 Isabelle Plo1,2,3
Isabelle Plo1,2,3 Christine Bellanné-Chantelot1,4
Christine Bellanné-Chantelot1,4 Matthieu Mosca1,2,3
Matthieu Mosca1,2,3 Stefania Mazzi1,2,5
Stefania Mazzi1,2,5 Caroline Marty1,2,3
Caroline Marty1,2,3 William Vainchenker1,2,3*
William Vainchenker1,2,3*
- 1INSERM UMR 1170, Gustave Roussy, Villejuif, France
- 2Université Paris-Saclay, UMR1170, Gustave Roussy, Villejuif, France
- 3Gustave Roussy, UMR1170, Villejuif, France
- 4Department of Genetics, AP-HP Hôpitaux Universitaires Pitié Salpêtrière - Charles Foix, UPMC Univ Paris 06, Paris, France
- 5Université Paris-Diderot, Paris, France
Megakaryopoiesis is an original and complex cell process which leads to the formation of platelets. The homeostatic production of platelets is mainly regulated and controlled by thrombopoietin (TPO) and the TPO receptor (MPL)/JAK2 axis. Therefore, any hereditary or acquired abnormality affecting this signaling axis can result in thrombocytosis or thrombocytopenia. Thrombocytosis can be due to genetic alterations that affect either the intrinsic MPL signaling through gain-of-function (GOF) activity (MPL, JAK2, CALR) and loss-of-function (LOF) activity of negative regulators (CBL, LNK) or the extrinsic MPL signaling by THPO GOF mutations leading to increased TPO synthesis. Alternatively, thrombocytosis may paradoxically result from mutations of MPL leading to an abnormal MPL trafficking, inducing increased TPO levels by alteration of its clearance. In contrast, thrombocytopenia can also result from LOF THPO or MPL mutations, which cause a complete defect in MPL trafficking to the cell membrane, impaired MPL signaling or stability, defects in the TPO/MPL interaction, or an absence of TPO production.
Introduction
Megakaryopoiesis is an original cell process which leads to the formation of platelets. The homeostatic production of platelets is mainly regulated by thrombopoietin (TPO) and the TPO receptor (MPL)/JAK2 axis. TPO regulates nearly all stages of the megakaryocyte (MK) differentiation, and this explains that numerous diseases characterized by an alteration in MK/platelet production leading to thrombocytopenia or thrombocytosis are due to either acquired or hereditary mutations in MPL, THPO, or JAK2.
Importance of the TPO/MPL/JAK2 Axis in Megakaryopoiesis
Megakaryopoiesis
Megakaryopoiesis is the process leading to the differentiation of bone marrow progenitors to MKs, giving rise to circulating platelets in the blood. The first step requires the commitment of a multipotent hematopoietic stem cell (HSC) to an MK progenitor followed by several proliferation and differentiation steps (1). There is increasing evidence that MK progenitors may arise directly from an HSC or from a MK/erythroid progenitor (2). This may explain that numerous transcription factors such as TAL1, GATA2, ERG/FLI-1, and RUNX1 regulate HSC properties and MK differentiation and that others, such as GATA1 and GFI1b, regulate also erythroid and MK differentiations. Moreover, TPO plays a central role not only in the MK/platelet lineage but also in HSCs (3). MK progenitors first proliferate before switching to endomitosis, which is a mitosis without cytokinesis (4). Polyploidization generates MKs with a modal distribution with a major peak at 16N. MKs further mature by increasing their cytoplasm and membrane to finally fragment to give rise to platelets. The fragmentation is a dynamic and regulated process. In the human adult, most of megakaryopoiesis takes place in the bone marrow, except the platelet release. Either MKs will send long pseudopods (proplatelets) through the endothelial barrier that will fragment into platelets in the blood flow (5) or the entire MKs will migrate into the circulation to fragment into platelets inside the lung microcirculation (6). It has also been underscored that mouse megakaryopoiesis may occur in the parenchymal lung (6).
TPO/MPL/JAK2 Axis
MPL is located on chromosome 1p34 and consists of 12 exons which encodes a protein of 70 kDa predicted weight (3). In human, MPL expression increases during megakaryopoiesis from HSCs and progenitors to MKs and platelets (7). MPL is a homodimeric class I receptor with three main domains: extracellular, transmembrane, and intracellular (Figure 1). The extracellular domain is composed of two consecutive cytokine receptor motifs composed of a fibronectin-III-like domain characterized by four conserved cysteine residues and a WSXWS motif. The transmembrane domain folds as an α-helix that is necessary for the insertion of the receptor in the membrane and is followed by a juxtamembrane or amphipathic domain composed of the RWQFP sequence (8). The intracellular domain is required to mediate the signaling through its binding via box1 to Janus tyrosine kinase JAK2 (9, 10). JAK2 was shown to be essential for MPL phosphorylation and for activation of downstream signaling pathways. TYK2, another member of the JAK family, can bind to MPL but can be activated by TPO only in the presence of JAK2. Thus, TYK2 was shown to be neither necessary nor sufficient for MPL phosphorylation and signaling (9).
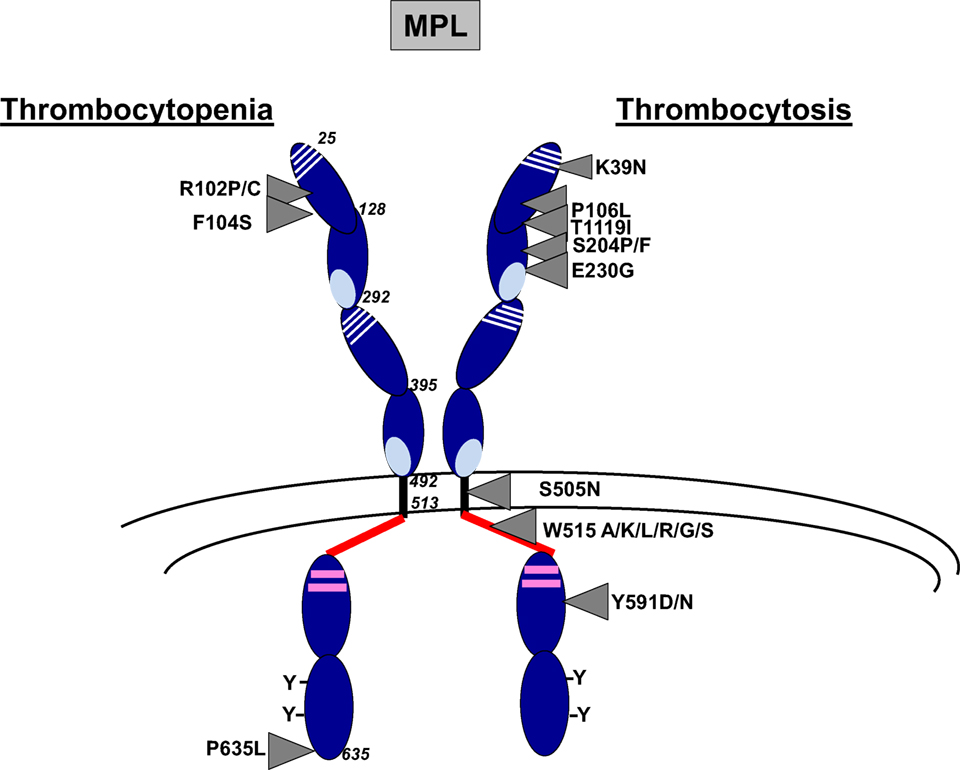
Figure 1. Main MPL mutations resulting in thrombocytosis or thrombocytopenia. MPL is composed of three main domains including extracellular (25–491), transmembrane, (492–513) and intracytoplasmic (514–635) domains. The extracellular domain is composed of two cytokine receptor domains (CRM) that correspond to an immunoglobulin fold of fibronectin type III. The first part of the CRM has four characteristically spaced cysteine residues, and the second part displays a WSXWS motif at the C-terminal. The intracellular domain contains box1 and box2 (in pink) and several conserved tyrosine (Y) residues. Main MPL mutations in thrombocytosis and thrombocytopenia are indicated by arrows.
The ligand of MPL is the glycoprotein hormone TPO encoded by the THPO gene at chromosome 3q27.1 and composed of five coding exons. It is organized in two domains: an N-terminal cytokine domain including the first 153 amino acids followed by a highly glycosylated C-terminal domain that regulates its production and increases its half-life in vivo (11) (Figure 2A). TPO is constitutively produced by the liver (12). However, its level is regulated by clearance through the platelet mass (13, 14) and its synthesis by inflammation. Kidney, spleen, and bone marrow have been found to contribute to the production of TPO at a lesser extent (12). It has been suggested that the TPO produced by bone marrow stromal cells and eventually MKs play a central role in the regulation of HSC quiescence. TPO synthesis in the liver can be regulated during inflammatory conditions by stimulation of IL-6 (15). Moreover, old desialylated platelets can bind to the Ashwell–Morell receptor on hepatocytes to induce TPO gene expression through activation of the JAK/STAT3 pathway via IL-6 receptor activation (16).
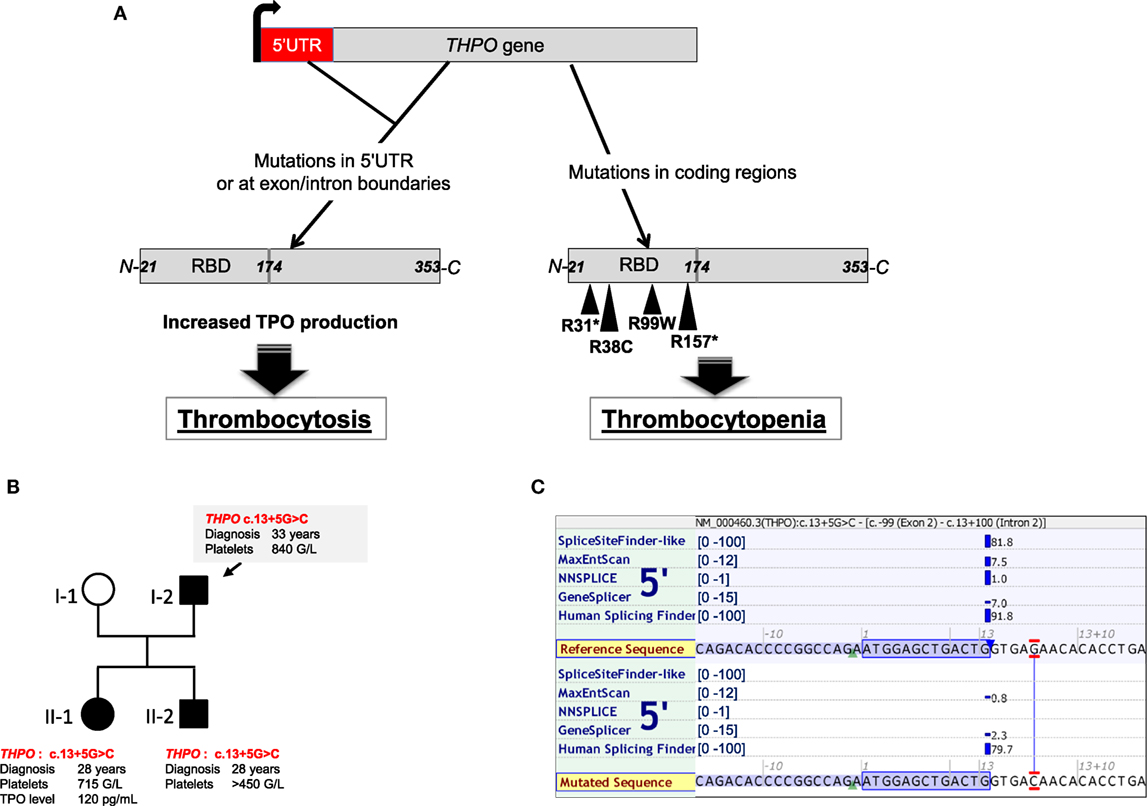
Figure 2. THPO mutations found in hereditary thrombocytosis, thrombocytopenia, and bone marrow aplasia. (A) Schemes of THPO gene and thrombopoietin (TPO) protein. Mutations in THPO gene affecting either 5′UTR or splice sites (defective alternative splicing) lead to increased THPO mRNA translation and increased TPO synthesis in hereditary thrombocytosis. Mutations in coding regions of THPO affect the RBD (receptor-binding domain) and are found in hereditary thrombocytopenia or bone marrow aplasia. (B) Illustration of a pedigree with THPO mutation from a family harboring a mild thrombocytosis. (C) In silico analysis of the THPO c.13+5G>C based on five predictive algorithms of pathogenicity for splice site defects. Analysis was run using the Alamut Visual version 2.9 software (Interactive Biosoftware, Rouen, France).
MPL is pre-associated with JAK2 and TYK2 in the endoplasmic reticulum. The two kinases play a role of chaperone molecules and increase MPL trafficking to the cell membrane (17). The MPL/JAK2 complex becomes mature through traffic in the Golgi apparatus, where MPL undergoes successive glycosylations on asparagine residues (N-glycosylations) and is addressed at cell surface as a partially pre-dimerized receptor. TPO binding to MPL involves not only two main residues, D261 and L265, on the extracellular domain of MPL but also a site close to residue F104 (18, 19) and induces an increased dimerization of MPL and its stabilization. TPO binding induces conformational changes of MPL, which move closer the intracellular domains of the homodimer and position the two associated JAK2/TYK2 in proximity leading to their transphosphorylation (20). Active JAK2 phosphorylates several conserved tyrosine residues in the intracellular domain of MPL, which serve as docking sites for a number of substrates including STAT1, STAT3, and STAT5 and adaptors such as CBL, VAV, and SHC leading to activation of mitogen-activated protein kinase (MAPK) and phosphatidylinositol 3-kinase/AKT (21). After MPL activation, the TPO/MPL/JAK2 is internalized to either be recycled or, more often, degraded through proteasomes and lysosomes induced after activation of negative regulators such as the ubiquitin ligase CBL (22). In addition, members of the SOCS family (SOCS1, SOCS2) bind to phosphorylated JAK2 and/or MPL and induce their degradation through the proteasome pathway (23). Furthermore, LNK, an adaptor protein that binds to JAK2, and other phosphatases can limit MPL activation (24). Finally, PIAS negatively regulates the JAK2/STAT pathway by directly inhibiting the binding of STAT to DNA or by causing sumoylation of STATs (25). This negative regulation plays a central role in limiting the stimulation of MPL.
TPO/MPL/JAK2 Axis As a Central Player in Megakaryopoiesis in Mice
The importance of the TPO/MPL/JAK2 axis in megakaryopoiesis is illustrated by several lines of evidence. Both the constitutive thpo−/− and mpl−/− knockout (KO) mice and the conditional jak2 KO mice induced in adulthood after crossing them with transgenic mice expressing tamoxifen-inducible Cre under SCL promoter showed marked alterations in the HSC compartment associated with a thrombocytopenia (150–400 × 109/L) (26–28). Of note, tyk2−/− KO mice did not harbor thrombocytopenia consistently with its dispensable role in MPL activation (29). The levels of TPO were highly elevated in mouse plasma from mpl−/− and conditional jak2−/− mice due to the loss of clearance by platelet mass through Mpl binding. The conditional mpl and jak2 KO mice were also crossed with PF4-Cre transgenic mice, which allowed the deletion of mpl or jak2 in MKs and platelets, thereby only in the late stages of megakaryopoiesis. In a counterintuitive manner, these mice presented a strong thrombocytosis (5,000–10,000 × 109/L) with normal TPO levels in the plasma (30, 31). Thrombocytosis in these models was explained by the stimulation of the MK progenitor proliferation still expressing Mpl. Therefore, these data showed that the proliferation of MK progenitors (first steps of megakaryopoiesis) is highly dependent on TPO in contrast to the platelet production from MKs. A strong thrombocytosis but with a partial defect in the HSC compartment was also observed in mpl transgenic mice performed in mpl−/− context which leads to subnormal Mpl expression in maturing MKs and platelets. These models demonstrate that low levels of MPL in MK progenitors are sufficient to induce their proliferation and highlighted the importance of MPL expression level during megakaryopoiesis (32, 33).
Effect of TPO/MPL/JAK2 Axis Alterations in the Stimulation of Megakaryopoiesis
THPO/MPL/JAK2 gain-of-function (GOF) or loss-of-function (LOF) mutations as well as LOF mutations of negative regulators have been identified in diseases leading to MK hyperplasia (Table 1).
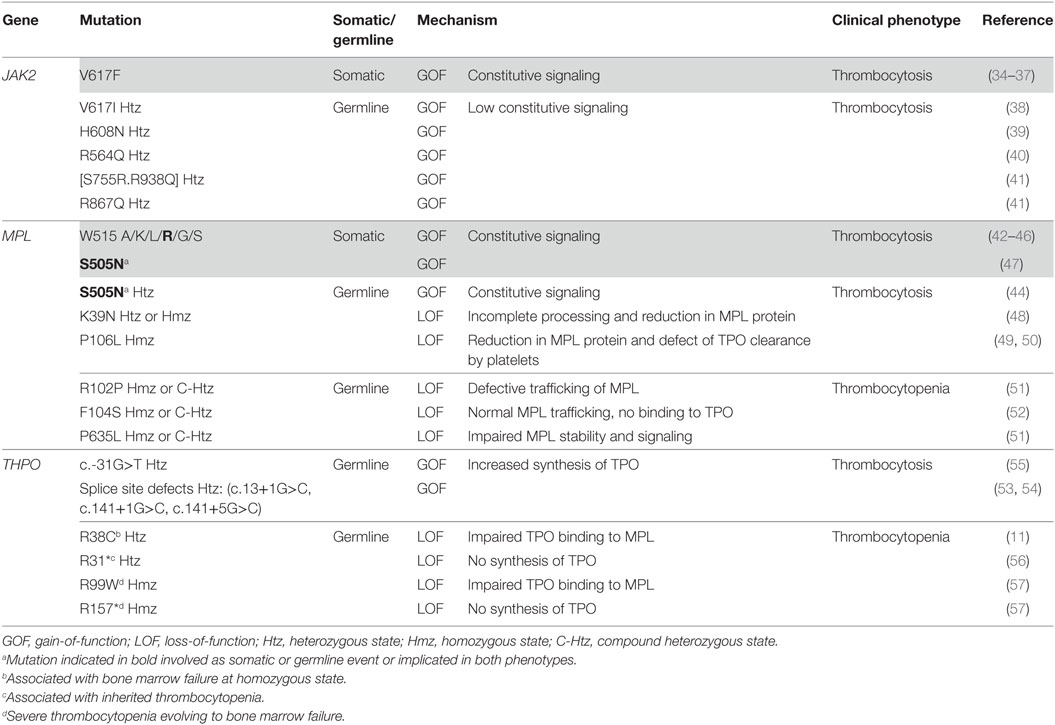
Table 1. Genetic alterations of the thrombopoietin (TPO)/MPL/JAK2 axis leading to thrombocytosis and thrombocytopenia.
Mutations on the TPO/MPL/JAK2 Axis
MPL Mutations
MPL mutations have been found in myeloproliferative neoplasms (MPNs) including essential thrombocythemia (ET) (3%) and primary myelofibrosis (PMF) (5%) and in hereditary thrombocytosis (Figure 1). The main mutations in MPNs are located in the exon 10 of MPL and affect the tryptophan at position 515 leading to amino acid changes such as MPL W515K/L/A/G/S/R that show a GOF activity (42–46). The tryptophan at position 515 is part of the amphipathic domain located at the junction between the transmembrane and the cytoplasmic domains of MPL (RWQFP) and has been shown to prevent the dimerization of MPL in the absence of cytokine (8). Its substitution for leucine, lysine, arginine, or alanine leads to constitutive activation of the receptor (58). MPL W515L retroviral mouse model after bone marrow transplantation is characterized by a strong thrombocytosis followed by a myelofibrosis recapitulating what is observed in human setting (46). Moreover, a few acquired MPL mutations outside the exon 10, leading to amino acid changes in the extra- and intra-cytoplasmic domains of MPL (S204P/F, E230G, Y591D/N, T119I), have been more recently described in sporadic MPN cases. They have been demonstrated to show a lower GOF activity than MPL W515 mutants (59, 60). They could be associated with other acquired or constitutional molecular events, including JAK2 mutations, to induce the disease phenotype. Finally, the MPL S505N mutation was initially described in familial cases of hereditary thrombocytosis, although it has also been described in sporadic cases of ET and PMF (44, 47, 61). S505 is localized in the transmembrane domain of the receptor and the mutation induces a constitutive dimerization of MPL in an active conformation (44).
Other very interesting MPL mutations affecting the extracellular domain of MPL (MPL P106L and MPL K39N) were discovered in hereditary thrombocytosis but are associated with high TPO levels in contrast to other MPL mutations.
MPL K39N (MPL Baltimore) was described as a polymorphism restricted to 7% of African Americans. Heterozygous cases present a mild thrombocytosis around 500 × 109/L platelets, whereas homozygous cases showed around 800 × 109/L platelets. It was shown that MPL K39N is expressed at low levels at the cell surface and in an incomplete maturation form suggesting a LOF mutation through an abnormal membrane expression of the receptor (48).
MPL P106L mutation was initially found in an Arab family, which segregates with strong thrombocytosis in homozygous cases suggesting autosomal recessive inheritance associated with high TPO levels (49). Sometimes, mild thrombocytosis was observed in heterozygous cases. Later on, it was reported that MPL P106L presents a defect in trafficking that prevents its translocation to the cell surface, but is still capable to bind TPO and to signal in the cytosol (62). This seems unlikely since the extracellular domain of MPL is localized in the lumen of endoplasmic reticulum. Our group has also identified such mutation in one Saudian and one Kuweitian family harboring strong thrombocytosis cases with high TPO levels with autosomal recessive inheritance. We confirmed that MPL P106L presents a defect of trafficking, but we demonstrated that low levels of this mutant are addressed at the membrane of MK progenitors. Moreover, there was no spontaneous growth of MKs from patients while MKs, but not platelets, responded to TPO. This study also showed that MPL P106L was better localized at the cell surface on immature than on mature MKs, explaining a proliferative response to TPO in MK progenitors and a defect in TPO clearance in platelets. Moreover, the retroviral MPL P106L mouse model performed in mpl−/− mice induced a thrombocytosis phenotype with a high TPO level (50). Overall, MPL P106L seems to be compatible with an LOF mutation (traffic defect), which induces a paradoxical thrombocytosis through proliferation of MK progenitors and a defect of TPO clearance by the platelet mass.
JAK2 Mutations
The recurrent JAK2 V617F mutation occurring at exon 14 of JAK2 was identified in MPNs (34–37) and refractory sideroblastic anemia with thrombocytosis (RARS-T) (63). In MPNs, JAK2 V617F is present in 50–60% of ET and PMF and 95% of polycythemia vera (PV). The replacement of a guanine to thymidine results in the substitution of a valine by a phenylalanine at position 617, in the pseudokinase JH2 domain of JAK2. The JAK2 V617F mutation leads to stiffening of the helix alpha C of this JH2 domain, favoring the transphosphorylation of JH1 and thus the constitutive activation of JAK2 (64). The JAK2 V617F mutation requires the presence of homodimeric type 1 receptors such as MPL, EPOR and G-CSFR to induce a constitutive signaling and a spontaneous proliferation (65). The mutation can be heterozygous or homozygous after a mitotic recombination, with heterozygous clones mainly found in ET while homozygous clones predominate in secondary MF showing a gene dosage effect contribution to the phenotype of the disease (66). In mice, all the models including retroviral, transgenic, and knock-in recapitulate an MPN-like disease (33, 67–70). Even if the most frequent phenotype is a PV progressing to MF, some differences have been observed depending on the species (human or mouse) and on the expression levels of JAK2 V617F. Particularly, low levels of human JAK2 V617F in hematopoiesis in inducible transgenic mice with a jak2 minimal promoter led to an ET, whereas higher expression resulted in PV (71). Moreover, inducible transgenic expression of human JAK2 V617F only in MK lineage (PF4-Cre) presents a specific ET-like phenotype (72). In knock-in mouse models, germline human JAK2 V617F led to an ET when heterozygous and to a PV when homozygous (69). The conditional heterozygous mouse jak2 V617F in hematopoiesis showed an ET/PV phenotype and a severe PV progressing to MF at homozygous state (67, 73). Therefore, as in human setting, the level of JAK2 V617F determines the disease phenotype.
Other JAK2 mutations have been highlighted in hereditary thrombocytosis with sometimes the same residue affected than in sporadic MPN cases but with a different amino acid changes, such as JAK2 V617I, or on different residue located in both the pseudokinase (JAK2 H608N, JAK2 R564Q, JAK2 S755R) and the kinase domain of the protein (JAK2 R938Q and JAK2 R867Q) (38–41). All these mutants show a low constitutive kinase activity, seem to trigger spontaneous signaling only in the presence of MPL, and activate more STAT1 than the other STATs in contrast to JAK2 V617F (41).
THPO Mutations
Germline GOF mutations in the THPO gene have been described in hereditary thrombocytosis. They are located in the 5′-untranslated region or affect splice donor sites of the gene and lead to an increased mRNA translation and synthesis of TPO (54, 55, 74). As an illustration, Figure 2B presents one family with THPO mutation predicted to affect the splice donor site of exon 2 (c.13+5G>C) in a hereditary thrombocytosis (Figures 2B,C).
Mutations on Important Regulators of the MPL/JAK2 Axis
Other molecules are important for the MPL/JAK2 signaling axis and the regulation of megakaryopoiesis.
CALR Mutations
CALR is a chaperone protein resident of the endoplasmic reticulum. It not only helps neosynthetized proteins and glycoproteins to fold before being addressed to the cell surface but also plays an essential role in calcium homeostasis (75). CALR mutations have been identified in ET and PMF in around 25–30% of the cases. These mutations are also, but rarely, found in RARS-T (76, 77). More than 50 different mutations have been described. All of them correspond to frameshift mutations in the exon 9 of the gene and lead to a new and quite similar positively charged C-terminal sequence and the loss of the retrieval KDEL motif into endoplasmic reticulum. The most frequent mutations accounting for more than 80–90% of cases are a 52 bp deletion (type 1 mutation or CALRdel52) and an insertion of 5 bp (type 2 mutation or CALRins5). These mutations do not have the same prevalence between ET and PMF with the CALRdel52 more frequently associated with PMF than with ET and conversely for CALRins5 (78–80). Recently, our laboratory and other groups showed that CALR mutants induce the JAK2/STAT pathway by specifically activating MPL and at a lower extent G-CSFR for the CALRdel52 mutation only (81). The mechanism is incompletely understood. However, we and others have shown that CALR mutants interact with the glycosylated asparagine in the extracellular part of MPL via its lectin domain present in its N-globular domain, but MPL activation requires the new C-terminus and its positive charges (81–84). The retroviral mouse models recapitulate a thrombocytosis progressing into MF more frequently for CALRdel52 mice than for CALRins5 mice (84) and the transgenic CALRdel52 mice recapitulate an ET (85).
LNK Mutations
Loss-of-function mutations of negative regulators of the MPL/JAK2 signaling axis have also been found to control megakaryopoiesis. SH2B3 (also named LNK) is an adaptor protein, inhibiting the JAK/STAT pathway. It binds to the phosphorylated tyrosine 813 of JAK2 via its SH2 domain and controls MPL-mediated signaling (24). SH2B3 mutations were described in rare cases of ET and PMF patients (86). Most of them occur in a hot spot encompassing residues E208 to G234 of the Pleckstrin (PH) domain, but other mutations are located outside this domain (87, 88). Since this PH domain could be involved in the membrane localization of the protein through the binding to phosphatidylinositol-3-phosphate, the hypothesis is that the mutations detected lead to an abnormal localization of the protein in the cytoplasm (89). However, since the N-terminal dimerization domain remains intact in the LNK mutants, they could bind and interact with the normal (wild-type) protein in the cytoplasm, thus leading to a dominant-negative effect (86). However, the mechanism is still debated since the study of different LNK mutants in the PH domain in cell lines showed a moderate LOF with no dominant-negative effect and an unaffected binding to JAK2 (90). lnk−/− mice recapitulate a thrombocytosis associated with a splenomegaly and a MF (91). In MPNs, it is presently unclear if mutations in LNK are sufficient to trigger the disease or if another driver mutation such as JAK2 V617F is absolutely required. Germline mutations in LNK have been associated with some primary erythrocytosis (92).
CBL Mutations
CBL is an E3 ligase, which induces the ubiquitination of MPL and JAK2 and leads to their degradation by the proteasome and the lysosome pathways (22). Mutations in c-CBL are nucleotide substitutions or small insertions/deletions in the exons 8 and 9 of the gene and lead to modification of the linker or the RING finger domain (93). They are found in rare cases of ET and PMF. These mutations frequently result in the loss of ubiquitin E3 ligase activity, thus inhibiting lysosome or ubiquitin/proteasome-mediated degradation of MPL/JAK2.
Thus, all the molecules of the TPO/MPL/JAK2 signaling axis are likely to be involved in the MK hyperplasia associated with hereditary thrombocytosis and sporadic classical MPNs. They include GOF mutations of THPO, MPL, JAK2, and LOF mutations of LNK and CBL. These mutations lead to constitutive activation of the downstream signaling pathways, including STAT1/3/5, PI3K/AKT, and MAPK, that control the proliferation/survival and differentiation. Surprisingly, MPL LOF can be found in hereditary thrombocytosis by inducing a paradoxical effect on MK progenitors and platelets. Finally, unexpected mutations in CALR gene, a chaperone of the endoplasmic reticulum that activates MPL, have also been implicated in sporadic ETs.
Mutations Involved in Defective Megakaryopoiesis
TPO/MPL axis alterations can also lead to defects in megakaryopoiesis like in some autosomal recessive aplastic anemia and in congenital amegakaryocytic thrombocytopenia (CAMT) (Table 1).
CAMT is a rare autosomal recessive bone marrow failure presenting an isolated thrombocytopenia at birth (94). These cases are associated with very high TPO levels. MPL mutations in CAMT are found at homozygous state or compound heterozygous mutations generally located throughout the MPL gene, but with a higher recurrence in the exons 2 and 3 encoding the first cytokine receptor motif (52). They can be missense, splicing, frameshift, and nonsense mutations (52, 95). They are LOF mutations, and the frameshift or nonsense mutations are generally more aggressive than missense or splicing mutations leading to early progression to bone marrow failure (52). This is probably due to a complete absence of protein due to truncated proteins or proteins without intracellular signaling. Missense mutations are classified into three groups (Figure 1): (i) those that cannot be addressed to the cell surface and that are blocked in the endoplasmic reticulum, such as MPL R102P, MPL R257C/L, and MPL W154R seem to behave in the same way (51, 95, 96); (ii) those without TPO binding activity due to a loss of stabilization of hydrogen bond, but the MPL mutants, are still normally addressed to the cell surface in a glycosylated mature form, such as MPL F104S (19, 52, 96); (iii) those that lead to an unstable protein that is highly degraded, such as MPL P635L (51, 97). In MPL F104S-like mutation, addition of eltrombopag, a non-peptide TPO mimetic that binds to the transmembrane domain of MPL at histine 499, may overcome the thrombocytopenia (98, 99). In contrast, romiplostim that is a TPO peptide mimetic that binds to MPL in a competitive manner could not be used in this case. In MPL P635L-like mutations, some proteasome inhibitors may show benefit by stabilizing the protein. However, in MPL R102P-like mutations, the only curative treatment remains the bone marrow transplantation.
THPO LOF mutations (Figure 2A) have also been found in families with autosomal recessive bone marrow failure. In one study, a homozygous missense THPO R38C mutation was found in two affected siblings localized in the receptor-binding domain that destabilized the interaction between TPO and MPL (11). A recent work has identified three other families carrying two novel mutations THPO R99W, THPO R157* (57). One case was efficiently treated with TPO mimetics such as romiplostim as a substitution therapy. Alternatively, eltrombopag could also be used (98, 100). Finally, another heterozygous THPO R31* was observed but this time in two families of inherited thrombocytopenia. These cases presented with various penetrance of the disease, low TPO levels, and mild thrombocytopenia (100 × 109/L) (56).
SRC GOF mutation (SRC E527K) has been identified in families with thrombocytopenia progressing into MF and displaying elevated TPO levels. Src family kinases such as BTK and SYK have been shown to interact with MPL at the residue Y591 and participate in its negative regulation since depletion of SRC induces an increase in MPL phosphorylation (101). SRC E527K mutation increases the kinase activity of SRC and may negatively regulate MPL activation, which could partially explain the defective platelet production.
Conclusion
Overall, thrombocytosis and thrombocytopenia have been shown to be due to mutations in molecules of the TPO/MPL/JAK2 signaling axis. In both disease cases, the same molecule can be affected leading to two opposite phenotypes by different mechanisms. Thrombocytosis is either due to GOF alterations that activate MPL signaling (MPL, JAK2, CALR) or due to LOF mutations of negative regulators of this pathway (CBL, LNK) (Figure 1). In certain cases, GOF mutations in THPO lead to increased TPO synthesis and thrombocytosis (Figure 2). Alternatively, thrombocytosis can paradoxically result from mutations in MPL that lead to defect in MPL trafficking and result in increased TPO levels by alteration of its clearance. Interestingly, these LOF mutations can thus functionally behave as GOF for platelet production in vivo.
Author Contributions
The authors IP, CBC, MM, SM, CM and WV wrote different parts of the manuscript.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
This work was supported by grants from l’Agence Nationale de la Recherche (ANR-13-JVSV1-GERMPN-01), the Laurette Fugain foundation, the GIS-Institute for rare diseases for high-throughput sequencing (AO9102LS), the Association de recherche sur la moelle osseuse (ARMO), the Association pour la Recherche contre le Cancer (ARC) (Fondation ARC libre 2012), and the regional PHRC AOR07014. IP, CM, WV are funded by the program “Investissements d’avenir” (Labex GR-Ex). MM is a recipient from la Ligue Nationale Contre le Cancer. SM is a recipient from MENRT grant.
Funding
Agence nationale pour la recherche (ANR-13-JVSV1-GERMPN-01) Association de recherche sur la moelle osseuse (ARMO) Association pour la Recherche contre le Cancer Regional PHRC AOR07014. IP, CM, WV are funded by the program “Investissements d’avenir” (Labex GR-Ex). MM is a recipient from la Ligue Nationale Contre le Cancer. SM is a recipient from MENRT grant.
References
1. Chang Y, Bluteau D, Debili N, Vainchenker W. From hematopoietic stem cells to platelets. J Thromb Haemost (2007) 5(Suppl 1):318–27. doi:10.1111/j.1538-7836.2007.02472.x
2. Notta F, Zandi S, Takayama N, Dobson S, Gan OI, Wilson G, et al. Distinct routes of lineage development reshape the human blood hierarchy across ontogeny. Science (2016) 351:aab2116. doi:10.1126/science.aab2116
3. Hitchcock IS, Kaushansky K. Thrombopoietin from beginning to end. Br J Haematol (2014) 165:259–68. doi:10.1111/bjh.12772
4. Lordier L, Jalil A, Aurade F, Larbret F, Larghero J, Debili N, et al. Megakaryocyte endomitosis is a failure of late cytokinesis related to defects in the contractile ring and Rho/Rock signaling. Blood (2008) 112:3164–74. doi:10.1182/blood-2008-03-144956
5. Scurfield G, Radley JM. Aspects of platelet formation and release. Am J Hematol (1981) 10:285–96. doi:10.1002/ajh.2830100308
6. Lefrancais E, Ortiz-Munoz G, Caudrillier A, Mallavia B, Liu F, Sayah DM, et al. The lung is a site of platelet biogenesis and a reservoir for haematopoietic progenitors. Nature (2017) 544:105–9. doi:10.1038/nature21706
7. Besancenot R, Roos-Weil D, Tonetti C, Abdelouahab H, Lacout C, Pasquier F, et al. JAK2 and MPL protein levels determine TPO-induced megakaryocyte proliferation vs differentiation. Blood (2014) 124:2104–15. doi:10.1182/blood-2014-03-559815
8. Staerk J, Lacout C, Sato T, Smith SO, Vainchenker W, Constantinescu SN. An amphipathic motif at the transmembrane-cytoplasmic junction prevents autonomous activation of the thrombopoietin receptor. Blood (2006) 107:1864–71. doi:10.1182/blood-2005-06-2600
9. Drachman JG, Millett KM, Kaushansky K. Thrombopoietin signal transduction requires functional JAK2, not TYK2. J Biol Chem (1999) 274:13480–4. doi:10.1074/jbc.274.19.13480
10. Tortolani PJ, Johnston JA, Bacon CM, McVicar DW, Shimosaka A, Linnekin D, et al. Thrombopoietin induces tyrosine phosphorylation and activation of the Janus kinase, JAK2. Blood (1995) 85:3444–51.
11. Dasouki MJ, Rafi SK, Olm-Shipman AJ, Wilson NR, Abhyankar S, Ganter B, et al. Exome sequencing reveals a thrombopoietin ligand mutation in a Micronesian family with autosomal recessive aplastic anemia. Blood (2013) 122:3440–9. doi:10.1182/blood-2012-12-473538
12. Qian S, Fu F, Li W, Chen Q, de Sauvage FJ. Primary role of the liver in thrombopoietin production shown by tissue-specific knockout. Blood (1998) 92:2189–91.
13. Fielder PJ, Gurney AL, Stefanich E, Marian M, Moore MW, Carver-Moore K, et al. Regulation of thrombopoietin levels by c-mpl-mediated binding to platelets. Blood (1996) 87:2154–61.
14. Fielder PJ, Hass P, Nagel M, Stefanich E, Widmer R, Bennett GL, et al. Human platelets as a model for the binding and degradation of thrombopoietin. Blood (1997) 89:2782–8.
15. Wolber EM, Jelkmann W. Interleukin-6 increases thrombopoietin production in human hepatoma cells HepG2 and Hep3B. J Interferon Cytokine Res (2000) 20:499–506. doi:10.1089/10799900050023915
16. Grozovsky R, Begonja AJ, Liu K, Visner G, Hartwig JH, Falet H, et al. The Ashwell-Morell receptor regulates hepatic thrombopoietin production via JAK2-STAT3 signaling. Nat Med (2015) 21:47–54. doi:10.1038/nm.3770
17. Royer Y, Staerk J, Costuleanu M, Courtoy PJ, Constantinescu SN. Janus kinases affect thrombopoietin receptor cell surface localization and stability. J Biol Chem (2005) 280:27251–61. doi:10.1074/jbc.M501376200
18. Chen WM, Yu B, Zhang Q, Xu P. Identification of the residues in the extracellular domain of thrombopoietin receptor involved in the binding of thrombopoietin and a nuclear distribution protein (human NUDC). J Biol Chem (2010) 285:26697–709. doi:10.1074/jbc.M110.120956
19. Fox NE, Lim J, Chen R, Geddis AE. F104S c-Mpl responds to a transmembrane domain-binding thrombopoietin receptor agonist: proof of concept that selected receptor mutations in congenital amegakaryocytic thrombocytopenia can be stimulated with alternative thrombopoietic agents. Exp Hematol (2010) 38:384–91. doi:10.1016/j.exphem.2010.02.007
20. Staerk J, Defour JP, Pecquet C, Leroy E, Antoine-Poirel H, Brett I, et al. Orientation-specific signalling by thrombopoietin receptor dimers. EMBO J (2011) 30:4398–413. doi:10.1038/emboj.2011.315
21. Drachman JG, Rojnuckarin P, Kaushansky K. Thrombopoietin signal transduction: studies from cell lines and primary cells. Methods (1999) 17:238–49. doi:10.1006/meth.1998.0734
22. Saur SJ, Sangkhae V, Geddis AE, Kaushansky K, Hitchcock IS. Ubiquitination and degradation of the thrombopoietin receptor c-Mpl. Blood (2010) 115:1254–63. doi:10.1182/blood-2009-06-227033
23. Starr R, Hilton DJ. Negative regulation of the JAK/STAT pathway. Bioessays (1999) 21:47–52. doi:10.1002/(SICI)1521-1878(199901)21:1<47::AID-BIES6>3.0.CO;2-N
24. Tong W, Lodish HF. Lnk inhibits TPO-MPL signaling and TPO-mediated megakaryocytopoiesis. J Exp Med (2004) 200:569–80. doi:10.1084/jem.20040762
25. Liu B, Liao J, Rao X, Kushner SA, Chung CD, Chang DD, et al. Inhibition of Stat1-mediated gene activation by PIAS1. Proc Natl Acad Sci U S A (1998) 95:10626–31. doi:10.1073/pnas.95.18.10626
26. Alexander WS, Roberts AW, Nicola NA, Li R, Metcalf D. Deficiencies in progenitor cells of multiple hematopoietic lineages and defective megakaryocytopoiesis in mice lacking the thrombopoietic receptor c-Mpl. Blood (1996) 87:2162–70.
27. de Sauvage FJ, Carver-Moore K, Luoh SM, Ryan A, Dowd M, Eaton DL, et al. Physiological regulation of early and late stages of megakaryocytopoiesis by thrombopoietin. J Exp Med (1996) 183:651–6. doi:10.1084/jem.183.2.651
28. Grisouard J, Hao-Shen H, Dirnhofer S, Wagner KU, Skoda RC. Selective deletion of Jak2 in adult mouse hematopoietic cells leads to lethal anemia and thrombocytopenia. Haematologica (2014) 99:e52–4. doi:10.3324/haematol.2013.100016
29. Shimoda K, Kato K, Aoki K, Matsuda T, Miyamoto A, Shibamori M, et al. Tyk2 plays a restricted role in IFN alpha signaling, although it is required for IL-12-mediated T cell function. Immunity (2000) 13:561–71. doi:10.1016/S1074-7613(00)00055-8
30. Meyer SC, Keller MD, Woods BA, LaFave LM, Bastian L, Kleppe M, et al. Genetic studies reveal an unexpected negative regulatory role for Jak2 in thrombopoiesis. Blood (2014) 124:2280–4. doi:10.1182/blood-2014-03-560441
31. Ng AP, Kauppi M, Metcalf D, Hyland CD, Josefsson EC, Lebois M, et al. Mpl expression on megakaryocytes and platelets is dispensable for thrombopoiesis but essential to prevent myeloproliferation. Proc Natl Acad Sci U S A (2014) 111:5884–9. doi:10.1073/pnas.1404354111
32. Lannutti BJ, Epp A, Roy J, Chen J, Josephson NC. Incomplete restoration of Mpl expression in the mpl-/- mouse produces partial correction of the stem cell-repopulating defect and paradoxical thrombocytosis. Blood (2009) 113:1778–85. doi:10.1182/blood-2007-11-124859
33. Tiedt R, Coers J, Ziegler S, Wiestner A, Hao-Shen H, Bornmann C, et al. Pronounced thrombocytosis in transgenic mice expressing reduced levels of Mpl in platelets and terminally differentiated megakaryocytes. Blood (2009) 113:1768–77. doi:10.1182/blood-2008-03-146084
34. Baxter EJ, Scott LM, Campbell PJ, East C, Fourouclas N, Swanton S, et al. Acquired mutation of the tyrosine kinase JAK2 in human myeloproliferative disorders. Lancet (2005) 365:1054–61. doi:10.1016/S0140-6736(05)74230-6
35. James C, Ugo V, Le Couedic JP, Staerk J, Delhommeau F, Lacout C, et al. A unique clonal JAK2 mutation leading to constitutive signalling causes polycythaemia vera. Nature (2005) 434:1144–8. doi:10.1038/nature03546
36. Kralovics R, Passamonti F, Buser AS, Teo SS, Tiedt R, Passweg JR, et al. A gain-of-function mutation of JAK2 in myeloproliferative disorders. N Engl J Med (2005) 352:1779–90. doi:10.1056/NEJMoa051113
37. Levine RL, Wadleigh M, Cools J, Ebert BL, Wernig G, Huntly BJ, et al. Activating mutation in the tyrosine kinase JAK2 in polycythemia vera, essential thrombocythemia, and myeloid metaplasia with myelofibrosis. Cancer Cell (2005) 7:387–97. doi:10.1016/j.ccr.2005.03.023
38. Mead AJ, Rugless MJ, Jacobsen SE, Schuh A. Germline JAK2 mutation in a family with hereditary thrombocytosis. N Engl J Med (2012) 366:967–9. doi:10.1056/NEJMc1200349
39. Rumi E, Harutyunyan AS, Casetti I, Pietra D, Nivarthi H, Moriggl R, et al. A novel germline JAK2 mutation in familial myeloproliferative neoplasms. Am J Hematol (2014) 89:117–8. doi:10.1002/ajh.23614
40. Etheridge SL, Cosgrove ME, Sangkhae V, Corbo LM, Roh ME, Seeliger MA, et al. A novel activating, germline JAK2 mutation, JAK2R564Q, causes familial essential thrombocytosis. Blood (2014) 123:1059–68. doi:10.1182/blood-2012-12-473777
41. Marty C, Saint-Martin C, Pecquet C, Grosjean S, Saliba J, Mouton C, et al. Germ-line JAK2 mutations in the kinase domain are responsible for hereditary thrombocytosis and are resistant to JAK2 and HSP90 inhibitors. Blood (2014) 123:1372–83. doi:10.1182/blood-2013-05-504555
42. Abe M, Suzuki K, Inagaki O, Sassa S, Shikama H. A novel MPL point mutation resulting in thrombopoietin-independent activation. Leukemia (2002) 16:1500–6. doi:10.1038/sj.leu.2402554
43. Chaligne R, Tonetti C, Besancenot R, Roy L, Marty C, Mossuz P, et al. New mutations of MPL in primitive myelofibrosis: only the MPL W515 mutations promote a G1/S-phase transition. Leukemia (2008) 22:1557–66. doi:10.1038/leu.2008.137
44. Ding J, Komatsu H, Wakita A, Kato-Uranishi M, Ito M, Satoh A, et al. Familial essential thrombocythemia associated with a dominant-positive activating mutation of the c-MPL gene, which encodes for the receptor for thrombopoietin. Blood (2004) 103:4198–200. doi:10.1182/blood-2003-10-3471
45. Pardanani AD, Levine RL, Lasho T, Pikman Y, Mesa RA, Wadleigh M, et al. MPL515 mutations in myeloproliferative and other myeloid disorders: a study of 1182 patients. Blood (2006) 108:3472–6. doi:10.1182/blood-2006-04-018879
46. Pikman Y, Lee BH, Mercher T, McDowell E, Ebert BL, Gozo M, et al. MPLW515L is a novel somatic activating mutation in myelofibrosis with myeloid metaplasia. PLoS Med (2006) 3:e270. doi:10.1371/journal.pmed.0030270
47. Beer PA, Campbell PJ, Scott LM, Bench AJ, Erber WN, Bareford D, et al. MPL mutations in myeloproliferative disorders: analysis of the PT-1 cohort. Blood (2008) 112:141–9. doi:10.1182/blood-2008-01-131664
48. Moliterno AR, Williams DM, Gutierrez-Alamillo LI, Salvatori R, Ingersoll RG, Spivak JL. Mpl Baltimore: a thrombopoietin receptor polymorphism associated with thrombocytosis. Proc Natl Acad Sci U S A (2004) 101:11444–7. doi:10.1073/pnas.0404241101
49. El-Harith el HA, Roesl C, Ballmaier M, Germeshausen M, Frye-Boukhriss H, von Neuhoff N, et al. Familial thrombocytosis caused by the novel germ-line mutation p.Pro106Leu in the MPL gene. Br J Haematol (2009) 144:185–94. doi:10.1111/j.1365-2141.2008.07430.x
50. Favale F, Messaoudi K, Varghese LN, Boukour S, Pecquet C, Gryshkova V, et al. An incomplete trafficking defect to the cell-surface leads to paradoxical thrombocytosis for human and murine MPL P106L. Blood (2016) 128:3146–58. doi:10.1182/blood-2016-06-722058
51. van den Oudenrijn S, Bruin M, Folman CC, Peters M, Faulkner LB, de Haas M, et al. Mutations in the thrombopoietin receptor, Mpl, in children with congenital amegakaryocytic thrombocytopenia. Br J Haematol (2000) 110:441–8. doi:10.1046/j.1365-2141.2000.02175.x
52. Germeshausen M, Ballmaier M, Welte K. MPL mutations in 23 patients suffering from congenital amegakaryocytic thrombocytopenia: the type of mutation predicts the course of the disease. Hum Mutat (2006) 27:296. doi:10.1002/humu.9415
53. Ghilardi N, Wiestner A, Kikuchi M, Ohsaka A, Skoda RC. Hereditary thrombocythaemia in a Japanese family is caused by a novel point mutation in the thrombopoietin gene. Br J Haematol (1999) 107:310–6. doi:10.1046/j.1365-2141.1999.01710.x
54. Wiestner A, Schlemper RJ, van der Maas AP, Skoda RC. An activating splice donor mutation in the thrombopoietin gene causes hereditary thrombocythaemia. Nat Genet (1998) 18:49–52. doi:10.1038/ng0198-49
55. Kondo T, Okabe M, Sanada M, Kurosawa M, Suzuki S, Kobayashi M, et al. Familial essential thrombocythemia associated with one-base deletion in the 5’-untranslated region of the thrombopoietin gene. Blood (1998) 92:1091–6.
56. Noris P, Marconi C, De Rocco D, Melazzini F, Pippucci T, Loffredo G, et al. A new form of inherited thrombocytopenia due to monoallelic loss of function mutation in the thrombopoietin gene. Br J Haematol (2017). doi:10.1111/bjh.14694
57. Seo A, Ben-Harosh M, Sirin M, Stein J, Dgany O, Kaplelushnik J, et al. Bone marrow failure unresponsive to bone marrow transplant is caused by mutations in THPO. Blood (2017) 130(7):875–80. doi:10.1182/blood-2017-02-768036
58. Defour JP, Chachoua I, Pecquet C, Constantinescu SN. Oncogenic activation of MPL/thrombopoietin receptor by 17 mutations at W515: implications for myeloproliferative neoplasms. Leukemia (2016) 30(5):1214–6. doi:10.1038/leu.2015.271
59. Cabagnols X, Favale F, Pasquier F, Messaoudi K, Defour JP, Ianotto JC, et al. Presence of atypical thrombopoietin receptor (MPL) mutations in triple-negative essential thrombocythemia patients. Blood (2016) 127:333–42. doi:10.1182/blood-2015-07-661983
60. Milosevic Feenstra JD, Nivarthi H, Gisslinger H, Leroy E, Rumi E, Chachoua I, et al. Whole-exome sequencing identifies novel MPL and JAK2 mutations in triple-negative myeloproliferative neoplasms. Blood (2016) 127:325–32. doi:10.1182/blood-2015-07-661835
61. Liu K, Martini M, Rocca B, Amos CI, Teofili L, Giona F, et al. Evidence for a founder effect of the MPL-S505N mutation in eight Italian pedigrees with hereditary thrombocythemia. Haematologica (2009) 94:1368–74. doi:10.3324/haematol.2009.005918
62. Stockklausner C, Klotter AC, Dickemann N, Kuhlee IN, Duffert CM, Kerber C, et al. The thrombopoietin receptor P106L mutation functionally separates receptor signaling activity from thrombopoietin homeostasis. Blood (2015) 125:1159–69. doi:10.1182/blood-2014-07-587170
63. Szpurka H, Tiu R, Murugesan G, Aboudola S, Hsi ED, Theil KS, et al. Refractory anemia with ringed sideroblasts associated with marked thrombocytosis (RARS-T), another myeloproliferative condition characterized by JAK2 V617F mutation. Blood (2006) 108:2173–81. doi:10.1182/blood-2006-02-005751
64. Bandaranayake RM, Ungureanu D, Shan Y, Shaw DE, Silvennoinen O, Hubbard SR. Crystal structures of the JAK2 pseudokinase domain and the pathogenic mutant V617F. Nat Struct Mol Biol (2012) 19:754–9. doi:10.1038/nsmb.2348
65. Lu X, Levine R, Tong W, Wernig G, Pikman Y, Zarnegar S, et al. Expression of a homodimeric type I cytokine receptor is required for JAK2V617F-mediated transformation. Proc Natl Acad Sci U S A (2005) 102:18962–7. doi:10.1073/pnas.0509714102
66. Scott LM, Scott MA, Campbell PJ, Green AR. Progenitors homozygous for the V617F mutation occur in most patients with polycythemia vera, but not essential thrombocythemia. Blood (2006) 108:2435–7. doi:10.1182/blood-2006-04-018259
67. Hasan S, Lacout C, Marty C, Cuingnet M, Solary E, Vainchenker W, et al. JAK2V617F expression in mice amplifies early hematopoietic cells and gives them a competitive advantage that is hampered by IFNalpha. Blood (2013) 122:1464–77. doi:10.1182/blood-2013-04-498956
68. Lacout C, Pisani DF, Tulliez M, Gachelin FM, Vainchenker W, Villeval JL. JAK2V617F expression in murine hematopoietic cells leads to MPD mimicking human PV with secondary myelofibrosis. Blood (2006) 108:1652–60. doi:10.1182/blood-2006-02-002030
69. Li J, Kent DG, Godfrey AL, Manning H, Nangalia J, Aziz A, et al. JAK2V617F homozygosity drives a phenotypic switch in myeloproliferative neoplasms, but is insufficient to sustain disease. Blood (2014) 123:3139–51. doi:10.1182/blood-2013-06-510222
70. Mullally A, Bruedigam C, Poveromo L, Heidel FH, Purdon A, Vu T, et al. Depletion of Jak2V617F myeloproliferative neoplasm-propagating stem cells by interferon-alpha in a murine model of polycythemia vera. Blood (2013) 121:3692–702. doi:10.1182/blood-2012-05-432989
71. Tiedt R, Hao-Shen H, Sobas MA, Looser R, Dirnhofer S, Schwaller J, et al. Ratio of mutant JAK2-V617F to wild-type Jak2 determines the MPD phenotypes in transgenic mice. Blood (2008) 111:3931–40. doi:10.1182/blood-2007-08-107748
72. Zhan H, Ma Y, Lin CH, Kaushansky K. JAK2V617F-mutant megakaryocytes contribute to hematopoietic stem/progenitor cell expansion in a model of murine myeloproliferation. Leukemia (2016) 30:2332–41. doi:10.1038/leu.2016.114
73. Akada H, Yan D, Zou H, Fiering S, Hutchison RE, Mohi MG. Conditional expression of heterozygous or homozygous Jak2V617F from its endogenous promoter induces a polycythemia vera-like disease. Blood (2010) 115:3589–97. doi:10.1182/blood-2009-04-215848
74. Ghilardi N, Skoda RC. A single-base deletion in the thrombopoietin (TPO) gene causes familial essential thrombocythemia through a mechanism of more efficient translation of TPO mRNA. Blood (1999) 94:1480–2.
75. Michalak M, Groenendyk J, Szabo E, Gold LI, Opas M. Calreticulin, a multi-process calcium-buffering chaperone of the endoplasmic reticulum. Biochem J (2009) 417:651–66. doi:10.1042/BJ20081847
76. Klampfl T, Gisslinger H, Harutyunyan AS, Nivarthi H, Rumi E, Milosevic JD, et al. Somatic mutations of calreticulin in myeloproliferative neoplasms. N Engl J Med (2013) 369:2379–90. doi:10.1056/NEJMoa1311347
77. Nangalia J, Massie CE, Baxter EJ, Nice FL, Gundem G, Wedge DC, et al. Somatic CALR mutations in myeloproliferative neoplasms with nonmutated JAK2. N Engl J Med (2013) 369:2391–405. doi:10.1056/NEJMoa1312542
78. Cabagnols X, Defour JP, Ugo V, Ianotto JC, Mossuz P, Mondet J, et al. Differential association of calreticulin type 1 and type 2 mutations with myelofibrosis and essential thrombocytemia: relevance for disease evolution. Leukemia (2015) 29:249–52. doi:10.1038/leu.2014.270
79. Tefferi A, Lasho TL, Finke C, Belachew AA, Wassie EA, Ketterling RP, et al. Type 1 vs type 2 calreticulin mutations in primary myelofibrosis: differences in phenotype and prognostic impact. Leukemia (2014) 28:1568–70. doi:10.1038/leu.2014.83
80. Tefferi A, Wassie EA, Guglielmelli P, Gangat N, Belachew AA, Lasho TL, et al. Type 1 versus Type 2 calreticulin mutations in essential thrombocythemia: a collaborative study of 1027 patients. Am J Hematol (2014) 89:E121–4. doi:10.1002/ajh.23743
81. Chachoua I, Pecquet C, El-Khoury M, Nivarthi H, Albu RI, Marty C, et al. Thrombopoietin receptor activation by myeloproliferative neoplasm associated calreticulin mutants. Blood (2016) 127:1325–35. doi:10.1182/blood-2015-11-681932
82. Araki M, Yang Y, Masubuchi N, Hironaka Y, Takei H, Morishita S, et al. Activation of the thrombopoietin receptor by mutant calreticulin in CALR-mutant myeloproliferative neoplasms. Blood (2016) 127:1307–16. doi:10.1182/blood-2015-09-671172
83. Elf S, Abdelfattah NS, Chen E, Perales-Paton J, Rosen EA, Ko A, et al. Mutant calreticulin requires both its mutant C-terminus and the thrombopoietin receptor for oncogenic transformation. Cancer Discov (2016) 6:368–81. doi:10.1158/2159-8290.CD-15-1434
84. Marty C, Pecquet C, Nivarthi H, El-Khoury M, Chachoua I, Tulliez M, et al. Calreticulin mutants in mice induce an MPL-dependent thrombocytosis with frequent progression to myelofibrosis. Blood (2016) 127:1317–24. doi:10.1182/blood-2015-11-679571
85. Shide K, Kameda T, Yamaji T, Sekine M, Inada N, Kamiunten A, et al. Calreticulin mutant mice develop essential thrombocythemia that is ameliorated by the JAK inhibitor ruxolitinib. Leukemia (2017) 31:1136–44. doi:10.1038/leu.2016.308
86. Oh ST, Simonds EF, Jones C, Hale MB, Goltsev Y, Gibbs KD Jr, et al. Novel mutations in the inhibitory adaptor protein LNK drive JAK-STAT signaling in patients with myeloproliferative neoplasms. Blood (2010) 116:988–92. doi:10.1182/blood-2010-02-270108
87. Ha JS, Jeon DS. Possible new LNK mutations in myeloproliferative neoplasms. Am J Hematol (2011) 86:866–8. doi:10.1002/ajh.22107
88. Hurtado C, Erquiaga I, Aranaz P, Migueliz I, Garcia-Delgado M, Novo FJ, et al. LNK can also be mutated outside PH and SH2 domains in myeloproliferative neoplasms with and without V617FJAK2 mutation. Leuk Res (2011) 35:1537–9. doi:10.1016/j.leukres.2011.07.009
89. Gery S, Koeffler HP. Role of the adaptor protein LNK in normal and malignant hematopoiesis. Oncogene (2013) 32:3111–8. doi:10.1038/onc.2012.435
90. Koren-Michowitz M, Gery S, Tabayashi T, Lin D, Alvarez R, Nagler A, et al. SH2B3 (LNK) mutations from myeloproliferative neoplasms patients have mild loss of function against wild type JAK2 and JAK2 V617F. Br J Haematol (2013) 161:811–20. doi:10.1111/bjh.12327
91. Velazquez L, Cheng AM, Fleming HE, Furlonger C, Vesely S, Bernstein A, et al. Cytokine signaling and hematopoietic homeostasis are disrupted in Lnk-deficient mice. J Exp Med (2002) 195:1599–611. doi:10.1084/jem.20011883
92. Spolverini A, Pieri L, Guglielmelli P, Pancrazzi A, Fanelli T, Paoli C, et al. Infrequent occurrence of mutations in the PH domain of LNK in patients with JAK2 mutation-negative ’idiopathic’ erythrocytosis. Haematologica (2013) 98:e101–2. doi:10.3324/haematol.2013.090175
93. Grand FH, Hidalgo-Curtis CE, Ernst T, Zoi K, Zoi C, McGuire C, et al. Frequent CBL mutations associated with 11q acquired uniparental disomy in myeloproliferative neoplasms. Blood (2009) 113:6182–92. doi:10.1182/blood-2008-12-194548
94. Geddis AE. Congenital amegakaryocytic thrombocytopenia. Pediatr Blood Cancer (2011) 57:199–203. doi:10.1002/pbc.22927
95. Fox NE, Chen R, Hitchcock I, Keates-Baleeiro J, Frangoul H, Geddis AE. Compound heterozygous c-Mpl mutations in a child with congenital amegakaryocytic thrombocytopenia: functional characterization and a review of the literature. Exp Hematol (2009) 37:495–503. doi:10.1016/j.exphem.2009.01.001
96. Varghese LN, Zhang JG, Young SN, Willson TA, Alexander WS, Nicola NA, et al. Functional characterization of c-Mpl ectodomain mutations that underlie congenital amegakaryocytic thrombocytopenia. Growth Factors (2014) 32:18–26. doi:10.3109/08977194.2013.874347
97. Tijssen MR, di Summa F, van den Oudenrijn S, Zwaginga JJ, van der Schoot CE, Voermans C, et al. Functional analysis of single amino-acid mutations in the thrombopoietin-receptor Mpl underlying congenital amegakaryocytic thrombocytopenia. Br J Haematol (2008) 141:808–13. doi:10.1111/j.1365-2141.2008.07139.x
99. Leroy E, Defour JP, Sato T, Dass S, Gryshkova V, Shwe MM, et al. His499 regulates dimerization and prevents oncogenic activation by asparagine mutations of the human thrombopoietin receptor. J Biol Chem (2016) 291:2974–87. doi:10.1074/jbc.M115.696534
100. Cines DB, Yasothan U, Kirkpatrick P. Romiplostim. Nat Rev Drug Discov (2008) 7:887–8. doi:10.1038/nrd2741
101. Sangkhae V, Saur SJ, Kaushansky A, Kaushansky K, Hitchcock IS. Phosphorylated c-Mpl tyrosine 591 regulates thrombopoietin-induced signaling. Exp Hematol (2014) 42(477–486):e474. doi:10.1016/j.exphem.2014.02.007
Keywords: thrombopoietin, MPL, JAK2, MPLR102P, thrombocytosis, thrombocytopenia
Citation: Plo I, Bellanné-Chantelot C, Mosca M, Mazzi S, Marty C and Vainchenker W (2017) Genetic Alterations of the Thrombopoietin/MPL/JAK2 Axis Impacting Megakaryopoiesis. Front. Endocrinol. 8:234. doi: 10.3389/fendo.2017.00234
Received: 30 June 2017; Accepted: 28 August 2017;
Published: 12 September 2017
Edited by:
Pierre De Meyts, de Duve Institute, BelgiumReviewed by:
Jean-Baptiste Demoulin, Catholic University of Louvain, BelgiumJames M. Murphy, Walter and Eliza Hall Institute of Medical Research, Australia
Copyright: © 2017 Plo, Bellanné-Chantelot, Mosca, Mazzi, Marty and Vainchenker. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: William Vainchenker, dmVycHJlQGlnci5mcg==