 Pia-Maria Wippert
Pia-Maria Wippert Michael Rector
Michael Rector Gisela Kuhn
Gisela Kuhn Karin Wuertz-Kozak
Karin Wuertz-Kozak- 1Department of Health Sciences, Institute of Sociology of Health and Physical Activity, University of Potsdam, Potsdam, Germany
- 2Department of Health Sciences and Technology, Institute for Biomechanics, ETH Zurich, Zurich, Switzerland
- 3Schön Klinik München Harlaching, Munich, Germany
- 4Spine Center, Academic Teaching Hospital and Spine Research Institute, Paracelsus Private Medical University Salzburg, Salzburg, Austria
- 5Competence Center for Applied Biotechnology and Molecular Medicine (CABMM), University of Zurich, Zurich, Switzerland
Decades of research have demonstrated that physical stress (PS) stimulates bone remodeling and affects bone structure and function through complex mechanotransduction mechanisms. Recent research has laid ground to the hypothesis that mental stress (MS) also influences bone biology, eventually leading to osteoporosis and increased bone fracture risk. These effects are likely exerted by modulation of hypothalamic–pituitary–adrenal axis activity, resulting in an altered release of growth hormones, glucocorticoids and cytokines, as demonstrated in human and animal studies. Furthermore, molecular cross talk between mental and PS is thought to exist, with either synergistic or preventative effects on bone disease progression depending on the characteristics of the applied stressor. This mini review will explain the emerging concept of MS as an important player in bone adaptation and its potential cross talk with PS by summarizing the current state of knowledge, highlighting newly evolving notions (such as intergenerational transmission of stress and its epigenetic modifications affecting bone) and proposing new research directions.
Introduction
Bones are an essential component of the musculoskeletal system, with their primary functions being protecting vital organs, supporting the body, assisting in movement, producing blood cells, and storing nutrients and minerals. To fulfill all this functions, bone mass and structure are regulated by a number of factors. Bone tissue is continuously remodeled and modeled to maintain a healthy matrix and to adapt to changing environmental factors. Disturbances in these mechanisms often result in reduced bone mass and an increased risk for fractures, with aging (especially in postmenopausal women), furthermore, impacting bone health. Natural aging leads to the accumulation of osteoporosis risk factors, including a gradual inability to cope with physical and mental stressors (with consequences on bone adaptation) as well as biochemical alterations elucidated in more detail within this perspective.
Physical Stress (PS): A Brief Summary
Mechanical loading, also often termed as PS, is an important trigger inducing structural adaptation in bone. Multitudes of studies have investigated the effects of PS on bone, including the identification of loading regimes supporting healthy tissue homeostasis (or reversely degeneration-associated matrix loss), analysis of underlying mechanotransduction mechanisms, as well as elucidation of load-induced molecular responses (1–5). Nowadays, it is widely accepted that osteocytes are the main sensors of mechanical forces and orchestrate the activity of osteoblasts (responsible for bone formation) and osteoclasts (responsible for bone resorption) by several signaling pathways (6).
Mental Stress (MS): A Brief Summary
Recent publications provide compelling evidence that psychosocial stress, defined here as MS, can disturb bone homeostasis. When studying signaling pathways and pathological consequences of MS different stressor characteristics are distinguished. Till now it is well known that specific social stress situations (e.g., social evaluative or threat components) provoke the strongest physiological responses (7). However, individuals respond differently to these situations depending on their interpretation, resources, and adaptation strategies, which refer also to their biographical time frames and the duration of the stress exposition (short term, such as acute or long term such as chronic, traumatic stress type). Thus, an understanding of physiological mechanisms behind the MS response is particularly complex (see Figure 1) (8). Biologically, both responses (short-/long-term MS) are driven by the autonomic nervous system and the hypothalamic–pituitary axis. Within the autonomic nervous system, the stress response proceeds to one of three peripheral catecholamine systems (sympathetic–nervous system, sympathetic–adrenal–medullary system, and dopamine systems), whereby their operation depends on stressor type and characteristic (9). In the anterior pituitary, the stress response is determined by hypothalamic nuclei interactions and neuroendocrine cell hormone regulation. It is influenced by the stressor characteristic, driven by synaptic input from different brain regions like the limbic system (hippocampus, amygdala), as well as the brainstem (locus coeruleus), and realized on five endocrine axes: hypothalamic–pituitary–adrenal (HPA), hypothalamic–pituitary–thyroid (HPT), hypothalamic–pituitary–gonadal (HPG), hypothalamic–pituitary–somatotropic (HPS), and hypothalamic–pituitary–prolactin (HPP).
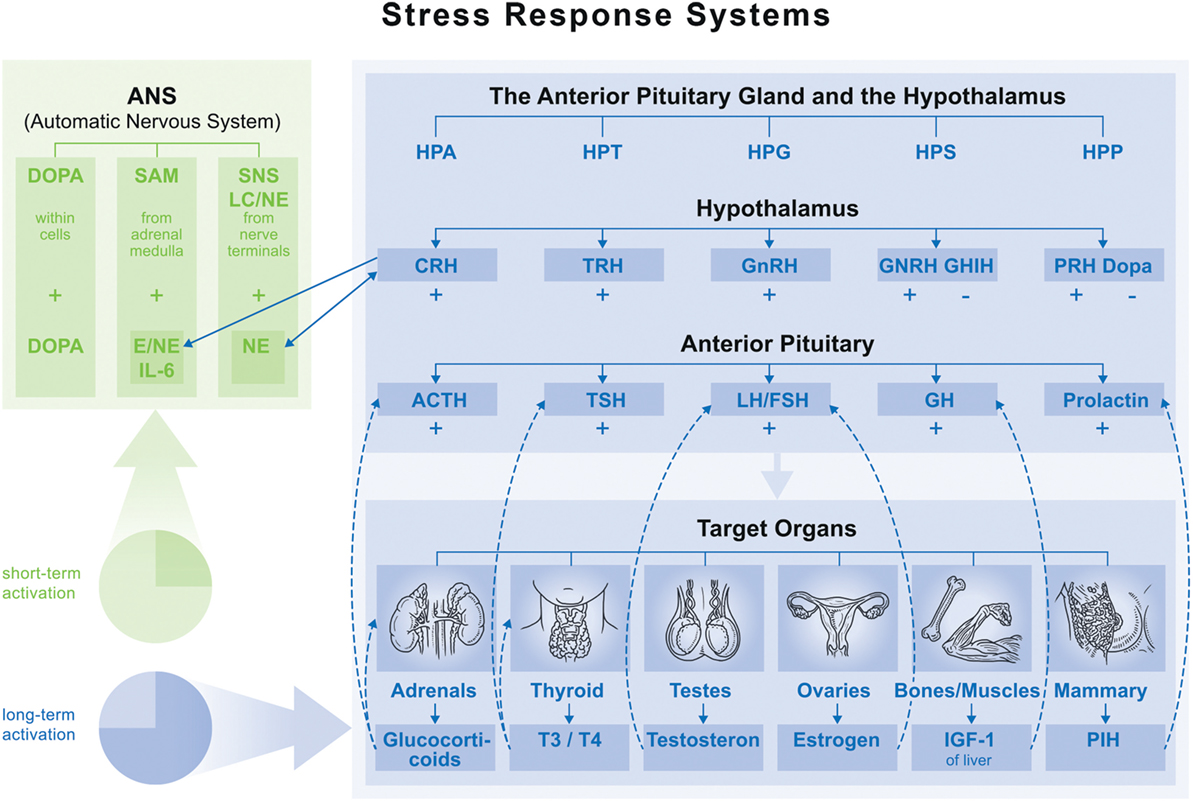
Figure 1. Overview of main endocrine axes. Normal function: feedback loop stops activation on each axis. Maladaptation of feedback function leads to fluctuation (accumulation) in one axis (hyper- or hypofunction), which influences function of the others. ⇢ = inhibition; → = stimulation. Abbreviations: HPA, hypothalamic–pituitary–adrenal; HPT, hypothalamic–pituitary–thyroid; HPG, hypothalamic–pituitary–gonadal; HPS, hypothalamic–pituitary–somatotropic; HPP, hypothalamic–pituitary–prolactin; CRH, corticotropin-releasing hormone; TRH, thyrotropin-releasing hormone; GnRH, gonadotropin-releasing hormone; GHRH, growth hormone-releasing hormone; GHIH, growth hormone-inhibiting hormone; DOPA, dopamine; ACTH, adrenocorticotropic hormone; TSH, thyroid-stimulating hormone; LH, luteinizing hormone; FSH, follicle-stimulating hormone; GH, growth hormone.
While these systems promote adaptation and allostasis (i.e., active maintenance of homeostasis) for performance in challenging situations, recent research shows that their repeated activation accumulates allostatic load leading to allostatic overload. This results in long-term maladaptation, which is often described in an HPA hyper- or hypofunction which then invokes immune, cardiovascular, or metabolic system maladaptation. The development of allostatic load is based on neural, neuroendocrine, and neuroendocrine-immune mechanisms and associated with a long list of pathologies [HPA hyperfunction: e.g., depression, Cushing’s syndrome, and type II diabetes; HPA hypofunction: e.g., multiple sclerosis and rheumatoid arthritis (8, 10)].
MS: Effects on Bone Structure and Function
Although the exact mechanisms remain to be elucidated and potential confounders (e.g., comorbidities, pharmacological therapies, physical inactivity, and sex) to be excluded, depression and anxiety have more recently been identified as additional risk factors for disturbed bone homeostasis, osteoporosis, and fractures in humans (11–14). For example, postmenopausal women suffering from depression exhibited decreased lumbar vertebra and femur DEXA scores compared to non-depressed controls, indicating a possible relationship between MS and bone mineral density (BMD) (15). A negative association between depression and BMD has been found in the majority of studies for both sexes, although many studies have not clearly defined MS (11, 13). Unfortunately, results on MS and BMD are often obtained from studies focusing on other topics (e.g., obesity/diabetes) and, furthermore, do not provide information discerning between acute or traumatic episodic stress effects. Therefore, future research in humans should differentiate specific stressor types and characteristics and improve patient selection and confounder controlling (smoking, medication, and physical activity) in studies. A systematical evaluation of pathways and mechanism linking MS and bone health (structure/function) could help establishing a conceptual framework to estimate whether stress-related effects will be naturally compensated for or can be blunted or even reversed naturally or therapeutically.
Compared to human studies, animal models allow for tighter control of experimental conditions, avoidance of confounders, and lower subject variability. A variety of methods exist to induce stress in animals, and these are often combined to complex regimes to prevent habituation effects: water deprivation, overnight illumination, stroboscopic illumination, cage tilt, housing in soiled cages, or exposure to noise (16, 17). Traumatic stress can be simulated by application of electric (foot) shocks (18, 19), exposure to predators (20) or physical restraint (21), or by exposure of pups to unpredictable maternal separation (22). Aside from selecting a suitable stress model to test the respective study hypothesis, choosing the most appropriate species is also crucial. Historically, a variety of animals have been used in bone research, but species differences, e.g., lack of a trabecular and Haversian remodeling in rodents, exaggerates the importance when choosing the most appropriate model (23–27). In rats, MS promotes disintegration of periodontal bone tissue (28). In mice, exposure to chronic stress activates bone resorption and suppresses bone formation shown by reduced BMD, deteriorated microarchitecture and/or altered biochemical markers (18, 29, 30). Further studies have investigated the role of stressor characteristics (social isolation/electroshock) on bone mass in mice, concluding that bone mass can be affected by different stressor characteristics (31). Future research would benefit from using newer techniques, such as in vivo micro-computer tomography imaging, to monitor changes in bone microarchitecture throughout an experiment (32, 33). Compelling evidence indicates early life stress has epigenetic consequences in humans and animals with similar findings emerging for chronic stress and depression in adults (34–38). Therefore, focusing on animal models would be useful in studying the consequences of intergenerational MS transmission on bone biology as well as the testing of therapeutic interventions, pharmaceutical as well as environmental (e.g., enriched living conditions) (39).
MS: Molecular Mechanism Leading to Bone Adaptation
Currently, the key molecular mediators between MS and bone health are considered to be growth hormones (GH), glucocorticoids, and inflammatory cytokines (see Figure 1). Levels of GH are altered by MS through modulation of the HPA axis and consequent upregulation of growth hormone-releasing hormone and growth hormone-inhibiting hormone (40). Recent in vitro studies demonstrated that GH can directly induce osteoblast proliferation and differentiation (41–43). For example, in an embryonal rat tibiae model, GH not only increased alkaline phosphatase levels but also induced local production of insulin-like growth factor-I (IGF-I) (41). In fact, numerous other studies have described the stimulatory effect of GH on IGF-I production and its role as an important growth factor in bone (44–48). Using IGF-I-overexpressing mice, Zhao et al. were able to demonstrate that IGF-I has anabolic effects by primarily promoting the activity of resident osteoblasts, but potentially also by prolonging the life span of osteocytes (46). Circulating IGF-I is then detected by the hypothalamus and pituitary gland, completing the negative feedback loop for gaining allostasis through suppressing GH secretion. When this feedback system is desensitized through chronic stress, GH deficiency can develop leading to catabolic effects on bone (49, 50). Mouse models of IGF-I and GH deficiencies present with (up to 87%) reduced postnatal bone mineral content (51), as well as (approximately 40%) reduced femoral length and bone size (52). Among other mechanisms, these outcomes are related to the fact that parathyroid hormone (PTH), which has well-known anabolic, bone-forming effects, is dependent on osteoblast-driven IGF-I production (53). Similarly, humans with GH deficiency acquired in adulthood show decreased BMD (54, 55). Furthermore, aging diminishes GH/IGF-I secretion and response and causes gradual deterioration of both the immune and endocrine systems (56).
The HPA axis is, furthermore, responsible for the release of glucocorticoids (predominately cortisol) (40), which in turn influences the basal HPA activity and termination of the stress response by acting on other regulatory centers, such as the hippocampus, frontal cortex, hypothalamus, and the pituitary gland (40, 57). Although exact mechanisms are not completely understood, glucocorticoids are believed to have a multifaceted and dose-dependent role in bone formation and homeostasis. At physiological levels, glucocorticoids promote bone formation through induction of osteogenic differentiation of progenitor cells (58). In contrast, elevated levels of cortisol directly inhibit osteoblast proliferation, differentiation, and apoptosis in various species (59–62), which could substantially blunt the bone formation process leading to lower bone density. Importantly, aging causes greater activation of the HPA axis, which results in elevated production of glucocorticoids and stronger feelings of stress, anxiousness, and depression as well as an overall detrimental shift (63).
Aside from direct effects on bone cells, glucocorticoids also inhibit GH and gonadal steroid production, further reducing bone mass. The effects of high cortisol levels are seen in patients suffering from Cushing’s syndrome, which typically present with decreased bone mass and quality (64, 65) as well as in certain types of depression and chronic anxiety disorders (see Mental Stress (MS): A Brief Summary) (16, 66–68).
Glucocorticoids further influence the transport and function of leukocytes and thus inhibit the production of pro-inflammatory cytokines [e.g., tumor necrosis factor alpha (TNF-α), interleukin (IL)-1β, and IL-6], e.g., via glucocorticoid receptor (GR)-induced suppression of nuclear factor kappa B and activator protein 1 (69). While these findings suggest an overall anti-inflammatory effect of stress, in vivo mechanisms are more complex. Recent research demonstrates chronic stress can induce GR resistance leading to decreased sensitivity of immune cells to glucocorticoids and a resultant inability to downregulate inflammatory responses (50, 70). GR resistance and impaired HPA responsiveness—relevant in numerous inflammatory diseases (e.g., rheumatoid arthritis)—play supposedly a role in the development of osteoporosis (71). Interestingly, patients suffering from chronic inflammatory diseases have a higher prevalence of osteoporotic fractures, providing an additional indirect link between GR resistance and bone pathologies (72). Aside from GR resistance, corticotropin-releasing hormone (CRH), also secreted during MS, induces the release of IL-6 (73–75). High levels of IL-6 and other cytokines, such as IL-1β and TNF-α, affect differentiation of mesenchymal stem cells, suppress osteoblast function, initiate osteoclastogenesis, and activate osteoclast function (76, 77). These findings indicate a delicate inflammatory balance exists to ensure appropriate bone formation in vivo.
Although the isolated pathways of these three mediators of bone mass are well evaluated, knowledge regarding their interaction and buffering effects due to environmental conditions in enriched living conditions is lacking. For this reason, future research should include genetically modified animals, such as conditional IGF-I knockout mice (78), opening the possibility for mechanistic investigations as well as environmental mediators like PS.
Interplay Between MS and PS
Despite no studies having been conducted to directly investigate the interplay between PS and MS, indirect indications of potential cross talk exist. Studies examining the loading of bone cells under different biochemical conditions (MS conditions or normal culture conditions) and/or with subsequent analysis of MS biomarkers have provided useful data.
One molecule that may allow interplay between stress types is IGF-I (see MS: Effects on Bone Structure and Function). In osteoblasts and osteocytes, IGF-I signaling is also activated in response to PS, leading to enhanced IGF production, IGF responsiveness, and inhibition of TNF-α-induced apoptosis, whereas inhibition of IGF-I abolishes loading-induced osteoblast proliferation (79–83). In osteoblasts, IGF-I synthesis is positively controlled by PTH via a cAMP-dependent mechanism (84). Interestingly, PTH was shown to enhance the PS-induced osteogenic response to PS, although this effect was found to be age dependent, indicating the complex interplay between MS, PS, and aging (85).
While mice overexpressing IGF-I reveal greater bone formation in response to PS than wild-type mice, knockout mice present with less periosteal bone formation as well as less trabecular bone volume, thickness, and density (82, 86). Similarly, GH has been found to modulate the levels of loading required to induce bone formation (87).
Inflammatory environments as found in patients exposed to MS can restrict osteocyte responses to PS, altering mechanotransduction mechanisms in bone (88). On the other hand, physiological PS prevents cytokine-induced osteoclast activation and bone loss exerting a protective role during inflammatory conditions and possibly MS (88, 89). MS, i.e., an inflammatory environment, could thus enhance osteocyte-to-osteoclast communication and osteoclastogenesis, whereas PS could be counteractive.
A further example is sustained physical conditioning that improves the performance of several allostatic mediators; for example, physically trained humans show a “trained HPA function” represented in a decreased HPA response under certain stress conditions (90–92). On the contrary, chronic strenuous exercise (CSE) can lead to overtraining, which is hallmarked by maladapted responses to excessive exercise without adequate rest, and perturbs multiple body systems (nervous, endocrine, and immune). CSE can increase basal glucocorticoid levels provoking mild hypercortisolism, diminishing the reactivity of adrenocorticotropic hormone and cortisol to CRH comparable to that of depressed persons. CSE also increases catecholamine levels, favoring a Th2 immunity profile, and inhibits long-term gonadal function (90, 93). To summarize, moderate PS provides health benefits, while CSE and overtraining can provoke biochemical and clinical abnormalities. This also influences bone quality, as already described in the “female athlete triad” (reproductive dysfunction, infertility, and osteoporosis) (90).
While current results point toward cross talk between PS and MS, further evidence for interaction, identification of dose–responses, and elucidation of molecular mechanisms is needed. Therefore, models of MS should initially be combined with existing animal models of PS, such as cyclic compression of caudal vertebrae, cyclic axial loading of the ulna or vibration platforms in rabbits, rats, or mice and later translated to human studies (94–98).
Conclusion
In addition to comprehensive mechanobiological concepts showing the importance of PS in bone health and disease, compelling evidence has recently emerged that biochemical and psychoneuroendocrinological maladaptations caused by MS are not only also relevant for bone quality, but may furthermore considerably interact with PS. Furthermore, it is unclear how age-related risk factors interplay and/or whether they can synergistically impair bone health.
Based on the highlighted limitations of previous research as well as current gaps in our knowledge, we propose several new research avenues in humans and animals including (1) the investigation of different types of MS (traumatic/chronic/acute) as well as their molecular mechanisms and dose-dependent effects on bone deformation and structure, (2) the incorporation of physical activity in models of MS, (3) investigation of genetically modified animals for evaluation of mechanistic effects of PS in environmental conditions, (4) epigenetics, and (5) the investigation of aging within the aforementioned studies.
Author Contributions
P-MW, MR, GK, and KW-K drafted, edited, and finalized the manuscript; approved the final version of manuscript; and agreed to be accountable for all aspects of the work.
Conflict of Interest Statement
The authors declare that this mini review was written in the absence of commercial or financial relationships that could be interpreted as possible conflicts of interest.
Funding
No funding was received for this work.
Abbreviations
ACTH, adrenocorticotropic hormone; AP-1, activator protein 1; BMD, bone mineral density; CRH, corticotropin-releasing hormone; CSE, chronic strenuous exercise; CT, computer tomography; GH, growth hormones; GHIH, growth hormone-inhibiting hormone; GHRH, growth hormone-releasing hormone; GR, glucocorticoid receptor; HPA, hypothalamic–pituitary–adrenal; HPG, hypothalamic–pituitary–gonadal; HPP, hypothalamic–pituitary–prolactin; HPS, hypothalamic–pituitary–somatotropic; HPT, hypothalamic–pituitary–thyroid; IGF-I, insulin-like growth factor-I; IL, interleukin; MS, mental stress; NF-κB, nuclear factor kappa B; PS, physical stress; PTH, parathyroid hormone; SAM, sympathetic–adrenal–medullary system; SNS, sympathetic–nervous system; TNF-α, tumor necrosis factor alpha.
References
1. Nomura S, Takano-Yamamoto T. Molecular events caused by mechanical stress in bone. Matrix Biol (2000) 19:91–6. doi:10.1016/S0945-053X(00)00050-0
2. Raggatt LJ, Partridge NC. Cellular and molecular mechanisms of bone remodeling. J Biol Chem (2010) 285:25103–8. doi:10.1074/jbc.R109.041087
3. Klein-Nulend J, Bacabac RG, Bakker AD. Mechanical loading and how it affects bone cells: the role of the osteocyte cytoskeleton in maintaining our skeleton. Eur Cell Mater (2012) 24:278–91. doi:10.22203/eCM.v024a20
4. Tan VPS, Macdonald HM, Kim S, Nettlefold L, Gabel L, Ashe MC, et al. Influence of physical activity on bone strength in children and adolescents: a systematic review and narrative synthesis. J Bone Miner Res (2014) 29:2161–81. doi:10.1002/jbmr.2254
5. Tobias JH. Editorial: Mechanical loading and bone. Front Endocrinol (2015) 6:184. doi:10.3389/fendo.2015.00184
7. Dickerson SS, Kemeny ME. Acute stressors and cortisol responses: a theoretical integration and synthesis of laboratory research. Psychol Bull (2004) 130:355–91. doi:10.1037/0033-2909.130.3.355
8. McEwen BS. Stress, adaptation, and disease: allostasis and allostatic load. Ann N Y Acad Sci (2006) 840:33–44. doi:10.1111/j.1749-6632.1998.tb09546.x
9. Levi L. Stress and Distress in Response to Psychosocial Stimuli: Laboratory and Real-Life Studies on Sympatho-Adrenomedullary and Related Reactions. Amsterdam: Elsevier (2016).
10. McEwen BS. Protection and damage from acute and chronic stress: allostasis and allostatic overload and relevance to the pathophysiology of psychiatric disorders. Ann N Y Acad Sci (2004) 1032:1–7. doi:10.1196/annals.1314.001
11. Mezuk B, Eaton WW, Golden SH. Depression and osteoporosis: epidemiology and potential mediating pathways. Osteoporos Int (2008) 19:1–12. doi:10.1007/s00198-007-0449-2
12. Cizza G, Primma S, Coyle M, Gourgiotis L, Csako G. Depression and osteoporosis: a research synthesis with meta-analysis. Horm Metab Res (2010) 42:467–82. doi:10.1055/s-0030-1252020
13. Azuma K, Adachi Y, Hayashi H, Kubo KY. Chronic psychological stress as a risk factor of osteoporosis. J UOEH (2015) 37:245–53. doi:10.7888/juoeh.37.245
14. Williams LJ, Pasco JA, Jackson H, Kiropoulos L, Stuart AL, Jacka FN, et al. Depression as a risk factor for fracture in women: a 10 year longitudinal study. J Affect Disord (2016) 192:34–40. doi:10.1016/j.jad.2015.11.048
15. Furlan PM, Ten Have T, Cary M, Zemel B, Wehrli F, Katz IR, et al. The role of stress-induced cortisol in the relationship between depression and decreased bone mineral density. Biol Psychiatry (2005) 57:911–7. doi:10.1016/j.biopsych.2004.12.033
16. Yirmiya R, Bab I. Major depression is a risk factor for low bone mineral density: a meta-analysis. Biol Psychiatry (2009) 66:423–32. doi:10.1016/j.biopsych.2009.03.016
17. Willner P. Chronic mild stress (CMS) revisited: consistency and behavioural-neurobiological concordance in the effects of CMS. Neuropsychobiology (2005) 52:90–110. doi:10.1159/000087097
18. Yu HR, Watt H, Kesavan C, Johnson PJ, Wergedal JE, Mohan S. Lasting consequences of traumatic events on behavioral and skeletal parameters in a mouse model for post-traumatic stress disorder (PTSD). PLoS One (2012) 7:e42684. doi:10.1371/journal.pone.0042684
19. Louvart H, Maccari S, Ducrocq F, Thomas P, Darnaudery M. Long-term behavioural alterations in female rats after a single intense footshock followed by situational reminders. Psychoneuroendocrinology (2005) 30:316–24. doi:10.1016/j.psyneuen.2004.09.003
20. Adamec R, Walling S, Burton P. Long-lasting, selective, anxiogenic effects of feline predator stress in mice. Physiol Behav (2004) 83:401–10. doi:10.1016/j.physbeh.2004.08.029
21. Schwab CL, Fan RP, Zheng Q, Myers LP, Hebert P, Pruett SB. Modeling and predicting stress-induced immunosuppression in mice using blood parameters. Toxicol Sci (2005) 83:101–13. doi:10.1093/toxsci/kfi014
22. Weiss IC, Franklin TB, Vizi S, Mansuy IM. Inheritable effect of unpredictable maternal separation on behavioral responses in mice. Front Behav Neurosci (2011) 5:3. doi:10.3389/fnbeh.2011.00003
23. Thompson DD, Simmons HA, Pirie CM, Ke HZ. FDA guidelines and animal models for osteoporosis. Bone (1995) 17:125S–33S. doi:10.1016/8756-3282(95)00285-L
24. Turner AS. Animal models of osteoporosis – necessity and limitations. Eur Cell Mater (2001) 1:66–81. doi:10.22203/eCM.v001a08
25. Pogoda P, Priemel M, Schilling AF, Gebauer M, Catala-Lehnen P, Barvencik F, et al. Mouse models in skeletal physiology and osteoporosis: experiences and data on 14,839 cases from the Hamburg Mouse Archives. J Bone Miner Metab (2005) 23(Suppl):97–102. doi:10.1007/BF03026332
26. Lelovas PP, Xanthos TT, Thoma SE, Lyritis GP, Dontasi IA. The laboratory rat as an animal model for osteoporosis research. Comp Med (2008) 58:424–30.
27. Zhang ZD, Ren H, Shen GY, Qiu T, Liang D, Yang ZD, et al. Animal models for glucocorticoid-induced postmenopausal osteoporosis: an updated review. Biomed Pharmacother (2016) 84:438–46. doi:10.1016/j.biopha.2016.09.045
28. Neporada KS, Leont’eva FS, Tarasenko LM. Chronic stress impairs structural organization of organic matrix in bone tissue of rat periodontium. Bull Exp Biol Med (2003) 135:543–4. doi:10.1023/A:1025464932135
29. Yirmiya R, Goshen I, Bajayo A, Kreisel T, Feldman S, Tam J, et al. Depression induces bone loss through stimulation of the sympathetic nervous system. Proc Natl Acad Sci U S A (2006) 103:16876–81. doi:10.1073/pnas.0604234103
30. Furuzawa M, Chen HY, Fujiwara S, Yamada K, Kubo K. Chewing ameliorates chronic mild stress-induced bone loss in senescence-accelerated mouse (SAMP8), a murine model of senile osteoporosis. Exp Gerontol (2014) 55:12–8. doi:10.1016/j.exger.2014.03.003
31. Seferos N, Kotsiou A, Petsaros S, Rallis G, Tesseromatis C. Mandibular bone density and calcium content affected by different kind of stress in mice. J Musculoskelet Neuronal Interact (2010) 10:231–6.
32. Schulte FA, Lambers FM, Webster DJ, Kuhn G, Muller R. In vivo validation of a computational bone adaptation model using open-loop control and time-lapsed micro-computed tomography. Bone (2011) 49:1166–72. doi:10.1016/j.bone.2011.08.018
33. Lambers FM, Kuhn G, Schulte FA, Koch K, Muller R. Longitudinal assessment of in vivo bone dynamics in a mouse tail model of postmenopausal osteoporosis. Calcif Tissue Int (2012) 90:108–19. doi:10.1007/s00223-011-9553-6
34. Franklin TB, Russig H, Weiss IC, Graff J, Linder N, Michalon A, et al. Epigenetic transmission of the impact of early stress across generations. Biol Psychiatry (2010) 68:408–15. doi:10.1016/j.biopsych.2010.05.036
35. Hammen C, Hazel NA, Brennan PA, Najman J. Intergenerational transmission and continuity of stress and depression: depressed women and their offspring in 20 years of follow-up. Psychol Med (2012) 42:931–42. doi:10.1017/S0033291711001978
36. Rodgers AB, Morgan CP, Bronson SL, Revello S, Bale TL. Paternal stress exposure alters sperm microRNA content and reprograms offspring HPA stress axis regulation. J Neurosci (2013) 33:9003–12. doi:10.1523/JNEUROSCI.0914-13.2013
37. Toepfer P, Heim C, Entringer S, Binder E, Wadhwa P, Buss C. Oxytocin pathways in the intergenerational transmission of maternal early life stress. Neurosci Biobehav Rev (2016) 73:293–308. doi:10.1016/j.neubiorev.2016.12.026
38. Bowers ME, Yehuda R. Intergenerational transmission of stress in humans. Neuropsychopharmacology (2016) 41:232–44. doi:10.1038/npp.2015.247
39. Gapp K, Bohacek J, Grossmann J, Brunner AM, Manuella F, Nanni P, et al. Potential of environmental enrichment to prevent transgenerational effects of paternal trauma. Neuropsychopharmacology (2016) 41:2749–58. doi:10.1038/npp.2016.87
40. Chrousos GP. Stressors, stress, and neuroendocrine integration of the adaptive response. The 1997 Hans Selye Memorial Lecture. Ann N Y Acad Sci (1998) 851:311–35. doi:10.1111/j.1749-6632.1998.tb09006.x
41. Stracke H, Schulz A, Moeller D, Rossol S, Schatz H. Effect of growth hormone on osteoblasts and demonstration of somatomedin-C/IGF I in bone organ culture. Acta Endocrinol (1984) 107:16–24.
42. Kassem M, Blum W, Ristelli J, Mosekilde L, Eriksen EF. Growth hormone stimulates proliferation and differentiation of normal human osteoblast-like cells in vitro. Calcif Tissue Int (1993) 52:222–6. doi:10.1007/BF00298723
43. Nilsson A, Swolin D, Enerback S, Ohlsson C. Expression of functional growth hormone receptors in cultured human osteoblast-like cells. J Clin Endocrinol Metab (1995) 80:3483–8. doi:10.1210/jc.80.12.3483
44. Birnbaum RS, Bowsher RR, Wiren KM. Changes in IGF-I and -II expression and secretion during the proliferation and differentiation of normal rat osteoblasts. J Endocrinol (1995) 144:251–9. doi:10.1677/joe.0.1440251
45. Rydziel S, Delany AM, Canalis E. Insulin-like growth factor I inhibits the transcription of collagenase 3 in osteoblast cultures. J Cell Biochem (1997) 67:176–83. doi:10.1002/(SICI)1097-4644(19971101)67:2<176::AID-JCB3>3.3.CO;2-D
46. Zhao G, Monier-Faugere MC, Langub MC, Geng Z, Nakayama T, Pike JW, et al. Targeted overexpression of insulin-like growth factor I to osteoblasts of transgenic mice: increased trabecular bone volume without increased osteoblast proliferation. Endocrinology (2000) 141:2674–82. doi:10.1210/en.141.7.2674
47. Zhang M, Xuan S, Bouxsein ML, von Stechow D, Akeno N, Faugere MC, et al. Osteoblast-specific knockout of the insulin-like growth factor (IGF) receptor gene reveals an essential role of IGF signaling in bone matrix mineralization. J Biol Chem (2002) 277:44005–12. doi:10.1074/jbc.M208265200
48. Olney RC. Regulation of bone mass by growth hormone. Med Pediatr Oncol (2003) 41:228–34. doi:10.1002/mpo.10342
49. Chrousos GP, Deterawadleigh SD, Karl M. Syndromes of glucocorticoid resistance. Ann Intern Med (1993) 119:1113–24. doi:10.7326/0003-4819-119-11-199312010-00009
50. Cohen S, Janicki-Deverts D, Doyle WJ, Miller GE, Frank E, Rabin BS, et al. Chronic stress, glucocorticoid receptor resistance, inflammation, and disease risk. Proc Natl Acad Sci U S A (2012) 109:5995–9. doi:10.1073/pnas.1118355109
51. Mohan S, Kesavan C. Role of insulin-like growth factor-1 in the regulation of skeletal growth. Curr Osteoporos Rep (2012) 10:178–86. doi:10.1007/s11914-012-0100-9
52. Mohan S, Richman C, Guo RQ, Amaar Y, Donahue LR, Wergedal J, et al. Insulin-like growth factor regulates peak bone mineral density in mice by both growth hormone-dependent and -independent mechanisms. Endocrinology (2003) 144:929–36. doi:10.1210/en.2002-220948
53. Bikle DD, Sakata T, Leary C, Elalieh H, Ginzinger D, Rosen CJ, et al. Insulin-like growth factor I is required for the anabolic actions of parathyroid hormone on mouse bone. J Bone Miner Res (2002) 17:1570–8. doi:10.1359/jbmr.2002.17.9.1570
54. Holmes SJ, Economou G, Whitehouse RW, Adams JE, Shalet SM. Reduced bone mineral density in patients with adult onset growth hormone deficiency. J Clin Endocrinol Metab (1994) 78:669–74. doi:10.1210/jcem.78.3.8126140
55. Degerblad M, Bengtsson BA, Bramnert M, Johnell O, Manhem P, Rosen T, et al. Reduced bone mineral density in adults with growth hormone (GH) deficiency: increased bone turnover during 12 months of GH substitution therapy. Eur J Endocrinol (1995) 133:180–8. doi:10.1530/eje.0.1330180
56. Corpas E, Harman SM, Blackman MR. Human growth hormone and human aging. Endocr Rev (1993) 14:20–39. doi:10.1210/edrv-14-1-20
57. Chrousos GP, Tsigos C, Kyrou I. Stress, endocrine manifestations, and diseases. Handbook of Stress Medicine and Health. Boca Raton, FL: CRC Press (2004). p. 101–29.
58. Zhou H, Cooper MS, Seibel MJ. Endogenous glucocorticoids and bone. Bone Res (2013) 1(2):107–9. doi:10.4248/BR201302001
59. Chyun YS, Kream BE, Raisz LG. Cortisol decreases bone formation by inhibiting periosteal cell proliferation. Endocrinology (1984) 114:477–80. doi:10.1210/endo-114-2-477
60. Delany AM, Dong Y, Canalis E. Mechanisms of glucocorticoid action in bone cells. J Cell Biochem (1994) 56:295–302. doi:10.1002/jcb.240560304
61. Patschan D, Loddenkemper K, Buttgereit F. Molecular mechanisms of glucocorticoid-induced osteoporosis. Bone (2001) 29:498–505. doi:10.1016/S8756-3282(01)00610-X
62. Pereira RM, Delany AM, Canalis E. Cortisol inhibits the differentiation and apoptosis of osteoblasts in culture. Bone (2001) 28:484–90. doi:10.1016/S8756-3282(01)00422-7
63. Bauer ME. Stress, glucocorticoids and ageing of the immune system. Stress (2005) 8:69–83. doi:10.1080/10253890500100240
64. Mancini T, Doga M, Mazziotti G, Giustina A. Cushing’s syndrome and bone. Pituitary (2004) 7:249–52. doi:10.1007/s11102-005-1051-2
65. Kaltsas G, Makras P. Skeletal diseases in Cushing’s syndrome: osteoporosis versus arthropathy. Neuroendocrinology (2010) 92:60–4. doi:10.1159/000314298
66. Dorn LD, Susman EJ, Pabst S, Huang B, Kalkwarf H, Grimes S. Association of depressive symptoms and anxiety with bone mass and density in ever-smoking and never-smoking adolescent girls. Arch Pediatr Adolesc Med (2008) 162:1181–8. doi:10.1001/archpedi.162.12.1181
67. Cizza G. Major depressive disorder is a risk factor for low bone mass, central obesity, and other medical conditions. Dialogues Clin Neurosci (2011) 13:73–87.
68. Calarge CA, Butcher BD, Burns TL, Coryell WH, Schlechte JA, Zemel BS. Major depressive disorder and bone mass in adolescents and young adults. J Bone Miner Res (2014) 29:2230–7. doi:10.1002/jbmr.2249
69. Cain DW, Cidlowski JA. Specificity and sensitivity of glucocorticoid signaling in health and disease. Best Pract Res Clin Endocrinol Metab (2015) 29:545–56. doi:10.1016/j.beem.2015.04.007
70. Marques AH, Silverman MN, Sternberg EM. Glucocorticoid dysregulations and their clinical correlates from receptors to therapeutics. Ann N Y Acad Sci (2009) 1179:1–18. doi:10.1111/j.1749-6632.2009.04987.x
71. Silverman MN, Sternberg EM. Glucocorticoid regulation of inflammation and its behavioral and metabolic correlates: from HPA axis to glucocorticoid receptor dysfunction. Ann N Y Acad Sci (2012) 1261:55–63. doi:10.1111/j.1749-6632.2012.06633.x
72. Hardy R, Cooper MS. Bone loss in inflammatory disorders. J Endocrinol (2009) 201:309–20. doi:10.1677/JOE-08-0568
73. Chrousos GP. The hypothalamic-pituitary-adrenal axis and immune-mediated inflammation. N Engl J Med (1995) 332:1351–62. doi:10.1056/NEJM199505183322008
74. Theoharides TC, Spanos C, Pang X, Alferes L, Ligris K, Letourneau R, et al. Stress-induced intracranial mast cell degranulation: a corticotropin-releasing hormone-mediated effect. Endocrinology (1995) 136:5745–50. doi:10.1210/endo.136.12.7588332
75. Chrousos GP. The stress response and immune function: clinical implications. The 1999 Novera H. Spector Lecture. Ann N Y Acad Sci (2000) 917:38–67. doi:10.1111/j.1749-6632.2000.tb05371.x
76. Lacey DC, Simmons PJ, Graves SE, Hamilton JA. Proinflammatory cytokines inhibit osteogenic differentiation from stem cells: implications for bone repair during inflammation. Osteoarthritis Cartilage (2009) 17:735–42. doi:10.1016/j.joca.2008.11.011
77. Croes M, Oner FC, Kruyt MC, Blokhuis TJ, Bastian O, Dhert WJ, et al. Proinflammatory mediators enhance the osteogenesis of human mesenchymal stem cells after lineage commitment. PLoS One (2015) 10:e0132781. doi:10.1371/journal.pone.0132781
78. Liu JL, Yakar S, LeRoith D. Conditional knockout of mouse insulin-like growth factor-1 gene using the Cre/loxP system. Proc Soc Exp Biol Med (2000) 223:344–51. doi:10.1046/j.1525-1373.2000.22349.x
79. Lean JM, Jagger CJ, Chambers TJ, Chow JW. Increased insulin-like growth factor I mRNA expression in rat osteocytes in response to mechanical stimulation. Am J Physiol (1995) 268:E318–27.
80. Reijnders CM, Bravenboer N, Tromp AM, Blankenstein MA, Lips P. Effect of mechanical loading on insulin-like growth factor-I gene expression in rat tibia. J Endocrinol (2007) 192:131–40. doi:10.1677/joe.1.06880
81. Triplett JW, O’Riley R, Tekulve K, Norvell SM, Pavalko FM. Mechanical loading by fluid shear stress enhances IGF-1 receptor signaling in osteoblasts in a PKCzeta-dependent manner. Mol Cell Biomech (2007) 4:13–25. doi:10.3970/mcb.2007.004.013
82. Kesavan C, Wergedal JE, Lau KHW, Mohan S. Conditional disruption of IGF-I gene in type 1 alpha collagen-expressing cells shows an essential role of IGF-I in skeletal anabolic response to loading. Am J Physiol Endocrinol Metab (2011) 301:E1191–7. doi:10.1152/ajpendo.00440.2011
83. Wasserman E, Webster D, Kuhn G, Attar-Namdar M, Muller R, Bab I. Differential load-regulated global gene expression in mouse trabecular osteocytes. Bone (2013) 53:14–23. doi:10.1016/j.bone.2012.11.017
84. Lombardi G, Di Somma C, Vuolo L, Guerra E, Scarano E, Colao A. Role of IGF-I on PTH effects on bone. J Endocrinol Invest (2010) 33:22–6.
85. Meakin LB, Todd H, Delisser PJ, Galea GL, Moustafa A, Lanyon LE, et al. Parathyroid hormone’s enhancement of bones’ osteogenic response to loading is affected by ageing in a dose- and time-dependent manner. Bone (2017) 98:59–67. doi:10.1016/j.bone.2017.02.009
86. Gross TS, Srinivasan S, Liu CC, Clemens TL, Bain SD. Noninvasive loading of the murine tibia: an in vivo model for the study of mechanotransduction. J Bone Miner Res (2002) 17:493–501. doi:10.1359/jbmr.2002.17.3.493
87. Forwood MR, Li L, Kelly WL, Bennett MB. Growth hormone is permissive for skeletal adaptation to mechanical loading. J Bone Miner Res (2001) 16:2284–90. doi:10.1359/jbmr.2001.16.12.2284
88. Kulkarni RN, Bakker AD, Everts V, Klein-Nulend J. Mechanical loading prevents the stimulating effect of IL-1beta on osteocyte-modulated osteoclastogenesis. Biochem Biophys Res Commun (2012) 420:11–6. doi:10.1016/j.bbrc.2012.02.099
89. Pathak JL, Bravenboer N, Luyten FP, Verschueren P, Lems WF, Klein-Nulend J, et al. Mechanical loading reduces inflammation-induced human osteocyte-to-osteoclast communication. Calcif Tissue Int (2015) 97:169–78. doi:10.1007/s00223-015-9999-z
90. Mastorakos G, Pavlatou M, Diamanti-Kandarakis E, Chrousos GP. Exercise and the stress system. Hormones (2005) 4:73–89.
91. McEwen BS. Physiology and neurobiology of stress and adaptation: central role of the brain. Physiol Rev (2007) 87:873–904. doi:10.1152/physrev.00041.2006
92. Rimmele U, Seiler R, Marti B, Wirtz PH, Ehlert U, Heinrichs M. The level of physical activity affects adrenal and cardiovascular reactivity to psychosocial stress. Psychoneuroendocrinology (2009) 34:190–8. doi:10.1016/j.psyneuen.2008.08.023
93. Kreher JB, Schwartz JB. Overtraining syndrome: a practical guide. Sports Health (2012) 4:128–38. doi:10.1177/1941738111434406
94. Guo XE, Eichler MJ, Takai E, Kim CH. Quantification of a rat tail vertebra model for trabecular bone adaptation studies. J Biomech (2002) 35:363–8. doi:10.1016/S0021-9290(01)00212-3
95. Webster D, Wasserman E, Ehrbar M, Weber F, Bab I, Muller R. Mechanical loading of mouse caudal vertebrae increases trabecular and cortical bone mass-dependence on dose and genotype. Biomech Model Mechanobiol (2010) 9:737–47. doi:10.1007/s10237-010-0210-1
96. Lambers FM, Schulte FA, Kuhn G, Webster DJ, Muller R. Mouse tail vertebrae adapt to cyclic mechanical loading by increasing bone formation rate and decreasing bone resorption rate as shown by time-lapsed in vivo imaging of dynamic bone morphometry. Bone (2011) 49:1340–50. doi:10.1016/j.bone.2011.08.035
97. Novotny SA, Mehta H, Lowe DA, Nuckley DJ. Vibration platform for mice to deliver precise, low intensity mechanical signals to the musculoskeleton. J Musculoskelet Neuronal Interact (2013) 13:412–7. doi:10.1371/journal.pone.0104339
Keywords: biomechanics, bone–brain–nervous system interactions, endocrine pathways, osteoporosis, exercise
Citation: Wippert P-M, Rector M, Kuhn G and Wuertz-Kozak K (2017) Stress and Alterations in Bones: An Interdisciplinary Perspective. Front. Endocrinol. 8:96. doi: 10.3389/fendo.2017.00096
Received: 03 March 2017; Accepted: 18 April 2017;
Published: 01 May 2017
Edited by:
Susan Miranda, University of Tennessee Health Science Center, USAReviewed by:
Jackson Cioni Bittencourt, University of São Paulo, BrazilRong Yuan, Southern Illinois University Carbondale, USA
Copyright: © 2017 Wippert, Rector, Kuhn and Wuertz-Kozak. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Pia-Maria Wippert, d2lwcGVydEB1bmktcG90c2RhbS5kZQ==