 Annett Helleskov1
Annett Helleskov1 Maria Melikyan2
Maria Melikyan2 Evgenia Globa3
Evgenia Globa3 Inna Shcherderkina4Fani Poertner1Anna-Maria Larsen1Karen Filipsen1
Inna Shcherderkina4Fani Poertner1Anna-Maria Larsen1Karen Filipsen1 Klaus Brusgaard5
Klaus Brusgaard5 Charlotte Dahl Christiansen1Lars Kjaersgaard Hansen1
Charlotte Dahl Christiansen1Lars Kjaersgaard Hansen1 Henrik T. Christesen1*
Henrik T. Christesen1*
- 1Hans Christian Andersen Children’s Hospital, Odense, Denmark
- 2Pediatric, Endocrinology Scientific Centre, Russian Academy of Science, Moscow, Russia
- 3Pediatric Endocrinology, Ukrainian Centre of Endocrine Surgery, Kyiv, Ukraine
- 4Pediatric, Endocrinology Scientific Centre, Moscow, Russia
- 5Clinical Genetic Department, Odense University Hospital, Odense, Denmark
Background/aims: Congenital hyperinsulinism (CHI) is a heterogeneous disease most frequently caused by KATP-channel (ABCC8 and KCNJ11) mutations, with neonatal or later onset, variable severity, and with focal or diffuse pancreatic involvement as the two major histological types. CHI confers a high risk of neurological impairment; however, sparsely studied in larger patient series. We assessed the neurodevelopmental outcome in children with CHI at follow-up in a mixed international cohort.
Methods: In two hyperinsulinism expert centers, 75 CHI patients were included (Russian, n = 33, referred non-Scandinavian, treated in Denmark n = 27, Scandinavian, n = 15). Hospital files were reviewed. At follow-up, neurodevelopmental impairment and neurodevelopmental, cognitive and motor function scores were assessed.
Results: Median (range) age at follow-up was 3.7 years (3.3 months–18.2 years). Neurodevelopmental impairment was seen in 35 (47%). Impairment was associated with abnormal brain magnetic resonance imaging (MRI); odds ratio (OR) (95% CI) 15.0 (3.0–74.3), p = 0.001; lowest recorded blood glucose ≤1 mmol/L; OR 3.8 (1.3–11.3), p = 0.015, being non-Scandinavian patient, OR 3.8 (1.2–11.9), p = 0.023; and treatment delay from first symptom to expert center >5 days; OR 4.0 (1.0–16.6), trend p = 0.05. In multivariate analysis (n = 31) for early predictors with exclusion of brain MRI, treatment delay from first symptom to expert center >5 days conferred a significantly increased risk of neurodevelopment impairment, adjusted OR (aOR) 15.6 (1.6–146.7), p = 0.016, while lowest blood glucose ≤1 mmol/L had a trend toward increased risk, aOR 3.5 (1.1–14.3), p = 0.058. No associations for early vs. late disease onset, KATP-channel mutations, disease severity, focal vs. diffuse disease, or age at follow-up were seen in uni- or multivariate analysis.
Conclusion: Not only very low blood glucose, but also insufficient treatment as expressed by delay until expert center hospitalization, increased the risk of neurodevelopmental impairment. This novel finding calls for improvements in spread of knowledge about CHI among health-care personnel and rapid contact with an expert CHI center on suspicion of CHI.
Introduction
Congenital hyperinsulinism (CHI) is a heterogeneous disease characterized by inappropriate hypersecretion of insulin from pancreatic islet β-cells resulting in hypoketotic hypoglycemia (1, 2). The incidence is estimated to 1:40,000–50,000 (3–5). Although rare, CHI is the most common form of persistent hypoglycemia of infancy and the most difficult to manage (6).
Mutations in at least ten genes have been identified to cause CHI (ABCC8, KCNJ11, GLUD1, GCK, HADH1, UCP2, HNF4A, HNF1A, SLC16A1, PHOX2B), most commonly in ABCC8 or KCNJ11 expressing the two subunits of the pancreatic β-cell KATP-channel, the sulfonylurea receptor 1, and the potassium inward rectifier 6.2 (Kir6.2) (7–11).
Focal CHI occurs in approximately 40% and is defined as a restricted pancreatic area with adenomatous β-cell hyperplasia, resulting in a lesion composed of confluent islet of Langerhans (9, 12). Focal CHI is strongly associated with a heterozygous, paternally inherited KATP-channel germline mutation combined with a focal loss of heterozygosity on chromosome 11p15 with loss of maternally expressed tumor suppressors, ABCC8 and KCNJ11 (2). 18F-DOPA PET/CT scan is the preferred method for preoperative localization of the focal lesion (13).
Diffuse CHI involves β-cells throughout the pancreas and is histologically characterized by hypertrophy of a few β-cells per islet. Recessive inactivating mutations in ABCC8 and KCNJ11 are the most common causes of medically unresponsive diffuse CHI, whereas dominantly inherited ABCC8/KCNJ11 mutations typically are medical responsive (2, 14). First line medical treatment after iv glucose and/or glucagon rescue is the KATP agonist diazoxide with the somatostatin analog octreotide as second line (15). Rapamycin, an mTOR inhibitor, has more recently been used with variable effect (16, 17).
The primary treatment goals are to increase and maintain blood glucose concentration above 3.5, or 3.9, mmol/L, depending on the reference (10). Although heterogeneous in phenotype, CHI often presents in the first days of life with severe and persistent hypoglycemia, representing a severe threat especially for brain metabolism. Furthermore, the elevated serum insulin level results in suppression of ketones bodies, which usually serve as alternative fuel to the brain in the absence of glucose (2, 18, 19) As glucose is required for the rapid growth of the brain, neonates and infants with CHI are especially susceptible to hypoglycemic damage (20, 21). Hypoglycemic convulsions and loss of consciousness are frequent and may be recurrent if not treated sufficiently. Abnormal neurodevelopment outcome has previously been reported in children with CHI, especially motor and speech delay in early childhood (22–30), but also epilepsy, visual impairment, and infantile spasms have been reported (31). Abnormal neurodevelopmental outcome has also been described for children with transient hyperinsulinism, comparable to persistent hyperinsulinism (32, 33).
It could be hoped that the increased recognition in the literature of CHI as a high-risk condition for brain damage has led to a higher prevalence of intact neurodevelopmental outcome today. However, systematic cohort follow-up studies of the neurodevelopmental outcome are sparse (27, 32, 34), especially studies that have clinically testing and neuro-pediatric assessments.
We, therefore, aimed to evaluate the neurodevelopmental outcomes for CHI patients from Scandinavia, Russia, and Eastern Europe including formal testing of cognitive development and motor function when possible.
Materials and Methods
An international cohort of children diagnosed with CHI was established in collaboration between the Endocrinology Scientific Centre, Russian Academy of Science, Moscow, Russia, and Hans Christian Andersen Children’s Hospital, Odense University Hospital, Denmark, the latter institution also treating patients from other Scandinavian countries, Eastern Europe, and Russia.
The cohort consisted of Scandinavian children, diagnosed, treated, and assessed at follow-up in Denmark at Hans Christian Andersen Children’s Hospital, Odense University Hospital; children from Russia who were diagnosed, treated, and assessed at follow-up in Russia, Endocrinology Scientific Centre, Russian Academy of Science, Moscow; and non-Scandinavians referred from Eastern Europe countries or Russia for further treatment in Denmark. The patients were treated according to international recommendations but with local variations in expertise (expert referral centers/local hospitals), drug availability, and preference. 18F-Fluoro-DOPA PET/CT was only performed in Odense, Denmark, in the period studied. Patients were recruited in the 4-year period January 2013 to December 2016.
The inclusion criteria were persistent (>1 month) CHI with inappropriately elevated p-insulin concentrations during hypoglycemia, defined as blood glucose below 2.5 mmol/L (age ≤3 days), or below 3.2 mmol/L (age >3 days). Exclusion criteria were prematurity [gestational age (GA) <32 weeks] and severe asphyxia.
From the hospital files, the following data were extracted: GA (weeks + days), birth weight, umbilical cord blood pH, and base excess, Apgar score at 1 and 5 min, age at first recorded hypoglycemia episode, blood glucose values and simultaneous beta cell peptides, maximal glucose infusion rate, age at CHI diagnosis, age at treatment start at a local hospital and at a CHI expert center, medical and surgical treatment, genetic investigations (rapid KATP-channel analysis; if negative followed by analysis of other CHI-related genes or next generation sequencing panel for nine known CHI genes), and 18F-Fluoro-DOPA PET/CT results.
Note was made of the treatment delay (in days) from first hypoglycemic sign to first hospital admission, and from first symptom to a CHI expert center. Neurological data were extracted from the hospital files including hypoglycemic seizures, electroencephalography (EEG) reports, diagnoses of epilepsy, head circumference (defining microcephaly <−2SD), blindness, hearing loss, cerebral palsy, neuropsychological development by clinical investigation, and magnetic resonance imaging (MRI) of the brain. In Odense University Hospital, all neurological investigations since 2011, except hearing and eye examination, were performed per protocol and not by indication. The latest reported data were used when appropriate.
After data collection, the children were categorized into severe or moderate disease. Severe CHI was a priori defined with one of the following criteria; maximal intravenous glucose >10 mL/kg/min; one or more episodes of hypoglycemia ≤1.0 mmol/L; or homozygous, compound heterozygous, or paternal heterozygous KATP-channel pathogenic mutations with no indication of dominant disease. If none of the above parameters were satisfied, the patients were categorized non-severe CHI.
Congenital hyperinsulinism was categorized as early onset when the first suspected hypoglycemic symptoms occurred <30 days of age; late onset >30 days.
Our cerebral clinical outcomes at follow-up included neurological examination by an expert pediatric neurologist, Bayley Scales of Infant and Toddler Development 3rd Edition (Bayley-III), Developmental Profile 3 (DP-3), Movement Assessment Battery for Children 2 (Movement ABC-2), and Wechsler’s Intelligence Scale for Children fourth edition (WISC-IV) when possible and appropriate. In addition to the follow-up investigations of CHI patients, we included Danish healthy siblings as controls when possible.
Pediatric neurologists, two physiotherapists, and a child psychologist performed the assessments. Bayley-III was used to assess fine and gross motor function, language (receptive and expressive), cognitive, and adaptive development at ages 1–42 months. The Russian children tested by a Russian pediatric neurologist tested all Bayley items. In Denmark, a child physiotherapist tested motor function only. Raw scores of successfully completed items were converted to scale scores and to composite scores (mean of 100) and transformed to a percentile score with a mean of 50.
Developmental Profile 3 was used for assessment of children in Russia from 2 to 6 years of age. DP-3 includes 180 items grouped into scores on adaptive behavior, social–emotional behavior, cognitive function, and communication. Scaled scores were transformed to mean standard scores. A DP-3 standard score of 100 is average and <70 denotes delayed development. A pediatric neurologist interviewed the parents to obtain this assessment.
Motor function was evaluated using Movement ABC-2. This test is suitable for examining 4–12 year-old children’s skills in three core areas: fine motor function, gross motor skills, and static/dynamic balance. The test provides a raw score and standard score, which is transformed to a percentile score with a mean of 50.
Cognitive function was evaluated in Danish children using WISC-IV, of which all 10 core subtests were administered. The full-scale IQ score and 4 index scores were derived (verbal comprehension, perceptual organization, processing speed, and working memory). These scales have population means of 100 and SDs of 15. WISC-IV is appropriate from the age of 6–16 years, 11 months. A single-blinded pediatric psychologist with experience in examining children conducted the WISC-IV testing and scoring.
After testing, the patients were categorized as with normal neurological development, e.g., Bayley-III or Movement ABC-2 ≥5 percentile, DP-3 score ≥70, or WISC-IV developmental quotient ≥70; or with neurodevelopmental impairment, e.g., Bayley-III or Movement ABC-2 score <5 percentile, DP-3 score <70, WISC-IV score <70, blindness, cerebral palsy, or epilepsy. Children who were not able to complete the testing due to severe neurological sequela, such as major intellectual disability or major motor impairment, were categorized to the neurodevelopmental impairment group.
Statistical Methods
Means were compared using an independent-sample t-test or Mann–Whitney, Mantel–Haenszel, or Fisher’s Exact test were appropriate. A p-value <0.05 was considered as statistical significant and a trend was considered for p-values 0.05–0.10. The association between potential predictive values and mild or severe neurodevelopmental impairment was analyzed. Covariates included lowest blood glucose value, treatment delay from first symptoms to admission at a local hospital or to an expert CHI center (analyzed as categorical values after search for optimal cut-offs), focal vs. diffuse, and CHI disease severity. Univariable regression analysis was followed by multivariable, backward logistic regression models to obtain adjusted odds ratios (aORs) for associations. Data were analyzed using Stata (StataCorp, College Station, TX, USA).
Ethics
The study was conducted according to the Helsinki Declaration. The Regional Committees on Health Research Ethics for Southern Denmark approved the study, protocol number S-20120119 and by the local Ethical Committee at the Endocrine Research Center of the Ministry of Health Care in Russian Federation.
Written informed consent was obtained from the parents/legal guardians of all research participants. The data were approved by the Danish Data Protection Agency (13/11991).
Results
The study included 75 children with CHI, Scandinavian group; n = 15, referred non-Scandinavians; n = 27, Russian group; n = 33. In non-Scandinavians, GA and disease severity were higher, and age at follow-up was lower, compared to Scandinavians, Table 1. In total, mean (SD) GA was 38 (2.3) weeks; birth weight 3,553 (730) g. The median (range) onset of first symptoms was ** (**-**) days. Early onset CHI (<1 months) was seen in 50 (67%; median ** days) and late onset in 25 (33%; median ** days). The mean (SD) lowest recorded blood glucose for the total cohort was 0.9 (0.7) mmol/L. Genetic analysis was performed in n = 67 patients and KATP-channel mutations were identified in 58%, most frequently in non-Scandinavians, trend p = 0.054. No genetic variations were found in 32% of investigated patients. Medical treatment at last hospital contact included diazoxide (n = 44), octreotide (n = 16), long-acting release octreotide (n = 2), and mTOR inhibitor (n = 1). Forty (54%) children underwent an 18F-DOPA PET/CT scan, of which 15 (38%) had a focal CHI diagnosis by scan. All 15 patients with a focal CHI by 18F-DOPA PET/CT had restricted focal surgery with histology-confirmed focal lesion and were normoglycemic after surgery.
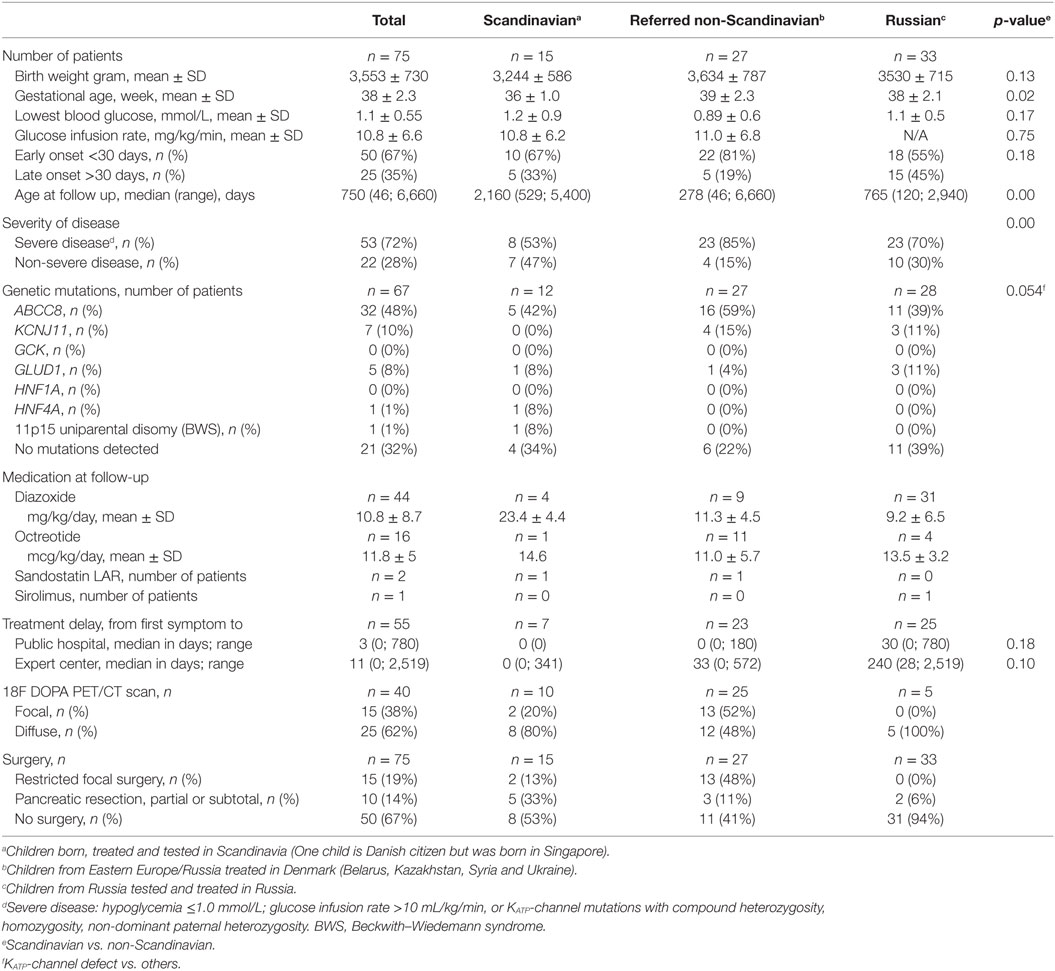
Table 1. Patient characteristics.
By clinical examination, six patients (8%) had microcephaly. Abnormal EEG was seen in 13 out of 55 (24%). MRI of the brain was performed in 40, of which in 19 (47.5%) had abnormal findings. White matter changes (hypoglycemic encephalopathy) was seen in 11 (27.5%) and cerebral atrophy in nine (22.5%) as the most frequent findings, Table 2. Infarctions were seen in two patients, probably after prolonged convulsions.
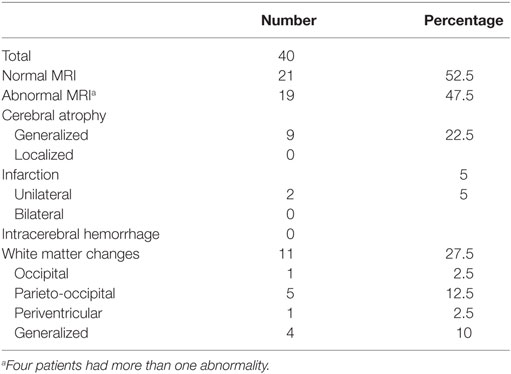
Table 2. Brain magnetic resonance imaging (MRI) findings in children with congenital hyperinsulinism.
Follow-up Investigations
The median age for the study group at the time of follow up was 3.7 years (range 3.3 months to 18.2 years). At the clinical follow-up, 10 patients (14%) had cerebral palsy, Table 3. Epilepsy was seen in 15 (20%), of which one was diagnosed with infantile spasms. Four (5%) had severe visual impairment; none had diagnosed hearing loss. Twenty-three (31%) had severe developmental delay based on clinical neurological examination. In total, 35 (41%) were categorized with severe neurodevelopmental impairment based on the above parameters.
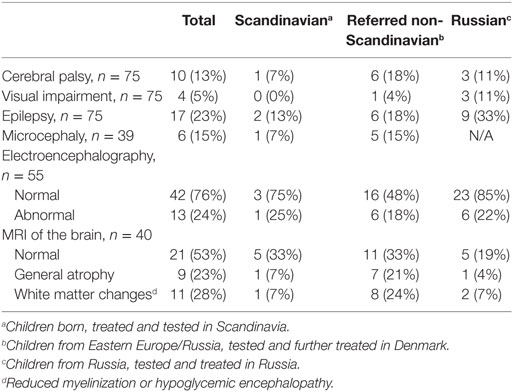
Table 3. Clinical neurological outcome for children with congenital hyperinsulinism.
Bayley Scales of Infant and Toddler Development
In the Bayley-III test, mean (SD) total motor function in 43 infants equaled the 29.5 (30) percentile, Table 4. The majority (n = 23, 58%) had a normal motor function ≥5 percentile with a mean (SD) of 53.4 (21.3) percentile. Fifteen (35%) were categorized as impaired by total motor score with a mean (SD) of 0.9 (1.4) percentile.
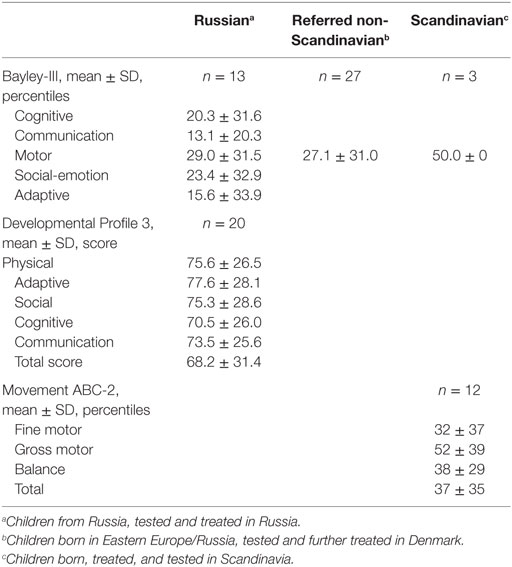
Table 4. Bayley-III, Developmental Profile 3 and Movement ABC-2 test results for children with congenital hyperinsulinism.
Mean (SD) cognitive Bayley-III score was 20 (31.6) in 13 infants, of which four had a mean <5 p denoting neurodevelopmental impairment. For communication, social–emotional behavior and adaptive behavior, mean (SD) percentile varied from 13 (20.3) to 23.4 (32.9). Based on all available Bayley-III data, 18 (42) were categorized as with neurodevelopmental impairment.
Developmental Profile 3
In the DP-3 test performed in Moscow in 20 children, mean (SD) full-scale score was 68 (31), which denoted to below average. Eight children (40%) were categorized as normally developed with a mean (SD) of 100 (23.8). Twelve children (60%) had a DP-3 score below 70 denoting neurodevelopmental impairment. Of these, nine had a score <50 denoting severe neurodevelopmental delay. The sub-items physical, adaptive, communication, cognitive, and social profile had a mean (SD) score from 77 (28) to 70 (26).
Movement ABC-2
Age-standardized motor function percentiles in the Danish patients aged >6 years could be compared to sibling controls (n = 5), thereby reducing the impact of parental factors. A significantly lower mean total motor function was observed in the CHI group, 38 ± 35 vs. 78 ± 35 percentile, p = 0.047, Table 5. This was driven by a significant lower balance function for the CHI patients, 38 ± 29 vs. 72 ± 26 percentile (p = 0.03), and to a less degree a reduced fine motor function, 32 ± 37 vs. 65 ± 34 percentile (trend p = 0.10). No significant difference was seen in gross motor function. Two children had a total motor function <5 p that denoted to the neurodevelopmental impairment group and two children had low normal total motor function score equaling 5–15 p.
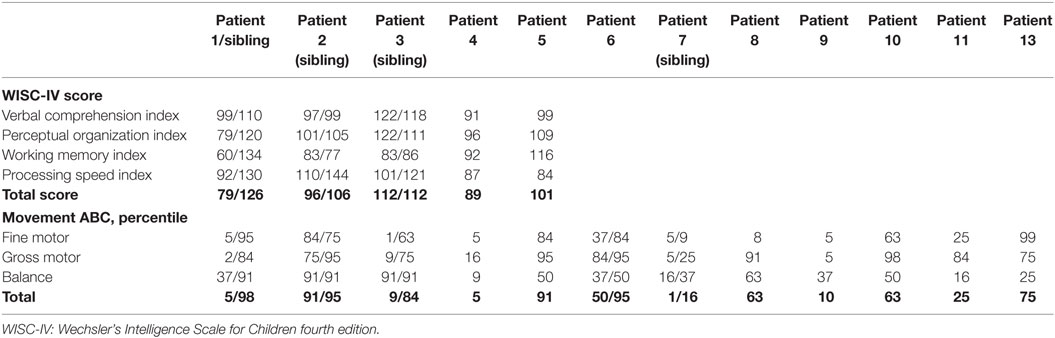
Table 5. Cognitive score and motor function for children with congenital hyperinsulinism above 6 years of age and healthy siblings as controls.
WISC-IV
Cognition was assessed by use of WISC-IV for five patients, of which three had a healthy sibling tested for control, Table 5. The mean (SD) full-scale score for the five patients were 95.6 (12.4) with a range of 79–112.
For the three tested sibling-pairs, a trend toward a nine score lower mean was observed for the CHI patients, 95 ± 12 vs. 114 ± 10 (p = 0.066). This trend was driven by a difference in the processing speed, 80 ± 14 vs. 131 ± 7, p = 0.054. All CHI children were on basis of this assessment denoted to the normally developed group.
Overall Outcome
The overall neurodevelopmental outcome is listed in Table 6. Of our total cohort, 47% (n = 35) had neurodevelopmental impairment. Low normal function by Bayley-III or movement ABC-2 equaling 5–15 percentile was detected in six (8%).
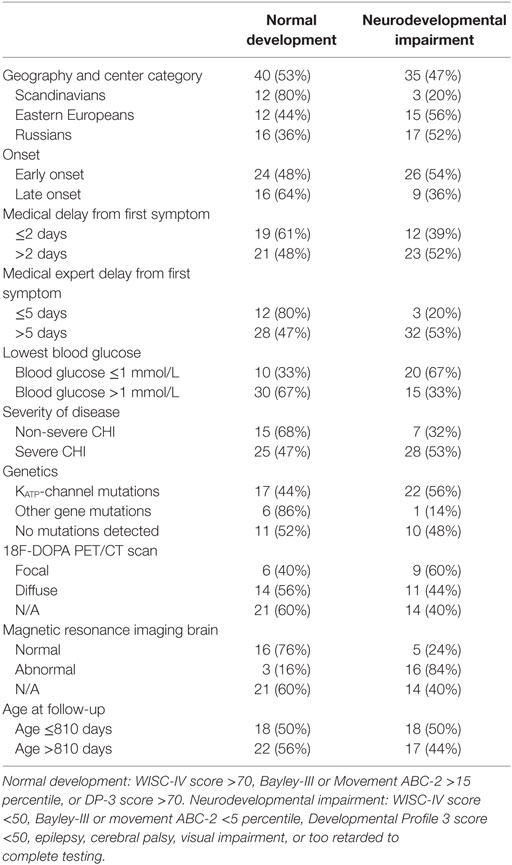
Table 6. Overall neurodevelopmental outcome for children with congenital hyperinsulinism (CHI) by patient data.
Split by nations, 20% of the Scandinavian patients had neurodevelopmental impairment, whereas 52% of the Russian patients, and 56% of the non-Scandinavian patients referred to Denmark were severely impaired. All six children with microcephaly were denoted to the neurodevelopmental impairment group. All of those belonging to the normally developed group had a normal EEG, whereas 45% of those with neurodevelopmental impairment had an abnormal EEG. Three children with occipital hypoglycemic encephalopathy by MRI scan showed normal development at follow-up.
In univariable analysis, neurodevelopmental impairment was associated with abnormal MRI of the brain; OR (95% CI) 15.0 (3.0–74.3), p = 0.001, non-Scandinavian patient, OR 3.8 (1.2–11.9), p = 0.023; treatment delay from first symptom to expert center >5 days; OR 4.0 (1.0–16.6), trend p = 0.05, and lowest blood glucose ≤1 mmol/L; OR 3.8 (1.3–11.3), p = 0.015, Tables 6 and 7. Patients with KATP-channel mutations and focal disease had marginal trends only toward higher odds of impairment. No associations were detected for early vs. late disease onset, disease severity, delay from first symptom to first treatment, or age at follow-up.
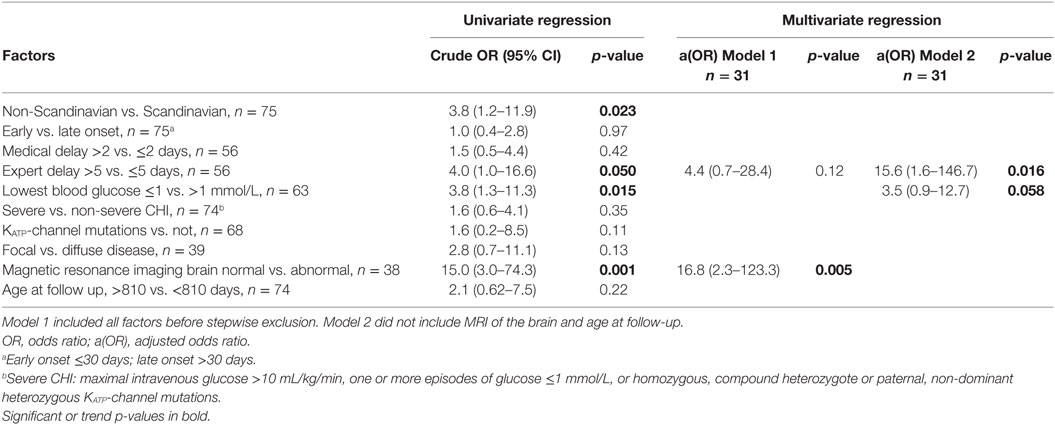
Table 7. Logistic regression for factors associated to neurodevelopmental impairment in congenital hyperinsulinism (CHI) patients.
In the multivariable analysis, only 31 patients could be included due to missing data. In Model 1, only MRI of the brain remained associated to neurodevelopmental impairment, aOR (95% CI) 16.8 (2.3–123.3), p = 0.005, while “expert delay” had a trend toward increased risk, OR 4.4 (0.7–28.4), p = 0.12. When analyzing associations to early predictors only by exclusion of age at follow-up and MRI of the brain (Model 2), delay from first symptom to expert center treatment significantly associated with neurological impairment, aOR 15.6 (1.6–146.7), p = 0.016, whereas lowest blood glucose ≤1 mmol/L conferred a trend toward increased risk, aOR 3.5 (0.9–12.7), p = 0.058.
Discussion
In our mixed international cohort of children with CHI, 47% of the patients had neurodevelopmental impairment at follow-up, despite huge advances in knowledge about CHI. Univariable factors associated with neurodevelopmental impairment were delayed from first symptom to expert treatment center (trend), lowest recorded blood glucose ≤1.0 mmol/L, non-Scandinavian patient, and abnormal MRI of the brain. By multivariable regression analysis, excluding brain MRI, “expert delay” became significantly associated, while lowest recorded blood glucose ≤1.0 mmol/L showed a trend toward increased risk. Compared to siblings, motor function and cognitive score were lower in a small subset of Danish patients.
Neonatal hypoglycemia is a well-known cause of neurodevelopmental impairment, depending on the hypoglycemia severity, duration, and comorbid factors (35–38). Neonatal hypoglycemia is, however, usually transient and predictable by perinatal risk factors, which confers opportunities for prevention and early management (39). Early detection and adequate treatment of transient neonatal hypoglycemia are associated with an un-increased risk of adverse neurological outcome (37), or lower fine motor function in boys and a higher internalizing score in girls, both within the normal range at school age follow-up. Other follow-up studies report of increased adjusted odds for developmental delay in late preterms with blood glucose <1.1 mmol/L (40), adverse neurodevelopmental outcome in diabetic mothers’ term neonates with neonatal blood glucose 0.0–1.5 mmol/L at 7–8 years’ follow-up (41), and lower school performance at 10 years after early transient hypoglycemia (<2.5 mmol/L < 3 h of age) in preterm and term neonates (42).
In CHI, an alarmingly high prevalence of neurodevelopmental delay, epilepsy, seizures, blindness, and microcephaly have been reported in other studies (22–24, 31, 34, 43–46). Of note, even children with transient CHI had a 30% risk of abnormal neurodevelopment in a recent UK study (32), with onset <7 days of age and disease severity defined by diazoxide dose as predictive factors. Other studies of various size and outcomes have shown a risk of adverse neurological outcome in CHI up to 50% (22, 26–28, 30, 34, 46–48).
Of larger follow-up studies, neurodevelopmental retardation was found in 44% of 114 CHI patients in Germany (23), psychomotor retardation in 26% of 90 patients in France (22), neurological deficits in 45% of 65 CHI patients in Australia (48), and neurodevelopmental delay in 18% of 43 Saudi Arabian CHI patients (47). Despite the huge improvements in knowledge and management possibilities in CHI, our multinational patient cohort showed no improvement in neurodevelopmental outcome compared to the former studies.
We also found that motor function, especially balance and fine motor function, and cognitive score were lower, although still in normal range, in a small subset of Danish patients compared to their siblings. Similar findings were seen in follow-up of Danish children with transient neonatal hypoglycemia (unpublished data). This indicate that even apparently normal functioning children with CHI should be fine-tested at follow-up, as some could benefit from interventions to improve especially balance and fine motor function.
In our study, abnormal MRI of the brain strongly associated to neurodevelopmental impairment, also in the multivariable analysis. Hypoglycemia of any kind in the neonatal period or infancy is associated to abnormal MRI findings, especially white matter changes also known as hypoglycemic encephalopathy (49–51). In CHI, posterior (parieto-occipital) white matter changes is especially prominent, which relate to timing of hypoglycemia to the neonatal period and lack of ketone bodies as alternative fuel (51) We also found a high prevalence of posterior white matter lesions, but in the most severe, undertreated patients, global cerebral atrophy, and intracerebral hemorrhage were seen.
In the Saudi study of 43 patients, only 14 out of 18 patients with developmental delay had cerebral atrophy on imaging (47), stating that not all with abnormal cerebral imaging will present with clinically overt impairment, as also seen in our study.
Among early predictors, recorded blood glucose ≤1.0 mmol/L significantly associated with neurodevelopmental impairment. In contrast to others (22, 23, 32), we detected no increased cerebral risk for early onset CHI, neither when using a cut-off of 30 days, nor 7 days (data not shown). We believe that untreated hypoketotic hypoglycemia is the major direct cause of brain damage in CHI. Although the neonatal brain is more susceptible to hypoglycemia than in later childhood, early onset may well be a proxy for severe hypoglycemia, which is strongly associated to early onset, but difficult to extract without information bias in a retrospective setting with data from many hospitals in many countries, where frequent and systematic blood glucose recordings may not have been the rule. Our a priori defined severe CHI category did not associate with the neurological outcome. We never the less succeeded in detecting an association to adverse outcome for lowest recorded blood glucose ≤1.0 mmol/L. The definition of severe CHI included an i.v. glucose demand >10 mg/kg/min, which we used as an alternative to lowest blood glucose <1.0 mmol/L, as the latter criteria was subjected to variations in treatment. However, initial i.v. glucose demand recordings were often missing and hence recorded if possible at follow up before 18F-DOPA PET/CT (performed without other medication).
In our mixed cohort, delay until expert center and non-Scandinavian patient status were also determining factors for neurodevelopmental impairment in univariable analysis. This was probably due to non-recorded low and/or long hypoglycemic periods in this group of patients. As hypoglycemia should be treated promptly and sufficiently, these factors may, therefore, reflect insufficient treatment. However, the multivariate analysis only had delay until expert center as a significant associated factor. This may be explained by a lower study number in the multivariate analysis, but also by the fact that non-Scandinavian patients had a higher disease severity than Scandinavians, possibly reflecting selection bias. Considering delay until expert treatment, this may also reflect both selection bias and unrecorded insufficient treatment (low blood glucose). However, selection bias could be bidirectional: Patients with mild CHI may not have been referred to an expert center, but on the other hand, some patients with very severe disease were referred promptly. This leaves insufficient treatment as the major likely explanatory factor. The range of documented insufficient treatment was in one non-Scandinavian patient 12 months with untreated, very severe CHI, and cerebral palsy and severe infantile spasms. The insufficient treatment in this patient reflected limited hospital resources and self-payment for medical treatment.
In the other end of the spectrum, a Danish term neonate was erroneously discharged untreated a few hours after birth despite an unmeasurable low blood glucose recording because of paucity of symptoms, but readmitted at the age of 2 days with loss of consciousness and convulsions and blood glucose of 0.0 mmol/L. Only 1 week later, this patient was referred to the national expert center. Despite adequate treatment from then on, this infant also showed neurodevelopmental impairment and infantile spasms.
The novel identification of insufficient treatment as a risk factor does not call for more knowledge about CHI, but for improvements in spread of knowledge about CHI among health care personnel, especially rapid contact with an expert CHI center on suspicion of CHI.
Already in year 2000, a workshop held by the European network for research into hyperinsulinism (ENRHI) advocated for early referral to a multidisciplinary expert center (15). National or international referral is nowadays frequent for this rare and complex disease with need of experts in 18F-DOPA PET, medical, and surgical management. However, our findings underscore the need of significant improvements in prompt diagnosis and adequate treatment of CHI in every birth clinic and hospital, as just a very short time without sufficient treatment can cause brain damage. Given the rarity and often surprising severity of CHI, this task remains a major challenge for health-care providers dealing with birth clinics, neonatal, and pediatric wards, for health-care authorities and for relevant medical societies. Education of health-care personnel and popular communication should be systematically enforced, potentially in the context of hypoglycemia in general. Force task groups should be set to develop strategies for the prevention of untreated or undertreated CHI. Last, rapid funding is urgently needed in order to avoid delay in international referrals when needed.
Limitations of our study included the observational, partially retrospective design, selection bias due to referral practice favoring more severely affected patients and those with genetic results suggesting focal CHI, and the cohort size, however, relatively large for a rare disease. Lack of systematic data included not only brain MRI but also initial blood glucose values, continuous glucose monitoring, and i.v. glucose demand recordings. The disease severity assessment was, therefore, subjected to limitations. Follow-up was performed at different ages by different investigators using different methods, why robust cut offs (normal/abnormal) were used for comparisons. At last follow-up, some of the patients had relatively low age, where neurodevelopmental adverse outcomes may not yet be detectable. Multivariable analyses were hampered by missing data and hence a lower study number, and residual confounding, including potential unmeasured factors related to local differences in our mixed cohort, e.g., socioeconomic factors.
Strengths included our multinational setting, hospital file review of blood glucose data, the detailed and systematical prospective neurological outcome testing at follow-up, and the relatively long median follow-up time.
In conclusion, 47% of our CHI patients had neurodevelopmental impairment at follow-up, despite huge advances in knowledge about CHI. Not only very low blood glucose but also insufficient treatment as expressed by delay until expert center hospitalization increased the risk of neurodevelopmental impairment. This novel finding calls for improvements in spread of knowledge about CHI among health-care personnel and rapid contact with an expert CHI center on suspicion of CHI.
Ethics Statement
The study was conducted according to the Helsinki declaration. The Regional Ethical Committee approved the study, protocol number S-20120119. The data were approved by the Danish Data Protection Agency (13/11991).
Author Contributions
AH collected and analyzed the data and wrote the manuscript. MM, EG, IS, FP, CC, KF, A-ML, LH, and KB have collected and interpreted data and written on the manuscript. HC initiated and supervised the study and supervised the manuscript writing.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1. Christesen HT, Brusgaard K, Hussain K. Recurrent spontaneous hypoglycaemia causes loss of neurogenic and neuroglycopaenic signs in infants with congenital hyperinsulinism. Clin Endocrinol (2012) 76(4):548–54. doi:10.1111/j.1365-2265.2011.04250.x
2. Arnoux JB, Verkarre V, Saint-Martin C, Montravers F, Brassier A, Valayannopoulos V, et al. Congenital hyperinsulinism: current trends in diagnosis and therapy. Orphanet J Rare Dis (2011) 6:63. doi:10.1186/1750-1172-6-63
3. Otonkoski T, Ammälä C, Huopio H, Cote GJ, Chapman J, Cosgrove K, et al. A point mutation inactivating the sulfonylurea receptor causes the severe form of persistent hyperinsulinemic hypoglycemia of infancy in Finland. Diabetes (1999) 48(2):408–15. doi:10.2337/diabetes.48.2.408
4. Bruining GJ. Recent advances in hyperinsulinism and the pathogenesis of diabetes mellitus. Curr Opin Pediatr (1990) 2(4):758–65. doi:10.1097/00008480-199008000-00024
5. Meissner T, Beinbrech B, Mayatepek E. Congenital hyperinsulinism: molecular basis of a heterogeneous disease. Hum Mutat (1999) 13(5):351–61. doi:10.1002/(sici)1098-1004(1999)13:5<351:aid-humu3>3.0.co;2-r
6. Glaser B, Thornton P, Otonkoski T, Junien C. Genetics of neonatal hyperinsulinism. Arch Dis Child Fetal Neonatal Ed (2000) 82(2):F79–86. doi:10.1136/fn.82.2.F79
7. Stanley CA. Perspective on the genetics and diagnosis of congenital hyperinsulinism disorders. J Clin Endocrinol Metab (2016) 101(3):815–26. doi:10.1210/jc.2015-3651
8. Nessa A, Rahman SA, Hussain K. Hyperinsulinemic hypoglycemia – the molecular mechanisms. Front Endocrinol (2016) 7:29. doi:10.3389/fendo.2016.00029
9. Rahier J, Guiot Y, Sempoux C. Morphologic analysis of focal and diffuse forms of congenital hyperinsulinism. Semin Pediatr Surg (2011) 20(1):3–12. doi:10.1053/j.sempedsurg.2010.10.010
10. Bruun MF, Christoffersen SH, Brusgaard K, Detlefsen S, Christesen HT. [Congenital hyperinsulinism – diagnosis and treatment]. Ugeskr Laeger (2011) 173(47):3026–31.
11. Hennewig U, Hadzik B, Vogel M, Meissner T, Goecke T, Peters H, et al. Congenital central hypoventilation syndrome with hyperinsulinism in a preterm infant. J Hum Genet (2008) 53(6):573–7. doi:10.1007/s10038-008-0275-1
12. Snider KE, Becker S, Boyajian L, Shyng SL, MacMullen C, Hughes N, et al. Genotype and phenotype correlations in 417 children with congenital hyperinsulinism. J Clin Endocrinol Metab (2013) 98(2):E355–63. doi:10.1210/jc.2012-2169
13. Laje P, States LJ, Zhuang H, Becker SA, Palladino AA, Stanley CA, et al. Accuracy of PET/CT Scan in the diagnosis of the focal form of congenital hyperinsulinism. J Pediatr Surg (2013) 48(2):388–93. doi:10.1016/j.jpedsurg.2012.11.025
14. Shah P, Demirbilek H, Hussain K. Persistent hyperinsulinaemic hypoglycaemia in infancy. Semin Pediatr Surg (2014) 23(2):76–82. doi:10.1053/j.sempedsurg.2014.03.005
15. Aynsley-Green A, Hussain K, Hall J, Saudubray JM, Nihoul-Fékété C, De Lonlay-Debeney P, et al. Practical management of hyperinsulinism in infancy. Arch Dis Child Fetal Neonatal Ed (2000) 82(2):F98–107. doi:10.1136/fn.82.2.F98
16. Méder Ü, Bokodi G, Balogh L, Körner A, Szabó M, Pruhova S, et al. Severe hyperinsulinemic hypoglycemia in a neonate: response to sirolimus therapy. Pediatrics (2015) 136(5):e1369–72. doi:10.1542/peds.2014-4200
17. Szymanowski M, Estebanez MS, Padidela R, Han B, Mosinska K, Stevens A, et al. mTOR inhibitors for the treatment of severe congenital hyperinsulinism: perspectives on limited therapeutic success. J Clin Endocrinol Metab (2016) 101(12):4719–29. doi:10.1210/jc.2016-2711
18. Senniappan S, Shanti B, James C, Hussain K. Hyperinsulinaemic hypoglycaemia: genetic mechanisms, diagnosis and management. J Inherit Metab Dis (2012) 35(4):589–601. doi:10.1007/s10545-011-9441-2
19. Evans M, Amiel SA. Carbohydrates as a cerebral metabolic fuel. J Pediatr Endocrinol Metab (1998) 11(Suppl 1):99–102. doi:10.1515/JPEM.1998.11.S1.99
20. Koh TH, Aynsley-Green A, Tarbit M, Eyre JA. Neural dysfunction during hypoglycaemia. Arch Dis Child (1988) 63(11):1353–8. doi:10.1136/adc.63.11.1353
21. Lucas A, Morley R, Cole TJ. Adverse neurodevelopmental outcome of moderate neonatal hypoglycaemia. BMJ (1988) 297(6659):1304–8. doi:10.1136/bmj.297.6659.1304
22. Menni F, de Lonlay P, Sevin C, Touati G, Peigné C, Barbier V, et al. Neurologic outcomes of 90 neonates and infants with persistent hyperinsulinemic hypoglycemia. Pediatrics (2001) 107(3):476–9. doi:10.1542/peds.107.3.476
23. Meissner T, Wendel U, Burgard P, Schaetzle S, Mayatepek E. Long-term follow-up of 114 patients with congenital hyperinsulinism. Eur J Endocrinol (2003) 149(1):43–51. doi:10.1530/eje.0.1490043
24. Ludwig A, Ziegenhorn K, Empting S, Meissner T, Marquard J, Holl R, et al. Glucose metabolism and neurological outcome in congenital hyperinsulinism. Semin Pediatr Surg (2011) 20(1):45–9. doi:10.1053/j.sempedsurg.2010.10.005
25. Mercimek-Mahmutoglu S, Rami B, Feucht M, Herle M, Rittinger O, Stoeckler-Ipsiroglu S, et al. Long-term follow-up of patients with congenital hyperinsulinism in Austria. J Pediatr Endocrinol Metab (2008) 21(6):523–32.
26. Mazor-Aronovitch K, Gillis D, Lobel D, Hirsch HJ, Pinhas-Hamiel O, Modan-Moses D, et al. Long-term neurodevelopmental outcome in conservatively treated congenital hyperinsulinism. Eur J Endocrinol (2007) 157(4):491–7. doi:10.1530/eje-07-0445
27. Levy-Shraga Y, Pinhas-Hamiel O, Kraus-Houminer E, Landau H, Mazor-Aronovitch K, Modan-Moses D, et al. Cognitive and developmental outcome of conservatively treated children with congenital hyperinsulinism. J Pediatr Endocrinol Metab (2013) 26(3–4):301–8. doi:10.1515/jpem-2012-0289
28. Dacou-Voutetakis C, Psychou F, Maniati-Christidis M. Persistent hyperinsulinemic hypoglycemia of infancy: long-term results. J Pediatr Endocrinol Metab (1998) 11(Suppl 1):131–41. doi:10.1515/JPEM.1998.11.S1.131
29. Cresto JC, Abdenur JP, Bergada I, Martino R. Long-term follow up of persistent hyperinsulinaemic hypoglycaemia of infancy. Arch Dis Child (1998) 79(5):440–4. doi:10.1136/adc.79.5.440
30. Steinkrauss L, Lipman TH, Hendell CD, Gerdes M, Thornton PS, Stanley CA. Effects of hypoglycemia on developmental outcome in children with congenital hyperinsulinism. J Pediatr Nurs (2005) 20(2):109–18. doi:10.1016/j.pedn.2004.12.009
31. Kumaran A, Kar S, Kapoor RR, Hussain K. The clinical problem of hyperinsulinemic hypoglycemia and resultant infantile spasms. Pediatrics (2010) 126(5):e1231–6. doi:10.1542/peds.2009-2775
32. Avatapalle HB, Banerjee I, Shah S, Pryce M, Nicholson J, Rigby L, et al. Abnormal neurodevelopmental outcomes are common in children with transient congenital hyperinsulinism. Front Endocrinol (2013) 4:60. doi:10.3389/fendo.2013.00060
33. Vercellino GF, Cremonte M, Carlando G, Colivicchi M, Crivelli S, Ricotti A, et al. Transient neonatal hyperinsulinemic hypoglycemia and neurological outcome: a case report. Minerva Pediatr (2011) 63(2):111–4.
34. Rother KI, Matsumoto JM, Rasmussen NH, Schwenk WF. Subtotal pancreatectomy for hypoglycemia due to congenital hyperinsulinism: long-term follow-up of neurodevelopmental and pancreatic function. Pediatr Diabetes (2001) 2(3):115–22. doi:10.1034/j.1399-5448.2001.002003115.x
35. Burns CM, Rutherford MA, Boardman JP, Cowan FM. Patterns of cerebral injury and neurodevelopmental outcomes after symptomatic neonatal hypoglycemia. Pediatrics (2008) 122(1):65–74. doi:10.1542/peds.2007-2822
36. Cornblath M, Reisner SH. Blood glucose in the neonate and its clinical significance. N Engl J Med (1965) 273(7):378–81. doi:10.1056/NEJM196508122730707
37. McKinlay CJ, Alsweiler JM, Ansell JM, Anstice NS, Chase JG, Gamble GD, et al. Neonatal glycemia and neurodevelopmental outcomes at 2 years. N Engl J Med (2015) 373(16):1507–18. doi:10.1056/NEJMoa1504909
38. Baron IS, Erickson K, Ahronovich MD, Baker R, Litman FR. Cognitive deficit in preschoolers born late-preterm. Early Hum Dev (2011) 87(2):115–9. doi:10.1016/j.earlhumdev.2010.11.010
39. Harris DL, Weston PJ, Harding JE. Incidence of neonatal hypoglycemia in babies identified as at risk. J Pediatr (2012) 161(5):787–91. doi:10.1016/j.jpeds.2012.05.022
40. Kerstjens JM, Bocca-Tjeertes IF, de Winter AF, Reijneveld SA, Bos AF. Neonatal morbidities and developmental delay in moderately preterm-born children. Pediatrics (2012) 130(2):e265–72. doi:10.1542/peds.2012-0079
41. Boluyt N, van Kempen A, Offringa M. Neurodevelopment after neonatal hypoglycemia: a systematic review and design of an optimal future study. Pediatrics (2006) 117(6):2231–43. doi:10.1542/peds.2005-1919
42. Kaiser JR, Bai S, Gibson N, Holland G, Lin TM, Swearingen CJ, et al. Association between transient newborn hypoglycemia and fourth-grade achievement test proficiency: a population-based study. JAMA Pediatr (2015) 169(10):913–21. doi:10.1001/jamapediatrics.2015.1631
43. Thomas CG Jr, Cuenca RE, Azizkhan RG, Underwood LE, Carney CN. Changing concepts of islet cell dysplasia in neonatal and infantile hyperinsulinism. World J Surg (1988) 12(5):598–609. doi:10.1007/BF01655860
44. Landau H, Perlman M, Meyer S, Isacsohn M, Krausz M, Mayan H, et al. Persistent neonatal hypoglycemia due to hyperinsulinism: medical aspects. Pediatrics (1982) 70(3):440–6.
45. Spitz L, Bhargava RK, Grant DB, Leonard JV. Surgical treatment of hyperinsulinaemic hypoglycaemia in infancy and childhood. Arch Dis Child (1992) 67(2):201–5. doi:10.1136/adc.67.2.201
46. Tyrrell VJ, Ambler GR, Yeow WH, Cowell CT, Silink M. Ten years’ experience of persistent hyperinsulinaemic hypoglycaemia of infancy. J Paediatr Child Health (2001) 37(5):483–8. doi:10.1046/j.1440-1754.2001.00748.x
47. Al-Nassar S, Sakati N, Al-Ashwal A, Bin-Abbas B. Persistent hyperinsulinaemic hypoglycaemia of infancy in 43 children: long-term clinical and surgical follow-up. Asian J Surg (2006) 29(3):207–11. doi:10.1016/s1015-9584(09)60089-0
48. Jack MM, Greer RM, Thomsett MJ, Walker RM, Bell JR, Choong C, et al. The outcome in Australian children with hyperinsulinism of infancy: early extensive surgery in severe cases lowers risk of diabetes. Clin Endocrinol (2003) 58(3):355–64. doi:10.1046/j.1365-2265.2003.01725.x
49. Wong DS, Poskitt KJ, Chau V, Miller SP, Roland E, Hill A, et al. Brain injury patterns in hypoglycemia in neonatal encephalopathy. AJNR Am J Neuroradiol (2013) 34(7):1456–61. doi:10.3174/ajnr.A3423
50. Kang EG, Jeon SJ, Choi SS, Song CJ, Yu IK. Diffusion MR imaging of hypoglycemic encephalopathy. AJNR Am J Neuroradiol (2010) 31(3):559–64. doi:10.3174/ajnr.A1856
Keywords: congenital hyperinsulinism, hypoglycemia, neurodevelopmental impairment, neurological outcome, treatment delay
Citation: Helleskov A, Melikyan M, Globa E, Shcherderkina I, Poertner F, Larsen A-M, Filipsen K, Brusgaard K, Christiansen CD, Hansen LK and Christesen HT (2017) Both Low Blood Glucose and Insufficient Treatment Confer Risk of Neurodevelopmental Impairment in Congenital Hyperinsulinism: A Multinational Cohort Study. Front. Endocrinol. 8:156. doi: 10.3389/fendo.2017.00156
Received: 26 January 2017; Accepted: 21 June 2017;
Published: 10 July 2017
Edited by:
Andrea Enzo Scaramuzza, Istituti Ospitalieri di Cremona, ItalyReviewed by:
Eli Hershkovitz, Soroka Medical University Center, IsraelVandana Jain, All India Institute of Medical Sciences, India
Copyright: © 2017 Helleskov, Melikyan, Globa, Shcherderkina, Poertner, Larsen, Filipsen, Brusgaard, Christiansen, Hansen and Christesen. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Henrik T. Christesen, aGVucmlrLmNocmlzdGVzZW5AcnN5ZC5kaw==