majority of literature supports the idea that transient ROS production and/or oxidative stress elicited through regular exercise regimes (e.g., accustomed and/or non-extreme muscle damaging exercise) is beneficial and a necessary requirement for optimal physiological functioning and adaptation to physiological stress (79). Samjoo et al. (132) reported that 12 weeks of endurance training (2–3 sessions per week of 30–60 min cycling at 50–70% VO2peak) in obese and sedentary men decreased basal skeletal muscle and urinary markers of oxidative stress (4-HNE, protein carbonyls, and 8-isoprostane), increased basal skeletal muscle MnSOD protein abundance, and improved indices of glycemic control. Thus, repetitive sessions of exercise-induced ROS (i.e., exercise training) can improve metabolic health through the upregulation of endogenous antioxidant defense and attenuation of basal chronic oxidative stress. Further support for the beneficial effect of exercise-induced ROS can be found in human and animal studies that have reported antioxidant compounds to impair exercise capacity (133, 134), adaptive gene expression and protein synthesis (133, 135–138), upregulation of antioxidant defense (10, 13, 133, 136, 139, 140), cardiovascular health (141, 142), skeletal muscle inflammatory response and repair capabilities (134, 139), and insulin sensitivity (13, 55, 143, 144). Not all studies have reported the blunting of the aforementioned exercise-mediated adaptations (145–149), with some reports indicating enhanced exercise-induced adaptation with antioxidant supplementation (150, 151). An overview of the diverse role of oxidative stress in metabolic health is presented in Figure 4.
Stress-Activated Protein Kinase (SAPK) and Mitogen-Activated Protein Kinase (MAPK) Signaling
Stress-activated protein kinase and MAPK signaling pathways include, but are not limited to, p38 MAPK (p38 MAPK), c-Jun N-terminal kinases (JNK), nuclear factor kappa-light-chain-enhancer of activated B cells (NF-κB), and PKC (152, 153). For the purpose of this review, both MAPK and SAPK are collectively referred to as SAPK.
Stress-activated protein kinase signaling pathways are associated with cellular proliferation, differentiation, survival, and cell death. Uncontrolled or sustained activation of SAPK signaling pathways are associated with the development and progression of cancer, neurodegenerative, and cardiometabolic disease (8, 57, 154). In contrast, controlled and/or transient SAPK activation is required for normal physiological functioning and reported to mediate many of the adaptations and health benefits received from regular exercise (12, 152).
Stress-activated protein kinase pathways are activated through numerous stimuli involving hormones, growth factors, cytokines, agents acting through G protein-coupled receptors, transforming growth factors, pathogens and danger-associated molecular patterns, and physical and chemical stresses (153, 155, 156). Relevant to the current review, however, is the inherent capacity of ROS to both directly and indirectly activate SAPK signaling pathways in skeletal muscle (157–161).
ROS-Induced SAPK Signaling
The direct oxidation of proteins on cysteine residues by ROS act as biological “switches” turning on the catalytic properties of numerous proteins and enzymes (162). Cysteine thiol oxidation produces sulfenic acids, which form irreversible oxidation products or, in many cases, react to form reversible disulfide and sulfenamide bonds. These bonds can later be reduced via enzymes or compounds such as thioredoxin and glutathione, acting as an “off switch” and inhibiting protein function and enzymatic activity (163, 164). ROS-induced SAPK signaling can occur through reversible oxidative modification processes that involve MAPK kinase kinases (MAP3K/MAP2Ks) (165) and oxidative inactivation of thioredoxin (166, 167) and MAPK phosphatases (168–171). In addition, SAPK activation can occur through ROS-induced inactivation of glutathione S-transferases (172), tyrosine phosphorylation of protein kinase D (173), tyrosine, and serine phosphorylation of upstream targets such as the nuclear factor of kappa light polypeptide gene enhancer in β-cells inhibitor alpha (174) and the interaction with growth factor and cytokine receptors (163, 175). Crosstalk also exists between SAPK signaling pathways, with activation of one pathway (e.g., JNK and p38 MAPK) often interacting with and activating other pathways (e.g., NF-κB) (176). Irrespective of the mechanisms, considerable research has reported increased SAPK signaling under conditions of elevated ROS production (135, 157–160).
Exercise-Induced SAPK Signaling
The mechanical and physiological stresses elicited by acute exercise are potent stimuli for the transient activation of SAPK signaling in human skeletal muscle in part through increased ROS production (12). Exercise-induced SAPK signaling activate important skeletal muscle transcription factors and coactivators, which include peroxisome proliferator-activated receptor gamma coactivator 1-alpha (PGC-1α), activating transcription factor 2, myocyte-enhancing factor 2, c-jun, c-fos, p53, and Elk-1 (12, 135, 177–185). Exercise-induced SAPK signaling is also associated with increased gene expression and the upregulation of antioxidant defenses such as SOD, iNOS, gamma-glutamylcysteine synthetase, GPx, and CAT (12, 135, 137, 161, 185–187).
Evidence supporting a role for exercise-induced ROS and SAPK signaling in exercise adaptation is primarily derived from research manipulating the redox environment to attenuate or enhance the exercise-induced ROS and protein signaling response. Henriquez-Olguin et al. (161) reported that inhibition of the ROS-producing enzyme complex NADPH oxidase 2 in rats attenuates the exercise-induced skeletal muscle phosphorylation of p38 MAPK and NF-κB p65 and gene expression of MnSOD, GPx, citrate synthase (CS), and mitochondrial transcription factor A (mtTFA). Similar findings have also been published using ROS inhibitors (e.g., antioxidant supplementation) in animals (10, 135, 188). Strobel et al. (189) reported that increased exercise-induced oxidative stress via skeletal muscle glutathione depletion in rats resulted in greater PGC-1α gene expression. In humans, antioxidant supplementation attenuates exercise-induced activation of p38 MAPK, NF-κB p65 and JNK protein signaling, and gene expression of SOD isoforms in skeletal muscle (10, 134, 137). Chronic inhibition of exercise-induced oxidative stress also impairs the training-induced upregulation of PGC-1α, nuclear respiratory factor (NRF)-1, and mtTFA in rats (135).
It is important to note that not all studies have reported an association between increased redox-sensitive protein kinase signaling and exercise adaptation. Wadley et al. (190) reported similar PGC-1α, NRF-2, and SOD gene expression after exercise in rats with allopurinol treatment, a xanthine oxidase inhibitor, despite decreased p38 MAPK phosphorylation and mtTFA gene expression. In addition, chronic allopurinol treatment was reported to have no effect on the training-induced upregulation of PGC-1α, mtTFA, cytochrome c, CS, and β-hydroxyacyl-CoA dehydrogenase (190). In humans, Morrison et al. (140) reported vitamin C and E supplementation to have little effect on exercise-induced gene expression of PGC-1α, mtTFA, and PGC-related coactivator, or training-induced improvements in VO2peak, CS activity, and expression of cytochrome oxidase subunit 4. However, SOD activity and protein abundance of SOD and mtTFA were attenuated by vitamin C and E supplementation (140). A summary of key findings from research investigating redox manipulation, exercise, and SAPK signaling are summarized in Table 1.
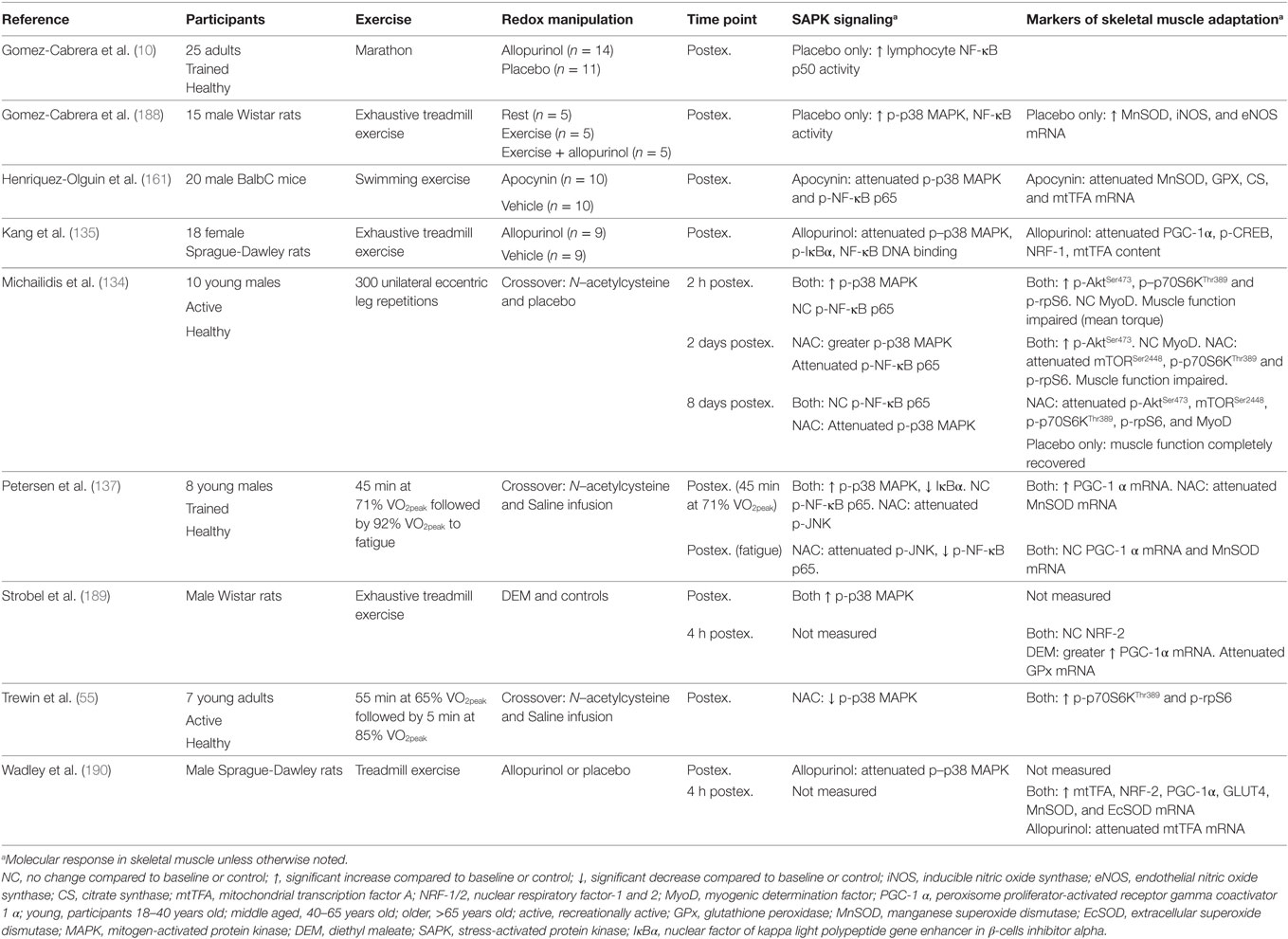
Table 1. Summary of key findings from research investigating the effect of redox state manipulation on acute exercise-induced protein signaling and molecular markers of skeletal muscle adaptation.
The discrepancy in findings are unclear, but likely include interstudy variations in the method and/or compounds used to modulate exercise-induced ROS, variations in the dose and treatment/supplementation time, and the often non-specific and/or ineffective action of antioxidant supplementation/treatment as a model for ROS inhibition (81, 191–195). Nevertheless, evidence provided so far supports a likely association between redox-sensitive SAPK signaling and skeletal muscle adaptation, specifically with that of mitochondrial biogenesis and endogenous antioxidant upregulation, which both participate in the regulation of glycemic control (6, 9).
Positive and Negative Regulation of Glycemic Control by ROS
Physical Inactivity, Excess Nutrient Intake, and Oxidative Stress
Chronic physical inactivity and overnutrition are associated with elevated systemic oxidative stress and the development of lifestyle disease in part through mitochondrial dysfunction (9). Metabolism of carbohydrate and lipids initiates the transfer of electrons from reducing equivalents (i.e., NADH, FADH2) into the mitochondrial electron transport system (ETS) (9). In the absence of energy demand, for example, physical inactivity, increased energy supply results in increased electron flow through the ETS and pumping of protons outside the mitochondrial membrane (9). When the membrane potential exceeds mitochondrial uncoupling capacity, electrons leak through complexes I and III reacting with O2 to form the free radical , where it is catalyzed by MnSOD to H2O2 (196–199). Providing there is sufficient antioxidant activity, H2O2 is further reduced to H2O by antioxidants such as GSH and/or CAT (200). In pathological conditions in which antioxidant defense is insufficient, H2O2 can accumulate in the mitochondrial matrix and intermembrane space or diffuse outside the permeable mitochondrial outer membrane (201). Excess ROS production results in oxidative stress and the signaling events leading to insulin resistance and chronic metabolic disease (59). This proposed mechanism for physical inactivity and excess nutrient intake-induced chronic disease is supported by reports that mitochondrial-specific antioxidants, which attenuate mitochondrial ROS production, reverse high-fat diet-induced insulin resistance in rodents (198).
Elevated basal and/or postprandial hyperglycemia elicited through excess nutrient intake, physical inactivity, and insulin resistance also increases oxidative stress through the formation of advanced glycation end products (AGEs) (202). Activation of the AGE receptor stimulates ROS production through NADPH oxidase (203), opening of the mitochondrial permeability transition pore (204), and through suppression of enzymatic antioxidant defenses (205–207). Therefore, hyperglycemia has the potential to elicit a potentially deleterious redox environment conducive to insulin resistance.
Numerous studies have reported increased biomarkers of systemic oxidative stress in humans for up to 4 h after the ingestion of pure carbohydrate (208, 209), fat, and protein meals (210); mixed macronutrient meals high in fat (211–214) and high in carbohydrate (215); and high-fat liquid meals (216, 217). Larger meals and meals higher in lipid content elicit greater postprandial oxidative stress (218, 219). This has led to many studies researching the effects of high-fat meal ingestion on postprandial oxidative stress (211–214, 220); however, meals adhering to national recommended dietary guidelines also induce systemic postprandial oxidative stress (5).
A single bout of low to moderate-intensity exercise in healthy males can attenuate the postprandial oxidative stress response to a meal ingested 1–2 h before exercise (5, 216) and 24 h after exercise (215), in part through improved glucose and triglyceride processing and clearance and increased antioxidant activity (214). Acute high-intensity exercise may also attenuate postprandial oxidative stress (212, 213); however findings are inconsistent and likely depend on whether exercise is performed before or after meal consumption (5, 214).
The divergent effects of postexercise oxidative stress (physiological) and postprandial oxidative stress (pathological) on metabolic health may stem from the mechanisms of ROS production (59, 79). The pathological effects of oxidative stress are reported to primarily occur through mitochondrial dysfunction and excess mitochondrial ROS production (9), whereas exercise-induced ROS production are reported to primarily occur through alternative mechanisms such as NADPH oxidase and xanthine oxidase (95). Furthermore, the effects of ROS on glycemic control appear to occur on a spatiotemporal paradigm that involve the concentration of ROS (221), the exposure time of ROS (160), the type of ROS, organs and organelles involved (79), the subcellular localization of redox-sensitive protein signaling (160), and the type of exercise and postexercise recovery timepoint (14, 128, 222).
Negative Regulation of Insulin Signaling by ROS
Sustained activation of redox-sensitive SAPK signaling pathways leads to impaired insulin signaling via increased phosphorylation of IRS-1 and IRS-2 on multiple serine and threonine residues, see the study by Copps and White (223) for a detailed review. Sustained IRS-1/2 serine phosphorylation impairs PI3K activity and downstream insulin signaling through attenuated tyrosine phosphorylation and IRS proteasomal degradation and subcellular relocalization (27, 160, 224–232) (Figure 5). The prevention of IRS-1 degradation through the inhibition of ROS and SAPK signaling restores insulin signaling and insulin-stimulated glucose uptake (8, 75, 181, 198, 233). Paradoxically, IRS serine phosphorylation may also be necessary for normal insulin signal transduction and glucose uptake (234). However, reports are contradictory (229, 231, 235) and depend largely on the length and degree of phosphorylation on specific serine residues (236). Previous studies have also reported that hyperinsulinemia initiates a negative feedback loop that inhibits insulin signaling and glucose uptake in part through SAPK-induced IRS serine phosphorylation (229, 231, 237–239).
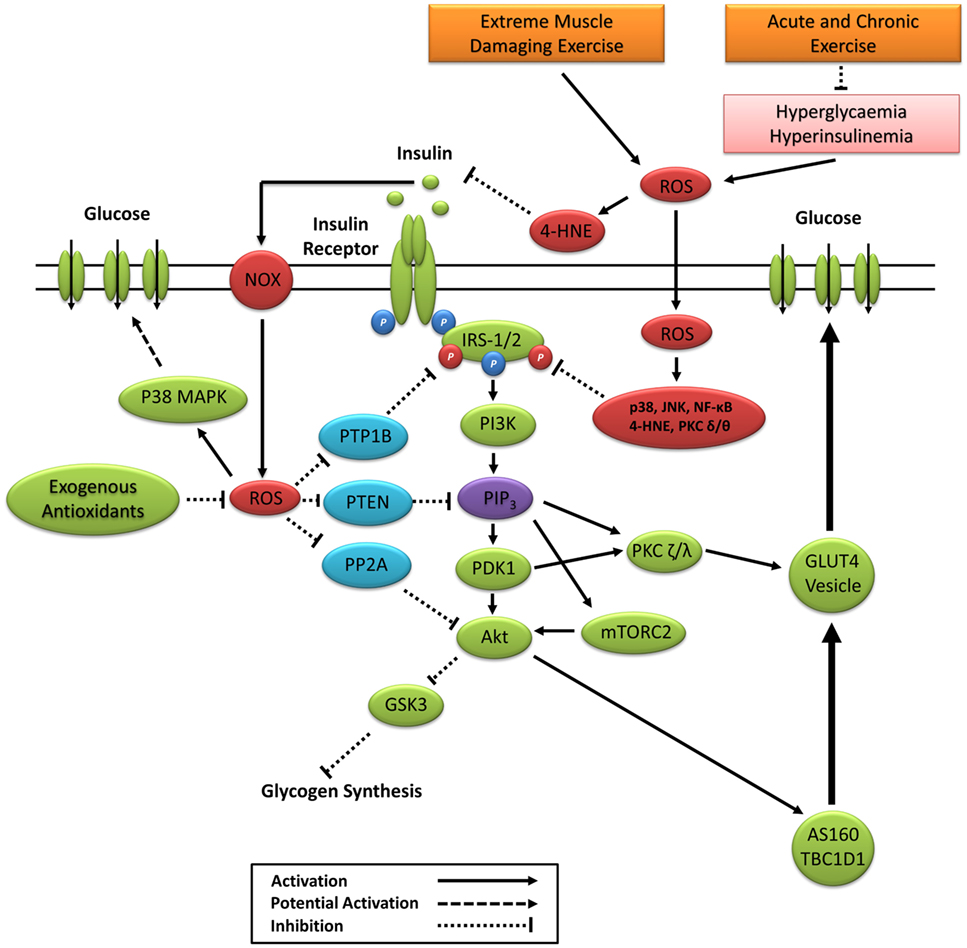
Figure 5. Primary ROS signaling pathways involved in positive and negative regulation of insulin signaling. 4-HNE, 4-hydroxynonenal; Akt, protein kinase B; AS160, Akt substrate of 160 kDa; GSK3, glycogen synthase kinase 3; IRS-1/2, insulin receptor substrates 1 and 2; JNK, c-Jun N-terminal kinases; mTORC2, mechanistic target of rapamycin complex 2; NF-κB, nuclear factor kappa-light-chain-enhancer of activated B cells; NOX, nicotinamide adenine dinucleotide phosphate oxidase; P38 MAPK, p38 mitogen-activated protein kinases; PDK1, phosphoinositide-dependent kinase-1; PI3K, phosphatidylinositol-3 kinase; PIP3, phosphatidylinositol (3,4,5)-trisphosphate; PKC, protein kinase C; PP2A, protein phosphatase 2; PTEN, phosphatase and tensin homolog; PTP1B, protein tyrosine phosphatase 1B; ROS, reactive oxygen species.
Positive Regulation of Insulin Signaling by ROS
The insulin receptor belongs to a subclass of the protein tyrosine kinase family. Positive regulation of the insulin signaling cascade is mediated in part through the oxidative inactivation of protein tyrosine phosphatases (PTP), which include protein tyrosine phosphatase 1B, phosphatase and tensin homolog, and protein phosphatase 2 (Figure 5). Insulin-induced inactivation of PTPs prevents the dephosphorylation of the insulin receptor (240), IRS-1 (241), and Akt proteins (242) and prevents the enzymatic degradation of PIP3 (243). The PTP superfamily signature motif contains an invariantly low-pKa catalytic cysteine residue making it highly susceptible to reversible oxidation by ROS (244). ROS inactivation of PTP activity is associated with numerous cellular processes, including the regulation of cell proliferation, differentiation, survival, metabolism, and motility (244). Under basal conditions, antioxidant defenses such as CAT and peroxiredoxin create a reduced intracellular redox environment prioritizing PTP activity. Increased PTP activity suppresses kinase activity and maintains a dephosphorylated state of the IR, IRS-1, and inhibition of the PI3K/Akt signaling pathway (243, 245). The binding of insulin to the insulin receptor signals a burst of endogenous superoxide production, which is reduced to H2O2 creating a local oxidative redox environment (246–248). This oxidative redox environment favors the oxidation of catalytic cysteine to sulphenic acid, suppressing PTP activity and enhancing kinase activity and propagation of the insulin signaling cascade (9).
Insulin can elicit ROS production through enzymatic activation of NADPH oxidases (246–249). Furthermore, insulin-induced receptor tyrosine phosphorylation inactivates the endogenous membrane-associated antioxidant peroxiredoxin I, allowing for increased ROS production (78). Mahadev et al. (246) reported that NADPH oxidase-induced H2O2 enhances insulin signaling via oxidative inhibition of PTPs. Furthermore, palmitate-induced insulin resistance in rat skeletal muscle occurs through increased activity of PTPs via JNK and NF-κB (250), which is reversed 16 h after acute exercise in rats (222). Loh et al. (54) revealed that the elevated H2O2 response to insulin in GPx1−/− mouse embryo fibroblasts coincided with elevated PI3K/Akt signaling, which can be suppressed by pretreating cells with ebselen, an NADPH oxidase inhibitor, or the antioxidant N-acetylcysteine. Subsequent experiments revealed that elevated H2O2 in GPx1−/− mice increased PI3K/Akt signaling and glucose uptake through decreased PTP activity, which was attenuated by the ingestion of n-acetylcysteine (NAC) (54). Thus, redox-mediated PTP activity appears to be associated with both positive and negative regulations of insulin signaling and glucose uptake.
Exercise-Induced ROS, SAPK Signaling, and Glycemic Control
Reactive oxygen species are readily induced through contraction of skeletal muscle (251–253). Importantly, contraction of skeletal muscle coincides with increased activation of redox-sensitive SAPK signaling pathways implicated in glucose metabolism (14, 160, 161, 254–256). Therefore, skeletal muscle SAPK signaling has emerged as a potential candidate for the postexercise enhancement of insulin sensitivity (14, 54, 182).
Loh et al. (54) reported that exercise-induced ROS in GPx1 knockout mice coincided with increased phosphorylation of Akt(Ser473) and AS160(Thr642) and enhanced insulin-stimulated glucose uptake 60 min after a single session of treadmill exercise. This beneficial effect on insulin sensitivity was reversed with NAC supplementation, suggesting that redox signaling is not only an important regulator of basal insulin signaling and glucose uptake but also postexercise enhancement of insulin sensitivity. Importantly, GPx1 knockout mice showed similar improvements in insulin sensitivity when measured immediately after exercise, supporting a growing consensus that the effects of postexercise-induced ROS on glycemic control are temporal (14, 222).
One of the first studies to indicate a regulatory role of redox signaling in exercise-induced enhancement of insulin sensitivity in humans was conducted by Ristow et al. (13). It was reported that vitamin C and E supplementation in humans attenuated the 4-week training-induced improvements in insulin sensitivity and gene expression of PGC-1α/β, SOD, GPx1, and CAT (13). Not all studies in humans and rodents have reported impaired exercise-induced improvements in insulin protein signaling and insulin sensitivity with antioxidant supplementation (146, 148, 257). Contradictory findings likely stem from variations in the type of antioxidant compound/s used, the dose used, the timing of supplementation, and the often non-specific and/or ineffective action of antioxidant supplementation for ROS inhibition in humans (81, 193, 194).
Enhanced glucose uptake approximately 4.5 h after one-legged knee extensor exercise in humans is reported to coincide with greater basal and insulin-stimulated p38 MAPK phosphorylation (182), highlighting SAPK signaling as a potential moderator of postexercise glucose metabolism. Trewin et al. (55) reported that NAC infusion attenuated whole-body insulin sensitivity approximately 5 h after exercise. Phosphorylation of p38 MAPK was lower immediately after exercise with NAC infusion; however, phosphorylation was not significantly different to baseline or the placebo after insulin stimulation. However, the null findings for p38 MAPK phosphorylation may be due to the timing of postexercise biopsies, the relatively small effect of NAC on insulin sensitivity (~6% reduction), and that NAC infusion was not maintained during the recovery period and subsequent insulin clamp (55). Interestingly, Parker et al. (14) demonstrated that a bout of high-intensity interval exercise prior to a 2-h euglycemic–hyperinsulinemic clamp in obese middle-aged males elicited greater insulin-stimulated p38 MAPK, JNK, NF-κB, and AS160Ser588 phosphorylation, which was associated with improved insulin sensitivity compared to a resting clamp. Equivocal findings in humans may stem from reports that postexercise skeletal muscle SAPK and insulin protein signaling are effected by training status and occur in an exercise intensity and postexercise time course-dependent manner (256, 258).
Berdichevsky et al. (160) reported similar JNK phosphorylation in C2C12 myoblasts and L6 myotubes treated with chronic oxidative stress (1 μM of H2O2 for 48 h) and acute oxidative stress conditions (500 μM of H2O2 for 3 h). Interestingly, chronic oxidative stress decreased insulin-stimulated Akt(Ser473) phosphorylation, whereas acute oxidative stress enhanced insulin-stimulated Akt(Ser473) and GSK3-α/β phosphorylation. Furthermore, acute oxidative stress exposure in insulin-resistant muscle cells rescues insulin-stimulated glucose uptake through increased IRS1 protein abundance; increased phosphorylation of JNK, Akt(Ser473), Akt(Thr308), and GSK3-α/β; and decreased IRS-1(Ser307) phosphorylation (160). In contrast, Ropelle et al. (222) reported that a single bout of exercise in male rats reverses diet-induced insulin resistance 16 h later via attenuation of JNK, NF-κB, and IRS-1(Ser307) signaling. It is possible that acute exercise enhances insulin signal transduction through the transient and immediate increase in ROS and SAPK signaling, which also leads to a delayed increase in antioxidant activity and subsequent attenuation of chronic oxidative stress and sustained SAPK signaling pathways associated with insulin resistance. Certainly, SOD protein content, SOD activity, and total antioxidant status increase and/or remain elevated for up to 16–24 h after exercise (116, 212, 259), whereas lipid-induced postprandial oxidative stress is attenuated (213).
Taken together, previous studies support a potential role for exercise-induced redox-sensitive protein signaling and glycemic control (Table 2); however, specific mechanisms remain to be elucidated (Figure 6).
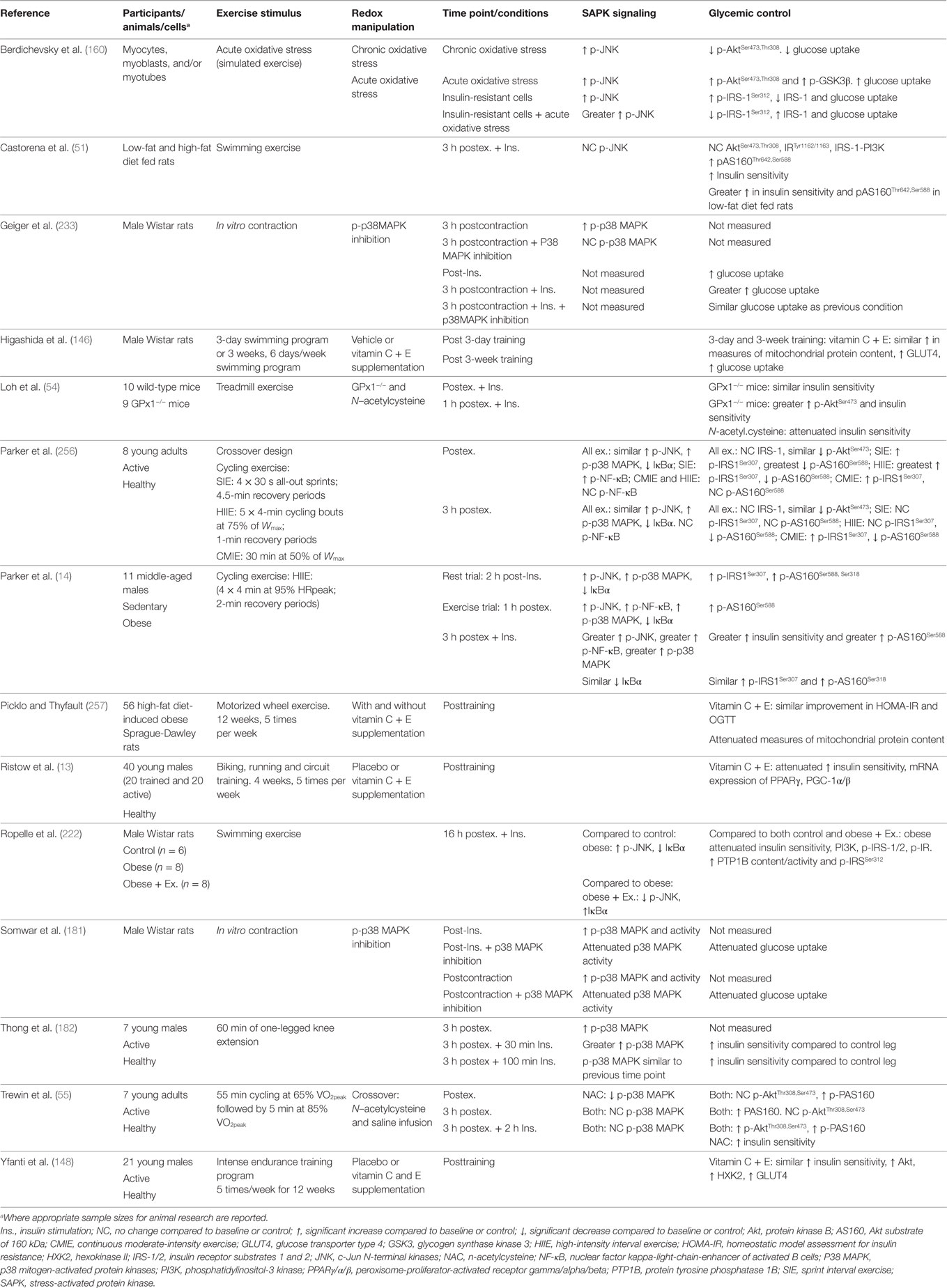
Table 2. Summary of key findings from research investigating exercise, redox state, and enhancement of glycemic control.
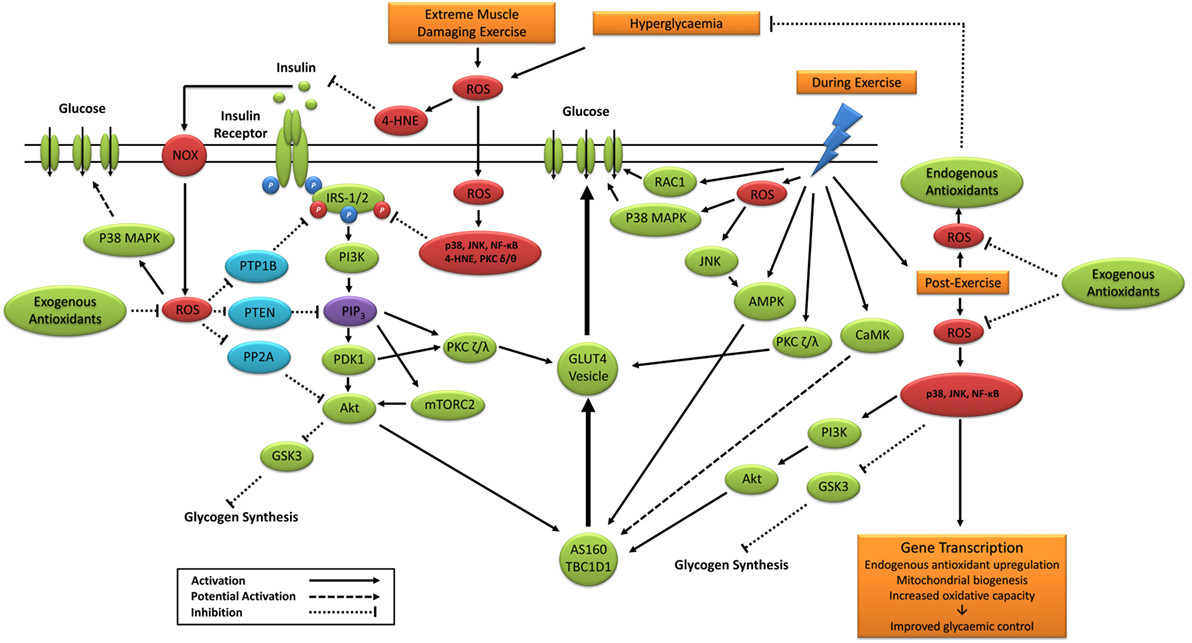
Figure 6. Primary signaling pathways involved in contraction and insulin-stimulated glucose uptake and the potential role of ROS. 4-HNE, 4-hydroxynonenal; Akt, protein kinase B; AMPK, 5′ adenosine monophosphate-activated protein kinase; AS160, Akt substrate of 160 kDa; CaMK, Ca2+/calmodulin-dependent protein kinase; GLUT4, glucose transporter 4; GSK3, glycogen synthase kinase 3; IRS-1/2, insulin receptor substrates 1 and 2; JNK, c-Jun N-terminal kinases; mTORC2, mechanistic target of rapamycin complex 2; NF-κB, nuclear factor kappa-light-chain-enhancer of activated B cells; NOX, nicotinamide adenine dinucleotide phosphate oxidase; P38 MAPK, p38 mitogen-activated protein kinases; PDK1, phosphoinositide-dependent kinase-1; PI3K, phosphatidylinositol-3 kinase; PIP3, phosphatidylinositol (3,4,5)-trisphosphate; PKC, protein kinase C; PP2A, protein phosphatase 2; PTEN, phosphatase and tensin homolog; PTP1B, protein tyrosine phosphatase 1B; RAC1, ras-related C3 botulinum toxin substrate 1; ROS, reactive oxygen species.
Potential Mechanisms Linking SAPK Signaling and Enhancement of Glycemic Control
Modulation of glycogen synthesis by oxidative stress-induced SAPK signaling has been associated with glucose metabolism and regulation (27, 160, 182, 260). Transient stimulation of C2C12 myoblasts with H2O2 increases JNK, Akt, and GSK3α/β phosphorylation (160), suggesting the short exposure to exercise-induced ROS may increase postexercise glycogen synthesis and skeletal muscle glucose uptake. Likewise, postexercise enhancement of insulin-stimulated p38 MAPK phosphorylation is associated with postexercise glycogen depletion (182). Chan et al. (261) established that low intramuscular glycogen was associated with greater phosphorylation of nuclear p38 MAPK after 60 min of cycle exercise. In contrast, insulin stimulation of rat skeletal muscle exposed to 1 h of H2O2 (~90 μM) exhibits impaired insulin protein signaling, glycogen synthesis, and glucose uptake, despite increased p38 MAPK phosphorylation (27). Diamond-Stanic et al. (260) reported similar findings and proposed that p38 MAPK and GSK3 were unlikely to play a beneficial role in insulin-stimulated glucose uptake. Activation of JNK in skeletal muscle of mice is also associated with increased insulin-stimulated glycogen synthesis via the RSK3/GSK3 signaling pathway (262); however, greater postexercise JNK phosphorylation and insulin sensitivity in human skeletal muscle do not coincide with greater insulin-stimulated phosphorylation of GSK3 α/β (14).
Other potential pathways include JNK-, NF-κB-, and p38 MAPK-stimulated secretion of the recently identified insulin-sensitizing interleukin-6 (IL-6) (255, 263–265). Carey et al. (264) reported that IL-6 infusion increases insulin-stimulated glucose uptake in humans. Furthermore, IL-6 treatment in L6 myotubes coincides with increased glucose uptake and GLUT4 translocation, likely through AMPK pathways independent of the canonical insulin signaling cascade (264). Importantly, IL-6 secretion is increased following muscular contraction, likely via activation of JNK, NF-κB, and/or p38MAPK (261, 266, 267). It has also been reported that p38 MAPK inhibiters, alongside expression of a dominant-negative p38 mutant, impairs insulin-stimulated glucose uptake without reductions in GLUT4 translocation (254). Researchers concluded that p38 MAPK may exert its insulin-sensitizing effect through increased activation of translocated GLUT4 (254), but not all findings are supportive (268) and have yet to be investigated in humans. The reported subcellular redistribution of phosphorylated JNK from the cytoplasm to the nucleus with acute hydrogen peroxide exposure in skeletal muscle cells highlights another potential mechanism for the postexercise insulin sensitizing effect of JNK (160). Future research is warranted to explore the subcellular localization and activation of SAPK proteins after exercise and insulin stimulation in humans.
The Future of Exercise-Induced Oxidative Stress, ROS, and Redox Signaling
Early studies, and the majority of current findings, rely primarily on associations and the assumption that increased/decreased ROS and/or markers of oxidative stress are reflective of, or are likely to lead to, increased/decreased redox signaling (91). Certainly, studies inhibiting or increasing ROS have been useful for establishing a relationship between ROS and certain biological outcomes such as glycemic control and exercise adaption (13, 14, 55, 135, 161). However, in the absence of specific redox signaling measurements such as protein cysteine oxidation or S-nitrosylation (162, 269), research studies are limited in their capacity to elucidate specific redox cellular signaling networks that are complex, compartmentalized, and spatiotemporally regulated (195). Future studies utilizing modern redox proteomics are required to establish the reversible and, in some cases, irreversible, redox regulation of kinases, phosphatases, transcription factors, and coactivators, thus establishing the “true” redox signaling role of exercise-induced ROS (195, 270–274). Furthermore, not all ROS are equal in their capacity to exert signaling effects (56). Future studies investigating exercise-induced oxidative stress should therefore strive to identify the specific ROS involved, which can be achieved through the use of robust techniques such as electron spin resonance, targeted fluorescent probes, and mass spectrometry (252, 275–278).
Despite their non-specificity and/or inability to adequately reflect redox signaling, the measurement of ROS, oxidative stress, and/or antioxidant activity in a biological sample provides insight into the effects of an intervention (e.g., exercise) on redox homeostasis and remains a useful biomarker of overall health and disease (91). As such, a combination of both traditional measures of redox biomarkers, the direct measurement of ROS, redox-sensitive protein signaling, and specific redox proteomics will likely provide a robust investigation of exercise-induced ROS and subsequent redox signaling.
Conclusion
Physical inactivity, excess energy consumption, and obesity are associated with elevated ROS production, systemic oxidative stress, and sustained activation of redox-sensitive protein signaling pathways. If left unchecked, this chronic state of physiological stress can lead to insulin resistance, which likely contributes toward the development of cardiometabolic disease. Paradoxically, a single session of exercise transiently increases ROS, oxidative stress, and redox-sensitive protein signaling, yet both acute and regular exercises elicit favorable improvements in glycemic control and skeletal muscle adaptation. It appears that exercise-induced redox-sensitive protein signaling is necessary for adaptation to physiological stress. However, the spatiotemporal interplay between physical activity/inactivity, ROS, PTP activity, SAPK and MAPK signaling, insulin protein signaling, and the subsequent effects on glycemic control and cardiometabolic health remain unclear. Future research would benefit by employing a combination of human primary cell culture, animal research, modern proteomics, and immunohistochemistry/subcellular analysis of human tissue to elucidate the physiological relevance of transient oxidative stress (exercise induced), chronic oxidative stress (physical inactivity/excess nutrition intake), and the role of redox-sensitive protein signaling in human health and disease.
Author Contributions
LP, CS, NS, and IL contributed to the conceptualization and overall design of the manuscript. LP drafted the initial version of the manuscript and figures. CS, NS, and IL critically revised the manuscript. All authors approved the final version of the manuscript.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1. Ng M, Fleming T, Robinson M, Thomson B, Graetz N, Margono C, et al. Global, regional, and national prevalence of overweight and obesity in children and adults during 1980-2013: a systematic analysis for the global burden of disease study 2013. Lancet (2014) 384:766–81. doi: 10.1016/S0140-6736(14)60460-8
2. Liubaoerjijin Y, Terada T, Fletcher K, Boule NG. Effect of aerobic exercise intensity on glycemic control in type 2 diabetes: a meta-analysis of head-to-head randomized trials. Acta Diabetol (2016) 53:769–81. doi:10.1007/s00592-016-0870-0
3. Frosig C, Richter EA. Improved insulin sensitivity after exercise: focus on insulin signaling. Obesity (Silver Spring) (2009) 17(Suppl 3):S15–20. doi:10.1038/oby.2009.383
4. Levinger I, Jerums G, Stepto NK, Parker L, Serpiello FR, Mcconell GK, et al. The effect of acute exercise on undercarboxylated osteocalcin and insulin sensitivity in obese men. J Bone Miner Res (2014) 29:2571–6. doi:10.1002/jbmr.2285
5. Parker L, Shaw CS, Banting L, Levinger I, Hill KM, Mcainch AJ, et al. Acute low-volume high-intensity interval exercise and continuous moderate-intensity exercise elicit a similar improvement in 24-h glycemic control in overweight and obese adults. Front Physiol (2016) 7:661. doi:10.3389/fphys.2016.00661
6. Hawley JA, Lessard SJ. Exercise training-induced improvements in insulin action. Acta Physiol (2008) 192:127–35. doi:10.1111/j.1748-1716.2007.01783.x
7. Richter EA, Hargreaves M. Exercise, GLUT4, and skeletal muscle glucose uptake. Physiol Rev (2013) 93:993–1017. doi:10.1152/physrev.00038.2012
8. Tiganis T. Reactive oxygen species and insulin resistance: the good, the bad and the ugly. Trends Pharmacol Sci (2011) 32:82–9. doi:10.1016/j.tips.2010.11.006
9. Fisher-Wellman KH, Neufer PD. Linking mitochondrial bioenergetics to insulin resistance via redox biology. Trends Endocrinol Metab (2012) 23:142–53. doi:10.1016/j.tem.2011.12.008
10. Gomez-Cabrera MC, Domenech E, Ji LL, Vina J. Exercise as an antioxidant: it up-regulates important enzymes for cell adaptations to exercise. Sci Sports (2006) 21:85–9. doi:10.1016/j.scispo.2005.06.012
11. Fisher-Wellman K, Bloomer RJ. Acute exercise and oxidative stress: a 30 year history. Dyn Med (2009) 8:1. doi:10.1186/1476-5918-8-1
12. Kramer HF, Goodyear LJ. Exercise, MAPK, and NF-kappaB signaling in skeletal muscle. J Appl Physiol (1985) (2007) 103:388–95. doi:10.1152/japplphysiol.00085.2007
13. Ristow M, Zarse K, Oberbach A, Kloting N, Birringer M, Kiehntopf M, et al. Antioxidants prevent health-promoting effects of physical exercise in humans. Proc Natl Acad Sci U S A (2009) 106:8665–70. doi:10.1073/pnas.0903485106
14. Parker L, Stepto NK, Shaw CS, Serpiello FR, Anderson M, Hare DL, et al. Acute high-intensity interval exercise-induced redox signaling is associated with enhanced insulin sensitivity in obese middle-aged men. Front Physiol (2016) 7:411. doi:10.3389/fphys.2016.00411
15. Fu Z, Gilbert ER, Liu D. Regulation of insulin synthesis and secretion and pancreatic beta-cell dysfunction in diabetes. Curr Diabetes Rev (2013) 9:25–53. doi:10.2174/157339913804143225
16. White MF. Insulin signaling in health and disease. Science (2003) 302:1710–1. doi:10.1126/science.1092952
17. Pessin JE, Saltiel AR. Signaling pathways in insulin action: molecular targets of insulin resistance. J Clin Invest (2000) 106:165–9. doi:10.1172/JCI10582
18. White MF. IRS proteins and the common path to diabetes. Am J Physiol Endocrinol Metab (2002) 283:E413–22. doi:10.1152/ajpendo.00514.2001
19. Sarbassov DD, Guertin DA, Ali SM, Sabatini DM. Phosphorylation and regulation of Akt/PKB by the rictor-mtor complex. Science (2005) 307:1098–101. doi:10.1126/science.1106148
20. Asano T, Fujishiro M, Kushiyama A, Nakatsu Y, Yoneda M, Kamata H, et al. Role of phosphatidylinositol 3-kinase activation on insulin action and its alteration in diabetic conditions. Biol Pharm Bull (2007) 30:1610–6. doi:10.1248/bpb.30.1610
21. Taylor EB, An D, Kramer HF, Yu H, Fujii NL, Roeckl KS, et al. Discovery of TBC1D1 as an insulin-, AICAR-, and contraction-stimulated signaling nexus in mouse skeletal muscle. J Biol Chem (2008) 283:9787–96. doi:10.1074/jbc.M708839200
22. Sano H, Kane S, Sano E, Miinea CP, Asara JM, Lane WS, et al. Insulin-stimulated phosphorylation of a Rab GTPase-activating protein regulates glut4 translocation. J Biol Chem (2003) 278:14599–602. doi:10.1074/jbc.C300063200
23. Eguez L, Lee A, Chavez JA, Miinea CP, Kane S, Lienhard GE, et al. Full intracellular retention of GLUT4 requires AS160 RAB GTPase activating protein. Cell Metab (2005) 2:263–72. doi:10.1016/j.cmet.2005.09.005
24. Cartee GD. Roles of TBC1D1 and TBC1D4 in insulin- and exercise-stimulated glucose transport of skeletal muscle. Diabetologia (2015) 58:19–30. doi:10.1007/s00125-014-3395-5
25. Ueki K, Yamamoto-Honda R, Kaburagi Y, Yamauchi T, Tobe K, Burgering BM, et al. Potential role of protein kinase B in insulin-induced glucose transport, glycogen synthesis, and protein synthesis. J Biol Chem (1998) 273:5315–22. doi:10.1074/jbc.273.9.5315
26. Takata M, Ogawa W, Kitamura T, Hino Y, Kuroda S, Kotani K, et al. Requirement for Akt (protein kinase B) in insulin-induced activation of glycogen synthase and phosphorylation of 4E-BP1 (PHAS-1). J Biol Chem (1999) 274:20611–8. doi:10.1074/jbc.274.29.20611
27. Dokken BB, Saengsirisuwan V, Kim JS, Teachey MK, Henriksen EJ. Oxidative stress-induced insulin resistance in rat skeletal muscle: role of glycogen synthase kinase-3. Am J Physiol Endocrinol Metab (2008) 294:E615–21. doi:10.1152/ajpendo.00578.2007
28. Standaert ML, Bandyopadhyay G, Perez L, Price D, Galloway L, Poklepovic A, et al. Insulin activates protein kinases C-zeta and C-lambda by an autophosphorylation-dependent mechanism and stimulates their translocation to GLUT4 vesicles and other membrane fractions in rat adipocytes. J Biol Chem (1999) 274:25308–16. doi:10.1074/jbc.274.36.25308
29. Frosig C, Sajan MP, Maarbjerg SJ, Brandt N, Roepstorff C, Wojtaszewski JF, et al. Exercise improves phosphatidylinositol-3,4,5-trisphosphate responsiveness of atypical protein kinase C and interacts with insulin signalling to peptide elongation in human skeletal muscle. J Physiol (2007) 582:1289–301. doi:10.1113/jphysiol.2007.136614
30. Jensen TE, Rose AJ, Jorgensen SB, Brandt N, Schjerling P, Wojtaszewski JFP, et al. Possible CaMKK-dependent regulation of AMPK phosphorylation and glucose uptake at the onset of mild tetanic skeletal muscle contraction. Am J Physiol Endocrinol Metab (2007) 292:E1308–17. doi:10.1152/ajpendo.00456.2006
31. Witczak CA, Jessen N, Warro DM, Toyoda T, Fujii N, Anderson ME, et al. Camkii regulates contraction- but not insulin-induced glucose uptake in mouse skeletal muscle. Am J Physiol Endocrinol Metab (2010) 298:E1150–60. doi:10.1152/ajpendo.00659.2009
32. Broberg S, Sahlin K. Adenine nucleotide degradation in human skeletal muscle during prolonged exercise. J Appl Physiol (1985) (1989) 67:116–22.
33. Stein SC, Woods A, Jones NA, Davison MD, Carling D. The regulation of AMP-activated protein kinase by phosphorylation. Biochem J (2000) 345(Pt 3):437–43. doi:10.1042/bj3450437
34. Hardie DG, Scott JW, Pan DA, Hudson ER. Management of cellular energy by the AMP-activated protein kinase system. FEBS Lett (2003) 546:113–20. doi:10.1016/S0014-5793(03)00560-X
35. Birk JB, Wojtaszewski JFP. Predominant α2/β2/γ3 AMPK activation during exercise in human skeletal muscle. J Physiol (2006) 577:1021–32. doi:10.1113/jphysiol.2006.120972
36. Sylow L, Nielsen IL, Kleinert M, Moller LL, Ploug T, Schjerling P, et al. Rac1 governs exercise-stimulated glucose uptake in skeletal muscle through regulation of GLUT4 translocation in mice. J Physiol (2016) 594(17):4997–5008. doi:10.1113/JP272039
37. Merry TL, Steinberg GR, Lynch GS, Mcconell GK. Skeletal muscle glucose uptake during contraction is regulated by nitric oxide and ROS independently of AMPK. Am J Physiol Endocrinol Metab (2010) 298:E577–85. doi:10.1152/ajpendo.00239.2009
38. Hawley JA, Hargreaves M, Zierath JR. Signalling mechanisms in skeletal muscle: role in substrate selection and muscle adaptation. Essays Biochem (2006) 42:1–12. doi:10.1042/bse0420001
39. Kramer HF, Witczak CA, Fujii N, Jessen N, Taylor EB, Arnolds DE, et al. Distinct signals regulate AS160 phosphorylation in response to insulin, AICAR, and contraction in mouse skeletal muscle. Diabetes (2006) 55:2067–76. doi:10.2337/db06-0150
40. Kramer HF, Witczak CA, Taylor EB, Fujii N, Hirshman MF, Goodyear LJ. The calmodulin-binding domain of the Akt substrate of 160 KDA (AS160) regulates contraction-but not insulin-stimulated glucose uptake. Diabetes (2007) 56:A37. doi:10.2337/db07-0681
41. Funai K, Cartee GD. Inhibition of contraction-stimulated AMP-activated protein kinase inhibits contraction-stimulated increases in PAS-TBC1D1 and glucose transport without altering PAS-AS160 in rat skeletal muscle. Diabetes (2009) 58:1096–104. doi:10.2337/db08-1477
42. Kjobsted R, Munk-Hansen N, Birk JB, Foretz M, Viollet B, Bjornholm M, et al. Enhanced muscle insulin sensitivity after contraction/exercise is mediated by AMPK. Diabetes (2016). doi:10.2337/db16-0530
43. Kingwell BA, Formosa M, Muhlmann M, Bradley SJ, Mcconell GK. Nitric oxide synthase inhibition reduces glucose uptake during exercise in individuals with type 2 diabetes more than in control subjects. Diabetes (2002) 51:2572–80. doi:10.2337/diabetes.51.8.2572
44. Vind BF, Pehmoller C, Treebak JT, Birk JB, Hey-Mogensen M, Beck-Nielsen H, et al. Impaired insulin-induced site-specific phosphorylation of TBC1 domain family, member 4 (TBC1D4) in skeletal muscle of type 2 diabetes patients is restored by endurance exercise-training. Diabetologia (2011) 54:157–67. doi:10.1007/s00125-010-1924-4
45. Middelbeek RJ, Chambers MA, Tantiwong P, Treebak JT, An D, Hirshman MF, et al. Insulin stimulation regulates AS160 and TBC1D1 phosphorylation sites in human skeletal muscle. Nutr Diabetes (2013) 3:e74. doi:10.1038/nutd.2013.13
46. Jelleyman C, Yates T, O’Donovan G, Gray LJ, King JA, Khunti K, et al. The effects of high-intensity interval training on glucose regulation and insulin resistance: a meta-analysis. Obes Rev (2015) 16:942–61. doi:10.1111/obr.12317
47. Garetto LP, Richter EA, Goodman MN, Ruderman NB. Enhanced muscle glucose metabolism after exercise in the rat: the two phases. Am J Physiol (1984) 246:E471–5.
48. Richter EA, Garetto LP, Goodman MN, Ruderman NB. Enhanced muscle glucose metabolism after exercise: modulation by local factors. Am J Physiol (1984) 246:E476–82.
49. Funai K, Schweitzer GG, Sharma N, Kanzaki M, Cartee GD. Increased AS160 phosphorylation, but not TBC1D1 phosphorylation, with increased postexercise insulin sensitivity in rat skeletal muscle. Am J Physiol Endocrinol Metab (2009) 297:E242–51. doi:10.1152/ajpendo.00194.2009
50. Treebak JT, Frosig C, Pehmoller C, Chen S, Maarbjerg SJ, Brandt N, et al. Potential role of TBC1D4 in enhanced post-exercise insulin action in human skeletal muscle. Diabetologia (2009) 52:891–900. doi:10.1007/s00125-009-1294-y
51. Castorena CM, Arias EB, Sharma N, Cartee GD. Postexercise improvement in insulin-stimulated glucose uptake occurs concomitant with greater AS160 phosphorylation in muscle from normal and insulin-resistant rats. Diabetes (2014) 63:2297–308. doi:10.2337/db13-1686
52. Iwabe M, Kawamoto E, Koshinaka K, Kawanaka K. Increased postexercise insulin sensitivity is accompanied by increased AS160 phosphorylation in slow-twitch soleus muscle. Physiol Rep (2014) 2:e12162. doi:10.14814/phy2.12162
53. Vendelbo MH, Moller AB, Treebak JT, Gormsen LC, Goodyear LJ, Wojtaszewski JF, et al. Sustained AS160 and TBC1D1 phosphorylations in human skeletal muscle 30 min after a single bout of exercise. J Appl Physiol (1985) (2014) 117:289–96. doi:10.1152/japplphysiol.00044.2014
54. Loh K, Deng H, Fukushima A, Cai X, Boivin B, Galic S, et al. Reactive oxygen species enhance insulin sensitivity. Cell Metab (2009) 10:260–72. doi:10.1016/j.cmet.2009.08.009
55. Trewin AJ, Lundell LS, Perry BD, Patil KV, Chibalin AV, Levinger I, et al. Effect of N-acetylcysteine infusion on exercise-induced modulation of insulin sensitivity and signaling pathways in human skeletal muscle. Am J Physiol Endocrinol Metab (2015) 309:E388–97. doi:10.1152/ajpendo.00605.2014
56. Holmstrom KM, Finkel T. Cellular mechanisms and physiological consequences of redox-dependent signalling. Nat Rev Mol Cell Biol (2014) 15:411–21. doi:10.1038/nrm3801
57. Valko M, Leibfritz D, Moncol J, Cronin MT, Mazur M, Telser J. Free radicals and antioxidants in normal physiological functions and human disease. Int J Biochem Cell Biol (2007) 39:44–84. doi:10.1016/j.biocel.2006.07.001
58. Moller P, Wallin H, Knudsen LE. Oxidative stress associated with exercise, psychological stress and life-style factors. Chem Biol Interact (1996) 102:17–36. doi:10.1016/0009-2797(96)03729-5
59. Wright E Jr, Scism-Bacon JL, Glass LC. Oxidative stress in type 2 diabetes: the role of fasting and postprandial glycaemia. Int J Clin Pract (2006) 60:308–14. doi:10.1111/j.1368-5031.2006.00825.x
60. Halliwell B. Reactive oxygen species in living systems: source, biochemistry, and role in human disease. Am J Med (1991) 91:14S–22S. doi:10.1016/0002-9343(91)90279-7
61. Patel RP, Mcandrew J, Sellak H, White CR, Jo H, Freeman BA, et al. Biological aspects of reactive nitrogen species. Biochim Biophys Acta (1999) 1411:385–400. doi:10.1016/S0005-2728(99)00028-6
62. Giles GI, Jacob C. Reactive sulfur species: an emerging concept in oxidative stress. Biol Chem (2002) 383:375–88. doi:10.1515/BC.2002.042
63. Levine RL, Stadtman ER. Oxidative modification of proteins during aging. Exp Gerontol (2001) 36:1495–502. doi:10.1016/S0531-5565(01)00135-8
64. Dhalla NS, Temsah RM, Netticadan T. Role of oxidative stress in cardiovascular diseases. J Hypertens (2000) 18:655–73. doi:10.1097/00004872-200018060-00002
65. Chakravarti B, Chakravarti DN. Oxidative modification of proteins: age-related changes. Gerontology (2007) 53:128–39. doi:10.1159/000097865
66. Davies MJ. The oxidative environment and protein damage. Biochim Biophys Acta (2005) 1703:93–109. doi:10.1016/j.bbapap.2004.08.007
67. Rimbach G, Hohler D, Fischer A, Roy S, Virgili F, Pallauf J, et al. Methods to assess free radicals and oxidative stress in biological systems. Arch Tierernahr (1999) 52:203–22. doi:10.1080/17450399909386163
68. Cooke MS, Evans MD, Dizdaroglu M, Lunec J. Oxidative DNA damage: mechanisms, mutation, and disease. FASEB J (2003) 17:1195–214. doi:10.1096/fj.02-0752rev
69. Kong Q, Lin CL. Oxidative damage to RNA: mechanisms, consequences, and diseases. Cell Mol Life Sci (2010) 67:1817–29. doi:10.1007/s00018-010-0277-y
70. Barciszewski J, Barciszewska MZ, Siboska G, Rattan SI, Clark BF. Some unusual nucleic acid bases are products of hydroxyl radical oxidation of DNA and RNA. Mol Biol Rep (1999) 26:231–8. doi:10.1023/A:1007058602594
71. Halliwell B, Chirico S. Lipid peroxidation: its mechanism, measurement, and significance. Am J Clin Nutr (1993) 57(5 Suppl):S715–24.
72. Sayre LM, Zelasko DA, Harris PL, Perry G, Salomon RG, Smith MA. 4-Hydroxynonenal-derived advanced lipid peroxidation end products are increased in Alzheimer’s disease. J Neurochem (1997) 68:2092–7. doi:10.1046/j.1471-4159.1997.68052092.x
73. Pillon NJ, Vella RE, Souleere L, Becchi M, Lagarde M, Soulage CO. Structural and functional changes in human insulin induced by the lipid peroxidation byproducts 4-hydroxy-2-nonenal and 4-hydroxy-2-hexenal. Chem Res Toxicol (2011) 24:752–62. doi:10.1021/tx200084d
74. Barrera G. Oxidative stress and lipid peroxidation products in cancer progression and therapy. ISRN Oncol (2012) 2012:1–21. doi:10.5402/2012/137289
75. Pillon NJ, Croze ML, Vella RE, Soulere L, Lagarde M, Soulage CO. The lipid peroxidation by-product 4-hydroxy-2-nonenal (4-HNE) induces insulin resistance in skeletal muscle through both carbonyl and oxidative stress. Endocrinology (2012) 153:2099–111. doi:10.1210/en.2011-1957
76. Kaur G, Cholia RP, Mantha AK, Kumar R. DNA repair and redox activities and inhibitors of apurinic/apyrimidinic endonuclease 1/redox effector factor 1 (APE1/REF-1): a comparative analysis and their scope and limitations toward anticancer drug development. J Med Chem (2014) 57:10241–56. doi:10.1021/jm500865u
77. Sen CK, Packer L. Thiol homeostasis and supplements in physical exercise. Am J Clin Nutr (2000) 72:653S–69S.
78. Woo HA, Yim SH, Shin DH, Kang D, Yu DY, Rhee SG. Inactivation of peroxiredoxin I by phosphorylation allows localized H(2)O(2) accumulation for cell signaling. Cell (2010) 140:517–28. doi:10.1016/j.cell.2010.01.009
79. Radak Z, Zhao Z, Koltai E, Ohno H, Atalay M. Oxygen consumption and usage during physical exercise: the balance between oxidative stress and ROS-dependent adaptive signaling. Antioxid Redox Signal (2013) 18:1208–46. doi:10.1089/ars.2011.4498
80. Li T, He S, Liu S, Kong Z, Wang J, Zhang Y. Effects of different exercise durations on KEAP1-NRF2-are pathway activation in mouse skeletal muscle. Free Radic Res (2015) 49:1269–74. doi:10.3109/10715762.2015.1066784
81. Mason SA, Morrison D, Mcconell GK, Wadley GD. Muscle redox signalling pathways in exercise. Role of antioxidants. Free Radic Biol Med (2016) 98:29–45. doi:10.1016/j.freeradbiomed.2016.02.022
82. Wang P, Li CG, Qi Z, Cui D, Ding S. Acute exercise stress promotes REF1/NRF2 signalling and increases mitochondrial antioxidant activity in skeletal muscle. Exp Physiol (2016) 101:410–20. doi:10.1113/EP085493
83. Packer L. Oxidants, antioxidant nutrients and the athlete. J Sports Sci (1997) 15:353–63. doi:10.1080/026404197367362
84. Atabek ME, Vatansev H, Erkul I. Oxidative stress in childhood obesity. J Pediatr Endocrinol Metab (2004) 17:1063–8. doi:10.1515/JPEM.2004.17.8.1063
85. Kamezaki F, Yamashita K, Kubara T, Suzuki Y, Tanaka S, Rkouzuma R, et al. Derivatives of reactive oxygen metabolites correlates with high-sensitivity C-reactive protein. J Atheroscler Thromb (2008) 15:206–12. doi:10.5551/jat.E538
86. Kotani K, Sakane N, Tsuzaki K, Matsuoka Y, Sano Y, Hamada T, et al. Lifestyles and oxidative stress in type 2 diabetic patients. Scand J Clin Lab Invest (2008) 68:516–8. doi:10.1080/00365510802023090
87. Fisher-Wellman K, Bell HK, Bloomer RJ. Oxidative stress and antioxidant defense mechanisms linked to exercise during cardiopulmonary and metabolic disorders. Oxid Med Cell Longev (2009) 2:43–51. doi:10.4161/oxim.2.1.7732
88. Fukui T, Yamauchi K, Maruyama M, Yasuda T, Kohno M, Abe Y. Significance of measuring oxidative stress in lifestyle-related diseases from the viewpoint of correlation between d-ROMs and BAP in Japanese subjects. Hypertens Res (2011) 34:1041–5. doi:10.1038/hr.2011.76
89. Naruse R, Suetsugu M, Terasawa T, Ito K, Hara K, Takebayashi K, et al. Oxidative stress and antioxidative potency are closely associated with diabetic retinopathy and nephropathy in patients with type 2 diabetes. Saudi Med J (2013) 34:135–41.
90. Kim JH, Baik HW, Yoon YS, Joung HJ, Park JS, Park SJ, et al. Measurement of antioxidant capacity using the biological antioxidant potential test and its role as a predictive marker of metabolic syndrome. Korean J Intern Med (2014) 29:31–9. doi:10.3904/kjim.2014.29.1.31
91. Margaritelis NV, Cobley JN, Paschalis V, Veskoukis AS, Theodorou AA, Kyparos A, et al. Going retro: oxidative stress biomarkers in modern redox biology. Free Radic Biol Med (2016) 98:2–12. doi:10.1016/j.freeradbiomed.2016.02.005
92. Parker L, Mcguckin TA, Leicht AS. Influence of exercise intensity on systemic oxidative stress and antioxidant capacity. Clin Physiol Funct Imaging (2014) 34:377–83. doi:10.1111/cpf.12108
93. Powers SK, Jackson MJ. Exercise-induced oxidative stress: cellular mechanisms and impact on muscle force production. Physiol Rev (2008) 88:1243–76. doi:10.1152/physrev.00031.2007
94. Powers SK, Nelson WB, Hudson MB. Exercise-induced oxidative stress in humans: cause and consequences. Free Radic Biol Med (2011) 51:942–50. doi:10.1016/j.freeradbiomed.2010.12.009
95. Jackson MJ, Vasilaki A, Mcardle A. Cellular mechanisms underlying oxidative stress in human exercise. Free Radic Biol Med (2016) 98:13–7. doi:10.1016/j.freeradbiomed.2016.02.023
96. Espinosa A, Leiva A, Pena M, Muller M, Debandi A, Hidalgo C, et al. Myotube depolarization generates reactive oxygen species through NAD(P)H oxidase; ROS-elicited Ca2+ stimulates ERK, CREB, early genes. J Cell Physiol (2006) 209:379–88. doi:10.1002/jcp.20745
97. Sakellariou GK, Vasilaki A, Palomero J, Kayani A, Zibrik L, Mcardle A, et al. Studies of mitochondrial and nonmitochondrial sources implicate nicotinamide adenine dinucleotide phosphate oxidase(s) in the increased skeletal muscle superoxide generation that occurs during contractile activity. Antioxid Redox Signal (2013) 18:603–21. doi:10.1089/ars.2012.4623
98. Radak Z, Asano K, Inoue M, Kizaki T, Oh-Ishi S, Suzuki K, et al. Superoxide dismutase derivative reduces oxidative damage in skeletal muscle of rats during exhaustive exercise. J Appl Physiol (1985) (1995) 79:129–35.
99. Stamler JS, Meissner G. Physiology of nitric oxide in skeletal muscle. Physiol Rev (2001) 81:209–37.
100. Gong MC, Arbogast S, Guo Z, Mathenia J, Su W, Reid MB. Calcium-independent phospholipase A2 modulates cytosolic oxidant activity and contractile function in murine skeletal muscle cells. J Appl Physiol (1985) (2006) 100:399–405. doi:10.1152/japplphysiol.00873.2005
101. St-Pierre J, Buckingham JA, Roebuck SJ, Brand MD. Topology of superoxide production from different sites in the mitochondrial electron transport chain. J Biol Chem (2002) 277:44784–90. doi:10.1074/jbc.M207217200
102. Kalyanaraman B, Felix CC, Sealy RC. Electron spin resonance-spin stabilization of semiquinones produced during oxidation of epinephrine and its analogues. J Biol Chem (1984) 259:354–8.
103. Lovlin R, Cottle W, Pyke I, Kavanagh M, Belcastro AN. Are indexes of free-radical damage related to exercise intensity. Eur J Appl Physiol (1987) 56:313–6. doi:10.1007/BF00690898
104. Mohazzab HK, Kaminski PM, Wolin MS. Lactate and PO2 modulate superoxide anion production in bovine cardiac myocytes: potential role of NADH oxidase. Circulation (1997) 96:614–20. doi:10.1161/01.CIR.96.2.614
105. McAnulty SR, Mcanulty L, Pascoe DD, Gropper SS, Keith RE, Morrow JD, et al. Hyperthermia increases exercise-induced oxidative stress. Int J Sports Med (2005) 26:188–92. doi:10.1055/s-2004-820990
106. Shikama K. The molecular mechanism of autoxidation for myoglobin and hemoglobin: a venerable puzzle. Chem Rev (1998) 98:1357–74. doi:10.1021/cr970042e
108. Vollaard NB, Shearman JP, Cooper CE. Exercise-induced oxidative stress: myths, realities and physiological relevance. Sports Med (2005) 35:1045–62. doi:10.2165/00007256-200535120-00004
109. Pedersen BK, Ostrowski K, Rohde T, Bruunsgaard H. The cytokine response to strenuous exercise. Can J Physiol Pharmacol (1998) 76:505–11. doi:10.1139/y98-055
110. Koyama K, Kaya M, Ishigaki T, Tsujita J, Hori S, Seino T, et al. Role of xanthine oxidase in delayed lipid peroxidation in rat liver induced by acute exhausting exercise. Eur J Appl Physiol Occup Physiol (1999) 80:28–33. doi:10.1007/s004210050554
111. Forman HJ, Torres M. Reactive oxygen species and cell signaling: respiratory burst in macrophage signaling. Am J Respir Crit Care Med (2002) 166:S4–8. doi:10.1164/rccm.2206007
112. O’Neill CA, Stebbins CL, Bonigut S, Halliwell B, Longhurst JC. Production of hydroxyl radicals in contracting skeletal muscle of cats. J Appl Physiol (1985) (1996) 81:1197–206.
113. Close GL, Kayani AC, Ashton T, Mcardle A, Jackson MJ. Release of superoxide from skeletal muscle of adult and old mice: an experimental test of the reductive hotspot hypothesis. Aging Cell (2007) 6:189–95. doi:10.1111/j.1474-9726.2007.00277.x
114. Silveira LR, Pereira-Da-Silva L, Juel C, Hellsten Y. Formation of hydrogen peroxide and nitric oxide in rat skeletal muscle cells during contractions. Free Radic Biol Med (2003) 35:455–64. doi:10.1016/S0891-5849(03)00271-5
115. Pattwell DM, Mcardle A, Morgan JE, Patridge TA, Jackson MJ. Release of reactive oxygen and nitrogen species from contracting skeletal muscle cells. Free Radic Biol Med (2004) 37:1064–72. doi:10.1016/j.freeradbiomed.2004.06.026
116. Elosua R, Molina L, Fito M, Arquer A, Sanchez-Quesada JL, Covas MI, et al. Response of oxidative stress biomarkers to a 16-week aerobic physical activity program, and to acute physical activity, in healthy young men and women. Atherosclerosis (2003) 167:327–34. doi:10.1016/S0021-9150(03)00018-2
117. Bloomer RJ, Fisher-Wellman KH. Blood oxidative stress biomarkers: influence of sex, exercise training status, and dietary intake. Gend Med (2008) 5:218–28. doi:10.1016/j.genm.2008.07.002
118. Jenkins RR. Exercise and oxidative stress methodology: a critique. Am J Clin Nutr (2000) 72:670S–4S.
119. Bloomer RJ. Effect of exercise on oxidative stress biomarkers. Adv Clin Chem (2008) 46:1–50. doi:10.1016/S0065-2423(08)00401-0
120. Michailidis Y, Jamurtas AZ, Nikolaidis MG, Fatouros IG, Koutedakis Y, Papassotiriou I, et al. Sampling time is crucial for measurement of aerobic exercise-induced oxidative stress. Med Sci Sports Exerc (2007) 39:1107–13. doi:10.1249/01.mss.0b013e318053e7ba
121. Forman HJ, Augusto O, Brigelius-Flohe R, Dennery PA, Kalyanaraman B, Ischiropoulos H, et al. Even free radicals should follow some rules: a guide to free radical research terminology and methodology. Free Radic Biol Med (2015) 78:233–5. doi:10.1016/j.freeradbiomed.2014.10.504
122. Nikolaidis MG, Paschalis V, Giakas G, Fatouros IG, Koutedakis Y, Kouretas D, et al. Decreased blood oxidative stress after repeated muscle-damaging exercise. Med Sci Sports Exerc (2007) 39:1080–9. doi:10.1249/mss.0b013e31804ca10c
123. Reid MB, Khawli FA, Moody MR. Reactive oxygen in skeletal muscle. III. Contractility of unfatigued muscle. J Appl Physiol (1985) (1993) 75:1081–7.
124. Palazzetti S, Richard MJ, Favier A, Margaritis I. Overloaded training increases exercise-induced oxidative stress and damage. Can J Appl Physiol (2003) 28:588–604. doi:10.1139/h03-045
125. Dosek A, Ohno H, Acs Z, Taylor AW, Radak Z. High altitude and oxidative stress. Respir Physiol Neurobiol (2007) 158:128–31. doi:10.1016/j.resp.2007.03.013
126. Rosa EF, Takahashi S, Aboulafia J, Nouailhetas VL, Oliveira MG. Oxidative stress induced by intense and exhaustive exercise impairs murine cognitive function. J Neurophysiol (2007) 98:1820–6. doi:10.1152/jn.01158.2006
127. Kuwahara H, Horie T, Ishikawa S, Tsuda C, Kawakami S, Noda Y, et al. Oxidative stress in skeletal muscle causes severe disturbance of exercise activity without muscle atrophy. Free Radic Biol Med (2010) 48:1252–62. doi:10.1016/j.freeradbiomed.2010.02.011
128. Aoi W, Naito Y, Tokuda H, Tanimura Y, Oya-Ito T, Yoshikawa T. Exercise-induced muscle damage impairs insulin signaling pathway associated with IRS-1 oxidative modification. Physiol Res (2012) 61:81–8.
129. Aoi W, Naito Y, Takanami Y, Kawai Y, Sakuma K, Ichikawa H, et al. Oxidative stress and delayed-onset muscle damage after exercise. Free Radic Biol Med (2004) 37:480–7. doi:10.1016/j.freeradbiomed.2004.05.008
131. Close GL, Ashton T, Mcardle A, Maclaren DP. The emerging role of free radicals in delayed onset muscle soreness and contraction-induced muscle injury. Comp Biochem Physiol A Mol Integr Physiol (2005) 142:257–66. doi:10.1016/j.cbpa.2005.08.005
132. Samjoo IA, Safdar A, Hamadeh MJ, Raha S, Tarnopolsky MA. The effect of endurance exercise on both skeletal muscle and systemic oxidative stress in previously sedentary obese men. Nutr Diabetes (2013) 3:e88. doi:10.1038/nutd.2013.30
133. Gomez-Cabrera MC, Domenech E, Romagnoli M, Arduini A, Borras C, Pallardo FV, et al. Oral administration of vitamin C decreases muscle mitochondrial biogenesis and hampers training-induced adaptations in endurance performance. Am J Clin Nutr (2008) 87:142–9.
134. Michailidis Y, Karagounis LG, Terzis G, Jamurtas AZ, Spengos K, Tsoukas D, et al. Thiol-based antioxidant supplementation alters human skeletal muscle signaling and attenuates its inflammatory response and recovery after intense eccentric exercise. Am J Clin Nutr (2013) 98:233–45. doi:10.3945/ajcn.112.049163
135. Kang C, O’Moore KM, Dickman JR, Ji LL. Exercise activation of muscle peroxisome proliferator-activated receptor-gamma coactivator-1alpha signaling is redox sensitive. Free Radic Biol Med (2009) 47:1394–400. doi:10.1016/j.freeradbiomed.2009.08.007
136. Strobel NA, Peake JM, Matsumoto A, Marsh SA, Coombes JS, Wadley GD. Antioxidant supplementation reduces skeletal muscle mitochondrial biogenesis. Med Sci Sports Exerc (2011) 43:1017–24. doi:10.1249/MSS.0b013e318203afa3
137. Petersen AC, Mckenna MJ, Medved I, Murphy KT, Brown MJ, Della Gatta P, et al. Infusion with the antioxidant N-acetylcysteine attenuates early adaptive responses to exercise in human skeletal muscle. Acta Physiol (2012) 204:382–92. doi:10.1111/j.1748-1716.2011.02344.x
138. Paulsen G, Cumming KT, Holden G, Hallen J, Ronnestad BR, Sveen O, et al. Vitamin C and E supplementation hampers cellular adaptation to endurance training in humans: a double-blind, randomised, controlled trial. J Physiol (2014) 592:1887–901. doi:10.1113/jphysiol.2013.267419
139. Olesen J, Gliemann L, Bienso R, Schmidt J, Hellsten Y, Pilegaard H. Exercise training, but not resveratrol, improves metabolic and inflammatory status in skeletal muscle of aged men. J Physiol (2014) 592:1873–86. doi:10.1113/jphysiol.2013.270256
140. Morrison D, Hughes J, Della Gatta PA, Mason S, Lamon S, Russell AP, et al. Vitamin C and E supplementation prevents some of the cellular adaptations to endurance-training in humans. Free Radic Biol Med (2015) 89:852–62. doi:10.1016/j.freeradbiomed.2015.10.412
141. Wray DW, Uberoi A, Lawrenson L, Bailey DM, Richardson RS. Oral antioxidants and cardiovascular health in the exercise-trained and untrained elderly: a radically different outcome. Clin Sci (Lond) (2009) 116:433–41. doi:10.1042/CS20080337
142. Gliemann L, Schmidt JF, Olesen J, Bienso RS, Peronard SL, Grandjean SU, et al. Resveratrol blunts the positive effects of exercise training on cardiovascular health in aged men. J Physiol (2013) 591:5047–59. doi:10.1113/jphysiol.2013.258061
143. Sharoff CG, Hagobian TA, Malin SK, Chipkin SR, Yu H, Hirshman MF, et al. Combining short-term metformin treatment and one bout of exercise does not increase insulin action in insulin-resistant individuals. Am J Physiol Endocrinol Metab (2010) 298:E815–23. doi:10.1152/ajpendo.00517.2009
144. Malin SK, Gerber R, Chipkin SR, Braun B. Independent and combined effects of exercise training and metformin on insulin sensitivity in individuals with prediabetes. Diabetes Care (2012) 35:131–6. doi:10.2337/dc11-0925
145. Yfanti C, Akerstrom T, Nielsen S, Nielsen AR, Mounier R, Mortensen OH, et al. Antioxidant supplementation does not alter endurance training adaptation. Med Sci Sports Exerc (2010) 42:1388–95. doi:10.1249/MSS.0b013e3181cd76be
146. Higashida K, Kim SH, Higuchi M, Holloszy JO, Han DH. Normal adaptations to exercise despite protection against oxidative stress. Am J Physiol Endocrinol Metab (2011) 301:E779–84. doi:10.1152/ajpendo.00655.2010
147. Lavie CJ, Milani JN. Do antioxidant vitamins ameliorate the beneficial effects of exercise training on insulin sensitivity? J Cardiopulm Rehabil Prev (2011) 31:211–6. doi:10.1097/HCR.0b013e318211e3d8
148. Yfanti C, Nielsen AR, Akerstrom T, Nielsen S, Rose AJ, Richter EA, et al. Effect of antioxidant supplementation on insulin sensitivity in response to endurance exercise training. Am J Physiol Endocrinol Metab (2011) 300:E761–70. doi:10.1152/ajpendo.00207.2010
149. Ostman B, Sjodin A, Michaelsson K, Byberg L. Coenzyme Q10 supplementation and exercise-induced oxidative stress in humans. Nutrition (2012) 28:403–17. doi:10.1016/j.nut.2011.07.010
150. Leelarungrayub D, Khansuwan R, Pothongsunun P, Klaphajone J. N-acetylcysteine supplementation controls total antioxidant capacity, creatine kinase, lactate, and tumor necrotic factor-alpha against oxidative stress induced by graded exercise in sedentary men. Oxid Med Cell Longev (2011) 2011:329643. doi:10.1155/2011/329643
151. Narotzki B, Reznick AZ, Navot-Mintzer D, Dagan B, Levy Y. Green tea and vitamin E enhance exercise-induced benefits in body composition, glucose homeostasis, and antioxidant status in elderly men and women. J Am Coll Nutr (2013) 32:31–40. doi:10.1080/07315724.2013.767661
152. Pahl HL. Activators and target genes of Rel/NF-kappaB transcription factors. Oncogene (1999) 18:6853–66. doi:10.1038/sj.onc.1203239
153. Kyriakis JM, Avruch J. Mammalian MAPK signal transduction pathways activated by stress and inflammation: a 10-year update. Physiol Rev (2012) 92:689–737. doi:10.1152/physrev.00028.2011
154. Muslin AJ. MAPK signalling in cardiovascular health and disease: molecular mechanisms and therapeutic targets. Clin Sci (Lond) (2008) 115:203–18. doi:10.1042/CS20070430
155. Karin M, Ben-Neriah Y. Phosphorylation meets ubiquitination: the control of NF-[kappa]B activity. Annu Rev Immunol (2000) 18:621–63. doi:10.1146/annurev.immunol.18.1.621
156. Katz M, Amit I, Yarden Y. Regulation of MAPKS by growth factors and receptor tyrosine kinases. Biochim Biophys Acta (2007) 1773:1161–76. doi:10.1016/j.bbamcr.2007.01.002
157. Schmidt KN, Amstad P, Cerutti P, Baeuerle PA. The roles of hydrogen peroxide and superoxide as messengers in the activation of transcription factor NF-κB. Chem Biol (1995) 2:13–22. doi:10.1016/1074-5521(95)90076-4
158. McCubrey JA, Lahair MM, Franklin RA. Reactive oxygen species-induced activation of the map kinase signaling pathways. Antioxid Redox Signal (2006) 8:1775–89. doi:10.1089/ars.2006.8.1745
159. Chambers MA, Moylan JS, Smith JD, Goodyear LJ, Reid MB. Stretch-stimulated glucose uptake in skeletal muscle is mediated by reactive oxygen species and p38 MAP-kinase. J Physiol (2009) 587:3363–73. doi:10.1113/jphysiol.2008.165639
160. Berdichevsky A, Guarente L, Bose A. Acute oxidative stress can reverse insulin resistance by inactivation of cytoplasmic JNK. J Biol Chem (2010) 285:21581–9. doi:10.1074/jbc.M109.093633
161. Henriquez-Olguin C, Diaz-Vegas A, Utreras-Mendoza Y, Campos C, Arias-Calderon M, Llanos P, et al. NOX2 inhibition impairs early muscle gene expression induced by a single exercise bout. Front Physiol (2016) 7:282. doi:10.3389/fphys.2016.00282
162. Miki H, Funato Y. Regulation of intracellular signalling through cysteine oxidation by reactive oxygen species. J Biochem (2012) 151:255–61. doi:10.1093/jb/mvs006
163. Klomsiri C, Karplus PA, Poole LB. Cysteine-based redox switches in enzymes. Antioxid Redox Signal (2011) 14:1065–77. doi:10.1089/ars.2010.3376
164. Schwertassek U, Haque A, Krishnan N, Greiner R, Weingarten L, Dick TP, et al. Reactivation of oxidized PTP1B and PTEN by thioredoxin 1. FEBS J (2014) 281:3545–58. doi:10.1111/febs.12898
165. Son Y, Cheong YK, Kim NH, Chung HT, Kang DG, Pae HO. Mitogen-activated protein kinases and reactive oxygen species: how can ROS activate MAPK pathways? J Signal Transduct (2011) 2011:792639. doi:10.1155/2011/792639
166. Ichijo H, Nishida E, Irie K, Tendijke P, Saitoh M, Moriguchi T, et al. Induction of apoptosis by ASK1, a mammalian MAPKKK that activates SAPK/JNK and p38 signaling pathways. Science (1997) 275:90–4. doi:10.1126/science.275.5296.90
167. Saitoh M, Nishitoh H, Fujii M, Takeda K, Tobiume K, Sawada Y, et al. Mammalian thioredoxin is a direct inhibitor of apoptosis signal-regulating kinase (ASK) 1. EMBO J (1998) 17:2596–606. doi:10.1093/emboj/17.9.2596
168. Farooq A, Zhou MM. Structure and regulation of MAPK phosphatases. Cell Signal (2004) 16:769–79. doi:10.1016/j.cellsig.2003.12.008
169. Kamata H, Honda S, Maeda S, Chang L, Hirata H, Karin M. Reactive oxygen species promote TNFalpha-induced death and sustained JNK activation by inhibiting map kinase phosphatases. Cell (2005) 120:649–61. doi:10.1016/j.cell.2004.12.041
170. Zhou JY, Liu Y, Wu GS. The role of mitogen-activated protein kinase phosphatase-1 in oxidative damage-induced cell death. Cancer Res (2006) 66:4888–94. doi:10.1158/0008-5472.CAN-05-4229
171. Nagai H, Noguchi T, Takeda K, Ichijo H. Pathophysiological roles of ASK1-MAP kinase signaling pathways. J Biochem Mol Biol (2007) 40:1–6.
172. Adler V, Yin ZM, Fuchs SY, Benezra M, Rosario L, Tew KD, et al. Regulation of JNK signaling by GSTp. EMBO J (1999) 18:1321–34. doi:10.1093/emboj/18.5.1321
173. Storz P, Toker A. Protein kinase D mediates a stress-induced NF-kappaB activation and survival pathway. EMBO J (2003) 22:109–20. doi:10.1093/emboj/cdg009
174. Kretz-Remy C, Mehlen P, Mirault ME, Arrigo AP. Inhibition of I kappa B-alpha phosphorylation and degradation and subsequent NF-kappa B activation by glutathione peroxidase overexpression. J Cell Biol (1996) 133:1083–93. doi:10.1083/jcb.133.5.1083
175. Guyton KZ, Liu Y, Gorospe M, Xu Q, Holbrook NJ. Activation of mitogen-activated protein kinase by HO: role in cell survival following oxidant injury. J Biol Chem (1996) 271:4138–42. doi:10.1074/jbc.271.8.4138
176. Kabe Y, Ando K, Hirao S, Yoshida M, Handa H. Redox regulation of NF-kappaB activation: distinct redox regulation between the cytoplasm and the nucleus. Antioxid Redox Signal (2005) 7:395–403. doi:10.1089/ars.2005.7.395
177. Cavigelli M, Dolfi F, Claret FX, Karin M. Induction of c-fos expression through JNK-mediated TCF/Elk-1 phosphorylation. EMBO J (1995) 14:5957–64.
178. Gupta S, Campbell D, Derijard B, Davis RJ. Transcription factor ATF2 regulation by the JNK signal-transduction pathway. Science (1995) 267:389–93. doi:10.1126/science.7824938
179. Aronson D, Boppart MD, Dufresne SD, Fielding RA, Goodyear LJ. Exercise stimulates c-Jun NH2 kinase activity and c-Jun transcriptional activity in human skeletal muscle. Biochem Biophys Res Commun (1998) 251:106–10. doi:10.1006/bbrc.1998.9435
180. Fuchs SY, Adler V, Pincus MR, Ronai Z. MEKK1/JNK signaling stabilizes and activates p53. Proc Natl Acad Sci U S A (1998) 95:10541–6. doi:10.1073/pnas.95.18.10541
181. Somwar R, Perreault M, Kapur S, Taha C, Sweeney G, Ramlal T, et al. Activation of p38 mitogen-activated protein kinase alpha and beta by insulin and contraction in rat skeletal muscle: potential role in the stimulation of glucose transport. Diabetes (2000) 49:1794–800. doi:10.2337/diabetes.49.11.1794
182. Thong FS, Derave W, Urso B, Kiens B, Richter EA. Prior exercise increases basal and insulin-induced p38 mitogen-activated protein kinase phosphorylation in human skeletal muscle. J Appl Physiol (1985) (2003) 94:2337–41. doi:10.1152/japplphysiol.00036.2003
183. Keren A, Tamir Y, Bengal E. The p38 MAPK signaling pathway: a major regulator of skeletal muscle development. Mol Cell Endocrinol (2006) 252:224–30. doi:10.1016/j.mce.2006.03.017
184. Lluis F, Perdiguero E, Nebreda AR, Munoz-Canoves P. Regulation of skeletal muscle gene expression by p38 MAP kinases. Trends Cell Biol (2006) 16:36–44. doi:10.1016/j.tcb.2005.11.002
185. Egan B, Zierath JR. Exercise metabolism and the molecular regulation of skeletal muscle adaptation. Cell Metab (2013) 17:162–84. doi:10.1016/j.cmet.2012.12.012
186. Ventura-Clapier R, Garnier A, Veksler V. Transcriptional control of mitochondrial biogenesis: the central role of PGC-1alpha. Cardiovasc Res (2008) 79:208–17. doi:10.1093/cvr/cvn098
187. Bartlett JD, Hwa Joo C, Jeong TS, Louhelainen J, Cochran AJ, Gibala MJ, et al. Matched work high-intensity interval and continuous running induce similar increases in PGC-1alpha mRNA, AMPK, p38, and p53 phosphorylation in human skeletal muscle. J Appl Physiol (1985) (2012) 112:1135–43. doi:10.1152/japplphysiol.01040.2011
188. Gomez-Cabrera M-C, Borrás C, Pallardó FV, Sastre J, Ji LL, Viña J. Decreasing xanthine oxidase-mediated oxidative stress prevents useful cellular adaptations to exercise in rats. J Physiol (2005) 567:113–20. doi:10.1113/jphysiol.2004.080564
189. Strobel NA, Matsumoto A, Peake JM, Marsh SA, Peternelj TT, Briskey D, et al. Altering the redox state of skeletal muscle by glutathione depletion increases the exercise-activation of PGC-1alpha. Physiol Rep (2014) 2:e12224. doi:10.14814/phy2.12224
190. Wadley GD, Nicolas MA, Hiam DS, Mcconell GK. Xanthine oxidase inhibition attenuates skeletal muscle signaling following acute exercise but does not impair mitochondrial adaptations to endurance training. Am J Physiol Endocrinol Metab (2013) 304:E853–62. doi:10.1152/ajpendo.00568.2012
191. Gomez-Cabrera MC, Ristow M, Vina J. Antioxidant supplements in exercise: worse than useless? Am J Physiol Endocrinol Metab (2012) 302:E476–7; author reply E478–9. doi:10.1152/ajpendo.00567.2011
192. Halliwell B. Free radicals and antioxidants: updating a personal view. Nutr Rev (2012) 70:257–65. doi:10.1111/j.1753-4887.2012.00476.x
193. Myung SK, Ju W, Cho B, Oh SW, Park SM, Koo BK, et al. Efficacy of vitamin and antioxidant supplements in prevention of cardiovascular disease: systematic review and meta-analysis of randomised controlled trials. BMJ (2013) 346:f10. doi:10.1136/bmj.f10
194. Mason S, Wadley GD. Skeletal muscle reactive oxygen species: a target of good cop/bad cop for exercise and disease. Redox Rep (2014) 19:97–106. doi:10.1179/1351000213Y.0000000077
195. Margaritelis NV, Cobley JN, Paschalis V, Veskoukis AS, Theodorou AA, Kyparos A, et al. Principles for integrating reactive species into in vivo biological processes: examples from exercise physiology. Cell Signal (2016) 28:256–71. doi:10.1016/j.cellsig.2015.12.011
196. Liu SS. Cooperation of a “reactive oxygen cycle” with the Q cycle and the proton cycle in the respiratory chain – superoxide generating and cycling mechanisms in mitochondria. J Bioenerg Biomembr (1999) 31:367–76. doi:10.1023/A:1018650103259
197. Nishikawa T, Edelstein D, Du XL, Yamagishi S, Matsumura T, Kaneda Y, et al. Normalizing mitochondrial superoxide production blocks three pathways of hyperglycaemic damage. Nature (2000) 404:787–90. doi:10.1038/35008121
198. Anderson EJ, Lustig ME, Boyle KE, Woodlief TL, Kane DA, Lin CT, et al. Mitochondrial H2O2 emission and cellular redox state link excess fat intake to insulin resistance in both rodents and humans. J Clin Invest (2009) 119:573–81. doi:10.1172/JCI37048
199. Drose S, Brandt U. Molecular mechanisms of superoxide production by the mitochondrial respiratory chain. Adv Exp Med Biol (2012) 748:145–69. doi:10.1007/978-1-4614-3573-0_6
200. Barbosa MR, Sampaio IH, Teodoro BG, Sousa TA, Zoppi CC, Queiroz AL, et al. Hydrogen peroxide production regulates the mitochondrial function in insulin resistant muscle cells: effect of catalase overexpression. Biochim Biophys Acta (2013) 1832:1591–604. doi:10.1016/j.bbadis.2013.04.029
201. Chance B, Sies H, Boveris A. Hydroperoxide metabolism in mammalian organs. Physiol Rev (1979) 59:527–605.
202. Bierhaus A, Humpert PM, Morcos M, Wendt T, Chavakis T, Arnold B, et al. Understanding rage, the receptor for advanced glycation end products. J Mol Med (2005) 83:876–86. doi:10.1007/s00109-005-0688-7
203. Wautier MP, Chappey O, Corda S, Stern DM, Schmidt AM, Wautier JL. Activation of NADPH oxidase by age links oxidant stress to altered gene expression via rage. Am J Physiol Endocrinol Metab (2001) 280:E685–94.
204. Coughlan MT, Thorburn DR, Penfold SA, Laskowski A, Harcourt BE, Sourris KC, et al. Rage-induced cytosolic ROS promote mitochondrial superoxide generation in diabetes. J Am Soc Nephrol (2009) 20:742–52. doi:10.1681/ASN.2008050514
205. Bierhaus A, Chevion S, Chevion M, Hofmann M, Quehenberger P, Illmer T, et al. Advanced glycation end product-induced activation of NF-kappaB is suppressed by alpha-lipoic acid in cultured endothelial cells. Diabetes (1997) 46:1481–90. doi:10.2337/diab.46.9.1481
206. Lander HM, Tauras JM, Ogiste JS, Hori O, Moss RA, Schmidt AM. Activation of the receptor for advanced glycation end products triggers a p21(ras)-dependent mitogen-activated protein kinase pathway regulated by oxidant stress. J Biol Chem (1997) 272:17810–4. doi:10.1074/jbc.272.28.17810
207. Yan LJ. Pathogenesis of chronic hyperglycemia: from reductive stress to oxidative stress. J Diabetes Res (2014) 2014:137919. doi:10.1155/2014/137919
208. Mohanty P, Hamouda W, Garg R, Aljada A, Ghanim H, Dandona P. Glucose challenge stimulates reactive oxygen species (ROS) generation by leucocytes. J Clin Endocrinol Metab (2000) 85:2970–3. doi:10.1210/jcem.85.8.6854
209. Miyazaki Y, Kawano H, Yoshida T, Miyamoto S, Hokamaki J, Nagayoshi Y, et al. Pancreatic B-cell function is altered by oxidative stress induced by acute hyperglycaemia. Diabet Med (2007) 24:154–60. doi:10.1111/j.1464-5491.2007.02058.x
210. Mohanty P, Ghanim H, Hamouda W, Aljada A, Garg R, Dandona P. Both lipid and protein intakes stimulate increased generation of reactive oxygen species by polymorphonuclear leukocytes and mononuclear cells. Am J Clin Nutr (2002) 75:767–72.
211. Tushuizen ME, Nieuwland R, Scheffer PG, Sturk A, Heine RJ, Diamant M. Two consecutive high-fat meals affect endothelial-dependent vasodilation, oxidative stress and cellular microparticles in healthy men. J Thromb Haemost (2006) 4:1003–10. doi:10.1111/j.1538-7836.2006.01914.x
212. Tyldum GA, Schjerve IE, Tjonna AE, Kirkeby-Garstad I, Stolen TO, Richardson RS, et al. Endothelial dysfunction induced by post-prandial lipemia: complete protection afforded by high-intensity aerobic interval exercise. J Am Coll Cardiol (2009) 53:200–6. doi:10.1016/j.jacc.2008.09.033
213. Gabriel B, Ratkevicius A, Gray P, Frenneaux MP, Gray SR. High-intensity exercise attenuates postprandial lipaemia and markers of oxidative stress. Clin Sci (Lond) (2012) 123:313–21. doi:10.1042/CS20110600
214. Canale RE, Farney TM, Mccarthy CG, Bloomer RJ. Influence of acute exercise of varying intensity and duration on postprandial oxidative stress. Eur J Appl Physiol (2014) 114:1913–24. doi:10.1007/s00421-014-2912-z
215. Takahashi M, Miyashita M, Park JH, Sakamoto S, Suzuki K. Effects of breaking sitting by standing and acute exercise on postprandial oxidative stress. Asian J Sports Med (2015) 6:e24902. doi:10.5812/asjsm.24902
216. Mc Clean CM, Mc Laughlin J, Burke G, Murphy MH, Trinick T, Duly E, et al. The effect of acute aerobic exercise on pulse wave velocity and oxidative stress following postprandial hypertriglyceridemia in healthy men. Eur J Appl Physiol (2007) 100:225–34. doi:10.1007/s00421-007-0422-y
217. Bloomer RJ, Ferebee DE, Fisher-Wellman KH, Quindry JC, Schilling BK. Postprandial oxidative stress: influence of sex and exercise training status. Med Sci Sports Exerc (2009) 41:2111–9. doi:10.1249/MSS.0b013e3181a9e832
218. Bloomer RJ, Kabir MM, Marshall KE, Canale RE, Farney TM. Postprandial oxidative stress in response to dextrose and lipid meals of differing size. Lipids Health Dis (2010) 9:79–79. doi:10.1186/1476-511X-9-79
219. Fisher-Wellman KH, Bloomer RJ. Exacerbated postprandial oxidative stress induced by the acute intake of a lipid meal compared to isoenergetically administered carbohydrate, protein, and mixed meals in young, healthy men. J Am Coll Nutr (2010) 29:373–81. doi:10.1080/07315724.2010.10719854
220. Bloomer RJ, Lee SR. Women experience lower postprandial oxidative stress compared to men. Springerplus (2013) 2:553. doi:10.1186/2193-1801-2-553
221. Iwakami S, Misu H, Takeda T, Sugimori M, Matsugo S, Kaneko S, et al. Concentration-dependent dual effects of hydrogen peroxide on insulin signal transduction in H4IIEC hepatocytes. PLoS One (2011) 6:e27401. doi:10.1371/journal.pone.0027401
222. Ropelle ER, Pauli JR, Prada PO, De Souza CT, Picardi PK, Faria MC, et al. Reversal of diet-induced insulin resistance with a single bout of exercise in the rat: the role of PTP1B and IRS-1 serine phosphorylation. J Physiol (2006) 577:997–1007. doi:10.1113/jphysiol.2006.120006
223. Copps KD, White MF. Regulation of insulin sensitivity by serine/threonine phosphorylation of insulin receptor substrate proteins IRS1 and IRS2. Diabetologia (2012) 55:2565–82. doi:10.1007/s00125-012-2644-8
224. Mothe I, Van Obberghen E. Phosphorylation of insulin receptor substrate-1 on multiple serine residues, 612, 632, 662, and 731, modulates insulin action. J Biol Chem (1996) 271:11222–7. doi:10.1074/jbc.271.19.11222
225. Aguirre V, Uchida T, Yenush L, Davis R, White MF. The c-Jun NH2-terminal kinase promotes insulin resistance during association with insulin receptor substrate-1 and phosphorylation of Ser307. J Biol Chem (2000) 275:9047–54. doi:10.1074/jbc.275.12.9047
226. Aguirre V, Werner ED, Giraud J, Lee YH, Shoelson SE, White MF. Phosphorylation of Ser307 in insulin receptor substrate-1 blocks interactions with the insulin receptor and inhibits insulin action. J Biol Chem (2002) 277:1531–7. doi:10.1074/jbc.M101521200
227. Gual P, Gremeaux T, Gonzalez T, Le Marchand-Brustel Y, Tanti JF. MAP kinases and mTOR mediate insulin-induced phosphorylation of insulin receptor substrate-1 on serine residues 307, 612 and 632. Diabetologia (2003) 46:1532–42. doi:10.1007/s00125-003-1223-4
228. de Alvaro C, Teruel T, Hernandez R, Lorenzo M. Tumor necrosis factor alpha produces insulin resistance in skeletal muscle by activation of inhibitor kappaB kinase in a p38 MAPK-dependent manner. J Biol Chem (2004) 279:17070–8. doi:10.1074/jbc.M312021200
229. Greene MW, Morrice N, Garofalo RS, Roth RA. Modulation of human insulin receptor substrate-1 tyrosine phosphorylation by protein kinase Cdelta. Biochem J (2004) 378:105–16. doi:10.1042/bj20031493
230. Li Y, Soos TJ, Li X, Wu J, Degennaro M, Sun X, et al. Protein kinase C Theta inhibits insulin signaling by phosphorylating IRS1 at Ser(1101). J Biol Chem (2004) 279:45304–7. doi:10.1074/jbc.C400186200
231. Werner ED, Lee J, Hansen L, Yuan M, Shoelson SE. Insulin resistance due to phosphorylation of insulin receptor substrate-1 at serine 302. J Biol Chem (2004) 279:35298–305. doi:10.1074/jbc.M405203200
232. Archuleta TL, Lemieux AM, Saengsirisuwan V, Teachey MK, Lindborg KA, Kim JS, et al. Oxidant stress-induced loss of IRS-1 and IRS-2 proteins in rat skeletal muscle: role of p38 MAPK. Free Radic Biol Med (2009) 47:1486–93. doi:10.1016/j.freeradbiomed.2009.08.014
233. Geiger PC, Wright DC, Han DH, Holloszy JO. Activation of p38 MAP kinase enhances sensitivity of muscle glucose transport to insulin. Am J Physiol Endocrinol Metab (2005) 288:E782–8. doi:10.1152/ajpendo.00477.2004
234. Danielsson A, Ost A, Nystrom FH, Stralfors P. Attenuation of insulin-stimulated insulin receptor substrate-1 serine 307 phosphorylation in insulin resistance of type 2 diabetes. J Biol Chem (2005) 280:34389–92. doi:10.1074/jbc.C500230200
235. Giraud J, Leshan R, Lee YH, White MF. Nutrient-dependent and insulin-stimulated phosphorylation of insulin receptor substrate-1 on serine 302 correlates with increased insulin signaling. J Biol Chem (2004) 279:3447–54. doi:10.1074/jbc.M308631200
236. Weigert C, Kron M, Kalbacher H, Pohl AK, Runge H, Haring HU, et al. Interplay and effects of temporal changes in the phosphorylation state of serine-302, -307, and -318 of insulin receptor substrate-1 on insulin action in skeletal muscle cells. Mol Endocrinol (2008) 22:2729–40. doi:10.1210/me.2008-0102
237. Rui LY, Aguirre V, Kim JK, Shulman GI, Lee A, Corbould A, et al. Insulin/IGF-1 and TNF-alpha stimulate phosphorylation of IRS-1 at inhibitory Ser(307) via distinct pathways. J Clin Invest (2001) 107:181–9. doi:10.1172/JCI10934
238. Lee YH, Giraud J, Davis RJ, White MF. c-Jun N-terminal kinase (JNK) mediates feedback inhibition of the insulin signaling cascade. J Biol Chem (2003) 278:2896–902. doi:10.1074/jbc.M208359200
239. Yi Z, Langlais P, De Filippis EA, Luo M, Flynn CR, Schroeder S, et al. Global assessment of regulation of phosphorylation of insulin receptor substrate-1 by insulin in vivo in human muscle. Diabetes (2007) 56:1508–16. doi:10.2337/db06-1355
240. Salmeen A, Andersen JN, Myers MP, Tonks NK, Barford D. Molecular basis for the dephosphorylation of the activation segment of the insulin receptor by protein tyrosine phosphatase 1B. Mol Cell (2000) 6:1401–12. doi:10.1016/S1097-2765(00)00137-4
241. Goldstein BJ, Bittner-Kowalczyk A, White MF, Harbeck M. Tyrosine dephosphorylation and deactivation of insulin receptor substrate-1 by protein-tyrosine phosphatase 1B. Possible facilitation by the formation of a ternary complex with the GRB2 adaptor protein. J Biol Chem (2000) 275:4283–9. doi:10.1074/jbc.275.6.4283
242. Tan PL, Shavlakadze T, Grounds MD, Arthur PG. Differential thiol oxidation of the signaling proteins Akt, PTEN or PP2A determines whether Akt phosphorylation is enhanced or inhibited by oxidative stress in C2C12 myotubes derived from skeletal muscle. Int J Biochem Cell Biol (2015) 62:72–9. doi:10.1016/j.biocel.2015.02.015
243. Myers MP, Pass I, Batty IH, Van Der Kaay J, Stolarov JP, Hemmings BA, et al. The lipid phosphatase activity of PTEN is critical for its tumor suppressor function. Proc Natl Acad Sci U S A (1998) 95:13513–8. doi:10.1073/pnas.95.23.13513
244. Tonks NK. Redox redux: revisiting PTPS and the control of cell signaling. Cell (2005) 121:667–70. doi:10.1016/j.cell.2005.05.016
245. Wright VP, Reiser PJ, Clanton TL. Redox modulation of global phosphatase activity and protein phosphorylation in intact skeletal muscle. J Physiol (2009) 587:5767–81. doi:10.1113/jphysiol.2009.178285
246. Mahadev K, Motoshima H, Wu X, Ruddy JM, Arnold RS, Cheng G, et al. The NAD(P)H oxidase homolog Nox4 modulates insulin-stimulated generation of H2O2 and plays an integral role in insulin signal transduction. Mol Cell Biol (2004) 24:1844–54. doi:10.1128/MCB.24.5.1844-1854.2004
247. Seo JH, Ahn Y, Lee SR, Yeol Yeo C, Chung Hur K. The major target of the endogenously generated reactive oxygen species in response to insulin stimulation is phosphatase and tensin homolog and not phosphoinositide-3 kinase (PI-3 kinase) in the PI-3 kinase/Akt pathway. Mol Biol Cell (2005) 16:348–57. doi:10.1091/mbc.E04-05-0369
248. Chen K, Kirber MT, Xiao H, Yang Y, Keaney JF Jr. Regulation of ROS signal transduction by NADPH oxidase 4 localization. J Cell Biol (2008) 181:1129–39. doi:10.1083/jcb.200709049
249. Bedard K, Krause KH. The NOX family of ROS-generating NADPH oxidases: physiology and pathophysiology. Physiol Rev (2007) 87:245–313. doi:10.1152/physrev.00044.2005
250. MohammadTaghvaei N, Taheripak G, Taghikhani M, Meshkani R. Palmitate-induced PTP1B expression is mediated by ceramide-JNK and nuclear factor kappaB (NF-kappaB) activation. Cell Signal (2012) 24:1964–70. doi:10.1016/j.cellsig.2012.04.019
251. Davies KJ, Quintanilha AT, Brooks GA, Packer L. Free radicals and tissue damage produced by exercise. Biochem Biophys Res Commun (1982) 107:1198–205. doi:10.1016/S0006-291X(82)80124-1
252. Jackson MJ, Edwards RH, Symons MC. Electron spin resonance studies of intact mammalian skeletal muscle. Biochim Biophys Acta (1985) 847:185–90. doi:10.1016/0167-4889(85)90019-9
253. Bailey DM, Lawrenson L, Mceneny J, Young IS, James PE, Jackson SK, et al. Electron paramagnetic spectroscopic evidence of exercise-induced free radical accumulation in human skeletal muscle. Free Radic Res (2007) 41:182–90. doi:10.1080/10715760601028867
254. Somwar R, Koterski S, Sweeney G, Sciotti R, Djuric S, Berg C, et al. A dominant-negative p38 MAPK mutant and novel selective inhibitors of p38 MAPK reduce insulin-stimulated glucose uptake in 3T3-L1 adipocytes without affecting GLUT4 translocation. J Biol Chem (2002) 277:50386–95. doi:10.1074/jbc.M205277200
255. Pal M, Febbraio MA, Whitham M. From cytokine to myokine: the emerging role of interleukin-6 in metabolic regulation. Immunol Cell Biol (2014) 92:331–9. doi:10.1038/icb.2014.16
256. Parker L, Trewin A, Levinger I, Shaw CS, Stepto NK. The effect of exercise-intensity on skeletal muscle stress kinase and insulin protein signaling. PLoS One (2017) 12:e0171613. doi:10.1371/journal.pone.0171613
257. Picklo MJ, Thyfault JP. Vitamin E and vitamin C do not reduce insulin sensitivity but inhibit mitochondrial protein expression in exercising obese rats. Appl Physiol Nutr Metab (2015) 40:343–52. doi:10.1139/apnm-2014-0302
258. Yu M, Stepto NK, Chibalin AV, Fryer LG, Carling D, Krook A, et al. Metabolic and mitogenic signal transduction in human skeletal muscle after intense cycling exercise. J Physiol (2003) 546:327–35. doi:10.1113/jphysiol.2002.034223
259. Hollander J, Fiebig R, Gore M, Ookawara T, Ohno H, Ji LL. Superoxide dismutase gene expression is activated by a single bout of exercise in rat skeletal muscle. Pflugers Arch (2001) 442:426–34. doi:10.1007/s004240100539
260. Diamond-Stanic MK, Marchionne EM, Teachey MK, Durazo DE, Kim JS, Henriksen EJ. Critical role of the transient activation of p38 MAPK in the etiology of skeletal muscle insulin resistance induced by low-level in vitro oxidant stress. Biochem Biophys Res Commun (2011) 405:439–44. doi:10.1016/j.bbrc.2011.01.049
261. Chan MH, Mcgee SL, Watt MJ, Hargreaves M, Febbraio MA. Altering dietary nutrient intake that reduces glycogen content leads to phosphorylation of nuclear p38 MAP kinase in human skeletal muscle: association with IL-6 gene transcription during contraction. FASEB J (2004) 18:1785–7. doi:10.1096/fj.03-1039fje
262. Moxham CM, Tabrizchi A, Davis RJ, Malbon CC. Jun N-terminal kinase mediates activation of skeletal muscle glycogen synthase by insulin in vivo. J Biol Chem (1996) 271:30765–73. doi:10.1074/jbc.271.48.30765
263. Febbraio MA, Pedersen BK. Muscle-derived interleukin-6: mechanisms for activation and possible biological roles. FASEB J (2002) 16:1335–47. doi:10.1096/fj.01-0876rev
264. Carey AL, Steinberg GR, Macaulay SL, Thomas WG, Holmes AG, Ramm G, et al. Interleukin-6 increases insulin-stimulated glucose disposal in humans and glucose uptake and fatty acid oxidation in vitro via amp-activated protein kinase. Diabetes (2006) 55:2688–97. doi:10.2337/db05-1404
265. Spangenburg EE, Brown DA, Johnson MS, Moore RL. Exercise increases SOCS-3 expression in rat skeletal muscle: potential relationship to IL-6 expression. J Physiol (2006) 572:839–48. doi:10.1113/jphysiol.2005.104315
266. Vella L, Caldow MK, Larsen AE, Tassoni D, Della Gatta PA, Gran P, et al. Resistance exercise increases NF-kappaB activity in human skeletal muscle. Am J Physiol Regul Integr Comp Physiol (2012) 302:R667–73. doi:10.1152/ajpregu.00336.2011
267. Whitham M, Chan MH, Pal M, Matthews VB, Prelovsek O, Lunke S, et al. Contraction-induced interleukin-6 gene transcription in skeletal muscle is regulated by c-Jun terminal kinase/activator protein-1. J Biol Chem (2012) 287:10771–9. doi:10.1074/jbc.M111.310581
268. Fujishiro M, Gotoh Y, Katagiri H, Sakoda H, Ogihara T, Anai M, et al. MKK6/3 and p38 MAPK pathway activation is not necessary for insulin-induced glucose uptake but regulates glucose transporter expression. J Biol Chem (2001) 276:19800–6. doi:10.1074/jbc.M101087200
269. Marozkina NV, Gaston B. S-nitrosylation signaling regulates cellular protein interactions. Biochim Biophys Acta (2012) 1820:722–9. doi:10.1016/j.bbagen.2011.06.017
270. Schoneich C, Sharov VS. Mass spectrometry of protein modifications by reactive oxygen and nitrogen species. Free Radic Biol Med (2006) 41:1507–20. doi:10.1016/j.freeradbiomed.2006.08.013
271. Winterbourn CC, Hampton MB. Thiol chemistry and specificity in redox signaling. Free Radic Biol Med (2008) 45:549–61. doi:10.1016/j.freeradbiomed.2008.05.004
272. Murray CI, Van Eyk JE. Chasing cysteine oxidative modifications: proteomic tools for characterizing cysteine redox status. Circ Cardiovasc Genet (2012) 5:591. doi:10.1161/CIRCGENETICS.111.961425
273. Marinho HS, Real C, Cyrne L, Soares H, Antunes F. Hydrogen peroxide sensing, signaling and regulation of transcription factors. Redox Biol (2014) 2:535–62. doi:10.1016/j.redox.2014.02.006
274. Gould NS, Evans P, Martinez-Acedo P, Marino SM, Gladyshev VN, Carroll KS, et al. Site-specific proteomic mapping identifies selectively modified regulatory cysteine residues in functionally distinct protein networks. Chem Biol (2015) 22:965–75. doi:10.1016/j.chembiol.2015.06.010
275. Ashton T, Rowlands CC, Jones E, Young IS, Jackson SK, Davies B, et al. Electron spin resonance spectroscopic detection of oxygen-centred radicals in human serum following exhaustive exercise. Eur J Appl Physiol Occup Physiol (1998) 77:498–502. doi:10.1007/s004210050366
276. Dickinson BC, Chang CJ. A targetable fluorescent probe for imaging hydrogen peroxide in the mitochondria of living cells. J Am Chem Soc (2008) 130:9638–9. doi:10.1021/ja802355u
277. Cocheme HM, Quin C, Mcquaker SJ, Cabreiro F, Logan A, Prime TA, et al. Measurement of H2O2 within living Drosophila during aging using a ratiometric mass spectrometry probe targeted to the mitochondrial matrix. Cell Metab (2011) 13:340–50. doi:10.1016/j.cmet.2011.02.003
Keywords: exercise, insulin signaling, stress kinase, glycemic control, oxidative stress, redox
Citation: Parker L, Shaw CS, Stepto NK and Levinger I (2017) Exercise and Glycemic Control: Focus on Redox Homeostasis and Redox-Sensitive Protein Signaling. Front. Endocrinol. 8:87. doi: 10.3389/fendo.2017.00087
Received: 31 January 2017; Accepted: 04 April 2017;
Published: 05 May 2017
Edited by:
Jonathan Peter Little, University of British Columbia, CanadaReviewed by:
Jeff Scott Coombes, The University of Queensland, AustraliaAdeel Safdar, Humber College, Canada
Copyright: © 2017 Parker, Shaw, Stepto and Levinger. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Lewan Parker, bGV3YW4ucGFya2VyQHZ1LmVkdS5hdQ==, bGV3YW4ucGFya2VyQGdtYWlsLmNvbQ==
†Co-senior authors.