 Jeff M. P. Holly
Jeff M. P. Holly Jessica Broadhurst
Jessica Broadhurst Rehanna Mansor1
Rehanna Mansor1 Amit Bahl
Amit Bahl Claire M. Perks
Claire M. Perks- 1IGFs & Metabolic Endocrinology Group, School of Clinical Sciences, Southmead Hospital, Bristol, United Kingdom
- 2Department of Clinical Oncology, Bristol Haematology and Oncology Centre, University Hospitals Bristol, Bristol, United Kingdom
Background: Epidemiologic evidence shows that obesity is associated with a greater risk of aggressive prostate cancer (PCa) and PCa-specific mortality and this is observed mainly in men with the TMPRSS2-ERG gene fusion. Obesity is often associated with comorbid conditions such as type 2 diabetes and hyperglycemia: we investigated whether some of the exposures associated with disturbed metabolism can also affect the frequency of this gene fusion.
Methods: Fusion was induced in LNCaP PCa cells in normal or high levels of glucose, with or without insulin-like growth factor binding protein-2 (IGFBP-2) silenced or the presence of insulin-like growth factor-1 (IGF-I), insulin, or epidermal growth factor (EGF). RNA was extracted for analysis by nested PCR. Abundance of IGFBP-2, γH2AX, DNA-dependent protein kinase catalytic subunit (DNAPKcs), and β-actin were analyzed by Western immunoblotting.
Results: Our data suggest that hyperglycemia-induced IGFBP-2 increased the frequency of the gene fusion that was accompanied by decreased levels of DNAPKcs implying that they were mediated by alterations in the rate of repair of double-strand breaks. In contrast insulin, IGF-I and EGF all decreased gene fusion events.
Conclusion: These novel observations may represent a further mechanism by which obesity can exert an effect aggravating PCa progression.
Introduction
The TMPRSS2-ERG fusion oncogene is thought to be important during tumor progression and development as it is found in approximately half of all prostate cancer (PCa) biopsies and also in metastases (1–3).
Joining of the 5′-untranslated region of TMPRSS2 with the oncogenic ETS transcription factor, ERG culminates in the TMPRSS2-ERG gene fusion. TMPRSS2 possesses androgen-responsive elements and so in response to androgens TMPRSS2 drives ERG overexpression. Antiandrogens can decrease ERG in patients carrying TMPRSS2-ERG through its ability to reduce the levels of androgen. In contrast, for patients whose PCa progresses and becomes resistant to antihormone therapy, the fusion oncogeneTMPRSS2-ERG can be reactivated and could thus contribute to tumor progression (4).
We are currently facing a global obesity epidemic that has been associated with a negative impact on PCa. There is strong epidemiologic evidence that obesity is associated with a greater risk of aggressive PCa and increased PCa-specific mortality (5–7). Furthermore, the negative impact of obesity on PCa prognosis has mainly been observed in men with the TMPRSS2-ERG gene fusion (8) implying an interaction.
Obesity is often associated with comorbid conditions such as insulin resistance, hyperglycemia, and type 2 diabetes. We have shown previously that hyperglycemia-induced chemoresistance in PCa cells and that this was mediated by an epigenetic upregulation of insulin-like growth factor binding protein-2 (IGFBP-2) (9). IGFBP-2 is one of the six high-affinity IGF-binding proteins, which bind to IGFs, acting as a carrier and protecting them from clearance, increasing their half-lives, and modulating their availability and activity. These IGFBPs, including IGFBP-2 can also regulate cell function independently of the insulin-like growth factor-1 (IGF-I) receptor (10, 11). IGFBP-2 is considered to be a key player in PCa progression (12) with IGFBP-2 levels being raised in the serum and in the tumors of patients with PCa (13, 14).
PTEN is a phosphoprotein that exhibits both protein- and lipid-phosphatase activity that inhibits the phosphatidylinositol 3-kinase/Akt and mitogen-activated protein kinase signaling pathways (15–17), thereby acting in an opposite manner to growth factors, which promote cell growth and survival. We identified that IGFBP-2 inhibited PTEN function in PCa cells by increasing its phosphorylation (18) and global expression profiling indicated that IGFBP-2 was the most important biomarker to indicate the status of PTEN in tumors (19). When PTEN is silenced, mice develop high grade prostatic intraepithelial neoplasia, but do not progress to develop cancer (20). 93% of ERG rearrangement positive samples showed either absent or reduced PTEN (21) and tumors lacking functional PTEN express higher levels of ERG rearrangement (22). IGFBP-2 has also been shown to translocate to the nucleus in neuroblastoma cells, via its nuclear localization sequence, where it directly associates with DNA and functions as a transcription factor, modulating specific tumorigenic genes (23, 24).
The high frequency of the TMPRSS2-ERG gene fusion in PCa is not due to random translocations but is promoted by the androgen receptor inducing changes in chromosomal architecture leading to the proximity of the TMPRSS2 and ERG genes that are then fused following double-strand breaks (DSB) and repair via the non-homologous end joining (NHEJ) pathway (25). There have been several studies examining the effects of androgen exposure on the formation of fusion products (26, 27), but little work examining potential effects of other exposures. In this study, we examined the effect of some of the exposures associated with disturbed metabolism on TMPRSS2-ERG gene fusion, in particular, hyperglycemia and the potential role of IGFBP-2 in the latter.
Materials and Methods
Materials
All chemicals, unless otherwise stated, were purchased from Sigma (Poole, UK). LNCaP cells were bought from ATCC and cultured as described previously (9). All cell lines tested negative for mycoplasma.
Fusion Induction
LNCaP cells were seeded in 6-well plates in DMEM growth media (Basel, Switzerland, GM: 5 mM glucose) with or without IGFBP-2 silenced for 24 h, serum starved for 24 h in the presence of aphidicolin (2 µg/ml) in DMEM and HAM’S Nutrient Mix F12 media containing charcoal-stripped serum (Invitrogen, Paisley, UK, SFM: 25 mM) and then dosed with dihydrotestosterone (DHT:0.1 μM) in the presence or absence of IGF-I, Gropep, Adelaide, SA, Australia (100 ng/ml), insulin, Novo Nordisk, West Sussex, UK (100 ng/ml), or epidermal growth factor (EGF), Merck, Hertfordshire, UK (20 ng/ml) for 2 h in fresh charcoal-stripped serum based media followed by the addition of etoposide (60 µM) for 1 h. We confirmed that dosing with etoposide at this dose for 1 h did not induce any consequent cell death (data not shown). Before assessing whether IGF-I, insulin, or EGF affected the rate of fusion in LNCaP cells, we initially assessed how responsive these cells were to the factors in relation to DNA proliferation. On performing dose responses, we found that 20 ng/ml EGF and 100 ng/ml IGF-I and insulin gave the greatest response in terms of growth and so used these doses for the fusion experiments. Optimum doses of DHT and etoposide that were used were selected from previous dose response curves (data not shown). Cells were incubated in fresh charcoal-stripped serum based media for a further 24 h prior to the extraction of RNA using Trizol reagent from Invitrogen (Carlsbad, CA, USA) according to manufacturer’s instructions and conversion to cDNA using a kit from Invitrogen (SuperScript III First-Strand Synthesis System). IGFBP-2 was silenced, parallel to non-silencing controls, using siRNA (100 nM) and a second siRNA for IGFBP-2 was also used to exclude off-target responses: sequences of siRNAs and methodology described previously (9, 28).
Quantitative Nested PCR
Each tube of cDNA was separated into 10 × 2 µl aliquots. These were used in 10 separate nested PCR reactions amplified using primer pair 1 (TMPRSS2 forward CAGGAGGCGGAGGCGGA: ERG reverse GGCGTTGTAGCTGGGGGTGAG). 2 μl of this PCR product was then taken and used to initiate the second round of PCR amplified using primer pair 2 (TMPRSS2 forward GGAGCGCCGCCTGGAG: ERG reverse CCATATTCTTTCACCGCCCACTCC) in a further 10 reactions as described previously (29). Each PCR product was run on a 1.7% agarose gel and the total number of PCR products from these 10 reactions counted and compared. This process was repeated for each treatment in triplicate.
Western Immunoblotting
Insulin-like growth factor binding protein-2, γH2AX, DNA-dependent protein kinase (DNAPK)cs, and β-actin were analyzed by Western immunoblotting as described previously (9).
Statistical Analysis
Data were analyzed with SPSS 12.0.1 for Windows using one-way ANOVA followed by least significant difference post hoc test. A statistically significant difference was present at *p < 0.05.
Results
Effect of Glucose on the Number of TMPRSS2:ERG Fusion Products: A Role for IGFBP-2
Figures 1A,B show that the average number of TMPRSS2:ERG fusion products was higher in 25 mM glucose (6.3) compared to 5 mM glucose (3), with the average rate over 2.1-fold higher at 25 mM than at 5 mM (p < 0.01). As we have shown previously, using ELISA and western blotting that high glucose increases the abundance of IGFBP-2 compared with levels observed in 5 mM glucose by 1.8-fold (p < 0.01) (9), these data suggested that the glucose-induced increase in IGFBP-2 may be related to the increase in TMPRSS2:ERG fusion products. Therefore, to examine this more specifically, we silenced IGFBP-2 using siRNA in high glucose conditions and observed a significant decrease in TMPRSS2:ERG fusion products (p < 0.05): effective silencing of IGFBP-2 is indicated by a western blot (Figures 1C,D).
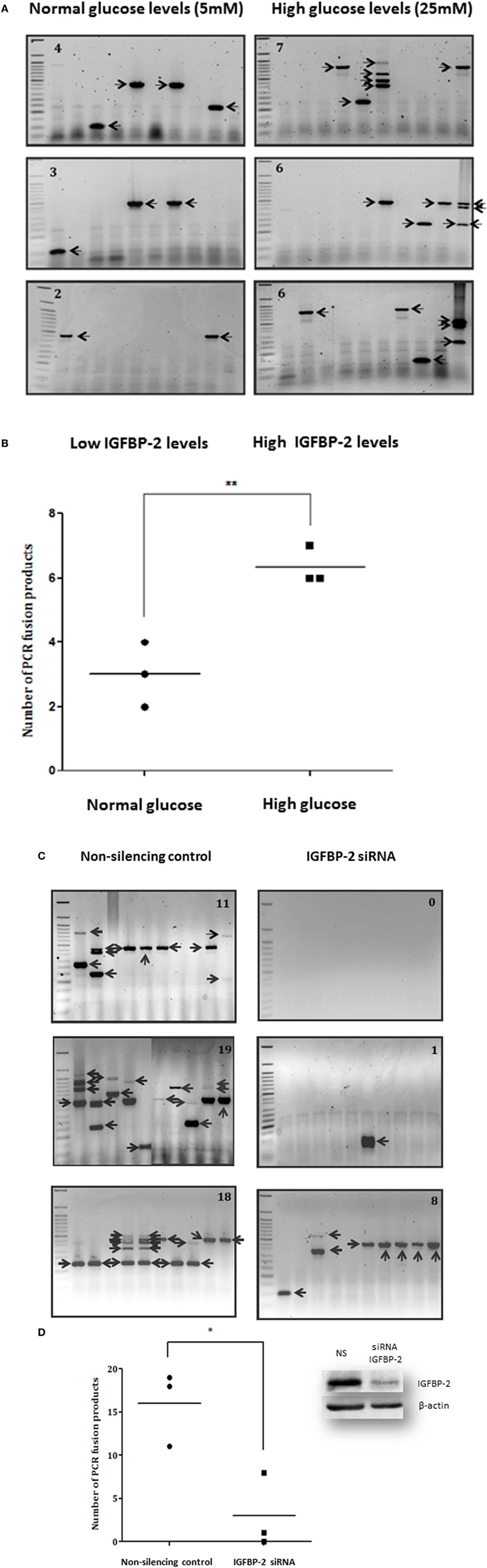
Figure 1. Effects of hyperglycemia on TMPRSS2:ERG fusion induction in LNCaP prostate cancer cells. LNCaP cells were seeded (0.7 × 106 cells/well) in 6-well plates in normal glucose-containing DMEM growth media (GM: 5 mM glucose) for 24 h, serum starved for 24 h in the presence of aphidicolin (2 µg/ml) in either normal or high glucose-containing DMEM and HAM’S Nutrient Mix F12 media containing charcoal-stripped serum (SFM: 25 mM), and then dosed with DHT (0.1 µM) for 2 h in fresh charcoal-stripped serum based media followed by the addition of etoposide (60 µM) for 1 h. Cells were incubated in fresh charcoal-stripped serum based media for a further 24 h. LNCaP cells were also seeded (0.7 × 106 cells/well) in high glucose-containing GM and insulin-like growth factor binding protein-2 (IGFBP-2) was silenced parallel to non-silencing controls using siRNA (100 nM). After 16 h cells were serum starved in high glucose-containing SFM as above for a further 24 h and treated with 0.1 µM DHT for 2 h and 60 µM etoposide for a further 1 h. Cells were incubated in fresh SFM for a further 24 h. Cells were extracted in trizol for performing nested PCR. (A,C) Illustrate three repeats of the gels showing PCR products (each indicated by an arrow) and number of PCR products in top corner of each blot (B,D) is the quantification of the PCR products. Insert to (D) is a representative western immunoblot for IGFBP-2 and β-actin (NS, non-silencing siRNA).
Effect of Insulin, EGF, and IGF-I on the Number of TMPRSS2-ERG Fusion Products
The average rate of fusion induction was decreased 3.5-fold by insulin (that does not bind IGFBPs) (Figures 2A,B,E,F,I,J) and over 2.5-fold by IGF-I (Figures 2A,D,E,H,I,L). We also observed a 3.5-fold decrease by an alternative growth factor, EGF (Figures 2A,C,E,G,I,K). While the decrease in fusion induction with insulin and EGF treatment were statistically significant (p < 0.05), the decrease observed in the presence of IGF-I was not statistically significant (Figure 2M).
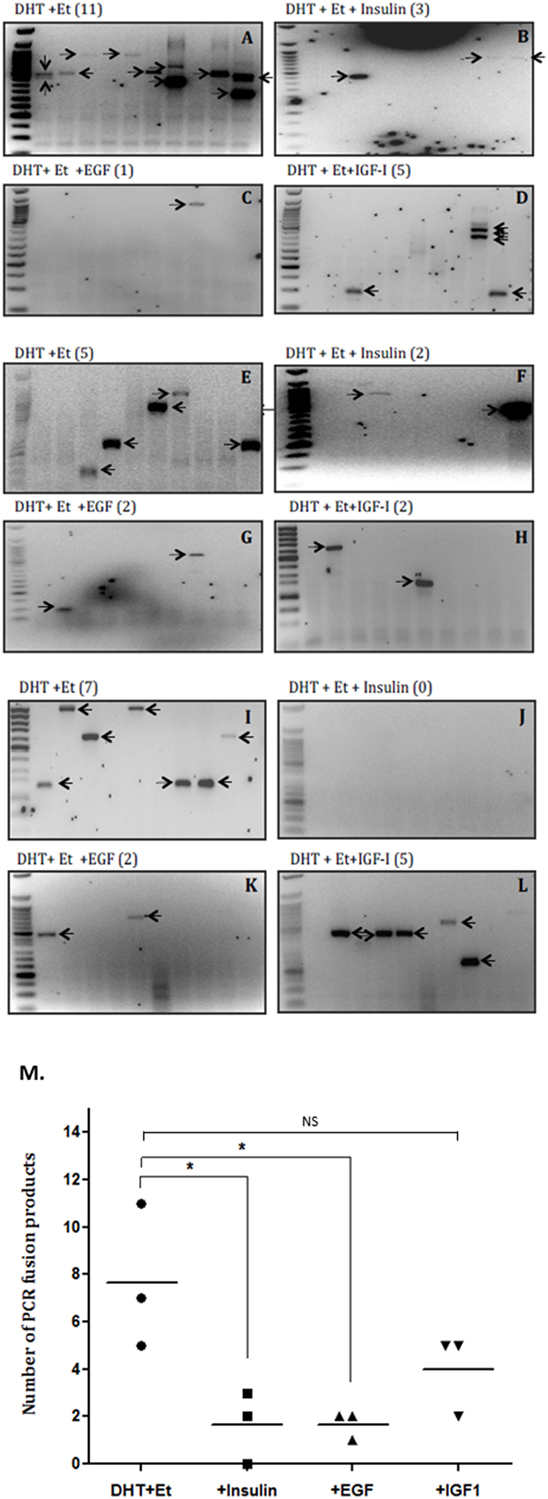
Figure 2. Effects of insulin, epidermal growth factor (EGF), and insulin-like growth factor-1 (IGF-I) on TMPRSS2:ERG fusion induction in LNCaP prostate cancer cells. LNCaP cells (0.7 × 106 cells/well) were seeded in 6-well plates and serum starved as described in figure legend 1 for a further 24 h and treated with 0.1 µM DHT for 2 h in the presence or absence of IGF-I (100 ng/ml), insulin (100 ng/ml), or EGF (20 ng/ml) followed by 60 µM etoposide for a further hour. A final 24 h incubation with fresh SFM was followed by RNA extraction in Trizol for analysis by nested PCR. (A–L) Illustrates three repeats of the gels showing PCR products with the number of PCR products in brackets after the title of each blot and (M) is the quantification of the PCR products.
Effects of Silencing IGFBP-2 or Adding IGF-I on Levels of γH2AX and DNA-Dependent Protein Kinase Catalytic Subunit (DNAPKcs)
Figure 3A shows an increase in γH2AX after etoposide treatment, corresponding to a dramatic increase in DSBs. At 3, 4, and 5 h the bands depicting the levels of γH2AX were substantially higher in cells in which IGFBP-2 had been knocked down compared to non-silencing controls. A decrease in the levels of DNAPKcs after IGFBP-2 knockdown was observed at 3, 4, and 5 h after etoposide and DHT dosing compared to non-silencing treated cells. This was verified by densitometry shown in Figure 3B. We repeated the experiment at the 4 h time point to confirm that levels of γH2AX were significantly increased (p = 0.003) and those of DNAPKcs were significantly (p = 0.011) decreased with IGFBP-2 silenced (p = 0.009) (Figure 3Ci,ii,iii). Figure 3Di,ii show that DHT and etoposide treatment alone increased levels of DNAPKcs and of γH2AX (p = 0.01 and p < 0.01, respectively). Although IGF-I alone increased DNAPKcs (p < 0.05), in the presence of DHT and etoposide, IGF-I acted in an opposite way and reduced DNAPKcs (p = 0.01). IGF-I, however, had no effect on γH2AX either alone or in combination with DHT and etoposide.
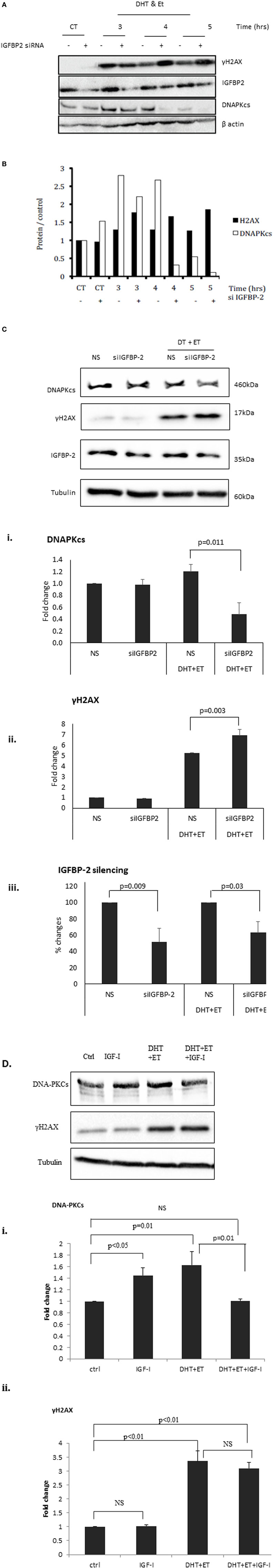
Figure 3. Effects of silencing insulin-like growth factor binding protein-2 (IGFBP-2) or adding insulin-like growth factor-1 (IGF-I) on levels of γH2AX and DNA-dependent protein kinase catalytic subunit (DNAPKcs) in LNCaP prostate cancer cells. Cells were seeded and dosed as in the legend for Figure 1 and following etoposide treatment were incubated in fresh SFM and lysed every 1 h from 3 h in SFM. (A) Shows a representative blot of levels of IGFBP-2, γH2AX, DNAPKcs, and β-actin analyzed by Western immunoblotting as described previously (9). (B) Densitometry of the western blot shown in (A) indicating levels of γH2AX, DNAPKcs normalized to β-actin levels relative to the non-silencing untreated control. Following etoposide, treatment cells were lysed after 4 h in fresh SFM and (C) shows a representative western blot that has been repeated three times showing of levels of IGFBP-2, γH2AX, DNAPKcs normalized to tubulin levels. [(C)i,ii,iii] densitometry showing the mean changes of three experiments showing levels ofγH2AX and DNAPKcs, respectively, normalized to tubulin levels relative to the non-silencing untreated control. Cells were seeded and dosed with IGF-I (100 ng/ml) as in the legend for Figure 2 and following etoposide treatment cells were lysed after 4 h in fresh SFM. (D) Shows a representative western blot that has been repeated three times showing of levels of γH2AX and DNAPKcs normalized to tubulin levels and [(D)i,ii] show densitometry of the mean changes of three experiments indicating levels of DNAPKcs and γH2AX, respectively, normalized to tubulin levels.
Discussion
Knowing that high glucose increases the abundance of IGFBP-2 in PCa cells (9), we used this model to assess whether increasing levels of glucose altered the number of TMPRSS2-ERG fusion products induced by exposure to DHT and etoposide and if this was mediated by IGFBP-2. Our data suggest that high glucose increases the number of TMPRSS2:ERG fusion products, and this was not seen when the accompanying increase in IGFBP-2 was prevented, consistent with IGFBP-2 playing a role in this effect. It would be interesting to investigate whether other inducers of IGFBP-2 also elicit such an effect and indeed whether adding exogenous IGFBP-2 to the cells in normo-glycemic conditions also increased the number of TMPRSS2-ERG fusion products induced by exposure to DHT and etoposide, as this would infer a more general role for IGFBP-2. The NHEJ pathway is a process that repairs DSB in DNA (30). Silencing components involved in the process of NHEJ prevents TMPRSS2:ERG gene fusions (25) indicating that NHEJ is a major method for generating fusions. DNAPK is a large protein complex that plays an important role in NHEJ in DNA-DSB repair and possesses a catalytic subunit called DNAPKcs. DNAPK is critical for controlling progression through the cell cycle and maintaining genomic stability (31). As well as DNAPK being modulated through its interactions with DNA, its activity can also be regulated by a variety of other mechanisms, including modulation of DNAPKcs. A study in HeLa cells (32) concluded that DNAPKcs plays an important role in the regulation of γH2AX phosphorylation in response to DNA damage. Phosphorylation of γH2AX is essential to mark the DSB allowing the DNA repair machinery to identify its location (33). Our data suggest that IGFBP-2 has a role in increasing the rate of repair of DSBs by increasing levels of DNAPKcs and this culminates in increased TMPRSS2:ERG gene fusion. An effect of IGFBP-2 on DNAPK has previously been observed: treatment of astrocytes with IGFBP-2 resulted in a direct induction of DNAPKcs (34). Additional studies provide further support suggesting a role of IGFBP-2 in facilitating DNA repair: in glioblastoma studies, IGFBP-2 alters the expression of the following DNA repair genes: X-ray repair complementing defective repair 2, cyclin-dependent kinase inhibitor 1A, and CDC28 protein kinase 2 (35). In addition, a large-scale study, also in glioblastomas, showed that both the DNA-DSB repair pathway and the homologous recombination pathway are associated with IGFBP-2 expression, altering a broad range of proteins including p53, GADD45, TOP2A, and BRCA1 (36). Of all the six similar IGFBPs, IGFBP-2 has most frequently been reported to be overexpressed in a range of human cancers and only IGFBP-2 has been linked to the DNA-DSB repair pathway. It would, however, be interesting to investigate whether other IGFBPs could have similar actions. In our model showing that adding or silencing any of the other IGFBPs had no effect on fusion induction would imply that this was a specific effect of IGFBP-2.
It has become increasingly clear that IGFBPs, including IGFBP-2, can exert effects that are both dependent and independent of its interactions with IGFs (11). To investigate if the effects of IGFBP-2 in promoting TMPRSS2:ERG gene fusions through facilitating DNA repair were dependent on IGF-I, we exposed LNCaP cells to IGF-I alone or following treatment with DHT and etoposide and compared this to the effects of insulin (that does not bind IGFBPs) and EGF (an alternative growth factor). The effects of IGF-I on DNAPKcs might suggest the effects of IGFBP-2 on DNAPKcs were dependent on binding to IGF-I and negating its effect. Further work is required to confirm the IGF-dependency of IGFBP-2 in DSB repair and the induction of fusion; as we did not observe any effect of IGF-I on γH2AX at this time point, although IGF-I induced a reduction in the frequency of fusion products, it was not statistically significant. It is possible that a potential reduction in fusion products induced by IGF-I was not due to an effect on DNA repair but could have been an effect on chromosomal architecture. It has been observed that, in LNCaP-LN3 cells (a derivative of the LNCaP cells that were used in our study), blocking the IGF-I receptor had no effect on γH2AX focus formation, suggesting that activation of the IGF-IR in this cell line has no effect on DSB repair (37).
In summary, our data suggest that exposure to insulin and potentially IGF-I reduced the frequency of formation of fusion products whereas both hyperglycemia and IGFBP-2 increased the number of TMPRSS2-ERG gene fusions and these factors could contribute to the negative impact that obesity has on PCa progression.
Author Contributions
JH and CP made substantial contributions to the design and with JB, RM, AB were responsible for the work, interpretation, and analysis of the data. All authors contributed to drafting and revising the manuscript and all approved the final version with an agreement of accountability for the work presented.
Conflict of Interest Statement
The authors declare that there is no conflict of interest that could be perceived as prejudicing the impartiality of the research reported.
Funding
We would like to thank the MRC as this work formed part of Jessica Broadhurst’s MRC-DTG supported Ph.D. We would also like to express our sincere thanks to Majlis Amanah Rakyat (MARA, Malaysia) and University Kuala Lumpur Royal College of Medicine Perak for supporting this work. CP and JH are supported by a Cancer Research UK (C18281/A19169) Programme Grant (the Integrative Cancer Epidemiology Programme).
References
1. Attard G, Clark J, Ambroisine L, Fisher G, Kovacs G, Flohr P, et al. Duplication of the fusion of TMPRSS2 to ERG sequences identifies fatal human prostate cancer. Oncogene (2008) 27:253–63. doi:10.1038/sj.onc.1210640
2. Hessels D, Schalken JA. Recurrent gene fusions in prostate cancer: their clinical implications and uses. Curr Urol Rep (2013) 14:214–22. doi:10.1007/s11934-013-0321-1
3. Mehra R, Tomlins SA, Yu J, Cao X, Wang L, Menon A, et al. Characterization of TMPRSS2-ETS gene aberrations in androgen-independent metastatic prostate cancer. Cancer Res (2008) 68:3584–90. doi:10.1158/0008-5472.CAN-07-6154
4. Cai C, Wang H, Xu Y, Chen S, Balk SP. Reactivation of androgen receptor-regulated TMPRSS2: ERG gene expression in castration-resistant prostate cancer. Cancer Res (2009) 69:6027–32. doi:10.1158/0008-5472.CAN-09-0395
5. Cao Y, Ma J. Body mass index, prostate cancer-specific mortality, and biochemical recurrence: a systematic review and meta-analysis. Cancer Prev Res (Phila) (2011) 4:486–501. doi:10.1158/1940-6207.CAPR-10-0229
6. Rodriguez C, Freedland SJ, Deka A, Jacobs EJ, McCullough ML, Patel AV, et al. Body mass index, weight change, and risk of prostate cancer in the Cancer Prevention Study II Nutrition Cohort. Cancer Epidemiol Biomarkers Prev (2007) 16:63–9. doi:10.1158/1055-9965.EPI-06-0754
7. Zhang X, Zhou G, Sun B, Zhao G, Liu D, Sun J, et al. Impact of obesity upon prostate cancer-associated mortality: a meta-analysis of 17 cohort studies. Oncol Lett (2015) 9:1307–12. doi:10.3892/ol.2014.2841
8. Pettersson A, Lis RT, Meisner A, Flavin R, Stack EC, Fiorentino M, et al. Modification of the association between obesity and lethal prostate cancer by TMPRSS2:ERG. J Natl Cancer Inst (2013) 105:1881–90. doi:10.1093/jnci/djt332
9. Biernacka KM, Uzoh CC, Zeng L, Persad RA, Bahl A, Gillatt D, et al. Hyperglycaemia-induced chemoresistance of prostate cancer cells due to IGFBP-2. Endocr Relat Cancer (2013) 20:741–51. doi:10.1530/ERC-13-0077
10. Jones JI, Clemmons DR. Insulin-like growth factors and their binding proteins: biological actions. Endocr Rev (1995) 16:3–34. doi:10.1210/edrv-16-1-3
11. Zeng L, Perks CM, Holly JM. IGFBP-2/PTEN: a critical interaction for tumours and for general physiology? Growth Horm IGF Res (2015) 25:103–7. doi:10.1016/j.ghir.2015.01.003
12. Degraff DJ, Aguiar AA, Sikes RA. Disease evidence for IGFBP-2 as a key player in prostate cancer progression and development of osteosclerotic lesions. Am J Transl Res (2009) 1:115–30.
13. Bubendorf L, Kolmer M, Kononen J, Koivisto P, Mousses S, Chen Y, et al. Hormone therapy failure in human prostate cancer: analysis by complementary DNA and tissue microarrays. J Natl Cancer Inst (1999) 91:1758–64. doi:10.1093/jnci/91.20.1758
14. Kanety H, Madjar Y, Dagan Y, Levi J, Papa MZ, Pariente C, et al. Serum insulin-like growth factor-binding protein-2 (IGFBP-2) is increased and IGFBP-3 is decreased in patients with prostate cancer: correlation with serum prostate-specific antigen. J Clin Endocrinol Metab (1993) 77:229–33. doi:10.1210/jc.77.1.229
15. Cully M, You H, Levine AJ, Mak TW. Beyond PTEN mutations: the PI3K pathway as an integrator of multiple inputs during tumorigenesis. Nat Rev Cancer (2006) 6:184–92. doi:10.1038/nrc1819
16. Dahia PL. PTEN, a unique tumor suppressor gene. Endocr Relat Cancer (2000) 7:115–29. doi:10.1677/erc.0.0070115
17. Tamura M, Gu J, Tran H, Yamada KM. PTEN gene and integrin signaling in cancer. J Natl Cancer Inst (1999) 91:1820–8. doi:10.1093/jnci/91.21.1820
18. Uzoh CC, Holly JM, Biernacka KM, Persad RA, Bahl A, Gillatt D, et al. Insulin-like growth factor-binding protein-2 promotes prostate cancer cell growth via IGF-dependent or -independent mechanisms and reduces the efficacy of docetaxel. Br J Cancer (2011) 104:1587–93. doi:10.1038/bjc.2011.127
19. Mehrian-Shai R, Chen CD, Shi T, Horvath S, Nelson SF, Reichardt JK, et al. Insulin growth factor-binding protein 2 is a candidate biomarker for PTEN status and PI3K/Akt pathway activation in glioblastoma and prostate cancer. Proc Natl Acad Sci U S A (2007) 104:5563–8. doi:10.1073/pnas.0609139104
20. Kwabi-Addo B, Giri D, Schmidt K, Podsypanina K, Parsons R, Greenberg N, et al. Haploinsufficiency of the Pten tumor suppressor gene promotes prostate cancer progression. Proc Natl Acad Sci U S A (2001) 98:11563–8. doi:10.1073/pnas.201167798
21. Carver BS, Tran J, Gopalan A, Chen Z, Shaikh S, Carracedo A, et al. Aberrant ERG expression cooperates with loss of PTEN to promote cancer progression in the prostate. Nat Genet (2009) 41:619–24. doi:10.1038/ng.370
22. King JC, Xu J, Wongvipat J, Hieronymus H, Carver BS, Leung DH, et al. Cooperativity of TMPRSS2-ERG with PI3-kinase pathway activation in prostate oncogenesis. Nat Genet (2009) 41:524–6. doi:10.1038/ng.371
23. Azar WJ, Azar SH, Higgins S, Hu JF, Hoffman AR, Newgreen DF, et al. IGFBP-2 enhances VEGF gene promoter activity and consequent promotion of angiogenesis by neuroblastoma cells. Endocrinology (2011) 152:3332–42. doi:10.1210/en.2011-1121
24. Azar WJ, Zivkovic S, Werther GA, Russo VC. IGFBP-2 nuclear translocation is mediated by a functional NLS sequence and is essential for its pro-tumorigenic actions in cancer cells. Oncogene (2014) 33:578–88. doi:10.1038/onc.2012.630
25. Lin C, Yang L, Tanasa B, Hutt K, Ju BG, Ohgi K, et al. Nuclear receptor-induced chromosomal proximity and DNA breaks underlie specific translocations in cancer. Cell (2009) 139:1069–83. doi:10.1016/j.cell.2009.11.030
26. Coll-Bastus N, Mao X, Young BD, Sheer D, Lu YJ. DNA replication-dependent induction of gene proximity by androgen. Hum Mol Genet (2015) 24:963–71. doi:10.1093/hmg/ddu508
27. Haffner MC, Aryee MJ, Toubaji A, Esopi DM, Albadine R, Gurel B, et al. Androgen-induced TOP2B-mediated double-strand breaks and prostate cancer gene rearrangements. Nat Genet (2010) 42:668–75. doi:10.1038/ng.613
28. Foulstone EJ, Zeng L, Perks CM, Holly JM. Insulin-like growth factor binding protein 2 (IGFBP-2) promotes growth and survival of breast epithelial cells: novel regulation of the estrogen receptor. Endocrinology (2013) 154:1780–93. doi:10.1210/en.2012-1970
29. Clark J, Merson S, Jhavar S, Flohr P, Edwards S, Foster CS, et al. Diversity of TMPRSS2-ERG fusion transcripts in the human prostate. Oncogene (2007) 26:2667–73. doi:10.1038/sj.onc.1210070
30. Kurimasa A, Kumano S, Boubnov NV, Story MD, Tung CS, Peterson SR, et al. Requirement for the kinase activity of human DNA-dependent protein kinase catalytic subunit in DNA strand break rejoining. Mol Cell Biol (1999) 19:3877–84. doi:10.1128/MCB.19.5.3877
31. Smith GC, Jackson SP. The DNA-dependent protein kinase. Genes Dev (1999) 13:916–34. doi:10.1101/gad.13.8.916
32. An J, Huang YC, Xu QZ, Zhou LJ, Shang ZF, Huang B, et al. DNA-PKcs plays a dominant role in the regulation of H2AX phosphorylation in response to DNA damage and cell cycle progression. BMC Mol Biol (2010) 11:18. doi:10.1186/1471-2199-11-18
33. Paull TT, Rogakou EP, Yamazaki V, Kirchgessner CU, Gellert M, Bonner WM. A critical role for histone H2AX in recruitment of repair factors to nuclear foci after DNA damage. Curr Biol (2000) 10:886–95. doi:10.1016/S0960-9822(00)00610-2
34. Becher OJ, Peterson KM, Khatua S, Santi MR, MacDonald TJ. IGFBP-2 is overexpressed by pediatric malignant astrocytomas and induces the repair enzyme DNA-PK. J Child Neurol (2008) 23:1205–13. doi:10.1177/0883073808321766
35. Wang H, Wang H, Shen W, Huang H, Hu L, Ramdas L, et al. Insulin-like growth factor binding protein 2 enhances glioblastoma invasion by activating invasion-enhancing genes. Cancer Res (2003) 63:4315–21.
36. Holmes KM, Annala M, Chua CY, Dunlap SM, Liu Y, Hugen N, et al. Insulin-like growth factor-binding protein 2-driven glioma progression is prevented by blocking a clinically significant integrin, integrin-linked kinase, and NF-κB network. Proc Natl Acad Sci U S A (2012) 109:3475–80. doi:10.1073/pnas.1120375109
Keywords: prostate cancer, insulin-like growth factor-binding protein-2, TMPRSS2-ERG, hyperglycemia, type II diabetes
Citation: Holly JMP, Broadhurst J, Mansor R, Bahl A and Perks CM (2017) Hyperglycemia Promotes TMPRSS2-ERG Gene Fusion in Prostate Cancer Cells via Upregulating Insulin-Like Growth Factor-Binding Protein-2. Front. Endocrinol. 8:305. doi: 10.3389/fendo.2017.00305
Received: 27 July 2017; Accepted: 20 October 2017;
Published: 06 November 2017
Edited by:
Haim Werner, Tel Aviv University, IsraelReviewed by:
Andrea Morrione, Thomas Jefferson University, United StatesMaximilian Bielohuby, Sanofi, France
Copyright: © 2017 Holly, Broadhurst, Mansor, Bahl and Perks. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Claire M. Perks, Y2xhaXJlLm0ucGVya3NAYnJpc3RvbC5hYy51aw==