 Paola Pelosi1
Paola Pelosi1 Elisabetta Lapi2
Elisabetta Lapi2 Loredana Cavalli3Alberto Verrotti4Marilena Pantaleo2Maurizio de Martino1
Loredana Cavalli3Alberto Verrotti4Marilena Pantaleo2Maurizio de Martino1 Stefano Stagi1*
Stefano Stagi1*
- 1Department of Health Sciences, University of Florence, Anna Meyer Children’s University Hospital, Florence, Italy
- 2Genetics and Molecular Medicine Unit, Anna Meyer Children’s University Hospital, Florence, Italy
- 3Department of Neuroscience, Neurorehabilitation Section, University of Pisa, Pisa, Italy
- 4Department of Paediatrics, University of L’Aquila, L’Aquila, Italy
Haploinsufficiency of the insulin-like growth factor (IGF)-1 receptor (IGF1R) gene is a rare, probably under-diagnosed, cause of short stature. However, the effects of IGF1R haploinsufficiency on glucose metabolism, bone status, and metabolism have rarely been investigated. We report the case of a patient referred to our center at the age of 18 months for short stature, failure to thrive, and Silver–Russell-like phenotype. Genetic analysis did not show hypomethylation of the 11p15.5 region or uniparental disomy of chromosome 7. Growth hormone (GH) stimulation tests revealed GH deficiency, whereas IGF-1 was 248 ng/mL. r-hGH treatment showed only a slight improvement (from −4.4 to −3.5 SDS). At 10 years of age, the child was re-evaluated: CGH-array identified a heterozygous de novo 4.92 Mb deletion in 15q26.2, including the IGF1R gene. Dual-energy X-ray absorptiometry showed a normal bone mineral density z-score, while peripheral quantitative computed tomography revealed reduced cortical and increased trabecular elements. A phalangeal bone quantitative ultrasonography showed significantly reduced amplitude-dependent speed of sound and bone transmission time values. The changes in bone architecture, quality, and metabolism in heterozygous IGF1R deletion patients, support the hypothesis that IGF-1 can be a key factor in bone modeling and accrual.
Introduction
Insulin-like growth factor (IGF)-1 (IGF-1) and 2 (IGF-2) are major regulators of cell growth, proliferation, and death (1). IGF plays an important role in several tissues through the IGF receptor type 1 (IGF1R), a heterotetramer (α2β2) with intrinsic tyrosine kinase activity (1).
Insulin-like growth factor-1 is a crucial factor in bone formation and remodeling via its actions on both osteoblasts and osteoclasts, and it is involved in the development of peak bone density during growth and bone loss in senile osteoporosis (2, 3). Furthermore, IGF-1 regulates glucose metabolism by modulating insulin sensitivity and secretion (4); it is also expressed in the central nervous system and is essential for brain development (5, 6).
Haploinsufficiency of the gene encoding the IGF1R may be a rare and under-diagnosed cause of short stature (7, 8). Both heterozygous deletion and point-inactivating mutations of the gene can lower IGF1R mRNA and protein expression, with partial IGF-1 resistance (7, 8).
Different clinical phenotypes, including microcephaly, mental retardation, and facial dysmorphic features, were observed in patients with IGF1R deletion, but these phenotypes are uncommon in IGF1R mutations (4, 6–21). Some of the phenotypical features described in the patients are likely to be attributable to the absence of one copy of the IGF1R gene, but other findings are linked to the haploinsufficiency of other genes included in the deletion (7, 14, 22). Heterozygous missense and nonsense mutations of IGF1R show similar effects on growth and development. However, IGF1R mutations and deletions seem to differ in vitro, probably because of a dominant-negative effect of the mutation, which could decrease the number of fully functional receptors by up to 25%, whereas haploinsufficiency would theoretically lead to a 50% reduction (15).
The first IGF1R mutation was described by Abuzzahab et al. These authors reported the case of two patients with a history of intrauterine growth restriction (IUGR), poor postnatal growth, and the biochemical features of IGF-1 resistance (9). One patient was a compound heterozygote for point mutations in exon 2 of the IGF1R gene and the other carried a nonsense mutation (Arg59stop) of the same gene (9).
Walenkamp et al. described the case of a girl carrying a terminal 15.q26.2 → qter deletion. She displayed growth retardation, microcephaly, short stature, and elevated IGF-1 levels. She was treated with growth hormone (GH) with a good growth response and her final height was within the normal range (14).
Various aspects of IGF1R defects have been analyzed to date, but the effects of IGF1R haploinsufficiency on bone status and metabolism have not been reported.
So, in this report, we describe a female patient with a terminal deletion of chromosome 15, involving the IGF1R gene, who has been treated with r-hGH from the age of 4.5 years with a long-term follow-up. In this patient, we evaluated glucose metabolism and bone metabolism and status using dual-energy X-ray absorptiometry (DXA), peripheral quantitative computed tomography (pQCT), and quantitative ultrasonography (QUS).
As per institutional and national guidelines, no ethics approval was needed. Written informed consent was obtained from the parents before publication of this case report and any accompanying images.
Case Report
The proband was a toddler referred to the Paediatric Auxoendocrinology Unit of Meyer Children’s University Hospital of Florence for short stature and failure to thrive.
The patient was the first child of healthy, unrelated parents and was born after a 36-week gestation, complicated by unexplained (IUGR), by cesarean section. No behavioral or dietary problems affected the proposita’s mother during pregnancy. The Apgar score was 7I-9V. Birth weight was 1,930 g (−1.46 SD), length 41.6 cm (−2.07 SD), and head circumference 29.2 cm (−2.16 SD).
She was breastfed, but did not catch up after birth, showing failure to thrive and suffering gastroesophageal reflux symptoms from the first months of life. Her psychomotor development was slightly delayed: she sat upright at 7 months, began to walk independently at 16 months, and began to use language at 17 months.
At 18 months of age, she was referred to our Hospital; at physical examination, she showed dysmorphic features suggestive of Silver–Russell Syndrome: a triangular face, with a prominent forehead and micrognathia, clinodactyly V, inferior limb asymmetry (near 1.5 cm), and multiple hyperpigmented lesions on the body. Her length was 67 cm (−4.27 SD), weight 6.600 kg (−3.77 SD), and head circumference 44.5 cm (−2.00 SD). Her mother’s height was 169 cm (1.09 SD) and the height of her father was 176 cm (−0.16 SD); consequently, her target height was 166.5 cm (0.67 SD). Screening blood tests, including celiac, disease serological markers, and thyroid function, were normal; basal IGF-1 was in the upper normal range (248 µg/L; 97th percentile 250 ng/mL). Bone age was delayed: 10 months at 18 months of age. She showed a normal female karyotype (46, XX), whereas the methylation study of the 11p15.5 region and the evaluation of uniparental disomy of chromosome 7 were both negative.
At the age of 4 years and 4 months, she was newly evaluated for a persistent and significant growth delay [length was 81.5 cm (−5.11 SD), weight 8.860 kg (−6.33 SD), and body mass index (BMI) 13.31 (−1.83 SD)].
An endocrine work-up was performed: free-thyroxin [(FT4) 1.32 ng/dL, n.v. 0.86–2.12 ng/dL], thyroid-stimulating hormone [(TSH) 3.03 μUI/dL, n.v. 0.4–4.0 μUI/dL], cortisol (11.34 µg/dL, n.v. 5–25 µg/dL), adrenocorticotropic hormone [(ACTH) 27 ng/L, n.v. 09–52 ng/L], glucose (78 mg/dL, n.v. 55–110 mg/dL), and prolactin [(PRL) 127 mUI/ml] were in the normal range. The electrolyte, venous blood gas, hemoglobin, total protein, serum albumin, coagulation profile, calcium, and phosphorous were also normal. The anti-tissue transglutaminase (tTG) test was negative.
Arginine [basal (GH) 1.99, peak 8.92 ng/mL] and clonidine (basal GH 0.48, peak 6.92 ng/mL) stimulation tests disclosed a GH deficiency; IGF-1 level was 243 µg/L. Bone age was significantly delayed: 1 year, 10 months at 4 years, 4 months of chronological age. r-hGH treatment was started at a dosage of 0.23 mg/kg/week; the auxological follow-up showed a slight improvement in the first year of r-hGH treatment (from −5.11 to −3.5 SD) (Figure 1).
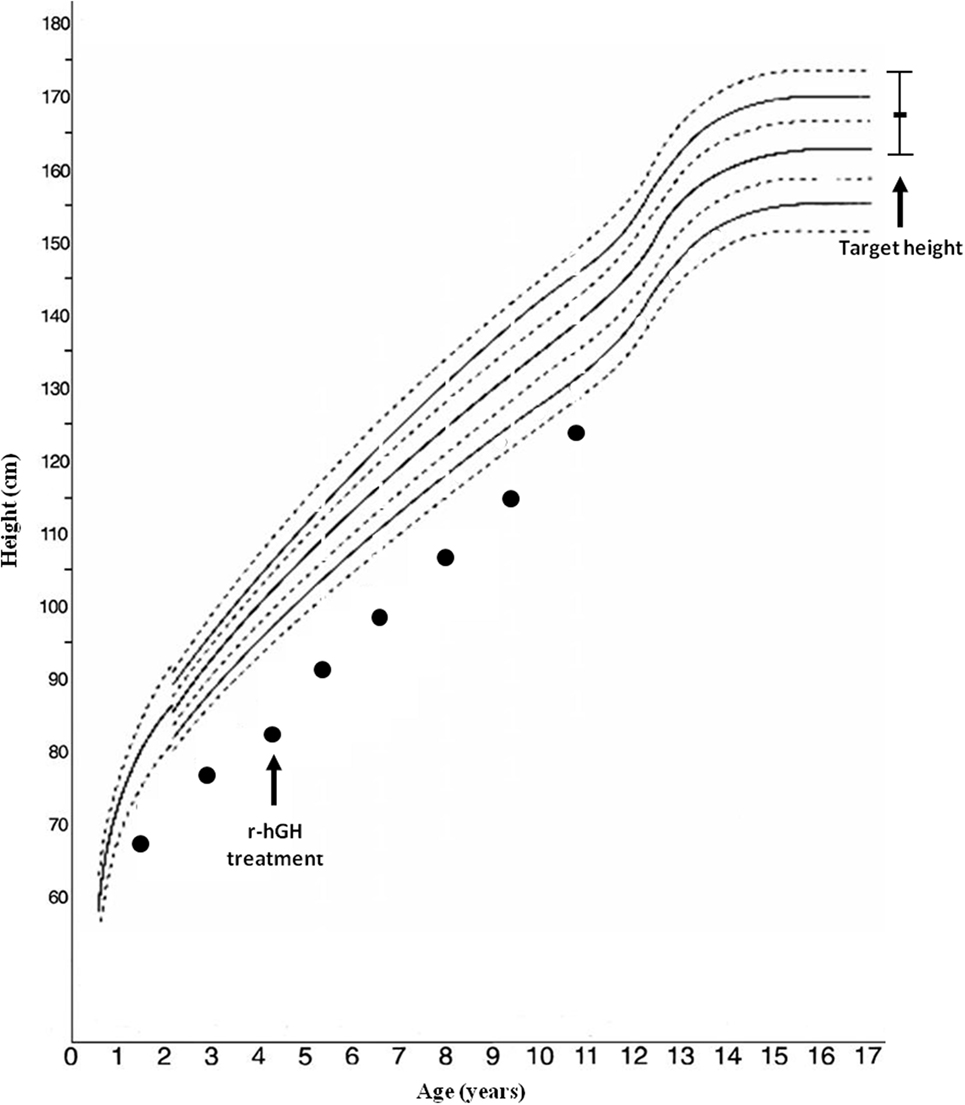
Figure 1. Growth chart of the patient.
During r-hGH therapy: IGF-I was persistently between the 90th and 97th percentile for age and sex, with glycemia, FT4, TSH, basal glycemia, basal insulin, and glycosylated hemoglobin (HbA1c) in normal ranges.
Therefore, in light of the unsatisfactory response to r-hGH treatment, a re-evaluation was performed at the age of 10 years and 10 months: height was 124.7 cm (−2.82 SDS), weight 21.900 kg (−2.93 SDS), BMI 14.08 (−2.14 SDS), and pubertal evaluation was B1 PH1 AH1. A re-testing of GH secretion confirmed low values of GH after arginine (basal GH 1.31, peak 7.76 ng/mL) and clonidine (basal GH 1.11, peak 7.23 ng/mL) testing; the IGF-I level was 371 µg/L. Bone age was still significantly delayed: 7 years, 11 months at 10 years, 10 months of chronological age. A new extensive endocrine work-up gave normal results. The tTG test was negative. Since the presence of HbA1c was in the upper limit of normality, we performed an oral glucose tolerance test that disclosed a reduced glucose tolerance: fasting glucose was 83 mg/dL, 2-h glucose 181 mg/dL, fasting insulin 2.19 μU/mL, peak 54.2 μU/mL 2-h insulin 54.0 μU/mL. The patient showed low leptin level (0.5 ng/mL, n.v. 1.0–12.0 ng/mL). At 11 years old, her intelligence quotient (IQ) was 108, even though the performance IQ was 85 and she exhibited some behavioral abnormalities.
Genetic Analysis
At 10 years and 10 months of age, CGH-array analysis was performed using the Agilent Human Genome CGH Microarray Kit 60 K (Agilent Technologies, Santa Clara, CA, USA). The CGH-array revealed a heterozygous deletion of chromosome 15, comprising 4.942 Mbp of the terminal part of its long arm (15q26.2-q26.3) involving several genes, such as IGF1R, ADAMTS17 (A Disintegrin-Like and Metalloproteinase with Thrombospondin Type 1 Motif, 17), CERS3 (Ceramide Synthase 3), ALDH1A3 (Aldehyde Dehydrogenase 1 Family, Member A3), and Chondroitin Sulfate Synthase 1 (CHSY1) (Figure 2; Table 1) The deletion was not present in the parents.
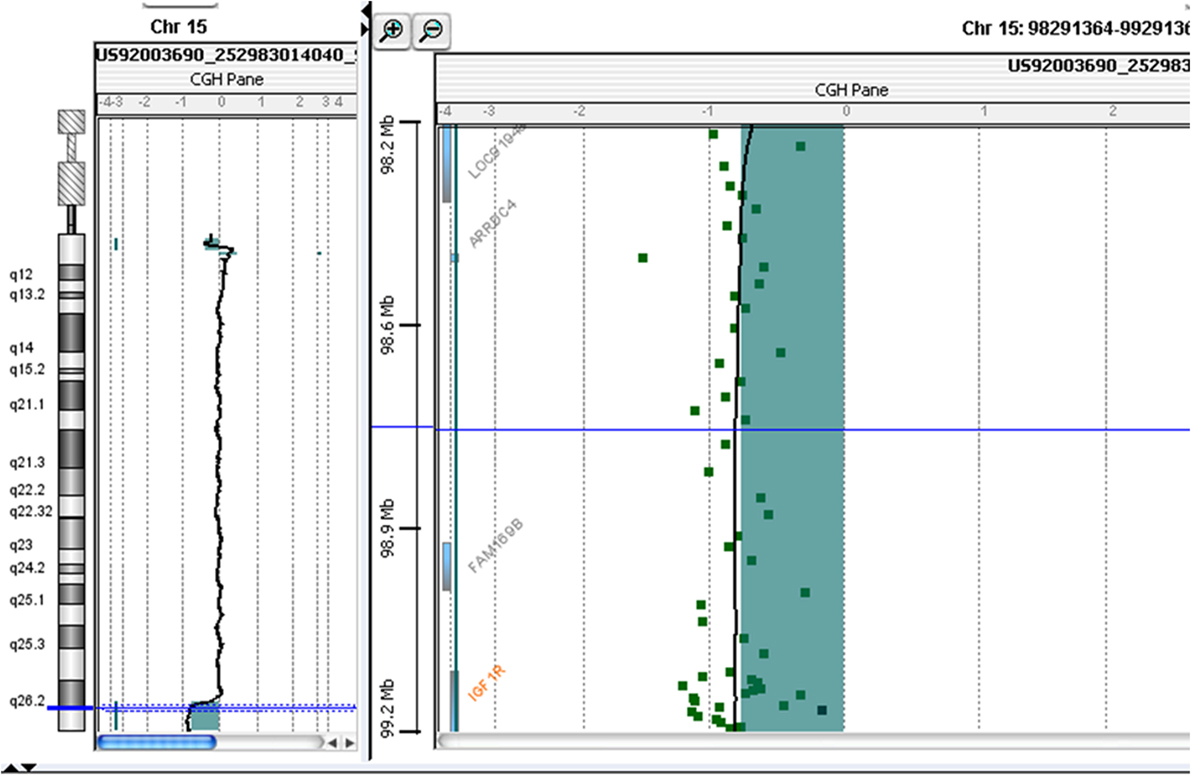
Figure 2. Molecular karyotyping was performed by array-CGH on the proband’s DNA using an Agilent 60 K array platform with a resolution of approximately 100 kb. Based on the physical mapping positions of the February 2009 Assembly (GRCh37/hg19) of the UCSC Genome Browser, this analysis showed a deletion of approximately 4,942 Mb that involved the 15q26.2-q26.3 region, with the breakpoint falling between 97,457,185 bp (first deleted oligomer) and 102,399,819 bp (last deleted oligomer).

Table 1. Genes involved in the 15q26.2-q26.3 deletion of the patient.
Bone Density and Structure Evaluation
At the age of 10 years and 11 months, the patient underwent an evaluation of bone metabolism, density, and structure. Bone mineral density (BMD, g/cm2) was measured by DXA at the lumbar spine (L2–L4) (Delphi-A System, Hologic, Inc., Waltham, MA, USA) and expressed as z-scores. To estimate the volumetric density (bone mineral apparent density or BMAD), we used the formula of Kröger et al. (23). The BMD z-score, corrected for height, was 0.67: BMD at the lumbar spine was 0.631 g/cm2 and the bone mineral content was 22.27 g.
Furthermore, we performed a pQCT of the left (non-dominant) radius at sites 4 and 66% using a Norland-Stratec XCT 3000 scanner (Stratec Medical, Pforzheim, Germany). As for the growth retardation of the patient, all bone size-dependent parameters (Total, Cortical, and MuscleCSA) were corrected for height (24, 25). We disclosed an imbalance between the trabecular and cortical bone, with an augmented trabecular component (318.4 mg/cm3, z-score 3.8) and a very low cortical density (727.8 mg/cm3, z-score −6.9) in relation to the age. The proband showed a normal total density value (321.3 mg/cm3, z-score 0.7) and a significantly reduced bone area for muscle area (31.2 mm2, z-score −4.0) and for height (28.9 mm2, z-score −4.1). The SSI polar (62.5 mm3, z-score −2.2) was significantly reduced. Fat and muscle components were also poorly represented (304 mm2, z-score −1.8; and 1,376 mm2, z-score −1.9, respectively) (Figures 3A–H).
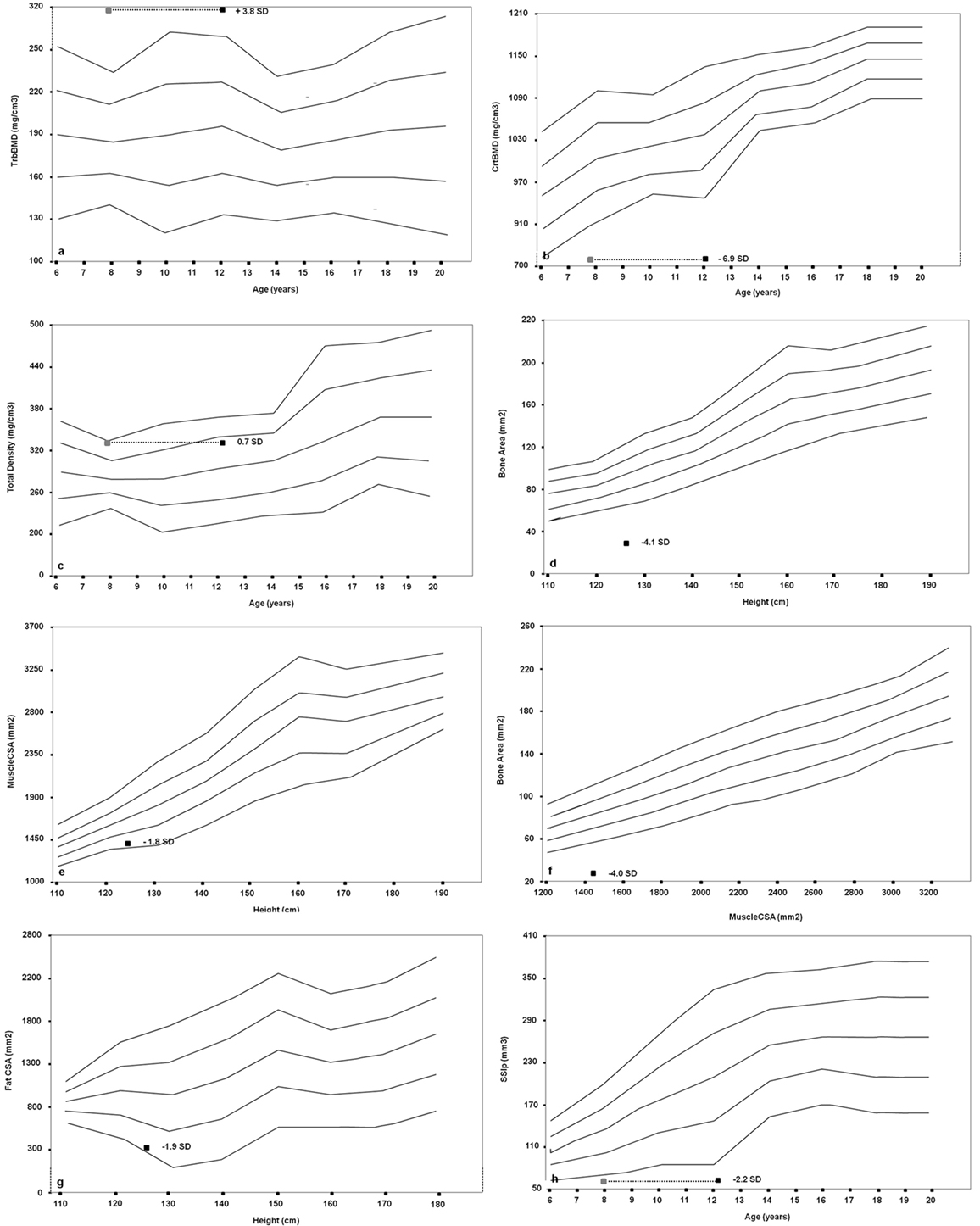
Figure 3. Cross-sectional evaluation of trabecular bone mineral density (TrabBMD) (A), cortical bone mineral density (CrtBMD) (B), total density corrected for age (C), bone area corrected for height (D), muscle cross-sectional area (MuscleCSA) corrected for height (E), bone area corrected for MuscleCSA (F), fat cross-sectional area (FatCSA) corrected for height (G), and density-weighted polar section modulus (SSIp) (H). The gray squares in the panels (A,B,C,H) represent the bone age-adjusted values.
Finally, the bone quality status was evaluated with a DBM Sonic 1200 device (IGEA Bone Profiler, Carpi, Italy) (24). The evaluation showed a very low amplitude-dependent speed of sound (AD-SoS, 1,791 m/s; z-score −3.85) and bone transmission time (BTT, 0.78 µs; z-score −2.18) values. Since bone size could also influence QUS parameters (26), we created a height-adjusted z-score for AD-SoS.
The study of the bone metabolism showed a low 25(OH) vitamin D [25(OH)D] level (14.3 ng/mL, n.v. >20 ng/mL) and a moderately high parathyroid hormone level (51 pg/mL; n.v. <43 pg/mL). Total protein, serum albumin, calcium, and phosphorous levels were normal; however, osteocalcin (34.3 mg/ml; n.v. 55–135 mg/ml), bone alkaline phosphatase (30 IU/L; n.v. 39.4–346.1 IU/L), and urinary deoxypyridinoline concentrations (23.45 nM/mM creatinine; n.v. 30.3–54.7 nM/mM creatinine) were lower than reference values for sex and age.
Discussion
We reported the case of a heterozygous, de novo 15q26.2-26.3 deletion involving the IGF1R gene. As in other previous reported cases of IGF1R deletion, our patient had a history of unexplained IUGR and severe short stature without catch-up growth (8, 10–14).
A recent study found that IGF1R haploinsufficiency was present in 2 out of 100 short small for gestational age (SGA) children with persistent short stature who benefited from GH therapy with moderate catch-up growth. The authors suggest that IUGR, microcephaly, micrognathia, relatively high IGF-1 levels, and developmental delays are the main predictors of IGF1R deletion (15). Our patient, treated with GH linear growth showed only a slight improvement, although she still remained persistently below her target height (Figure 1). Treatment response seems to be variable across patients, but all cases reported in the literature benefited from therapy, through the increase of GH and IGF-1 levels and most likely overcoming IGF-1 partial resistance (14, 15, 27).
Furthermore, our study provides very interesting data about bone structure and metabolism in IGF1R deletion patients, assessed by using three different approaches and sites scanned: DXA in lumbar spine, typically rich in trabecular bone (28); pQCT at radius, distinguishing cortical and trabecular components, as well as fat and muscle areas (24); quantitative ultrasonography at non-dominant hand phalanges, providing information not only on the density but also on structure and mechanical properties of the segment in question, whose composition is similar to femoral one (24, 29).
The data obtained in this IGF1R haploinsufficient patient seem to suggest an impaired cortical bone density (low z-scores at pQCT) and an increased trabecular component, despite normal BMD evaluated by DXA. As we described in a previous work (29), in fact, pQCT allows us to differentiate trabecular and cortical components, unlike the projective methods, such as DXA. The impaired bone quality of cortical component, well represented in phalanges, also resulted in the low z-score values related to density and stiffness measured at QUS. Moreover, pQCT has the advantage of measuring the real density of bone in a given volume without the superposition of other tissues, reducing the effect of auxological parameters, such as height and BMI. Therefore, the apparently normal outcome of lumbar DXA z-score is probably due to the prevalence of trabecular bone in vertebral site, component that may be less affected in IGF-1 disorders. The impairment of cortical compartment is instead shown by radial pQCT, which assess it separately from trabecular one, and by QUS, which evaluate a site where cortical bone is well represented, i.e., phalanges.
Several studies have demonstrated that humans lacking a functional IGF-1 gene suffer from severe osteopenia (30); however, mice that are deficient in liver-derived IGF-1 (LID), acid labile subunit (ALS) knockout mice (KO), and double gene disruption LID + ALSKO mice have a reduced femoral periosteal circumference, a smaller cross-sectional area, and a thinner cortical bone, when compared to control mice (31), which may be partially explained by the dramatic growth retardation in IGF-1-deficient mice (32). However, altered parameters were observed in both the trabecular and cortical compartments of IGF-I-null mice femurs, compared to wild-type mice (32). In rats, combined rhGH and rhIGF-1 treatment appeared to stimulate cortical bone mass, as evaluated by pQCT, more than rhGH alone does (33). Nevertheless, another study conducted in young adult mice with an IGF-I gene deletion showed significant alterations in bone mass and bone structure in both the cortical and trabecular compartments (32). Previous studies in IGF-I-deficient mice of a different background showed a decrease in cortical bone formation but an increase of several trabecular parameters in the tibia (34, 35). These data indicate that circulating IGF-1 is critical for bone modeling, quality, and structure, and it has been hypothesized that bone regional differences in response to IGF-I deficiency might be a consequence of the dual effect of IGF-1 on both osteoblastogenesis and osteoclastogenesis (36).
Circulating IGF-1 exerts its anabolic effects on the periosteal surface. Studies have shown that circulating IGF-1 stimulates periosteal bone growth along the cortex (37), whereas reduced serum levels of IGF-1 in mice lacking liver-specific IGF-1 were associated with impaired periosteal apposition, leading to the development of slender bones during growth (38). When subjected to a loading regimen, periosteal bone formation was substantially elevated in the IGF-1-overexpressing mice but not in wild-type littermates (39), suggesting that circulating IGF-1 enhances bone responses to mechanical loading.
We can observe that the haploinsufficiency of IGF1R may cause an imbalance of bone quality, structure, and modeling, probably leading to inferior mechanical properties and an increased risk of fracture, as showed by the SSIp value.
However, although IGF-1 undoubtedly affects GH action, it is now clear that: GH can have direct effects on multiple tissues; IGF-1 has endocrine, paracrine, and autocrine effects that are, in part, GH-independent; and GH and IGF-1 are likely to have overlapping, counteractive, and/or collaborative effects (40). However, we cannot know the effects of the performed r-hGH treatment on the bone quality and structure of our patient. More data are necessary to better understand this aspect in patients with IGF1R deletion.
Using current knowledge, it is likely that the only other gene involved in this patient’s deletion that could have affected the patient’s bone is CHSY1. Mice lacking Chsy1 for homozygous mutations display chondrodysplasia and decreased bone density, but no such data are available for humans.
Furthermore, IGF-1 also exerts an anabolic action on muscle, increasing the protein synthesis and decreasing the protein breakdown (41, 42). In fact, IGF-1 elicits skeletal muscle cell proliferation and myocytes differentiation (42). These data may explain the poorly represented muscle component in our patient.
Finally, another interesting aspect is the role of the IGF-system on glucose metabolism and the possible effects of IGF1R haploinsuficiency on carbohydrate homeostasis, as shown by our case report. The IGF1R gene has sequence homology with the insulin receptor gene; both are transmembrane tyrosine kinase receptors. IGF-I also has structural homology with pro-insulin and has insulin-like metabolic effects, while GH has some effects that are antagonistic to those of insulin (43). IGF-1 is important in maintaining beta-cell mass by stimulating their proliferation and can enhance peripheral insulin sensitivity (44). Mohn et al. studied four members of a family carrying a novel nonsense mutation of the IGF1R gene. The defect was associated with a variable impairment of prenatal and postnatal growth. The authors also reported alterations in carbohydrate metabolism, ranging from normal glucose tolerance in the presence of insulin resistance to IGT and fasting hyperglycemia in association with both insulin resistance and impaired beta-cell function (4).
In conclusions, our study showed the presence of changes in bone architecture, quality, and metabolism in a heterozygous IGF1R deletion patient. The changes in bone metabolism due to the lack of action of IGF-1, a key in bone modeling and accrual, as occurring in a heterozygous IGF1R deletion patient, can be well evaluated through three different techniques (DXA, p-QCT, and QUS) assessing its effects on bone density and quality.
Author Contributions
PP carried out the endocrinological evaluation, conceived the study, and participated in its design. EL carried out the clinical genetic diagnosis, conceived the study, and participated in its design. LC and AV participated in the endocrinological evaluation and in the design of the study. MM participated in the endocrinological evaluation and in the coordination of the study. SS carried out the endocrinological evaluation, conceived the study, and participated in its design and coordination. All authors have read and approved the final manuscript.
Conflict of Interest Statement
The authors do not have any financial or non-financial competing interests in relation to this manuscript.
References
1. LeRoith D, Werner H, Beitner-Johnson D, Roberts CT Jr. Molecular and cellular aspects of the insulin-like growth factor I receptor. Endocr Rev (1995) 16:143–63. doi:10.1210/edrv-16-2-143
2. Baylink D, Lau KH, Mohan S. The role of IGF system in the rise and fall in bone density with age. J Musculoskelet Neuronal Interact (2007) 7:304–5.
3. Mohan S, Richman C, Guo R, Amaar Y, Donahue LR, Wergedal J, et al. Insulin-like growth factor regulates peak bone mineral density in mice by both growth hormone-dependent and -independent mechanisms. Endocrinology (2003) 144:929–36. doi:10.1210/en.2002-220948
4. Mohn A, Marcovecchio ML, de Giorgis T, Pfaeffle R, Chiarelli F, Kiess W. An insulin-like growth factor-I receptor defect associated with short stature and impaired carbohydrate homeostasis in an Italian pedigree. Horm Res Paediatr (2011) 76:136–43. doi:10.1159/000324957
5. Joseph D’Ercole A, Ye P. Expanding the mind: insulin-like growth factor I and brain development. Endocrinology (2008) 149:5958–62. doi:10.1210/en.2008-0920
6. Netchine I, Azzi S, Le Bouc Y, Savage MO. IGF1 molecular anomalies demonstrate its critical role in fetal, postnatal growth and brain development. Best Pract Res Clin Endocrinol Metab (2011) 25:181–90. doi:10.1016/j.beem.2010.08.005
7. Savage MO, Hwa V, David A, Rosenfeld RG, Metherell LA. Genetic defects in the growth hormone-IGF-I axis causing growth hormone insensitivity and impaired linear growth. Front Endocrinol (2011) 2:95. doi:10.3389/fendo.2011.00095
8. Choi JH, Kang M, Kim GH, Hong M, Jin HY, Lee BH, et al. Clinical and functional characteristics of a novel heterozygous mutation of the IGF1R gene and IGF1R haploinsufficiency due to terminal 15q26.2->qter deletion in patients with intrauterine growth retardation and postnatal catch-up growth failure. J Clin Endocrinol Metab (2011) 96:E130–4. doi:10.1210/jc.2010-1789
9. Abuzzahab MJ, Schneider A, Goddard A, Grigorescu F, Lautier C, Keller E, et al. IGF-I receptor mutations resulting in intrauterine and postnatal growth retardation. N Engl J Med (2003) 349:2211–22. doi:10.1056/NEJMoa010107
10. Peoples R, Milatovich A, Francke U. Hemizygosity at the insulin-like growth factor I receptor (IGF1R) locus and growth failure in the ring chromosome 15 syndrome. Cytogenet Cell Genet (1995) 70:228–34. doi:10.1159/000134040
11. Rogan PK, Seip JR, Driscoll DJ, Papenhausen PR, Johnson VP, Raskin S, et al. Distinct 15q genotypes in Russell-Silver and ring 15 syndromes. Am J Med Genet (1996) 62:10–5. doi:10.1002/(SICI)1096-8628(19960301)62:1<10:AID-AJMG3>3.0.CO;2-#
12. de Lacerda L, Carvalho JA, Stannard B, Werner H, Boguszewski MC, Sandrini R, et al. In vitro and in vivo responses to short-term recombinant human insulin-like growth factor-1 (IGF-I) in a severely growth-retarded girl with ring chromosome 15 and deletion of a single allele for the type 1 IGF receptor gene. Clin Endocrinol (Oxf) (1999) 51:541–50. doi:10.1046/j.1365-2265.1999.00799.x
13. Tönnies H, Schulze I, Hennies H, Neumann LM, Keitzer R, Neitzel H. De novo terminal deletion of chromosome 15q26.1 characterised by comparative genomic hybridisation and FISH with locus specific probes. J Med Genet (2001) 38:617–21. doi:10.1136/jmg.38.9.617
14. Walenkamp MJ, de Muinck Keizer-Schrama SM, de Mos M, Kalf ME, van Duyvenvoorde HA, Boot AM, et al. Successful long-term growth hormone therapy in a girl with haploinsufficiency of the insulin-like growth factor-I receptor due to a terminal 15q26.2->qter deletion detected by multiplex ligation probe amplification. J Clin Endocrinol Metab (2008) 93:2421–5. doi:10.1210/jc.2007-1789
15. Ester WA, van Duyvenvoorde HA, de Wit CC, Broekman AJ, Ruivenkamp CA, Govaerts LC, et al. Two short children born small for gestational age with insulin-like growth factor 1 receptor haploinsufficiency illustrate the heterogeneity of its phenotype. J Clin Endocrinol Metab (2009) 94:4717–27. doi:10.1210/jc.2008-1502
16. Veenma DC, Eussen HJ, Govaerts LC, de Kort SW, Odink RJ, Wouters CH, et al. Phenotype-genotype correlation in a familial IGF1R microdeletion case. J Med Genet (2010) 47:492–8. doi:10.1136/jmg.2009.070730
17. Dateki S, Fukami M, Tanaka Y, Sasaki G, Moriuchi H, Ogata T. Identification of chromosome 15q26 terminal deletion with telomere sequences and its bearing on genotype-phenotype analysis. Endocr J (2011) 58:155–9. doi:10.1507/endocrj.K10E-251
18. Rudaks LI, Nicholl JK, Bratkovic D, Barnett CP. Short stature due to 15q26 microdeletion involving IGF1R: report of an additional case and review of the literature. Am J Med Genet A (2011) 155A:3139–43. doi:10.1002/ajmg.a.34310
19. Caliebe J, Broekman S, Boogaard M, Bosch CA, Ruivenkamp CA, Oostdijk W, et al. IGF1, IGF1R and SHOX mutation analysis in short children born small for gestational age and short children with normal birth size (idiopathic short stature). Horm Res Paediatr (2012) 77:250–60. doi:10.1159/000338341
20. Jezela-Stanek A, Kucharczyk M, Pelc M, Chrzanowska KH, Krajewska-Walasek M. Minimal clinical findings in a patient with 15qter microdeletion syndrome: delineation of the associated phenotype. Am J Med Genet A (2012) 158A:922–6. doi:10.1002/ajmg.a.34440
21. Witsch J, Szafranski P, Chen CA, Immken L, Patel GS, Hixson P, et al. Intragenic deletions of the IGF1 receptor gene in five individuals with psychiatric phenotypes and developmental delay. Eur J Hum Genet (2013) 21:1304–7. doi:10.1038/ejhg.2013.42
22. Walenkamp MJ, van der Kamp HJ, Pereira AM, Kant SG, van Duyvenvoorde HA, Kruithof MF, et al. A variable degree of intrauterine and postnatal growth retardation in a family with a missense mutation in the insulin-like growth factor I receptor. J Clin Endocrinol Metab (2006) 91:3062–70. doi:10.1210/jc.2005-1597
23. Kröger H, Kotaniemi A, Vainio P, Alhava E. Bone densitometry of the spine and femur in children by dual-energy x-ray absorptiometry. Bone Miner (1992) 17:75–85. doi:10.1016/0169-6009(92)90712-M
24. Stagi S, Cavalli L, Signorini C, Bertini F, Cerinic MM, Brandi ML, et al. Bone mass and quality in patients with juvenile idiopathic arthritis: longitudinal evaluation of bone-mass determinants by using dual-energy x-ray absorptiometry, peripheral quantitative computed tomography, and quantitative ultrasonography. Arthritis Res Ther (2014) 16:R83. doi:10.1186/ar4525
25. Martin DD, Schweizer R, Schönau E, Binder G, Ranke MB. Growth hormone-induced increases in skeletal muscle mass alleviates the associated insulin resistance in short children born small for gestational age, but not with growth hormone deficiency. Horm Res (2009) 72:38–45. doi:10.1159/000224339
26. Drozdzowska B, Pluskiewicz W, Halaba Z, Misiolek H, Beck B. Quantitative ultrasound at the hand phalanges in 2850 females aged 7 to 77 yr: a cross-sectional study. J Clin Densitom (2005) 8:216–21. doi:10.1385/JCD:8:2:216
27. Okubo Y, Siddle K, Firth H, O’Rahilly S, Wilson LC, Willatt L, et al. Cell proliferation activities on skin fibroblasts from a short child with absence of one copy of the type 1 insulin-like growth factor receptor (IGF1R) gene and a tall child with three copies of the IGF1R gene. J Clin Endocrinol Metab (2003) 88:5981–8. doi:10.1210/jc.2002-021080
28. Frost ML, Siddique M, Blake GM, Moore AE, Schleyer PJ, Dunn JT, et al. Differential effects of teriparatide on regional bone formation using (18)F-fluoride positron emission tomography. J Bone Miner Res (2011) 26:1002–11. doi:10.1002/jbmr.305
29. Stagi S, Cavalli L, Iurato C, Seminara S, Brandi ML, de Martino M. Bone health in children and adolescents: the available imaging techniques. Clin Cases Miner Bone Metab (2013) 10:166–71.
30. Woods KA, Camacho-Hübner C, Savage MO, Clark AJ. Intrauterine growth retardation and postnatal growth failure associated with deletion of the insulin-like growth factor I gene. N Engl J Med (1996) 335:1363–7. doi:10.1056/NEJM199610313351805
31. Yakar S, Rosen CJ, Beamer WG, Ackert-Bicknell CL, Wu Y, Liu JL, et al. Circulating levels of IGF-1 directly regulate bone growth and density. J Clin Invest (2002) 110:771–81. doi:10.1172/JCI15463
32. Rodríguez-de la Rosa L, López-Herradón A, Portal-Núñez S, Murillo-Cuesta S, Lozano D, Cediel R, et al. Treatment with N- and C-terminal peptides of parathyroid hormone-related protein partly compensate the skeletal abnormalities in IGF-I deficient mice. PLoS One (2014) 9:e87536. doi:10.1371/journal.pone.0087536
33. Sundström K, Cedervall T, Ohlsson C, Camacho-Hübner C, Sävendahl L. Combined treatment with GH and IGF-I: additive effect on cortical bone mass but not on linear bone growth in female rats. Endocrinology (2014) 155:4798–807. doi:10.1210/en.2014-1160
34. Wang Y, Nishida S, Elalieh HZ, Long RK, Halloran BP, Bikle DD. Role of IGF-I signaling in regulating osteoclastogenesis. J Bone Miner Res (2006) 21:1350–8. doi:10.1359/jbmr.060610
35. Bikle D, Majumdar S, Laib A, Powell-Braxton L, Rosen C, Beamer W, et al. The skeletal structure of insulin-like growth factor I-deficient mice. J Bone Miner Res (2001) 16:2320–9. doi:10.1359/jbmr.2001.16.12.2320
36. Yakar S, Courtland HW, Clemmons D. IGF-1 and bone: new discoveries from mouse models. J Bone Miner Res (2010) 25:2543–52. doi:10.1002/jbmr.234
37. Elis S, Courtland HW, Wu Y, Rosen CJ, Sun H, Jepsen KJ, et al. Elevated serum levels of IGF-1 are sufficient to establish normal body size and skeletal properties even in the absence of tissue IGF-1. J Bone Miner Res (2010) 25:1257–66. doi:10.1002/jbmr.20
38. Yakar S, Rosen CJ, Bouxsein ML, Sun H, Mejia W, Kawashima Y, et al. Serum complexes of insulin-like growth factor-1 modulate skeletal integrity and carbohydrate metabolism. FASEB J (2009) 23:709–19. doi:10.1096/fj.08-118976
39. Gross TS, Srinivasan S, Liu CC, Clemens TL, Bain SD. Noninvasive loading of the murine tibia: an in vivo model for the study of mechanotransduction. J Bone Miner Res (2002) 17:493–501. doi:10.1359/jbmr.2002.17.3.493
40. Gan Y, Paterson AJ, Zhang Y, Jiang J, Frank SJ. Functional collaboration of insulin-like growth factor-1 receptor (IGF-1R), but not insulin receptor (IR), with acute GH signaling in mouse calvarial cells. Endocrinology (2014) 155:1000–9. doi:10.1210/en.2013-1732
41. Widdowson WM, Gibney J. The effect of growth hormone (GH) replacement on muscle strength in patients with GH-deficiency: a meta-analysis. Clin Endocrinol (Oxf) (2010) 72:787–92. doi:10.1111/j.1365-2265.2009.03716.x
42. Improda N, Capalbo D, Esposito A, Salerno M. Muscle and skeletal health in children and adolescents with GH deficiency. Best Pract Res Clin Endocrinol Metab (2016) 30:771–83. doi:10.1016/j.beem.2016.11.012
43. Holt RI, Simpson HL, Sönksen PH. The role of the growth hormone-insulin-like growth factor axis in glucose homeostasis. Diabet Med (2003) 20:3–15. doi:10.1046/j.1464-5491.2003.00827.x
Keywords: insulin-like growth factor-I receptor, insulin-like growth factor-I, bone metabolism, quantitative ultrasonography, peripheral quantitative computed tomography
Citation: Pelosi P, Lapi E, Cavalli L, Verrotti A, Pantaleo M, de Martino M and Stagi S (2017) Bone Status in a Patient with Insulin-Like Growth Factor-1 Receptor Deletion Syndrome: Bone Quality and Structure Evaluation Using Dual-Energy X-Ray Absorptiometry, Peripheral Quantitative Computed Tomography, and Quantitative Ultrasonography. Front. Endocrinol. 8:227. doi: 10.3389/fendo.2017.00227
Received: 07 January 2017; Accepted: 21 August 2017;
Published: 05 September 2017
Edited by:
Basem M. Abdallah, University of Southern Denmark Odense, DenmarkReviewed by:
Fátima Baptista, Universidade de Lisboa, PortugalBronwen Evans, Cardiff University, United Kingdom
Copyright: © 2017 Pelosi, Lapi, Cavalli, Verrotti, Pantaleo, de Martino and Stagi. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Stefano Stagi, c3RlZmFuby5zdGFnaUB5YWhvby5pdA==