 Takanori Utsumi1*
Takanori Utsumi1* Satoshi Okada2
Satoshi Okada2 Kazushi Izawa3Yoshitaka Honda3Gen Nishimura4Ryuta Nishikomori3Rika Okano1Masao Kobayashi2
Kazushi Izawa3Yoshitaka Honda3Gen Nishimura4Ryuta Nishikomori3Rika Okano1Masao Kobayashi2
- 1Department of Pediatrics, Onomichi General Hospital, Hiroshima, Japan
- 2Department of Pediatrics, Hiroshima University Graduate School of Biomedical & Health Sciences, Hiroshima, Japan
- 3Department of Pediatrics, Kyoto University Graduate School of Medicine, Kyoto, Japan
- 4Department of Pediatric Imaging, Tokyo Metropolitan Children’s Medical Center, Tokyo, Japan
Spondyloenchondrodysplasia (SPENCD) is an autosomal recessive skeletal dysplasia caused by loss of function mutations in acid phosphatase 5, tartrate resistant (ACP5). Hypomorphic ACP5 mutations impair endochondral bone growth and create an interferon (INF) signature, which lead to distinctive spondylar and metaphyseal dysplasias, and extraskeletal morbidity, such as neurological involvement and immune dysregulation, respectively. We report an affected boy with novel ACP5 mutations, a splice-site mutation (736-2 A>C) and a nonsense mutation (R176X). He presented with postnatal short stature, which led to a diagnosis of partial growth hormone (GH) deficiency at 3 years of age. GH therapy was beneficial in accelerating his growth velocity. At 6 years of age, however, metaphyseal abnormalities of the knee attracted medical attention, and subsequent assessment ascertained the typical skeletal phenotype of SPENCD, brain calcifications, and an INF signature. This anecdotal experience indicates the potential efficacy of GH for growth failure in SPENCD.
Introduction
Spondyloenchondrodysplasia (SPENCD, OMIM #607944) is a rare autosomal recessive skeletal dysplasia characterized by vertebral dysplasia and enchondroma-like radiolucent metaphyseal lesions of the long bones (1). Affected individuals manifest with short-trunked short stature. In addition, they show a variety of extraskeletal abnormalities, including neurological symptoms (intracranial calcification, developmental delay, spasticity, clumsy movements), and immune dysregulation (autoimmune diseases, immunodeficiency) (2–10). Recently, two groups reported biallelic hypomorphic mutations in the Acid Phosphatase 5, Tartrate Resistant (ACP5) gene as a genetic cause of SPENCD (11, 12). ACP5 encodes tartrate-resistant acid phosphatase (TRAP). TRAP deficiency increases levels of phosphorylated osteopontin, which causes dysregulated endochondral ossification and other manifestations, and also an interferon (INF) signature (increased expression of type I INF regulated genes) leading to extraskeletal complications (11–13). Very little is documented concerning medical intervention for short stature and INF signature in SPENCD. Some SPENCD reports have mentioned attempts of growth hormone (GH) therapy, but the effect of GH has not been fully detailed (4, 10).
We report here an affected Japanese boy with novel compound heterozygous ACP5 mutations, who had the typical skeletal phenotype of SPENCD, brain calcifications, absence of TRAP activity, and laboratory findings of an INF signature, but no immunodeficiency or autoimmune disorders. He had developed short stature in early childhood, leading to a diagnosis of partial GH deficiency (GHD) and introduction of GH therapy prior to the definitive diagnosis of SPENCD. Interestingly, his growth velocity was significantly accelerated in response to GH treatment.
Case Report
The patient is a 6-year-old Japanese boy who was born to non-consanguineous healthy parents. The patient’s mid-parental target height was 180 cm (final height of his father and mother was 177 and 170 cm, respectively). His older brother was healthy (Figure 1A). He was born at term after an uncomplicated pregnancy and delivery. Birth weight was 3,055 g (−0.64 SD), length 51.5 cm (+1.06 SD), and head circumference 34.0 cm (+0.40 SD). At 19 months, he was referred to us because of his inability to walk unaided. At that time brain MRI yielded a normal finding. He entered a rehabilitation program that improved his ambulation. His linear growth started to slow down from 5 months of age. At 38 months of age his height was 84.5 cm (−2.89 SD) (Figure 1B). Thyroid function was normal (TSH 4.520 μU/mL, FT4 1.06 ng/dL). The level of baseline insulin-like growth factor I (IGF-I) was low (18 ng/mL). These clinical manifestations and results of a GH stimulation test led to the diagnosis of partial GHD (peak GH response to arginine was 4.60 ng/mL and to clonidine was 2.41 ng/mL; the cut-off point was <6 ng/mL to define partial GHD and <3 ng/mL to define severe GHD) (Table S1 in Supplementary Material). GH therapy (0.175 mg/kg/week) was introduced, and it significantly accelerated his growth velocity (Figure 1B).
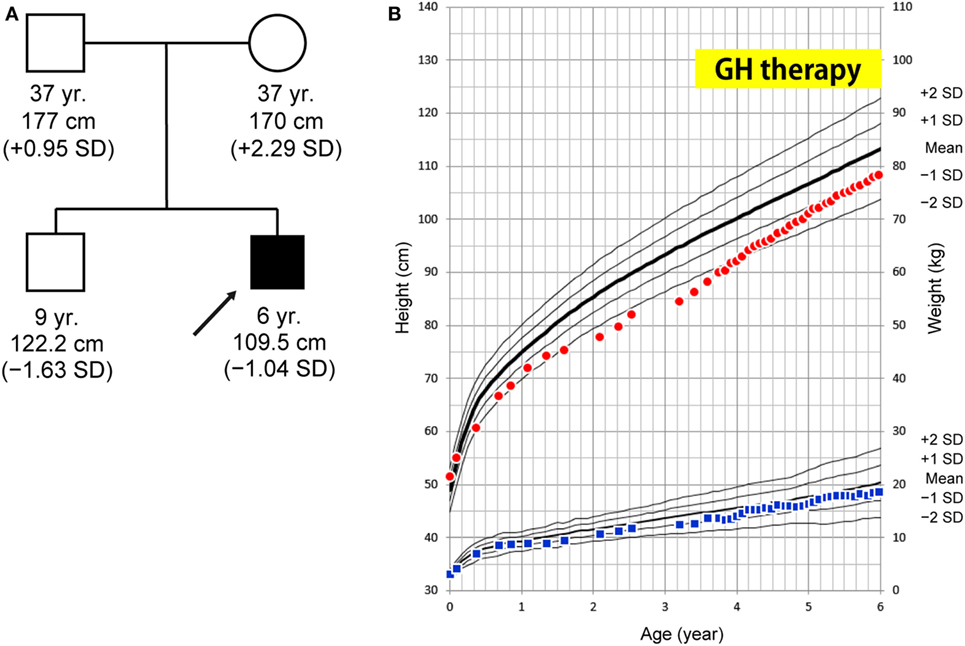
Figure 1. (A) Family tree. (B) Growth curve. At age 38 months, the patient’s height was 84.5 cm (−2.89 SD) and growth hormone (GH) therapy was commenced. The patient’s growth curve significantly improved with GH treatment over time. Height curve; red circles, weight curve; blue squares.
At 6 years of age, he presented with painful knee joints. Height was 109.5 cm (−1.04 SD), and arm span was 111 cm. Facial features and body proportion were normal (Figure 2A). Development (language and cognitive skills, and fine and gross motor skills) was appropriate for his age, other than some clumsiness in motion. A radiograph of the knee showed mild metaphyseal dysplasia of the knee (shaved contour and marginal spur in the distal femoral metaphyses and to a lesser extent in the proximal tibial metaphyses, and mild cupping proximal fibular metaphyses) (Figure 2C). A subsequent skeletal survey revealed generalized metaphyseal dysplasia involving the hip and wrist as well as the knee (Figures 2D,E). The spine showed generalized platyspondyly with irregular endplates. The posterior vertebral bodies were irregularly ossified (Figure 2B). These radiographic findings raised a suspicion of SPENCD. Laboratory data showed normal levels of serum calcium, phosphate, alkaline phosphatase, intact PTH, IGF-I, and thyroid function (Table S1 in Supplementary Material). No immunological abnormality was found, including normal levels of complements and no detectable autoantibodies. TRAP activity was undetectable (TRAP-5b < 0.1 U/L). Bone mineral density on dual-energy x-ray absorptiometry was normal (L1–L4: 0.942 g/cm2). Brain CT of the patient showed bilateral calcifications of the basal ganglia (Figure 2F).
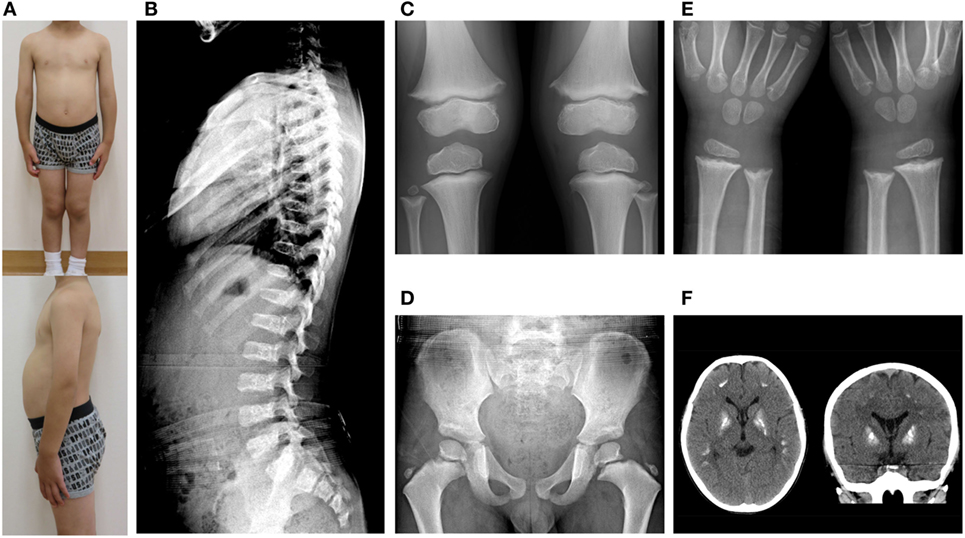
Figure 2. (A) Clinical photographs. (B) Spinal radiograph showing generalized platyspondyly with irregular endplates and irregular ossifications in the posterior vertebral bodies. (C) Radiograph of the knees showing irregular and sclerotic splayed metaphyses of the distal femur, proximal tibia, and fibula. (D,E) Radiograph of the hip and wrist showing similar metaphyseal changes as the knee. (F) Brain CT showing bilateral calcification of the basal ganglia.
Materials and Methods
Genetic Analysis
The genetic study of the patient was performed using a candidate gene approach with Sanger sequencing of the ACP5 gene. A familial study was not performed because consent was not given.
INF Analysis
The expression of INF-stimulated genes in the patient’s whole blood cells were measured by qPCR as described previously (14). Whole blood was collected into PAXgene tubes (PreAnalytix, Hombrechtikon, Swizerland) and frozen at −20°C until extraction using the manufacturer’s protocol. Total RNA was extracted with a PAXgene RNA Blood kit (PreAnalytix) and reverse-transcribed to cDNA using an Omniscript RT kit (Qiagen, Hilden, Germany). Real time quantitative PCR was performed in triplicate using a 7900HT Fast Real-Time PCR system (Applied Biosystems, Waltham, MA, USA) and a kit for each INF-stimulated gene (IFI27, Hs01086370_m1; IFIT1, Hs00356631_g1; RSAD2, Hs01057264_m1; SIGLEC1, Hs00988063_m1; ISG15, Hs00192713_m1; IFI44L, Hs00199115_m1; BACT, Hs01060665_g1) (Life Technologies, Carlsbad, CA, USA). The expression levels were normalized to those of β-actin. Results are shown relative to a single calibrator (control 1).
Results
Identification of ACP5 Mutations
The sequence analysis revealed novel ACP5 mutations, a splice-site mutation (c.736-2 A>C) and a nonsense mutation (c.526 C>T; p.R176X) (Figures 3A,B). Neither 736-2 A>C nor R176X were found in the NCBI, Ensembl, dbSNP, or ExAc databases.
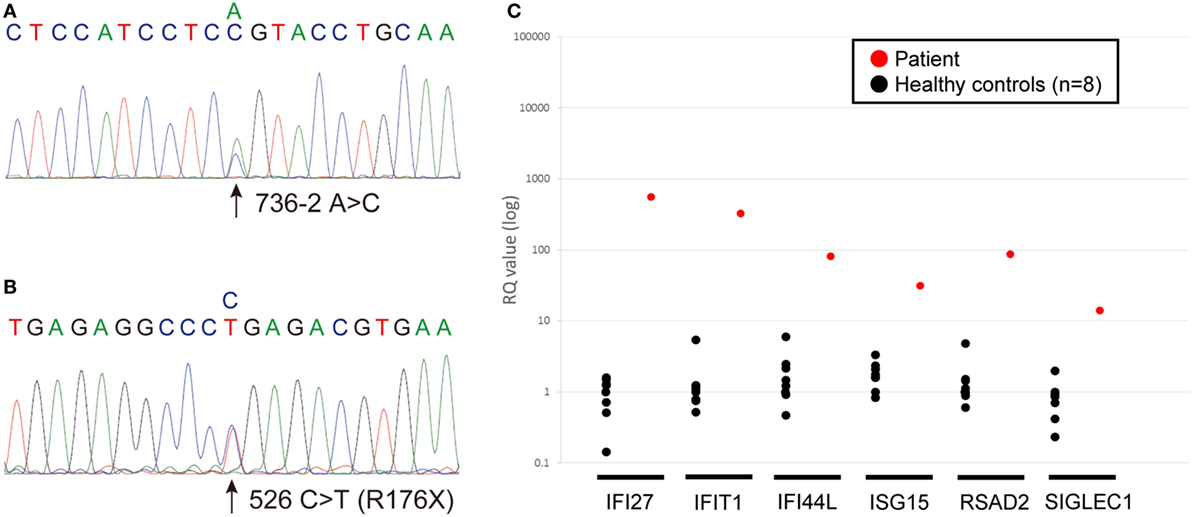
Figure 3. (A,B) Chromatograms showing the novel heterozygous mutations in ACP5 identified in the patient; a splice-site mutation (c.736-2 A>C) (A) and a nonsense mutation (c.526 C>T; p.R176X) (B). (C) Quantitative RT-PCR of INF-stimulated gene expression for, IFI27, IFIT1, IFI44L, ISG15, RSAD2, SIGLEC1 in peripheral blood cells. The relative abundance of each transcript was normalized to the expression of β-actin. The experiment was performed in triplicate. Healthy control (n = 8); black circles, patient; red circles.
Detection of INF Signature
We identified marked upregulation of IFI27, IFIT1, IFI44L, ISG15, RSAD2, and SIGLEC1 in the patient compared with healthy controls (n = 8) (Figure 3C).
Discussion
We report a boy with a typical SPENCD skeletal phenotype, who presented with postnatal short stature and brain calcifications. He has not developed immune dysregulation despite laboratory evidence of a type I INF signature. An IFN signature in SPENCD plays a major role in neurological and immune involvement. We summarized the clinical manifestations of 48 patients with SPENCD by reviewing previous reports, as presented in Table 1 (1–9, 15–22). Height ranged from −1.5 to −7.5 SD (only cases with SD notation were included). The common features that first prompt the seeking of medical attention include musculoskeletal symptoms (60.4%), such as short stature, short limbs, kyphoscoliosis, and bowlegs, followed by neurological symptoms (27.1%) and immune dysregulation (18.8%). On the other hand, Briggs et al. (10) reviewed 26 patients, including 12 previously reported patients, and identified that up to 50% initially presented with immune dysregulation, followed by musculoskeletal symptoms (46.2%) and neurological symptoms (23.1%). This difference between our review and Briggs’ may represent the clinical diversity of SPENCD.
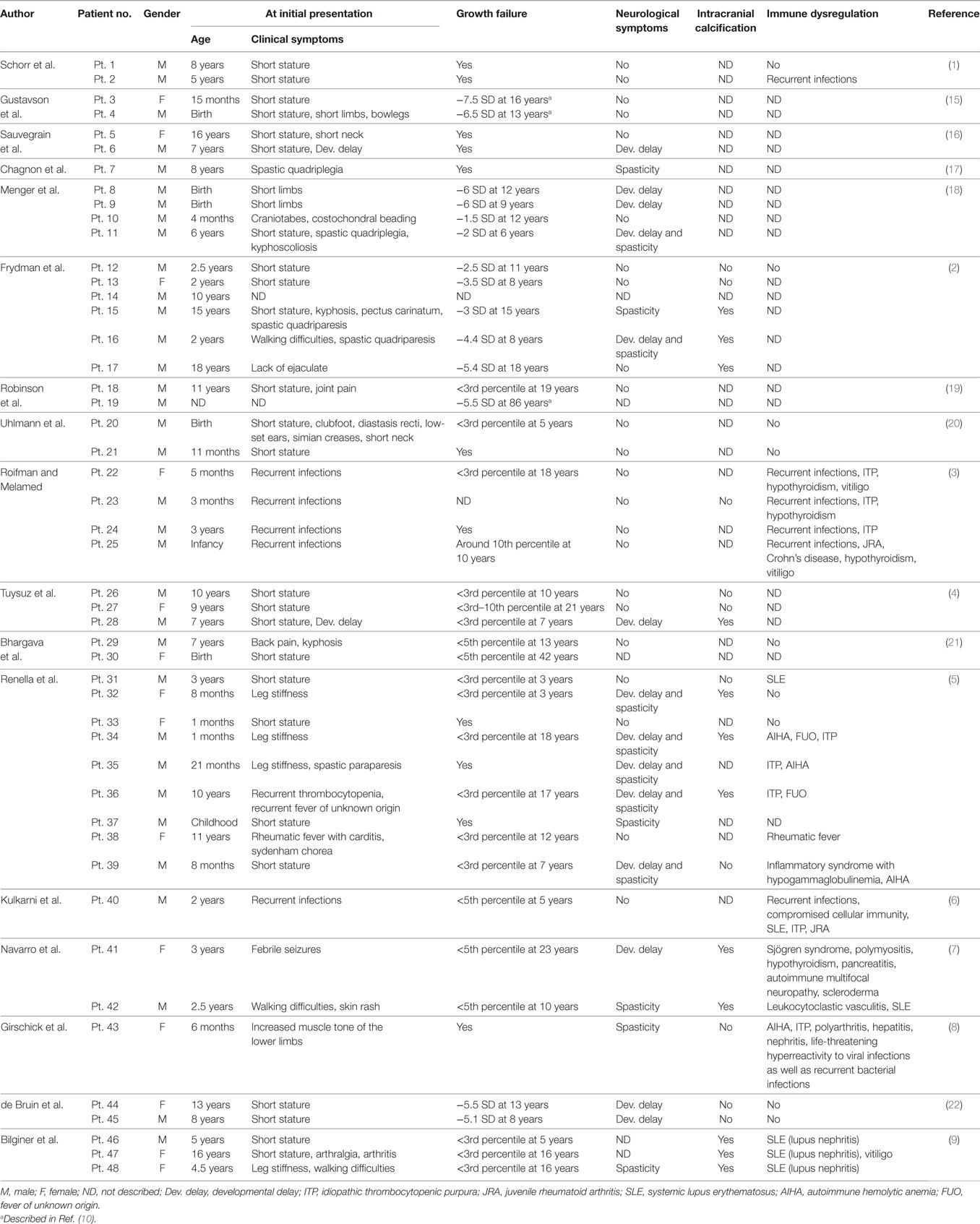
Table 1. Literature review of SPENCD case reports.
In the present case, postnatal growth failure was ameliorated with GH therapy; height increased from −2.89 SD to −1.04 SD over 3 years. The patient’s IGF-I levels increased to within the normal range after commencing GH therapy (Figure S1 in Supplementary Material). In addition, GH therapy did not accelerate bone age; bone ages were 2.3 and 5.2 years at the chronological ages of 3 years 4 months and 6 years 11 months, respectively. These findings indicate the potential therapeutic benefit of GH to improve short stature in patients with SPENCD.
Two reports have been published describing three SPENCD patients who underwent GH therapy (4, 10). Briggs et al. (10) described two patients, including one patient with GHD who responded well to GH therapy. Tuysuz et al. (4) reported a 21-year-old woman with SPENCD who received GH therapy for 2 years from the age of 9 years such that she achieved a normal height (153 cm: 3–10 centile). It remains unclear whether a good response to GH therapy in these SPENCD patients is universal or not. Lack of TRAP leads to an increase in phosphorylated osteopontin, which is responsible for disordered endochondral ossification and reduced resorption of calcified cartilage matrix and primary spongiosa as a result of impaired adhesion, migration, and activation of osteoclasts (11, 13, 23, 24). Meanwhile, GH advances longitudinal bone growth directly by stimulating prechondrocyte differentiation, and indirectly by clonal expansion of differentiated chondrocytes through upregulation of IGF-I (25–29). Moreover, GH stimulates resorption of cartilage matrix and immature bone through both its direct and indirect actions on osteoclast formation and differentiation and through indirect activation of mature osteoclasts via osteoblasts. Upregulation of IGF-I associated with GH therapy also supports osteoclastic activities (30, 31). These effects of GH may efficiently counteract the negative effects on endochondral bone growth in SPENCD.
Growth hormone therapy is associated with adverse risks in some patients, such as femoral head necrosis, slipped capital femoral epiphysis, exostosis, and the progression of bone deformities, particularly spinal deformities, such as scoliosis. We have not observed any obvious adverse events associated with GH therapy in the present case up to this time. However, because only 3 years has passed since the patient commenced GH therapy, the patient will require regular follow-up to evaluate the potential long-term effects of GH therapy. In addition, there are some other limitations in our study. First, the patient has partial GHD as well as SPENCD, and therefore the potential benefit of GH therapy for short stature in patients with SPENCD might be overestimated. Second, the patient is 6 years old and is too young to evaluate the effect of GH therapy on his final height. Despite these limitations, however, the improvement in growth velocity found in the present case is encouraging and may suggest the potential therapeutic benefit of GH in patients with SPENCD.
Concluding Remarks
This case report described the treatment of a patient who presented progressive growth failure associated with SPENCD and partial GHD. After commencing GH therapy, the patient’s height SD score improved from −2.89 SD to −1.04 SD over 3 years without accelerating the bone age. Considering the improvement in growth velocity in the present case, it is reasonable to suggest that GH therapy is warranted in patients with SPENCD, at least for those who show evidence of a partial GHD. Further studies are recommended to evaluate the benefit of GH therapy in improving the height of patients with SPENCD, even in the absence of GHD.
Ethics Statement
This study was approved by the Institutional Review Board of Kyoto University. We obtained written informed consent for genomic and interferon analysis of the patient from the parents in accordance with the Declaration of Helsinki. Also, the parents of the patient provided written informed consent for the publication of the patient’s identifiable information.
Author Contributions
Patient workup: TU. Interpretation of radiographs: GN. Genetic and IFN signature analysis: KI, YH, and RN. Interpretation of data, drafting the manuscript or critical revision, final approval of the version to be published, and agreement to be accountable for all aspects of the work: TU, SO, KI, YH, GN, RN, RO, and MK.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We sincerely appreciate our patient and his family for their kind cooperation. We also thank Dr. Y. Sumen (Department of Orthopaedic Surgery, Onomichi General Hospital, Hiroshima, Japan) and Dr. T. Shimura (Department of Orthopaedic Surgery, Hiroshima Prefectural Rehabilitation Center, Hiroshima, Japan) for diagnostic advice.
Funding
This study was supported in part by the Practical Research Project for Rare/Intractable Diseases from the Japan Agency for Medical Research and Development, AMED.
Supplementary Material
The Supplementary Material for this article can be found online at http://journal.frontiersin.org/article/10.3389/fendo.2017.00157/full#supplementary-material.
References
1. Schorr S, Legum C, Ochshorn M. Spondyloenchondrodysplasia. Enchondromatomosis with severe platyspondyly in two brothers. Radiology (1976) 118(1):133–9. doi:10.1148/118.1.133
2. Frydman M, Bar-Ziv J, Preminger-Shapiro R, Brezner A, Brand N, Ben-Ami T, et al. Possible heterogeneity in spondyloenchondrodysplasia: quadriparesis, basal ganglia calcifications, and chondrocyte inclusions. Am J Med Genet (1990) 36(3):279–84. doi:10.1002/ajmg.1320360306
3. Roifman CM, Melamed I. A novel syndrome of combined immunodeficiency, autoimmunity and spondylometaphyseal dysplasia. Clin Genet (2003) 63(6):522–9. doi:10.1034/j.1399-0004.2003.00033.x
4. Tuysuz B, Arapoglu M, Ungur S. Spondyloenchondrodysplasia: clinical variability in three cases. Am J Med Genet A (2004) 128A(2):185–9. doi:10.1002/ajmg.a.30078
5. Renella R, Schaefer E, LeMerrer M, Alanay Y, Kandemir N, Eich G, et al. Spondyloenchondrodysplasia with spasticity, cerebral calcifications, and immune dysregulation: clinical and radiographic delineation of a pleiotropic disorder. Am J Med Genet A (2006) 140(6):541–50. doi:10.1002/ajmg.a.31081
6. Kulkarni ML, Baskar K, Kulkarni PM. A syndrome of immunodeficiency, autoimmunity, and spondylometaphyseal dysplasia. Am J Med Genet A (2007) 143A(1):69–75. doi:10.1002/ajmg.a.31526
7. Navarro V, Scott C, Briggs TA, Barete S, Frances C, Lebon P, et al. Two further cases of spondyloenchondrodysplasia (SPENCD) with immune dysregulation. Am J Med Genet A (2008) 146A(21):2810–5. doi:10.1002/ajmg.a.32518
8. Girschick H, Wolf C, Morbach H, Hertzberg C, Lee-Kirsch MA. Severe immune dysregulation with neurological impairment and minor bone changes in a child with spondyloenchondrodysplasia due to two novel mutations in the ACP5 gene. Pediatr Rheumatol Online J (2015) 13(1):37. doi:10.1186/s12969-015-0035-7
9. Bilginer Y, Duzova A, Topaloglu R, Batu ED, Boduroglu K, Gucer S, et al. Three cases of spondyloenchondrodysplasia (SPENCD) with systemic lupus erythematosus: a case series and review of the literature. Lupus (2016) 25(7):760–5. doi:10.1177/0961203316629000
10. Briggs TA, Rice GI, Adib N, Ades L, Barete S, Baskar K, et al. Spondyloenchondrodysplasia due to mutations in ACP5: a comprehensive survey. J Clin Immunol (2016) 36(3):220–34. doi:10.1007/s10875-016-0252-y
11. Lausch E, Janecke A, Bros M, Trojandt S, Alanay Y, De Laet C, et al. Genetic deficiency of tartrate-resistant acid phosphatase associated with skeletal dysplasia, cerebral calcifications and autoimmunity. Nat Genet (2011) 43(2):132–7. doi:10.1038/ng.749
12. Briggs TA, Rice GI, Daly S, Urquhart J, Gornall H, Bader-Meunier B, et al. Tartrate-resistant acid phosphatase deficiency causes a bone dysplasia with autoimmunity and a type I interferon expression signature. Nat Genet (2011) 43(2):127–31. doi:10.1038/ng.748
13. Behrens TW, Graham RR. TRAPing a new gene for autoimmunity. Nat Genet (2011) 43(2):90–1. doi:10.1038/ng0211-90
14. Oda H, Nakagawa K, Abe J, Awaya T, Funabiki M, Hijikata A, et al. Aicardi-Goutieres syndrome is caused by IFIH1 mutations. Am J Hum Genet (2014) 95(1):121–5. doi:10.1016/j.ajhg.2014.06.007
15. Gustavson KH, Holmgren G, Probst F. Spondylometaphyseal dysplasia in two sibs of normal parents. Pediatr Radiol (1978) 7(2):90–6. doi:10.1007/BF00975677
16. Sauvegrain J, Maroteaux P, Ribier J, Garel L, Tato L, Rochiccioli P, et al. [Multiple chondroma affecting the spine: spondylo-enchondroplasia and other forms (author’s transl)]. J Radiol (1980) 61(8–9):495–501.
18. Menger H, Kruse K, Spranger J. Spondyloenchondrodysplasia. J Med Genet (1989) 26(2):93–9. doi:10.1136/jmg.26.2.93
19. Robinson D, Tieder M, Copeliovitch L, Halperin N. Spondyloenchondrodysplasia. A rare cause of short-trunk syndrome. Acta Orthop Scand (1991) 62(4):375–8.
20. Uhlmann D, Rupprecht E, Keller E, Hormann D. Spondyloenchondrodysplasia: several phenotypes – the same syndrome. Pediatr Radiol (1998) 28(8):617–21. doi:10.1007/s002470050431
21. Bhargava R, Leonard NJ, Chan AK, Spranger J. Autosomal dominant inheritance of spondyloenchondrodysplasia. Am J Med Genet A (2005) 135(3):282–8. doi:10.1002/ajmg.a.30732
22. de Bruin C, Orbak Z, Andrew M, Hwa V, Dauber A. Severe short stature in two siblings as the presenting sign of ACP5 deficiency. Horm Res Paediatr (2016) 85(5):358–62. doi:10.1159/000443684
23. Hayman AR, Jones SJ, Boyde A, Foster D, Colledge WH, Carlton MB, et al. Mice lacking tartrate-resistant acid phosphatase (Acp 5) have disrupted endochondral ossification and mild osteopetrosis. Development (1996) 122(10):3151–62.
24. Janckila AJ, Yam LT. Biology and clinical significance of tartrate-resistant acid phosphatases: new perspectives on an old enzyme. Calcif Tissue Int (2009) 85(6):465–83. doi:10.1007/s00223-009-9309-8
25. Isaksson OG, Jansson JO, Gause IA. Growth hormone stimulates longitudinal bone growth directly. Science (1982) 216(4551):1237–9. doi:10.1126/science.7079756
26. Green H, Morikawa M, Nixon T. A dual effector theory of growth-hormone action. Differentiation (1985) 29(3):195–8. doi:10.1111/j.1432-0436.1985.tb00316.x
27. Zezulak KM, Green H. The generation of insulin-like growth factor-1 – sensitive cells by growth hormone action. Science (1986) 233(4763):551–3. doi:10.1126/science.3726546
28. Isaksson OG, Lindahl A, Nilsson A, Isgaard J. Mechanism of the stimulatory effect of growth hormone on longitudinal bone growth. Endocr Rev (1987) 8(4):426–38. doi:10.1210/edrv-8-4-426
29. Isgaard J, Moller C, Isaksson OG, Nilsson A, Mathews LS, Norstedt G. Regulation of insulin-like growth factor messenger ribonucleic acid in rat growth plate by growth hormone. Endocrinology (1988) 122(4):1515–20. doi:10.1210/endo-122-4-1515
30. Nishiyama K, Sugimoto T, Kaji H, Kanatani M, Kobayashi T, Chihara K. Stimulatory effect of growth hormone on bone resorption and osteoclast differentiation. Endocrinology (1996) 137(1):35–41. doi:10.1210/endo.137.1.8536635
Keywords: spondyloenchondrodysplasia, ACP5, growth hormone deficiency, growth hormone therapy, skeletal dysplasia
Citation: Utsumi T, Okada S, Izawa K, Honda Y, Nishimura G, Nishikomori R, Okano R and Kobayashi M (2017) A Case with Spondyloenchondrodysplasia Treated with Growth Hormone. Front. Endocrinol. 8:157. doi: 10.3389/fendo.2017.00157
Received: 10 March 2017; Accepted: 22 June 2017;
Published: 10 July 2017
Edited by:
Silvano Bertelloni, S. Chiara University-Hospital, ItalyReviewed by:
Benjamin Udoka Nwosu, University of Massachusetts Medical School, United StatesMarek Niedziela, Poznan University of Medical Sciences, Poland
Copyright: © 2017 Utsumi, Okada, Izawa, Honda, Nishimura, Nishikomori, Okano and Kobayashi. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Takanori Utsumi, dC51dHN1bWkuczE1QGdtYWlsLmNvbQ==