 Michele L. Semeraro
Michele L. Semeraro Lindsey M. Glenn
Lindsey M. Glenn Margaret A. Morris
Margaret A. Morris- Department of Internal Medicine, Strelitz Diabetes Center, Eastern Virginia Medical School, Norfolk, VA, United States
Natural killer (NK) cells represent an important effector arm against viral infection, and mounting evidence suggests that viral infection plays a role in the development of type 1 diabetes (T1D) in at least a portion of patients. NK cells recognize their target cells through a delicate balance of inhibitory and stimulatory receptors on their surface. If unbalanced, NK cells have great potential to wreak havoc in the pancreas due to the beta cell expression of the as-yet-defined NKp46 ligand through interactions with the activating NKp46 receptor found on the surface of most NK cells. Blocking interactions between NKp46 and its ligand protects mice from STZ-induced diabetes, but differential expression non-diabetic and diabetic donor samples have not been tested. Additional studies have shown that peripheral blood NK cells from human T1D patients have altered phenotypes that reduce the lytic and functional ability of the NK cells. Investigations of humanT1D pancreas tissues have indicated that the presence of NK cells may be beneficial despite their infrequent detection. In non-obese diabetic (NOD) mice, we have noted that NK cells express high levels of the proinflammatory mediator 12/15-lipoxygenase (12/15-LO), and decreased levels of stimulatory receptors. Conversely, NK cells of 12/15-LO deficient NOD mice, which are protected from diabetes development, express significantly higher levels of stimulatory receptors. Furthermore, the human NK92 cell line expresses the ALOX12 protein [human 12-lipoxygenase (12-LO), related to mouse 12/15-LO] via Western blotting. Human 12-LO is upregulated in the pancreas of both T1D and T2D human donors with insulin-containing islets, showing a link between 12-LO expression and diabetes progression. Therefore, our hypothesis is that NK cells in those susceptible to developing T1D are unable to function properly during viral infections of pancreatic beta cells due to increased 12-LO expression and activation, which contributes to increased interferon-gamma production and an imbalance in activating and inhibitory NK cell receptors, and may contribute to downstream autoimmune T cell responses. The work presented here outlines evidence from our lab, as well as published literature, supporting our hypothesis, including novel data.
Introduction
Autoimmune destruction of the pancreatic beta cells leads to the development of Type 1 diabetes (T1D). The number of T1D cases is on the rise, with the relative risk for developing the disease ranging from 0.1% [no family history, protective human leukocyte antigen (HLA)] to up to 70% (monozygotic twin with susceptible HLA), and is dependent largely upon genetic susceptibility (1). Importantly, the strongest genetic link to the development of T1D is the expression of certain HLA haplotypes. Class II HLA genes, especially DR3, DR4, and DQ8, are the strongest links; however, HLA Class I molecules also play a role in diabetes development (2, 3). Expression of both Class I and Class II molecules is the largest contributing factor in determining the immune response to a given pathogen, as peptides are processed and presented to T and natural killer (NK) cells via the proteins of the major histocompatibility complex (MHC) locus (4). Therefore, these molecules play a key role in directing immune responses, be they beneficial or detrimental. However, the genetic contributions to T1D development are unable to fully account for the increased rates, supporting the idea that environmental factors play a role in the development of T1D. Furthermore, susceptible siblings of T1D patients who are closely monitored frequently show signs of autoimmunity in the form of autoantibodies prior to metabolic dysfunction. Many believe, based on this evidence, that development of full-blown diabetes requires multiple insults to the system in order to manifest itself.
Patients with T1D currently depend upon treatment options that are limited to methods that replace the deficit in insulin production, either via injection or transplantation [reviewed in Ref. (5), in press]. While technological advances have helped improve these methods, they still do not provide a cure for the disease. Therefore, determining the mechanisms leading to immune damage of pancreatic beta (β) cells, and treatments to maintain β cell mass, are of the utmost importance.
Recently, perceptions of T1D development have evolved, with a greater attention being paid to islet inflammation as an important event propagating autoimmunity and further loss of β cell mass (6–8). Debates persist as to whether islets are independently inflamed prior to the autoimmune response or the autoimmune response brings about the islet inflammation. One of these recent studies described the incorrect processing of the insulin protein that led to the generation of abnormal peptides recognized by circulating CD8+ T cells in T1D patients (8). This line of evidence certainly points to β cell defects contributing to diabetes pathogenesis; however, this study does not address what might cause β cells to produce this incorrectly processed protein. One study in non-obese diabetic (NOD) mice has suggested that incorrect protein processing in these mice causes an increase in endoplasmic reticulum (ER) stress, and results in the development of autoimmunity (9). Given the lack of complete concordance among monozygotic twins, many believe external environmental factors, such as viruses, strongly influence the development of islet inflammation leading to T1D. Trying to understand all of these data in concert brings researchers in the field to ponder the chicken and egg scenario. Are either islets or immune cells in susceptible individuals causing the initial insults that spark diabetes development, or does an environmental factor trigger the disease? Do we see signs of virus infections in patients with T1D because the infection is what precipitates diabetes development, or are patients with diabetes more susceptible to developing virus infections because of defects in their bodies’ defense systems? With the data that are currently available, the order of events in the precipitation of T1D is unclear.
A New Hypothesis
As we gather more evidence, it is becoming clear that we must look at the integrated physiology to fully understand the mechanism(s) of T1D development. Here, we outline an idea that incorporates early antiviral immune effectors, NK cells, with proinflammatory processes involving 12-lipoxygenase (12-LO) occurring in the pancreatic beta cells. We hypothesize that the activation of NK cell 12/15-LO (Alox15, in mice) or 12-LO (ALOX12, in humans) through environmental triggers, such as Coxsackievirus infection, contributes to T1D initiation by affecting the normal innate immune interplay between NK cells and islets, which primes downstream autoimmune responses leading to islet destruction. This may occur, in part, due to the effects of inflammation (including 12-LO) on the balance of NK cell receptor expression (10). Below, we will describe the evidence supporting this hypothesis, beginning with one of the suspected environmental triggers, enteroviruses.
Direct Evidence for Virus Infections in T1D
Over the past fifty years, there has been accumulating evidence linking viruses, and the patients’ responses to these viruses, to the initiation of T1D. This idea that viruses contribute to the initiation and development of T1D was first introduced in the 1960s (11, 12). This is, of course, difficult to fully pinpoint, as the infection may occur long before disease onset, and scientists with access to human pancreas tissues are granted only a snapshot of the patient’s final day as their window into the disease process. Additionally, as mentioned previously, it is unclear whether or not patients susceptible to developing T1D are also more susceptible to developing virus-mediated infections in the pancreatic islets, which might increase the viral signature in the islets of patients with T1D. Therefore, this might not be a causal relationship, but merely coincidental. Since we cannot directly test whether viruses initiate T1D in humans, researchers have used animal models to test this theory. NOD mouse models have been used to show that Coxsackievirus B1 and B4 (CVB1, CVB4) infection speeds diabetes pathogenesis (13), and is dependent upon host sensors of virus (14–16). Others have shown that these effects are highly age dependent, as infection at before 10 weeks of age can prevent diabetes development (17, 18). Additionally, studies of immunodeficient mice engrafted with human islets have shown that human islets can become infected with CVB4, which causes direct damage to the β cells, and results in diabetes. Gene expression profiles of these infected islets indicated significant increases in genes related to the Type 1 interferon (T1-IFN) pathway, as well as genes related to ER stress (19). While these data support the idea of viruses contributing to diabetes development, they do not answer the question about which occurs first: islet dysfunction or immune activation.
To address the role of virus infections in human T1D, groups such as Persistent Virus Infection in Diabetes Network and the Network for Pancreatic Organ Donors with Diabetes—Viral Working Group (nPOD-V) have approached the question with great coordination across multiple platforms (20) assessing the same donor samples (PCR, immunohistochemistry, proteomics, and ISH). These team science efforts have yielded results estimating that Coxsackievirus infections might contribute to diabetes development in over 50% of cases (21, 22). While certainly not causal, pancreas tissues, and specifically β cells, from T1D donors have been found to express viral VP1 proteins more frequently than non-diabetic (ND) donors (23, 24). These studies continue to progress, generating a wealth of information from human donor samples.
Mechanistically, enteroviruses can infect via a fecal/oral route, thereby implicating intestinal involvement during the infection process. Mounting evidence has shown a role for the gut microbiome as a contributing factor in autoimmune diabetes development. Viruses and microbiota are known to interact with one another, and shape the response of both parties, which may influence the development of T1D (25). Recent human studies of closely matched control and T1D experimental groups demonstrate both increased inflammation in the duodenum of T1D patients (26), and direct detection of enteroviruses (27). In the first study, donors were tested for markers of inflammation using histological techniques and PCR array, indicating significant inflammatory processes in T1D donors, including increased macrophage numbers in the duodenum of T1D (26). In the second study, T1D donors were much more likely to have markers of enterovirus infection than control donors independent of HLA haplotypes, as tested by in situ hybridization and histological techniques (27). This work could not conclude whether T1D patients were more susceptible to the virus infections, or the infections are persistent. Coxsackievirus infection of β cells with strains B1 (28) and B4 (13) may occur via β cell expression of the Coxsackie Adenovirus Receptor (29, 30) following viral migration from the duodenum to the pancreas through the common bile duct or affiliated vessels (27). Pursuant to the role of islet inflammation following environmental insult, in vitro studies indicate that infection could lead to ER stress in β cells, contributing to islet dysfunction that activates the autoimmune response (9, 31). Alternatively, the infection could also directly activate immune responses that become uncontrolled due to inherent immune defects. Until imaging of live T1D patients affords the ability to detect virus infection in real-time, other experimental avenues must be explored, including the use of cultured islets and mouse models.
Indirect Evidence for Virus Infections in T1D
While viruses themselves may be difficult to detect in our snapshot views of human T1D, there is ample “circumstantial” evidence that exists in the form of immune cells and mediators. Both mouse models and organ donors with T1D have provided clear evidence that islet inflammation is a key hallmark of this disease. Immune cells infiltrate the islets, albeit at different intensities, in both species. Many patients show signs of adaptive immunity against the pancreatic islets in the form of autoantibodies and islet-specific T cell clones.
Beyond cellular responses, cytokines and chemokines also contribute to islet demise and can stem from both innate and adaptive responses. T1-IFNs have recently gained more respect as effectors in the development of T1D (see the review by Newby and Mathews in this issue). Indeed, virus infections are strong stimulators of T1-IFN production, which leads to a subsequent upregulation of MHC Class I expression, another hallmark of T1D (32).
Downstream of this response, numerous proinflammatory cytokines and chemokines have been detected in patients with diabetes (33, 34). One of these, IFN-gamma (IFN-γ), has been shown to play an important, albeit controversial, role in T1D pathogenesis (7, 35). While absence of the cytokine itself leads to delayed disease development (36), absence of the receptor protects against the development of insulitis (37). IFN-γ has many points at which it can act in the development of T1D, from altering MHC/HLA expression on involved cells to altering endothelial cell function and signaling to immune cells to activate cytotoxic effectors (37). Diminished IFN-γ responses can prevent the recruitment of insulitic T cells, as well as their ability to respond to antigens, which might prevent diabetes progression. However, increased IFN-γ production by CD4+ T cells can actually contribute to the resolution of CD8+ T cell responses (35). While it is appreciated that CD4+ T cells contribute to the IFN-γ production during T1D pathogenesis, this does not exclude the idea that NK cells may be the first producers of IFN-γ present in the islets. Interestingly, IFN-γ is also frequently detected following virus infections, and is used by the immune system to combat viral replication. Given these data, it is unclear whether IFN-γ is serving in a proinflammatory capacity or an unsuccessful attempt at tolerance induction during the development of T1D (35).
NK Cells and Their Role in T1D
Natural killer cells are large granular lymphocytes that are considered part of the innate immune system. While they do not react as quickly as neutrophils and macrophages against invading pathogens, they mount a response more quickly than do T cells from the adaptive arm of the immune system. NK cells are known as key players in fighting off both tumor cells and virus-infected cells. Despite their small number (only 5–10% of leukocytes in the spleen and 1–6% in peripheral blood) (38), NK cells are powerful cytolytic effectors. Upon stimulation by a variety of cytokines, including T1-IFNs and IL-12 (39), NK cells utilize several different mechanisms to lyse their targets: the combination of perforin and granzymes, signaling through death receptors (i.e., Fas/FasL), and antibody-dependent cellular cytotoxicity leading to either apoptosis or necrosis (40). NK cells can also produce potent cytokines, such as IFN-γ and TNF-α (41). IFN-γ production by NK cells might also serve an antigen presenting capacity (42–44), which, along with their potent cytokine production abilities, would give them the power to stimulate immune responses downstream of their own activation.
In order to recognize their targets, NK cells have developed an intricate system of check and balances. As NK cells are expected to determine aberrant “self” cells (tumors and virus-infected cells), they must be able to distinguish which cells are healthy, and which are not. NK cells respond to virus infections in both mouse and man (4) through signaling mechanisms involving a delicate balance of inhibitory and stimulatory receptors expressed by NK cells. Normal expression of MHC Class I molecules (HLA in humans) send “self” signals to NK cells, inhibiting lytic responses (45). Virus infection can lead to the downregulation of MHC Class I molecules on the surface of infected cells. While this prevents CD8+ T cells from responding to viral peptides, it also diminishes the inhibitory signal transmitted to NK cells [reviewed in Ref. (46)]. Subsequently, stimulatory signals to the NK cell are able to override inhibitory signals, leading to lysis of affected cells. In some instances, including during infection of pancreatic islets, viruses push the cellular machinery into overdrive and promote Type 1 IFN production (47), causing hyperexpression of the MHC Class I molecules (32). To circumvent this tactic, NK cells utilize receptors that recognize the upregulation of ligands for the natural cytotoxicity receptors, like NKp46, on the surface of infected cells (48, 49). Thus, NK cells can become “licensed to kill” through several mechanisms that allow them to detect alterations in MHC Class I molecules, as well as increased expression of stimulatory ligands, making them versatile effectors during virus infections (50).
Typically, T1D is thought to be dominated by autoimmune T cell responses; however, growing evidence suggests that NK cells are also involved (51). NK cells take up residence throughout the body (52), providing immune surveillance and protection against viruses wherever they enter the body. NK cells are plentiful in the intestines as compared to other organs (53), comprising 20–40% of Intestinal Epithelial Lymphocytes in healthy children (54), as compared to about 10% of the blood and spleen. This provides NK cells ample opportunity to respond to Coxsackievirus infections, as well as others (Salmonella, Toxoplasma gondii, other parasites, viruses, and bacteria) (55), transmitted via the fecal/oral route. Paired with the evidence of increased inflammation in duodenum of T1D patients, these data support our hypothesis. Furthermore, NK cells have been detected in the pancreas of both diabetic mice and humans. In mice, the cells appear shortly after macrophages (56). In human pancreatic samples, although NK cells are not frequently detected (57), they have been found in insulitic lesions, and show indications of having a protective effect (58). When one considers the frequency of NK cells in lymphocyte-rich organs (5–10% of leukocytes in the spleen), and also accounts for the number of cells required to define insulitis in humans [six or more CD3+ cells in at least three islets (59)], then perhaps it is not surprising that NK cells are rarely detected in donor samples. Alternatively, it is possible that NK cells may prime the pancreatic environment for the entry of diabetogenic T cells, and subsequently depart. As we only have access to one time point for each human donor, we cannot currently distinguish these hypotheses. However, the use of mouse models made aid in this differentiation.
Natural killer cells themselves have recently been directly implicated in the development of T1D through additional expression quantitative trait loci analysis following genome-wide association studies, which further suggests that NK cells play a key role in T1D pathogenesis (60). Interestingly, this study indicates that NK cells may impact T1D development more than CD8+ T cells. The carefully designed and executed study is limited to only 105 Japanese subjects, which might not apply to other ethnic backgrounds. However, it is comprehensive, and provides a solid approach for other ethnic backgrounds to be tested. Another data set investigating NK cell phenotypes from patients with T1D showed that NK cells from these patients express significantly reduced levels of activating receptors on their surface as compared to healthy controls (10).
It is unlikely that NK cells act independently in T1D development. Macrophages recognize environmental signals, and have been shown to enter pancreatic islets at 3–4 weeks of age in NOD mice (61, 62). Macrophage production of IL-12 and IL-18 can strongly activate NK cells, which are found in the pancreas of diabetes-prone NOD mice as early as 4 weeks of age (56, 63), to produce high levels of IFN-γ (64). Indeed, serum levels of both IL-12 and IL-18 are higher patients with T1D (65, 66), and IL-18 has been shown to participate in T1D pathogenesis of NOD mice (67). Despite a defect in IL-15 signaling in NOD mice (68), which affects NK cell development and function, others have shown in IL-15-deficient mice that increased IL-12 signaling may allow NK cells to overcome this deficit when faced with pathogenic stimuli (69, 70). The early appearance of NK cells in the pancreas may enable them to activate diabetogenic T cell responses.
Perhaps most importantly, NK cells can directly interact with pancreatic islets through expression of ligands for the NK activating receptors NKG2D and NKp46. Both of these receptors have been implicated in NK-mediated self-aggression in human NK cells that can be triggered by signaling through NKG2D and NKp46 (71). The NKG2D ligand, RAE-1 (72), is one of these ligands. Some speculate that NKG2D ligands cause down-modulation of the receptors, thereby making the NK cells less active (73); however, others have failed to validate this hypothesis (74). They instead showed that differential expression of NKG2D ligands did not hinder NK cytotoxicity through methodical assessment of receptor and ligand levels using genetic tools to dictate the alteration of expression.
Pancreatic islets also broadly express ligands for the NKp46 natural cytotoxicity receptor. NKp46 is a Type I transmembrane protein with two extracellular Ig-like domains followed by a short stalk region, a transmembrane domain containing a positively charged amino acid residue, and a short cytoplasmic tail (75, 76). However, the cellular ligands for NKp46 have not been identified or characterized. The only NKp46 ligands identified so far are the hemagglutinin of influenza virus and the hemagglutinin-neuraminidase of parainfluenza virus (48), suggesting a role for sugars in NKp46 ligand recognition. Studies of NKp46 ligands have utilized the NKp46 Fc chimeric protein in flow cytometry and histological techniques to examine expression over time in the islets of mice and humans (63). Functional studies from the same group showed that blockade of NKp46 receptor/ligand interactions protects against streptozotocin-induced diabetes (63). Although these studies have assessed expression over time, differential expression of the NKp46 ligands in human ND controls versus T1D donor samples has not been tested.
We recently studied ND, autoantibody positive (AAb+), and T1D donor samples from nPOD in order to determine whether there were expression differences in NKp46 ligands using the NKp46 Fc chimeric protein. Figure 1A highlights representative images from donors with different health statuses. Islet images from two ND, three AAb+, and three T1D donors were analyzed by NIH Image J to quantify the density of NKp46 Fc staining (red) within the islet area as determined by glucagon staining (Figure 1B). Islets from T1D donors frequently retain alpha cell mass longer than insulin-positive beta cell mass (77, 78). Therefore, we used glucagon staining to more accurately determine the islet area for each donor in order to calculate the intensity of NKp46 Fc staining in islets. Interestingly, NKp46 Fc levels were significantly higher in the AAb+ donors as compared to ND donors. Although the difference was not statistically significant when comparing T1D and AAb+ donors, there was a trend toward higher expression in the AAb+ donors. These data suggest that NKp46 ligands are upregulated during the development of T1D, and diminish as the islet health and mass decrease over the course of the disease. While this may be the result of ongoing immune responses in these tissues, insulitis has only been detected in one each of the AAb+ and T1D donors. General characteristics of donors tested are listed in Table 1.
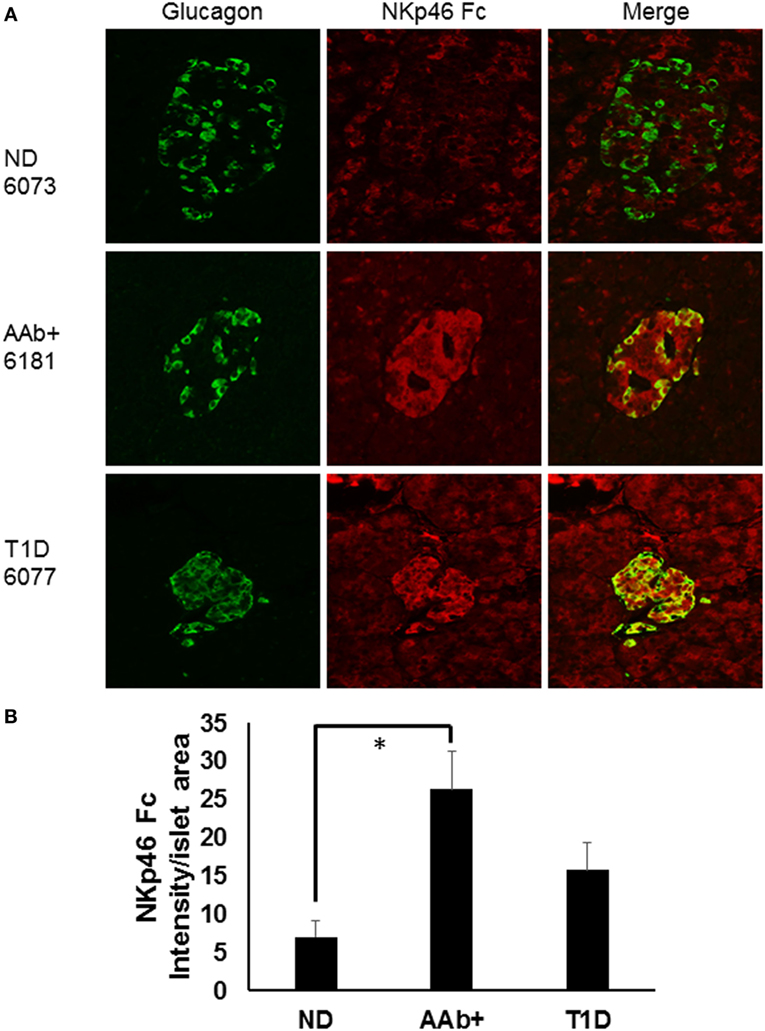
Figure 1. Human islets express NKp46 ligands. (A) Representative images comparing donor pancreas tissues from Network for Pancreatic Organ Donors with Diabetes (nPOD) biorepository samples. Sections were stained with antibodies against glucagon (green) and the NKp46 Fc chimeric receptor (red). (B) Quantification of NKp46 Fc staining per islet area. Density of NKp46 Fc staining was determined for each islet area. Islets for each donor were assessed, and donors of the same group were averaged. N = 2 for non-diabetic (ND); N = 3 for both autoantibody positive (AAb+) and T1D. *p < 0.05 by one-way ANOVA.
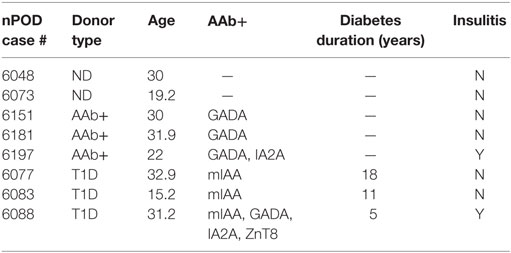
Table 1. Donor profiles for NKp46 ligand staining.
Mechanisms of NK cell action in diabetes are not well understood, and phenotypic differences in NK cells residing in different tissues may confound the results reported to date (40). Increased expression of stimulatory receptors on NK cells has been reported in both diabetic mice (79) and humans (80), while others maintain that a lack of NK cell activation contributes to diabetes development (10, 68, 73, 81). This could be due in part to the type of analysis, as genomic studies do not always translate to protein expression. Several groups have studied NK cells in NOD mice. Both found that murine pancreatic NK cells exhibit a different phenotype from those found in the spleen and lymph nodes and have increased proliferative capacity (56, 82); however, there is not a consensus on levels of IFN-γ production, as one indicates lower levels ex vivo, but normal levels in vivo (82). Depletion of NK1.1+ cells in NOD.NK1.1 congenic mice did not significantly affect disease onset, but the depletion protocol also removed NK/T cells (82). These tissue-specific phenotypic differences may also alter the detectability of the NK cells residing within the human pancreas using standard methodologies, but this has not yet been studied in humans. NOD mice have been shown to have a defect in IL-15 production, which contributes to NK cell dysfunction (68). IL-15 is required for NK cell maturation, and although NOD mice are not completely IL-15 deficient, they do show impaired NK cell development. Taken in concert with other systemic alterations in the NOD strain, the impact of this IL-15 defect has not been fully explored. As mentioned previously, NK cells functionally adjust to the absence of IL-15 by responding to IL-12 and IL-18 in order to produce IFN-γ (83), and by responding to pathogenic stimuli in the presence of IL-12 (69).
The Proinflammatory Mediator, 12-LO, in T1D
12-Lipoxygenase [(12-LO) gene name ALOX12S in humans; 12/15-lipoxygenase (12/15-LO), gene name Alox15 in mice] converts arachidonic acid to the proinflammatory 12(S)-hydroxyeicosatetraenoic acid (12(S)-HETE) through a 12-S-hydroperoxy-eicosatetraenoic acid (12-HPETE) intermediate (84, 85). IL-12 signaling downstream of 12(S)-HETE production (86, 87) activates STAT4 [reviewed in Ref. (88)], contributing to additional inflammation. IL-12 signaling through STAT4 strongly activates NK cells and T cells and is also known to be a strong contributor to autoimmune conditions in general [reviewed in Refs. (89, 90)]. In addition to T1D, 12/15-LO has been implicated in many inflammatory processes, including cancers (91, 92), asthma (93), and Type 2 diabetes (94).
Several lines of evidence indicate a critical role for 12/15-LO in the pathogenesis of T1D. It has been shown that deletion of STAT4 signaling molecules, which are downstream of 12/15-LO activation, in NOD mice protects the NOD strain from developing T1D (95). Subsequently, we published that the NOD mouse line congenic for the global Alox15 (NOD-Alox15null) deletion is >98% protected from developing spontaneous T1D (61). This line boasts a narrow congenic region delineated through extensive microsatellite mapping, and shows significantly reduced disease incidence (~2%). To understand the origin of the protection seen in these mice, wild-type NOD mice were tested for their expression 12/15-LO in islets, macrophages, and lymphocytes. Islets and macrophages expressed the enzyme in appreciable amounts, while lymphocytes had either low or undetectable amounts (96). Additionally, Alox15null mice have been shown to express decreased levels of IL-12 (96) and IL-18 (97), which are cytokines that contribute to NK cell IFN-γ production (83). Since publication, this strain has been shipped to additional vivaria and maintained this phenotype. Subsequent studies of human islets have also indicated 12-LO expression under inflamed conditions (98, 99), which feeds into the detrimental cycle of inflammation.
Natural killer cell expression of 12/15-LO has not been extensively studied, although historical data suggest that NK cells expressed a member of the lipoxygenase family (100). Using more modern information and methods, we discovered that both mouse and the human NK92 cell line express 12/15-LO and 12-LO, respectively (Figure 2). Surprisingly, this expression of 12/15-LO in freshly isolated NOD mouse NK cells is significantly higher (3.3-fold more) than that seen in thioglycollate-induced NOD macrophages (61, 96). Since NK cells are closely related to T cells, we expected that expression levels would be similar to those seen in T cells, which is almost undetectable (96). While investigating the downstream effects of 12/15-LO expression in our NOD-Alox15null mice, we found that the pancreatic lymph node NK cells of the NOD-Alox15null mice had an increased percentage of NK cells expressing activating markers (Figure 3), which are similar phenotypically to peripheral blood NK cells from ND human controls (10). Taken together, it appears that activation of 12/15-LO, which leads to increased IL-12 levels (96), increases the inflammatory nature of the NK cells, presumably through the 12/15-LO pathway. We are currently testing whether NK cells are better able to resolve infections without contributing to the chronic inflammatory milieu in the absence of 12/15-LO.
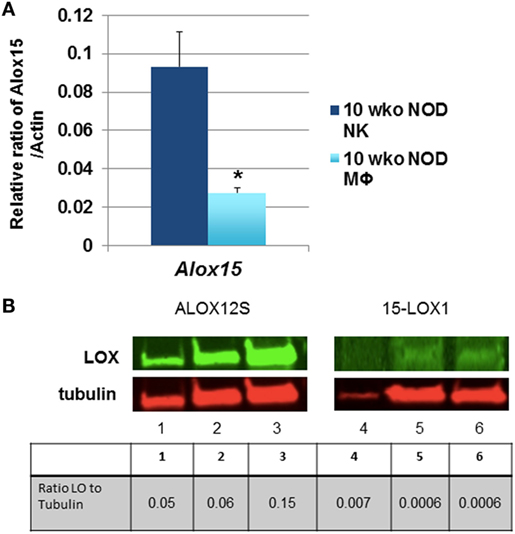
Figure 2. Natural killer (NK) cells express 12/15-lipoxygenase (12/15-LO). (A) Murine non-obese diabetic (NOD) natural killer cells express Alox15. mRNA levels of the Alox15 gene were tested in NK cells from NOD mice. These levels were compared to thioglycollate-induced peritoneal macrophages from 10-week-old NOD mice using the relative ratio of Alox15/Actb. *p < 0.05 using a two-tailed Student’s t-test to compare NK vs. macrophages in age-matched NOD mice, n = 6 mice per group. (B) 12-Lipoxygenase (12-LO) protein expression in human NK92 cells. The human NK92 cell line was tested for protein expression of 12-LO by western blotting. ALOX12S expression was most abundant in the cell line, which is the most abundant form found in human islets. Lanes 1 and 4 are nuclear proteins from the two pooled NK92 samples; lanes 2 and 5 are cytoplasmic proteins from one NK92 sample; lanes 3 and 6 are cytoplasmic proteins from different NK92 samples.
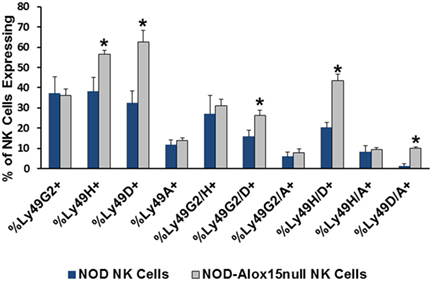
Figure 3. Pancreatic lymph node natural killer (NK) cell expression of NK cell markers. Ly49 receptors determine which targets NK cells recognize and respond to during interactions with potential targets. Several of the expression patterns are altered in the absence of Alox15, with Alox15null cells expressing higher proportions of the activating Ly49 receptors [*p < 0.05 using a two-tailed Student’s t-test to compare receptor expression in non-obese diabetic (NOD) vs. NOD-Alox15null NK cells for each receptor or receptor pair]. n = 4 mice/group.
Several additional lines of evidence suggest that NK cells are strongly influenced by 12/15-LO activity and that they have the capacity to play an important role in the development of diabetes. As mentioned above, activation of 12/15-LO leads to IL-12 production [reviewed in Ref. (94)], which activates the STAT4 signaling cascade that is required for NK cells to respond functionally (101–103), including production of IFN-γ (104, 105). This combination can exacerbate T1D, although IL-12 can also trigger the activation of different cytokine pathways in the absence of IFN-γ (106). The involvement of IL-12 in T1D pathogenesis is not without controversy. Deletion of the IL-12p40 subunit, which can heterodimerize with either IL-12p35 to form IL-12 or the p19 subunit to form IL-23, did not protect against T1D development (107). This may be due to the effect of inhibiting both IL-12 and IL-23 generation simultaneously, although IL-23 had not been discovered at the time these results were published. Subsequently, the same group published work indicating that administration of exogenous IL-12 exacerbated diabetes development (106). Importantly, when key molecules in the 12/15-LO pathway (i.e., 12/15-LO or STAT4), upstream of IL-12, are disrupted in NOD mice, diabetes is prevented (61, 95).
A New Model of T1D Development
By bringing NK cell expression of 12/15-LO into the equation of diabetes initiation following virus infection, one can envision a model in which duodenal NK cells encounter some sort of pathogen- or virus-infected cells, such as Coxsackievirus-infected cells. As the pathogen is transmitted through the bile duct or related vasculature on the way to the pancreas, NK cells and macrophages are alerted. Under normal conditions, NK cells and macrophages quickly dispense of the virus, removing only the infected cells. In a patient susceptible to developing diabetes, the interactions are altered, perhaps due to activation of 12/15-LO either by the virus directly or due to increased stress placed upon the beta cells upon infection (19). This leads to abnormal interactions between the innate immune cells with islets expressing NK cell ligands and prevents the resolution of the infection. 12/15-LO activation is known to feed into a vicious cycle of chronic inflammation, which in this instance, may be perpetuated by macrophages and NK cells (shown graphically in Figure 4). As mentioned earlier, in individuals with susceptible HLA haplotypes, or in mice (in was left out inadvertently) with susceptible MHC haplotypes, chronic inflammation signals the diabetogenic T cells to join the fight. This leads to significant islet destruction. Both HLA Class I and II molecules strongly influence the T cell responses in humans, as they dictate the ability of the T cells to recognize and react to autoantigens during the T cell development process, as well as in the periphery (108). This autoreactivity can be precipitated by stress placed on the islets, perhaps due to inflammatory processes. Such stress may lead to HLA Class II-mediated recognition of hybrid insulin (109) or posttranslationally modified (110) peptides by CD4+ T cells, thereby breaking peripheral tolerance to neoantigens and furthering the disease progression.
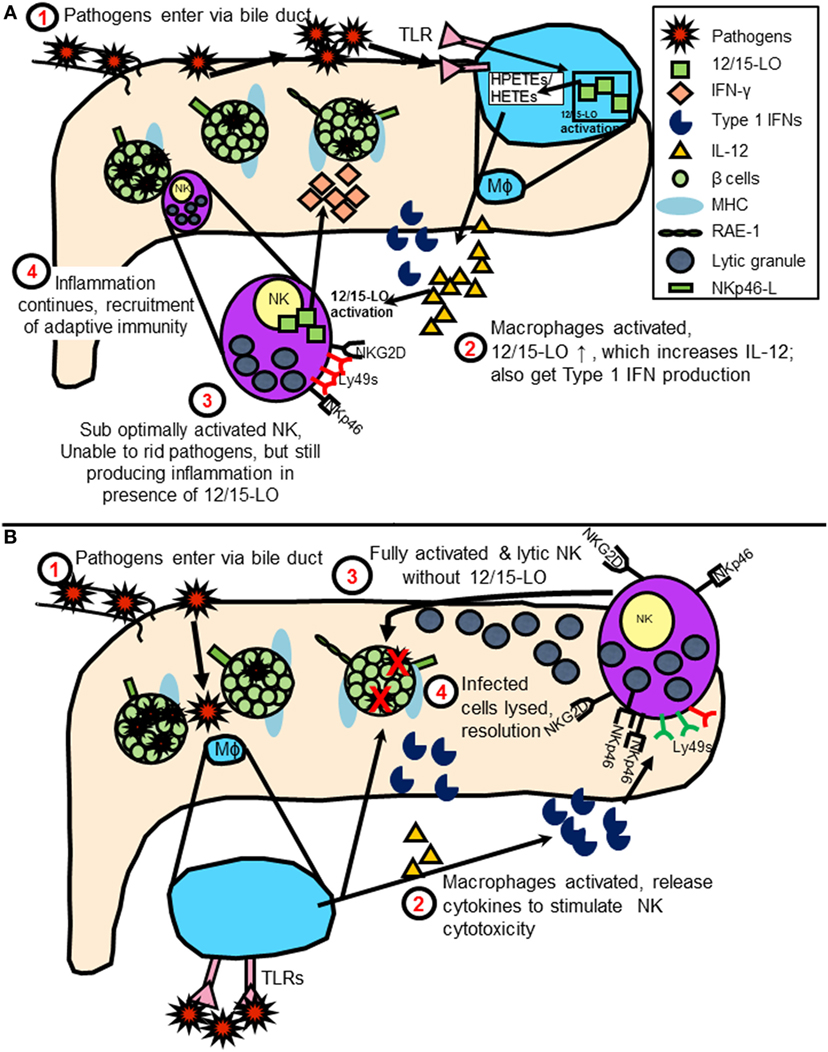
Figure 4. (A) Model of natural killer (NK) cell activation leading to type 1 diabetes (T1D) in the presence of 12/15-lipoxygenase (12/15-LO) following virus infection. IL-12 production stimulates NK interferon-gamma (IFN-γ) response, feeds cycle of 12/15-LO activation and inflammation. (B) Model of NK cell activation in absence of 12/15-lipoxygenase following virus infection. NK cells respond to macrophage T1 IFN production following TLR signaling by using lytic mechanisms to rid virus-infected cells. Reduced inflammation prevents T1D development.
Conclusion
We have hypothesized that activation of NK cell 12/15-LO (or 12-LO, ALOX12, in humans) contributes to T1D initiation by affecting the normal innate immune interplay between NK cells and islets, which primes downstream autoimmune responses leading to islet destruction.
At best, our current understanding of T1D initiation is murky. It is appreciated that there is a role for cells of the mucosal-associated lymphoid tissues, including NK cells, and it is quite likely that infectious initiation of T1D would occur through fecal-oral routes. However, T1D progression also requires the presence of macrophages, which produce 12/15-LO. Following the appearance of the macrophages in the pancreas of NOD mice, NK cells, which also produce 12/15-LO, are found. This process in humans has not yet been delineated, and, therefore, it is unclear if NK cells are migrating from the gut to the pancreas following an infection, or if they are recruited by other means. Macrophages have both the ability to respond to virus infections through TLR signaling, as well as activate NK cells through IL-12 and Type 1 IFN production. It is known that IL-12 production in macrophages is increased following 12/15-LO activation and that IL-12 signaling can feedback into the 12/15-LO signaling cascade (84). NK cell 12/15-LO is then a target for activation following IL-12 stimulation. IL-12, in concert with IL-18, is also known to drive IFN-γ production by NK cells (83). Both IL-12 and IL-18 are increased in mouse models of T1D (67, 96), as well as in patients with T1D (65, 66), and both are increased in the presence of 12/15-LO (96, 97). NK cell-derived IFN-γ could aid in expanding the effector T cell population (111). Conversely, in the absence of 12/15-LO, normal IL-12 levels [as generated through TLR signaling (112)] and Type 1 IFNs from activated macrophages might play a stronger role in influencing pancreatic NK cell function, leading to upregulation of activating receptors, optimal cytotoxic activation, and clearance of viral pathogens with minimal residual inflammation.
Moving forward, it is important to understand mechanisms by which environmental factors might spark the activation of 12-LO in NK cells, macrophages, and islets, leading to the development of T1D. While some pieces of this puzzle remain to be placed, there is striking evidence that our hypothesis and model is possible. Many of the remaining questions can be answered in part by the use of novel global and conditional knock-outs of 12/15-LO on the NOD background in experiments with Coxsackievirus infections. By understanding the order in which these events occur, we will be better able to design selective therapies that might prevent the disease development and progression without resorting to global immunosuppression.
Methods
Mice
Female NOD/ShiLtJ (NOD) mice were ordered from Jackson Laboratory (Bar Harbor, ME, USA); global NOD-Alox15null mice (bred on-site at EVMS) were housed in SPF conditions and treated in accordance with the AAALAC and IACUC guidelines at the Eastern Virginia Medical Center. Mice were euthanized by asphyxiation with CO2. Blood glucose levels were assessed following euthanasia at 4 and 10 weeks of age. Spleens, lymph nodes, and islets were removed.
Cell Isolations
Natural killer cells were isolated from spleen using cell isolation kits from Stem Cell Technologies (Vancouver, BC, Canada) per the manufacturer’s instructions. Purity of the isolated populations was assessed by flow cytometry (see below) after staining with antibodies against cell surface markers including anti-CD3, anti-CD19, anti-NKp46, and anti-CD11b. Cells were generally 85–90% pure.
Flow Cytometry
Cells isolated from the pancreatic draining lymph nodes of NOD and NOD-Alox15null mice at 10 weeks of age were stained with antibodies against cell surface markers for T (CD3), B (CD19), and NK cells (NKp46, Ly49A, Ly49G2, Ly49D, Ly49H). Gates were determined by using fluorescence minus one controls. All antibodies were purchased from Biolegend (San Diego, CA, USA).
qRT-PCR
mRNA was isolated from indicated cells and tissues using the RNeasy Kit from QIAgen (Germantown, MD, USA), and used to generate cDNA for use in qRT-PCR assays as described (96). 12/15-LO expression in mouse cells was assessed by a SYBR green protocol, and compared to a newly available Alox15 Taqman probe (Thermofisher Scientific, Waltham, MA, USA). Expression was tested in two independent experiments using five mice per group for each experiment.
Western Blotting
Cell lysates from the NK92 human NK cell line was used as a source of proteins to measure level human 12-LO levels using the Odyssey LI-COR system (Lincoln, NE, USA) as previously described (61). Nuclear and cytosolic proteins were fractionated and tested separately. Duplicate samples were stained with antibodies recognizing tubulin, ALOX12S, and ALOX15-1.
Immunofluorescence
Formalin-fixed, paraffin-embedded tissues sections from human pancreas tissues (obtained through nPOD) were stained as described with antibodies to glucagon (DAKO, Copenhagen, Denmark), and the NKp46 Ligand (using the NKp46 Fc chimera, R&D Systems, Minneapolis, MN, USA) as described previously (48), with a modification using tyramide amplification (Perkin Elmer, Waltham, MA, USA) to amplify NKp46 Fc staining. Islet area was determined, and the intensity of NKp46 Fc staining within the islet area was calculated using NIH Image J. Data are expressed as density of NKp46 Fc staining per islet area for ND, autoantibody positive, and T1D donor (n = 3 donors per group), which is calculated with the following equation: [Integrated density − (area of selected cell × mean fluorescence of background readings)]/total islet area = average fluorescence per islet.
Statistical Procedures
Statistically significant differences were determined by the use of Student’s t-test where appropriate, or ANOVA followed by post hoc testing. Significant differences in all cases were determined by p < 0.05.
Ethics Statement
This study was carried out in accordance with the recommendations of “Principles of laboratory animal care” (NIH publication no. 85–23), AAALAC, and IACUC guidelines at the Eastern Virginia Medical Center. The protocol was approved by the IACUC at Eastern Virginia Medical Center. The studies of the nPOD human donor tissues were considered to be exempt and deemed “Non-Human Subjects Research” by the Eastern Virginia Medical School Institutional Review Board due to the nature of the donors. All donors are deceased and de-identified.
Author Contributions
MM conceived the hypothesis outlined. MS and MM wrote the manuscript. MM, LG, and MS contributed to experimental design and data analysis. MS and LG critically reviewed the manuscript prior to submission.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
The authors wish to acknowledge the support of the Network for Pancreatic Organ Donors with Diabetes (nPOD), including Irina Kusmartseva. Additionally, this work was supported by a Junior Faculty Award from the American Diabetes Association (ADA #7-11-JF-33) and the Helmsley Charitable Trust George S. Eisenbarth nPOD Award for Team Science (#2015PG-TID052-668894).
References
1. Ziegler A-G, Nepom GT. Prediction and pathogenesis in type 1 diabetes. Immunity (2010) 32:468–78. doi:10.1016/j.immuni.2010.03.018
2. Howson JMM, Walker NM, Clayton D, Todd JA, Type 1 Diabetes Genetics Consortium. Confirmation of HLA class II independent type 1 diabetes associations in the major histocompatibility complex including HLA-B and HLA-A. Diabetes Obes Metab (2009) 11:31–45. doi:10.1111/j.1463-1326.2008.01001.x
3. Noble JA, Erlich HA. Genetics of type 1 diabetes. Cold Spring Harb Perspect Med. (2012) 2:a007732. doi:10.1101/cshperspect.a007732
4. Abbas AK, Lichtman AH, Pillai S. Basic immunology: functions and disorders of the immune system. Basic Immunol (2012):303–5. doi:10.1007/978-1-62703-589-7_1
5. Morris MA, Satin-Smith M. Type 1 diabetes mellitus: is a cure in sight? In: Khardori R, Ullal J, Lieb DC, editors. Diabetes Mellitus – Clinical Focus Series in Endocrinology. Jaypee Brothers Medical Publishing (in press).
6. Størling J, Pociot F. Type 1 diabetes candidate genes linked to pancreatic islet cell inflammation and beta-cell apoptosis. Genes (Basel) (2017) 8:1–12. doi:10.3390/genes8020072
7. Waugh K, Snell-Bergeon J, Michels A, Dong F, Steck AK, Frohnert BI, et al. Increased inflammation is associated with islet autoimmunity and type 1 diabetes in the Diabetes Autoimmunity Study in the Young (DAISY). PLoS One (2017) 12:e0174840. doi:10.1371/journal.pone.0174840
8. Kracht MJ, van Lummel M, Nikolic T, Joosten AM, Laban S, van der Slik AR, et al. Autoimmunity against a defective ribosomal insulin gene product in type 1 diabetes. Nat Med (2017) 23:501–7. doi:10.1038/nm.4289
9. Tersey SA, Nishiki Y, Templin AT, Cabrera SM, Stull ND, Colvin SC, et al. Islet β-cell endoplasmic reticulum stress precedes the onset of type 1 diabetes in the nonobese diabetic mouse model. Diabetes (2012) 61:818–27. doi:10.2337/db11-1293
10. Qin H, Lee IF, Panagiotopoulos C, Wang X, Chu AD, Utz PJ, et al. Natural killer cells from children with type 1 diabetes have defects in NKG2D-dependent function and signaling. Diabetes (2011) 60:857–66. doi:10.2337/db09-1706
11. Gamble DR, Taylor KW. Seasonal incidence of diabetes mellitus. Br Med J (1969) 3:631–3. doi:10.1136/bmj.3.5671.631
12. Gamble DR, Kinsley ML, FitzGerald MG, Bolton R, Taylor KW. Viral antibodies in diabetes mellitus. Br Med J (1969) 3:627–30. doi:10.1136/bmj.3.5671.627
13. Horwitz MS, Bradley LM, Harbertson J, Krahl T, Lee J, Sarvetnick N. Diabetes induced by Coxsackie virus: initiation by bystander damage and not molecular mimicry. Nat Med (1998) 4:781–5. doi:10.1038/nm0798-781
14. Lincez PJ, Shanina I, Horwitz MS. Reduced expression of the MDA5 gene IFIH1 prevents autoimmune diabetes. Diabetes (2015) 64:2184–93. doi:10.2337/db14-1223
15. McCall KD, Thuma JR, Courreges MC, Benencia F, James CB, Malgor R, et al. Toll-like receptor 3 is critical for coxsackievirus B4-induced type 1 diabetes in female nod mice. Endocrinology (2015) 156:453–61. doi:10.1210/en.2013-2006
16. Richer MJ, Lavallée DJ, Shanina I, Horwitz MS. Toll-like receptor 3 signalling on macrophages is required for survival following coxsackievirus B4 infection. PLoS One (2009) 4:e4127. doi:10.1371/journal.pone.0004127
17. Davydova B, Härkönen T, Kaialainen S, Hovi T, Vaarala O, Roivainen M. Coxsackievirus immunization delays onset of diabetes in non-obese diabetic mice. J Med Virol (2003) 69:510–20. doi:10.1002/jmv.10339
18. Serreze DV, Wasserfall C, Ottendorfer EW, Stalvey M, Pierce MA, Gauntt C, et al. Diabetes acceleration or prevention by a coxsackievirus B4 infection: critical requirements for both interleukin-4 and gamma interferon. J Virol (2005) 79:1045–52. doi:10.1128/JVI.79.2.1045-1052.2005
19. Gallagher GR, Brehm MA, Finberg RW, Barton BA, Shultz LD, Greiner DL, et al. Viral infection of engrafted human islets leads to diabetes. Diabetes (2015) 64:1358–69. doi:10.2337/db14-1020
20. Laiho JE, Oikarinen M, Richardson SJ, Frisk G, Nyalwidhe J, Burch TC, et al. Relative sensitivity of immunohistochemistry, multiple reaction monitoring mass spectrometry, in situ hybridization and PCR to detect Coxsackievirus B1 in A549 cells. J Clin Virol (2016) 77:21–8. doi:10.1016/j.jcv.2016.01.015
21. Hyöty H. Viruses in type 1 diabetes. Pediatr Diabetes (2016) 17(Suppl 22):56–64. doi:10.1111/pedi.12370
22. Honkanen H, Oikarinen S, Nurminen N, Laitinen OH, Huhtala H, Lehtonen J, et al. Detection of enteroviruses in stools precedes islet autoimmunity by several months: possible evidence for slowly operating mechanisms in virus-induced autoimmunity. Diabetologia (2017) 60:424–31. doi:10.1007/s00125-016-4177-z
23. Krogvold L, Edwin B, Buanes T, Frisk G, Skog O, Anagandula M, et al. Detection of a low-grade enteroviral infection in the islets of langerhans of living patients newly diagnosed with type 1 diabetes. Diabetes (2015) 64:1682–7. doi:10.2337/db14-1370
24. Richardson SJ, Willcox A, Bone AJ, Foulis AK, Morgan NG. The prevalence of enteroviral capsid protein vp1 immunostaining in pancreatic islets in human type 1 diabetes. Diabetologia (2009) 52:1143–51. doi:10.1007/s00125-009-1276-0
25. Robinson CM, Pfeiffer JK. Viruses and the microbiota. Annu Rev Virol (2014) 1:55–69. doi:10.1146/annurev-virology-031413-085550
26. Pellegrini S, Sordi V, Bolla AM, Saita D, Ferrarese R, Canducci F, et al. Duodenal mucosa of patients with type 1 diabetes shows distinctive inflammatory profile and microbiota. J Clin Endocrinol Metab (2017) 102:1468–77. doi:10.1210/jc.2016-3222
27. Oikarinen M, Tauriainen S, Oikarinen S, Honkanen T, Collin P, Rantala I, et al. Type 1 diabetes is associated with enterovirus infection in gut mucosa. Diabetes (2012) 61:687–91. doi:10.2337/db11-1157
28. Laitinen OH, Honkanen H, Pakkanen O, Oikarinen S, Hankaniemi MM, Huhtala H, et al. Coxsackievirus B1 is associated with induction of β-cells autoimmunity that portends type 1 diabetes. Diabetes (2014) 63:446–55. doi:10.2337/db13-0619
29. Hodik M, Anagandula M, Fuxe J, Krogvold L, Dahl-Jørgensen K, Hyöty H, et al. Coxsackie–adenovirus receptor expression is enhanced in pancreas from patients with type 1 diabetes. BMJ Open Diabetes Res Care (2016) 4:e000219. doi:10.1136/bmjdrc-2016-000219
30. Ylipaasto P, Klingel K, Lindberg AM, Otonkoski T, Kandolf R, Hovi T, et al. Enterovirus infection in human pancreatic islet cells, islet tropism in vivo and receptor involvement in cultured islet beta cells. Diabetologia (2004) 47:225–39. doi:10.1007/s00125-003-1297-z
31. Marré ML, Profozich JL, Coneybeer JT, Geng X, Bertera S, Ford MJ, et al. Inherent ER stress in pancreatic islet β cells causes self-recognition by autoreactive T cells in type 1 diabetes. J Autoimmun (2016) 72:33–46. doi:10.1016/j.jaut.2016.04.009
32. Richardson SJ, Rodriguez-Calvo T, Gerling IC, Mathews CE, Kaddis JS, Russell MA, et al. Islet cell hyperexpression of HLA class I antigens: a defining feature in type 1 diabetes. Diabetologia (2016) 59:2448–58. doi:10.1007/s00125-016-4067-4
33. Padgett LE, Broniowska KA, Hansen PA, Corbett JA, Tse HM. The role of reactive oxygen species and proinflammatory cytokines in type 1 diabetes pathogenesis. Ann N Y Acad Sci (2013) 1281:16–35. doi:10.1111/j.1749-6632.2012.06826.x
34. Fallahi P, Corrado A, Di Domenicantonio A, Frenzilli G, Antonelli A, Ferrari SM. CXCR3, CXCR5, CXCR6, and CXCR7 in diabetes. Curr Drug Targets (2016) 17:515–9. doi:10.2174/1389450115666141229153949
35. Driver JP, Racine JJ, Ye C, Lamont DJ, Newby BN, Leeth CM, et al. Interferon-γ limits diabetogenic CD8+ T-cell effector responses in type 1 diabetes. Diabetes (2017) 66:710–21. doi:10.2337/db16-0846
36. Hultgren B, Huang X, Dybdal N, Stewart TA. Genetic absence of gamma-interferon delays but does not prevent diabetes in NOD mice. Diabetes (1996) 45:812–7. doi:10.2337/diab.45.6.812
37. Wang B, André I, Gonzalez A, Katz JD, Aguet M, Benoist C, et al. Interferon-gamma impacts at multiple points during the progression of autoimmune diabetes. Proc Natl Acad Sci U S A (1997) 94:13844–9. doi:10.1073/pnas.94.25.13844
38. Angelo LS, Banerjee PP, Monaco-Shawver L, Rosen JB, Makedonas G, Forbes LR, et al. Practical NK cell phenotyping and variability in healthy adults. Immunol Res (2015) 62:341–56. doi:10.1007/s12026-015-8664-y
39. Marçais A, Viel S, Grau M, Henry T, Marvel J, Walzer T. Regulation of mouse NK cell development and function by cytokines. Front Immunol (2013) 4:1–4. doi:10.3389/fimmu.2013.00450
40. Zhang C, Tian Z. NK cell subsets in autoimmune diseases. J Autoimmun (2017) 83:22–30. doi:10.1016/j.jaut.2017.02.005
41. Reefman E, Kay JG, Wood SM, Offenhäuser C, Brown DL, Roy S, et al. Cytokine secretion is distinct from secretion of cytotoxic granules in NK cells. J Immunol (2010) 184:4852–62. doi:10.4049/jimmunol.0803954
42. Spits H, Lanier LL. Natural killer or dendritic: what’s in a name? Immunity (2007) 26:11–6. doi:10.1016/j.immuni.2007.01.004
43. Homann D, Jahreis A, Wolfe T, Hughes A, Coon B, van Stipdonk MJ, et al. CD40L blockade prevents autoimmune diabetes by induction of bitypic NK/DC regulatory cells. Immunity (2002) 16:403–15. doi:10.1016/S1074-7613(02)00290-X
44. Hanna J, Gonen-Gross T, Fitchett J, Rowe T, Daniels M, Arnon TI, et al. Novel APC-like properties of human NK cells directly regulate T cell activation. J Clin Invest (2004) 114:1612–23. doi:10.1172/JCI22787
45. Ljunggren HG, Kärre K. In search of the ‘missing self’: MHC molecules and NK cell recognition. Immunol Today (1990) 11:237–44. doi:10.1016/0167-5699(90)90097-S
46. Seliger B, Ritz U, Ferrone S. Molecular mechanisms of HLA class I antigen abnormalities following viral infection and transformation. Int J Cancer (2006) 118:129–38. doi:10.1002/ijc.21312
47. Foulis A, Farquharson M, Meager A. Immunoreactive α-interferon in insulin-secreting β cells in type 1 diabetes mellitus. Lancet (1987) 2(8573):1423–7. doi:10.1016/S0140-6736(87)91128-7
48. Mandelboim O, Lieberman N, Lev M, Paul L, Arnon TI, Bushkin Y, et al. Recognition of haemagglutinins on virus-infected cells by NKp46 activates lysis by human NK cells. Nature (2001) 409:1055–60. doi:10.1038/35059110
49. Magri G, Muntasell A, Romo N, Sáez-Borderías A, Pende D, Geraghty DE, et al. NKp46 and DNAM-1 NK cell receptors drive the response to human cytomegalovirus infected myeloid dendritic cells overcoming viral immune evasion strategies. Blood (2011) 117:848–56. doi:10.1182/blood-2010-08-301374
50. Pegram HJ, Andrews DM, Smyth MJ, Darcy PK, Kershaw MH. Activating and inhibitory receptors of natural killer cells. Immunol Cell Biol (2011) 89:216–24. doi:10.1038/icb.2010.78
51. Flodström M, Maday A, Balakrishna D, Cleary MM, Yoshimura A, Sarvetnick N. Target cell defense prevents the development of diabetes after viral infection. Nat Immunol (2002) 3:373–82. doi:10.1038/ni771
52. Morris MA, Ley K. Trafficking of natural killer cells. Curr Mol Med (2004) 4:431–8. doi:10.2174/1566524043360609
53. Pang G, Buret A, Batey RT, Chen QY, Couch L, Cripps A, et al. Morphological, phenotypic and functional characteristics of a pure population of CD56+ CD16- CD3- large granular lymphocytes generated from human duodenal mucosa. Immunology (1993) 79:498–505.
54. Eiras P, Leon F, Camarero C, Lombardia M, Roldan E, Bootello A, et al. Intestinal intraepithelial lymphocytes contain a CD3- CD7+ subset expressing natural killer markers and a singular pattern of adhesion molecules. Scand J Immunol (2000) 52:1–6. doi:10.1046/j.1365-3083.2000.00761.x
55. Ivanova D, Krempels R, Ryfe J, Weitzman K, Stephenson D, Gigley JP. NK cells in mucosal defense against infection. Biomed Res Int (2014) 2014:413982. doi:10.1155/2014/413982
56. Brauner H, Elemans M, Lemos S, Broberger C, Holmberg D, Flodström-Tullberg M, et al. Distinct phenotype and function of NK cells in the pancreas of nonobese diabetic mice. J Immunol (2010) 184:2272–80. doi:10.4049/jimmunol.0804358
57. Willcox A, Richardson SJ, Bone AJ, Foulis AK, Morgan NG. Analysis of islet inflammation in human type 1 diabetes. Clin Exp Immunol (2009) 155:173–81. doi:10.1111/j.1365-2249.2008.03860.x
58. Dotta F, Censini S, van Halteren AG, Marselli L, Masini M, Dionisi S, et al. Coxsackie B4 virus infection of β cells and natural killer cell insulitis in recent-onset type 1 diabetic patients. Proc Natl Acad Sci U S A (2007) 104:5115–20. doi:10.1073/pnas.0700442104
59. Campbell-Thompson M, Fu A, Kaddis JS, Wasserfall C, Schatz DA, Pugliese A, et al. Insulitis and β-cell mass in the natural history of type 1 diabetes. Diabetes (2016) 65:719–31. doi:10.2337/db15-0779
60. Ishigaki K, Kochi Y, Suzuki A, Tsuchida Y, Tsuchiya H, Sumitomo S, et al. Polygenic burdens on cell-specific pathways underlie the risk of rheumatoid arthritis. Nat Genet (2017) 49(7):1120–5. doi:10.1038/ng.3885
61. McDuffie M, Maybee NA, Keller SR, Stevens BK, Garmey JC, Morris MA, et al. Nonobese diabetic (NOD) mice congenic for a targeted deletion of 12/15-lipoxygenase are protected from autoimmune diabetes. Diabetes (2008) 57:199–208. doi:10.2337/db07-0830
62. Jansen A, Homo-Delarche F, Hooijkaas H, Leenen PJ, Dardenne M, Drexhage HA. Immunohistochemical characterization of monocytes-macrophages and dendritic cells involved in the initiation of the insulitis and beta-cell destruction in NOD mice. Diabetes (1994) 43:667–75. doi:10.2337/diab.43.5.667
63. Gur C, Porgador A, Elboim M, Gazit R, Mizrahi S, Stern-Ginossar N, et al. The activating receptor NKp46 is essential for the development of type 1 diabetes. Nat Immunol (2010) 11:121–8. doi:10.1038/ni.1834
64. Zhang C, Zhang J, Niu J, Zhou Z, Zhang J, Tian Z. Interleukin-12 improves cytotoxicity of natural killer cells via upregulated expression of NKG2D. Hum Immunol (2008) 69:490–500. doi:10.1016/j.humimm.2008.06.004
65. Ryba-Stanislawowska M, Rybarczyk-Kapturska K, Mysliwiec M, Mysliwska J. Elevated levels of serum IL-12 and IL-18 are associated with lower frequencies of CD4(+)CD25 (high)FOXP3 (+) regulatory t cells in young patients with type 1 diabetes. Inflammation (2014) 37:1513–20. doi:10.1007/s10753-014-9878-1
66. Altinova AE, Yetkin I, Akbay E, Bukan N, Arslan M. Serum IL-18 levels in patients with type 1 diabetes: relations to metabolic control and microvascular complications. Cytokine (2008) 42:217–21. doi:10.1016/j.cyto.2008.02.006
67. Frigerio S, Holländer GA, Zumsteg U. Functional IL-18 is produced by primary pancreatic mouse islets and NIT-1 beta cells and participates in the progression towards destructive insulitis. Horm Res (2002) 57:94–104.
68. Suwanai H, Wilcox MA, Mathis D, Benoist C. A defective Il15 allele underlies the deficiency in natural killer cell activity in nonobese diabetic mice. Proc Natl Acad Sci U S A (2010) 107:9305–10. doi:10.1073/pnas.1004492107
69. Sun JC, Ma A, Lanier LL. Cutting edge: IL-15-independent NK cell response to mouse cytomegalovirus infection. J Immunol (2009) 183:2911–4. doi:10.4049/jimmunol.0901872
70. Lieberman LA, Villegas EN, Hunter CA. Interleukin-15-deficient mice develop protective immunity to Toxoplasma gondii. Infect Immun (2004) 72:6729–32. doi:10.1128/IAI.72.11.6729-6732.2004
71. Poggi A, Zocchi MR. Antigen presenting cells and stromal cells trigger human natural killer lymphocytes to autoreactivity: evidence for the involvement of natural cytotoxicity receptors (NCR) and NKG2D. Clin Dev Immunol (2006) 13:325–36. doi:10.1080/17402520600578194
72. Ogasawara K, Hamerman JA, Ehrlich LR, Bour-Jordan H, Santamaria P, Bluestone JA, et al. NKG2D blockade prevents autoimmune diabetes in NOD mice. Immunity (2004) 20:757–67. doi:10.1016/j.immuni.2004.05.008
73. Ogasawara K, Hamerman JA, Hsin H, Chikuma S, Bour-Jordan H, Chen T, et al. Impairment of NK cell function by NKG2D modulation in NOD mice. Immunity (2003) 18:41–51. doi:10.1016/S1074-7613(02)00505-8
74. Maier LM, Howlett SK, Rainbow KM, Clark J, Howson JM, Todd JA, et al. NKG2D-RAE-1 receptor-ligand variation does not account for the NK cell defect in nonobese diabetic mice. J Immunol (2008) 181:7073–80. doi:10.4049/jimmunol.181.10.7073
75. Foster CE, Colonna M, Sun PD. Crystal structure of the human natural killer (NK) cell activating receptor NKp46 reveals structural relationship to other leukocyte receptor complex immunoreceptors. J Biol Chem (2003) 278:46081–6. doi:10.1074/jbc.M308491200
76. Ponassi M, Cantoni C, Biassoni R, Conte R, Spallarossa A, Pesce A, et al. Structure of the human NK cell triggering receptor NKp46 ectodomain. Biochem Biophys Res Commun (2003) 309:317–23. doi:10.1016/j.bbrc.2003.08.007
77. Rahier J, Goebbels RM, Henquin JC. Cellular composition of the human diabetic pancreas. Diabetologia (1983) 24:366–71. doi:10.1007/BF00251826
78. Stefan Y, Orci L, Malaisse-Lagae F, Perrelet A, Patel Y, Unger RH. Quantitation of endocrine cell content in the pancreas of nondiabetic and diabetic humans. Diabetes (1982) 31:694–700. doi:10.2337/diab.31.8.694
79. Belanger S, Tai L-H, Anderson SK, Makrigiannis AP. Ly49 cluster sequence analysis in a mouse model of diabetes: an expanded repertoire of activating receptors in the NOD genome. Genes Immun (2008) 9:509–21. doi:10.1038/gene.2008.43
80. van der Slik AR, Koeleman BP, Verduijn W, Bruining GJ, Roep BO, Giphart MJ. KIR in type 1 diabetes: disparate distribution of activating and inhibitory natural killer cell receptors in patients versus HLA-matched control subjects. Diabetes (2003) 52:2639–42. doi:10.2337/diabetes.52.10.2639
81. Rodacki M, Svoren B, Butty V, Besse W, Laffel L, Benoist C, et al. Altered natural killer cells in type 1 diabetic patients. Diabetes (2007) 56:177–85. doi:10.2337/db06-0493
82. Beilke JN, Meagher CT, Hosiawa K, Champsaur M, Bluestone JA, Lanier LL. NK cells are not required for spontaneous autoimmune diabetes in NOD mice. PLoS One (2012) 7:e36011. doi:10.1371/journal.pone.0036011
83. Keppel MP, Saucier N, Mah AY, Vogel TP, Cooper MA. Activation-specific metabolic requirements for NK Cell IFN-γ production. J Immunol (2015) 194:1954–62. doi:10.4049/jimmunol.1402099
84. Dobrian AD, Lieb DC, Cole BK, Taylor-Fishwick DA, Chakrabarti SK, Nadler JL. Functional and pathological roles of the 12- and 15-lipoxygenases. Prog Lipid Res (2011) 50:115–31. doi:10.1016/j.plipres.2010.10.005
85. Kuhn H, O’Donnell VB. Inflammation and immune regulation by 12/15-lipoxygenases. Prog Lipid Res (2006) 45:334–56. doi:10.1016/j.plipres.2006.02.003
86. Cole BK, Kuhn NS, Green-Mitchell SM, Leone KA, Raab RM, Nadler JL, et al. 12/15-Lipoxygenase signaling in the endoplasmic reticulum stress response. Am J Physiol Endocrinol Metab (2012) 302:E654–65. doi:10.1152/ajpendo.00373.2011
87. Dioszeghy V, Rosas M, Maskrey BH, Colmont C, Topley N, Chaitidis P, et al. 12/15-Lipoxygenase regulates the inflammatory response to bacterial products in vivo. J Immunol (2008) 181:6514–24. doi:10.4049/jimmunol.181.9.6514
88. Liang Y, Pan H-F, Ye D-Q. Therapeutic potential of STAT4 in autoimmunity. Expert Opin Ther Targets (2014) 18:945–60. doi:10.1517/13543776.2014.971754
89. Nepom GT, Ehlers M, Mandrup-Poulsen T. Anti-cytokine therapies in T1D: concepts and strategies. Clin Immunol (2013) 149:279–85. doi:10.1016/j.clim.2013.02.003
90. Pope RM, Shahrara S. Possible roles of IL-12-family cytokines in rheumatoid arthritis. Nat Rev Rheumatol (2013) 9:252–6. doi:10.1038/nrrheum.2012.170
91. Gondek T, Szajewski M, Szefel J, Aleksandrowicz-Wrona E, Skrzypczak-Jankun E, Jankun J, et al. Evaluation of 12-lipoxygenase (12-LOX) and plasminogen activator inhibitor 1 (pai-1) as prognostic markers in prostate cancer. Biomed Res Int (2014) 2014:102478. doi:10.1155/2014/102478
92. Kang KH, Ling TY, Liou HH, Huang YK, Hour MJ, Liou HC, et al. Enhancement role of host 12/15-lipoxygenase in melanoma progression. Eur J Cancer (2013) 49:2747–59. doi:10.1016/j.ejca.2013.03.030
93. Mabalirajan U, Rehman R, Ahmad T, Kumar S, Leishangthem GD, Singh S, et al. 12/15-lipoxygenase expressed in non-epithelial cells causes airway epithelial injury in asthma. Sci Rep (2013) 3:1540. doi:10.1038/srep01540
94. Imai Y, Dobrian AD, Weaver JR, Butcher MJ, Cole BK, Galkina EV, et al. Interaction between cytokines and inflammatory cells in islet dysfunction, insulin resistance and vascular disease. Diabetes Obes Metab (2013) 15:117–29. doi:10.1111/dom.12161
95. Yang Z, Chen M, Ellett JD, Fialkow LB, Carter JD, McDuffie M, et al. Autoimmune diabetes is blocked in Stat4-deficient mice. J Autoimmun (2004) 22:191–200. doi:10.1016/j.jaut.2003.08.006
96. Green-Mitchell SM, Tersey SA, Cole BK, Ma K, Kuhn NS, Cunningham TD, et al. Deletion of 12/15-lipoxygenase alters macrophage and islet function in NOD-Alox15null mice, leading to protection against type 1 diabetes development. PLoS One (2013) 8:e56763. doi:10.1371/journal.pone.0056763
97. Martínez-Clemente M, Ferré N, Titos E, Horrillo R, González-Périz A, Morán-Salvador E, et al. Disruption of the 12/15-lipoxygenase gene (Alox15) protects hyperlipidemic mice from nonalcoholic fatty liver disease. Hepatology (2010) 52:1980–91. doi:10.1002/hep.23928
98. Ma K, Nunemaker CS, Wu R, Chakrabarti SK, Taylor-Fishwick DA, Nadler JL. 12-Lipoxygenase products reduce insulin secretion and {beta}-cell viability in human islets. J Clin Endocrinol Metab (2010) 95:887–93. doi:10.1210/jc.2009-1102
99. Grzesik WJ, Nadler JL, Machida Y, Nadler JL, Imai Y, Morris MA. Expression pattern of 12-lipoxygenase in human islets with type 1 diabetes and type 2 diabetes. J Clin Endocrinol Metab (2015) 100:E387–95. doi:10.1210/jc.2014-3630
100. Leung KH, Ip MM, Koren HS. Regulation of human natural killing. IV. Role of lipoxygenase in regulation of natural killing activity. Scand J Immunol (1986) 24(4):371–80. doi:10.1111/j.1365-3083.1986.tb02124.x
101. Lee S-H, Fragoso MF, Biron CA. Cutting edge: a novel mechanism bridging innate and adaptive immunity: IL-12 induction of CD25 to form high-affinity IL-2 receptors on NK cells. J Immunol (2012) 189:2712–6. doi:10.4049/jimmunol.1201528
102. Chang HC, Lewis D, Tung CY, Han L, Henriquez SM, Voiles L, et al. Soypeptide lunasin in cytokine immunotherapy for lymphoma. Cancer Immunol Immunother (2014) 63:283–95. doi:10.1007/s00262-013-1513-8
103. Sun JC, Madera S, Bezman NA, Beilke JN, Kaplan MH, Lanier LL. Proinflammatory cytokine signaling required for the generation of natural killer cell memory. J Exp Med (2012) 209:947–54. doi:10.1084/jem.20111760
104. Duluc D, Tan F, Scotet M, Blanchard S, Frémaux I, Garo E, et al. PolyI:C plus IL-2 or IL-12 induce IFN-gamma production by human NK cells via autocrine IFN-beta. Eur J Immunol (2009) 39:2877–84. doi:10.1002/eji.200838610
105. Jaime-Ramirez AC, Mundy-Bosse BL, Kondadasula S, Jones NB, Roda JM, Mani A, et al. IL-12 enhances the antitumor actions of trastuzumab via NK cell IFN- production. J Immunol (2011) 186:3401–9. doi:10.4049/jimmunol.1000328
106. Trembleau S, Penna G, Bosi E, Mortara A, Gately MK, Adorini L. Interleukin 12 administration induces T helper type 1 cells and accelerates autoimmune diabetes in NOD mice. J Exp Med (1995) 181:817–21. doi:10.1084/jem.181.2.817
107. Trembleau S, Penna G, Gregori S, Chapman HD, Serreze DV, Magram J, et al. Pancreas-infiltrating Th1 cells and diabetes develop in IL-12-deficient nonobese diabetic mice. J Immunol (1999) 163:2960–8.
108. Tsai S, Santamaria P. MHC class II polymorphisms, autoreactive T-cells, and autoimmunity. Front Immunol (2013) 4(321):1–7. doi:10.3389/fimmu.2013.00321
109. Delong T, Wiles TA, Baker RL, Bradley B, Barbour G, Reisdorph R, et al. Pathogenic CD4 T cells in type 1 diabetes recognize epitopes formed by peptide fusion. Science (2016) 351:711–4. doi:10.1126/science.aad2791
110. Marré ML, James EA, Piganelli JD. β cell ER stress and the implications for immunogenicity in type 1 diabetes. Front Cell Dev Biol (2015) 3:67. doi:10.3389/fcell.2015.00067
111. Cook KD, Waggoner SN, Whitmire JK. NK cells and their ability to modulate T cells during virus infections. Crit Rev Immunol (2014) 34:359–88. doi:10.1615/CritRevImmunol.2014010604
Keywords: coxsackievirus infections, islets, natural killer cells, 12-lipoxygenase, type 1 diabetes
Citation: Semeraro ML, Glenn LM and Morris MA (2017) The Four-Way Stop Sign: Viruses, 12-Lipoxygenase, Islets, and Natural Killer Cells in Type 1 Diabetes Progression. Front. Endocrinol. 8:246. doi: 10.3389/fendo.2017.00246
Received: 03 August 2017; Accepted: 08 September 2017;
Published: 25 September 2017
Edited by:
Marc S. Horwitz, University of British Columbia, CanadaReviewed by:
Maria Bettini, Baylor College of Medicine, United StatesZhichao Feng, Albert Einstein College of Medicine, United States
Jon D. Piganelli, University of Pittsburgh, United States
Copyright: © 2017 Semeraro, Glenn and Morris. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Margaret A. Morris, bW9ycmlzbWFAZXZtcy5lZHU=