 Pierre-Damien Denechaud
Pierre-Damien Denechaud Lluis Fajas
Lluis Fajas Albert Giralt
Albert Giralt- Center for Integrative Genomics, University of Lausanne, Lausanne, Switzerland
In the past years, several lines of evidence have shown that cell cycle regulatory proteins also can modulate metabolic processes. The transcription factor E2F1 is a central player involved in cell cycle progression, DNA-damage response, and apoptosis. Its crucial role in the control of cell fate has been extensively studied and reviewed before; however, here, we focus on the participation of E2F1 in the regulation of metabolism. We summarize recent findings about the cell cycle-independent roles of E2F1 in various tissues that contribute to global metabolic homeostasis and highlight that E2F1 activity is increased during obesity. Finally, coming back to the pivotal role of E2F1 in cancer development, we discuss how E2F1 links cell cycle progression with different metabolic adaptations required for cell growth and survival.
Introduction: A Cell Cycle Protein with New Skills
The E2F transcription factors were first identified as proteins that were able to bind to the promoter of the adenoviral gene E2 (1). Eight E2F genes (E2F1-8) have been described to date, which can be classified based on their protein structures, their interaction partners, and their transcriptional properties (2). E2F1 was the first member of the E2F family to be identified because of its ability to bind the retinoblastoma protein (pRB), a tumor suppressor mutated in many types of cancer (3, 4). The activity of E2F1 is dependent on its binding partners, which include dimerization proteins (DP) and the retinoblastoma family proteins (also known as “pocket proteins”), composed by pRB (RB1), p107 (RBL1), and p130 (RBL2) (5). E2F1–pRB interaction blocks the transcriptional activation domain of the E2F1–DP complex and prevents the recruitment of transcriptional co-activators to the promoters of its target genes (6). During cell cycle progression, cyclin-dependent kinases (CDKs) phosphorylate pRB, releasing E2F1, which is then available to promote the expression of genes involved in S-phase entry, DNA synthesis, and mitosis (7–9).
Three decades after its discovery, it is now clear that the control of cell cycle represents only a subset of the E2F1 roles, which include the regulation of apoptosis (10), senescence (11), and DNA-damage response (12). Indeed, genome-wide location studies have revealed that E2F1 binds to hundreds of promoter regions of genes involved in a myriad of cellular pathways (13–16). What ultimately determines E2F1 distinct biological functions are its protein levels, the combination of several posttranscriptional modifications and its interaction with different partners (17). The intricate role of E2F1 as a master regulator of cell fate has been extensively examined before and is out of scope for this review (17, 18). Instead, here, we want to focus on the recent research evidencing that E2F1 is a master regulator of metabolism both in normal and pathological conditions.
E2F1 Regulates Metabolism in Non-Proliferative Conditions and Contributes to Global Metabolic Homeostasis
Role of E2F1 in Normal Physiology
Despite the critical function of E2F1 in cell proliferation, E2f1−/− mice undergo normal development, likely due to the compensation by other E2F family members (19, 20). However, E2f1−/− mice present some metabolic perturbations that highlight its specific role in the regulation of metabolism independently from cell cycle control. E2F1 participates in the development and the differentiation of several tissues involved in global metabolic homeostasis, but it is also implicated in specific metabolic functions of fully differentiated organs like pancreas, adipose tissues, muscle and liver (Figure 1).
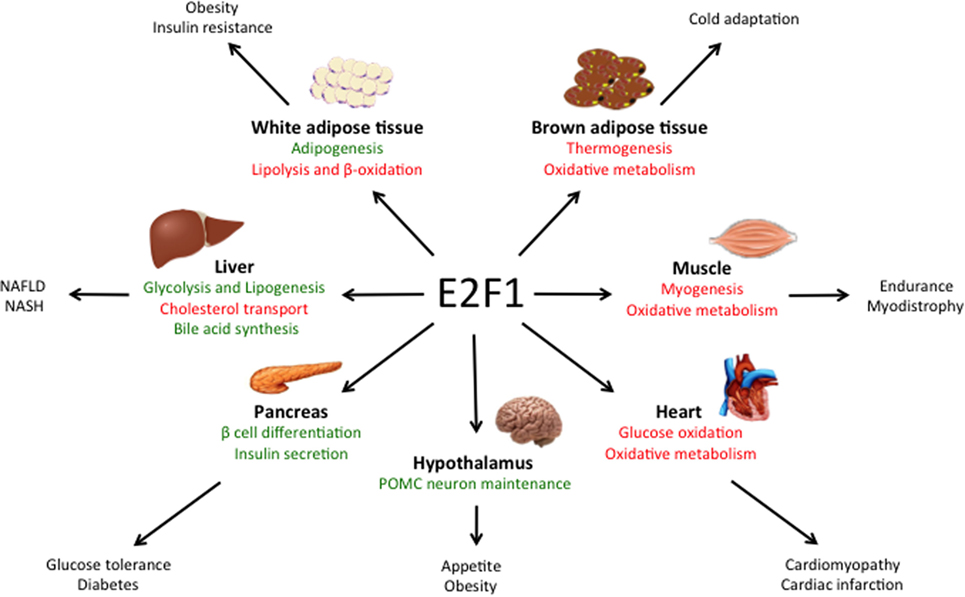
Figure 1. Main roles of E2F1 in metabolic tissues. E2F1 participates in the differentiation of several tissues, but also in the regulation of specific metabolic functions in fully differentiated organs, thus contributing to global metabolic homeostasis. Moreover, during obesity, E2F1 activity is increased and it contributes to some of the comorbidities of this pathological condition. Pathways activated by E2F1 are represented in green while pathways repressed by E2F1 are in red.
E2f1/E2f2 mutant mice show severe exocrine atrophy of pancreatic β cells, primarily resulting from E2F1 mutation, which leads to insulin-dependent diabetes (21). E2F1 promotes β cell proliferation and differentiation through the regulation of the endocrine markers PDX-1 and Neurogenin 3 (22, 23). In addition, in fully differentiated β cells, E2F1 directly controls the expression of the major subunit of the ATP-sensitive K+ channel Kir6.2, hence promoting glucose-stimulated insulin secretion (24). These studies show that E2F1 participates in pancreas development, maintenance, and endocrine function, hence contributing to global glucose homeostasis.
In the adipose tissue, E2F1 promotes adipogenesis though the regulation of PPARG and RIP140 gene expression, two master regulators of adipocyte fate and differentiation (25, 26). Moreover, in mature adipocytes E2F1 can form a repressor complex with TRIP-Br2—a transcriptional co-regulator—which inhibits lipolysis and mitochondrial β-oxidation (27). Interestingly, CDK4, the main E2F1 upstream activator during cell cycle progression, also promotes adipogenesis though PPARG activation and in mature adipocytes it sustains insulin signaling by phosphorylation of the Insulin Receptor Substrate 2(28, 29). Altogether, these findings show that the canonical CDK4-pRB-E2F1 axis is essential for adipogenesis and to maintain adipocyte function.
In contrast to white adipose tissue, E2F1 represses mouse myogenic differentiation by inhibiting the transcription factors MyoD and Myogenin (30, 31). MyoD in turn, promotes the expression of the Kelch Repeat and BTB Domain Containing Protein 5 (Kbtbd5), which represses E2F1 activity in a negative feedback loop by the ubiquitination and degradation of DP1 (32). Conversely, in Drosophila, depletion of the dE2F1 blunts the expression of late myogenic markers during muscle differentiation, which is critical for survival (33). The differences between the two organisms are puzzling and require further exploration, but they may rely on the fact that in Drosophila there are only two E2F isoforms, dE2F1 and dE2F2, which work as activators and repressors of transcription, respectively.
Chromatin immunoprecipitation (ChIP) analysis revealed that in basal conditions E2F1 and pRB form a repressor complex in the promoters of several genes involved in oxidative metabolism and mitochondrial biogenesis in muscle, but also in brown adipose tissue (34). In response to exercise or cold exposure, pRB is phosphorylated in muscle and brown adipose tissue, respectively, and mitochondrial and thermogenic genes are expressed (34, 35). As a consequence, deletion of E2F1 in mice results in better resistance to fatigue during exercise and higher body temperature upon cold stimulation due to increased oxidative metabolism (34). Strikingly, E2F1 depletion in a dystrophic mouse model significantly improved muscle performance by increasing muscle oxidative metabolism (36).
Some studies using pRB lack of function models support the role of the E2F1–pRB complex as a negative regulator of oxidative metabolism. For instance, adipose-specific RB1-deficient mice are resistant to high-fat diet (HFD)-induced obesity and display increased mitochondrial activity in white and brown adipose tissues (37). This was reproduced in RB1-haplosufficient mice (38). However, the HFD-resistant phenotype of RB1-deficient mice could also be attributed to the role of pRB in promoting white versus brown fat cell differentiation (35, 39), as evidenced by the increased expression of the thermogenic protein UCP1 in both white and brown adipose tissue depots (37, 38). Additionally, acute loss of pRB or depletion of p170 increased mitochondrial content and activity in muscle cells (40, 41). Conversely, other studies report that pRB may in fact promote mitochondrial biogenesis. Deletion of RB1 led to impaired mitochondrial function in myocytes (42) and erythrocytes (43). More recently, it was shown that acute pRB loss in adult mice results in a decreased content of oxidative phosphorylation proteins in the lung and in the colon (44), while RB1 depletion blocked muscle differentiation due to an impairment in oxidative metabolism (45). The above confounding studies evidence the relevance of the E2F1-pRB complex in the control of oxidative metabolism in highly metabolic tissues, but they highlight that its specific function may be context dependent. It should also be taken into account that pRB loss of function also leads to multiple E2F1-independent effects (4). Moreover, the fact that E2F1 can activate or repress its target genes often complicates the understanding of the phenotype of E2f1 knockout models.
Role of E2F1 in Metabolic Diseases
Obesity is associated with increased risk of developing cardiovascular diseases, type 2 diabetes, and cancer (46). As we will discuss in this section, E2F1 expression and activity are increased during obesity in several tissues involved in metabolic homeostasis, suggesting that E2F1 could contribute to some of the comorbidities of this condition.
E2f1 mRNA and protein levels are increased in the visceral white adipose tissue of obese human subjects and positively correlated with insulin resistance and circulating free-fatty acids (47). E2F1 expression was also increased in the visceral adipose tissue of two widely used mouse models of obesity: mice fed a HFD and leptin-deficient (ob/ob) mice (48). This effect was reversed when HFD-fed mice were treated with resveratrol, which in parallel decreased body weight gain and the levels of pro-inflammatory cytokines levels in white adipose tissue (49). In addition, pRB levels and repressor activity decrease in white adipose tissue during obesity both in rats and in humans (50), which is consistent with increased E2F1 activity. These evidences are supported by ChIP analysis in human white adipose tissue that revealed increased E2F1 binding to the promoters of stress signaling genes during the progression of obesity (51). Interestingly, E2F1 has been shown to enhance NF-κB-mediated inflammatory response (52, 53). However, the contribution of E2F1 to the inflammation of white adipose tissue during insulin resistance remains to be explored.
Obesity is a well-known inducer of cardiac hypertrophy, which often contributes to heart failure (54). Pathological cardiac hypertrophy occurs in parallel with the development of metabolic inflexibility and a re-activation of the cell cycle machinery (55). Similar to the effects observed in the white adipose tissue, HFD increased E2F1 levels and increased RB phosphorylation in mouse heart. This correlated with elevated expression of the E2F1 transcriptional target pyruvate dehydrogenase kinase 4 (PDK4) (56, 57). PDKs inhibit pyruvate dehydrogenase, blocking pyruvate conversion into acetyl-CoA, which results in decreased glucose oxidation. Hence, upregulation of the E2F1–PDK4 axis during obesity may account for the impairment in glucose oxidation that characterizes cardiomyopathy. Moreover, through the regulation of PINK1 translation via miR-421 expression, E2F1 promotes mitochondrial fragmentation in cardiomyocytes, which can lead to myocardial infarction (58). Additionally, E2F1 has been shown to suppress cardiac neovascularization by downregulating VEGF and PIGF expression. Consequently, E2f1−/− mice present better cardiac function after myocardial infarction than their wild-type littermates (59). Altogether, these studies suggest that increased E2F1 activity occurring during obesity contributes to the development of cardiomyopathy through the re-entry in the cell cycle and the re-wiring of cardiac metabolism.
Some laboratories, including ours, have recently demonstrated the importance of E2F1 in the physiopathological context of non-alcoholic fatty liver disease (NAFLD), which is highly related to the epidemic of obesity. NAFLD is a progressive disease that starts with a benign accumulation of lipids in the liver (hepatic steatosis) that can develop to non-alcoholic steatohepatitis (NASH) which, in its worst prognosis, can lead to liver fibrosis, cirrhosis, and hepatocarcinoma (60). Hepatic E2F1 levels are increased in steatotic liver in mice but also in humans (16). Additionally, NAFLD correlated with the phosphorylation of pRB in the liver in different mouse models of obesity and diabetes (16, 61), altogether consistent with increased E2F1 activity in these conditions. One major contributor to NAFLD is an increase in hepatic de novo lipogenesis, a process in which E2F1 plays an important role. Indeed, E2F1 directly activates the expression of key glycolytic and lipogenic genes and E2F1 depletion protects against NAFLD (16). Remarkably, hepatic E2F1 expression is increased in patients with NASH and in different mouse models of liver fibrosis (62, 63). Perturbed bile acid metabolism and/or cholesterol homeostasis are major contributors to NASH. The importance of E2F1 in bile acid synthesis was found in a mouse model of NASH—bile duct ligation and 3, 5- diethoxycarbonyl-1, 4-dihydrocollidine (DCC) feeding—in which bile acid accumulation in the liver contributes to fibrosis. Indeed, knockout of E2F1 in mice reduced bile acid synthesis, which protected from the development of biliary fibrosis under DCC feeding (62). We also recently revealed that E2F1 participates in cholesterol homeostasis by enhancing the expression of PCSK9, a negative regulator of the LDL receptor and cholesterol uptake (63). Importantly, anti PCSK9 antibodies were recently approved for the treatment of cardiovascular diseases due to their capacity to lower LDL cholesterol levels (64). E2f1−/− mice present decreased circulating levels of cholesterol as a consequence of increased cholesterol uptake by several tissues, including the liver. However, when subjected to a high cholesterol diet, E2f1−/− mice presented increased liver fibrosis, likely due to the combination of exacerbated cholesterol uptake and a defect in bile acid secretion (63). Taken together, these studies imply that the convenience of targeting E2F1 to treat liver fibrosis could be context dependent and that this approach requires further investigation. Nevertheless, in humans, the increase of E2F1 during NASH was more substantial than the induction of standard fibrosis markers such as α-SMA and α1-collagen, which suggest that E2F1 could be potentially used as a new diagnostic marker for increased risk of developing liver fibrosis and cirrhosis (62).
Long-term HFD also increased E2F1 protein levels and pRB phosphorylation in hypothalamic Arcuate nucleus neurons, which are involved in global energy balance (65). This in turn led to a de-repression of E2F1-target genes involved in cell cycle regulation and apoptosis. Lu et al. found that the E2F1–pRB repressor complex is necessary for POMC neuron maintenance, whereas specific RB1 depletion in these neurons led to hyperphagia, obesity and diabetic syndrome in an E2F1-dependent manner (65). These results indicated that dysregulation of E2F1 at the central level also contributes to the development of the metabolic syndrome during the progression of obesity.
Altogether, recent work has highlighted the importance of the pRB-E2F1 pathway in the pathophysiology of obesity.
E2F1 Contributes to the Metabolic Reprogramming of Cancer Cells
Cancer cells adapt their metabolism in order to promote growth, proliferation, survival, and metastasis. The specific metabolic profile of a tumor ultimately depends on the tissue of origin, the oncogenic alterations, the tumor stage, and the tumor microenvironment. Metabolic reprogramming is now considered one of the hallmarks of cancer and selectively targeting tumor metabolism has been proposed in the recent years as a therapeutic strategy to treat cancer (66, 67). Remarkably, some oncogenes such as p53 and Myc regulate cancer metabolism (68, 69) and, as we will discuss in this section, so does E2F1 (Table 1).

Table 1. E2F1 contributes to the metabolic reprograming of cancer cells.
E2F1 Contributes to the Warburg Effect
One metabolic feature of many cancer cells is the so-called Warburg effect, which consists on increased aerobic glycolysis and decreased glucose oxidation, resulting in high rates of glucose utilization and lactate production (66, 70). It has been shown that, against the assumption of Otto Warburg, who first described this phenomenon almost a century ago, in most cancers this is not caused by defective mitochondria. Several hypotheses have been proposed on how the Warburg effect benefits cancer cells, including higher rates of ATP synthesis, the generation of glycolytic intermediates for biosynthetic reactions or the remodeling of the tumor microenvironment; however, this phenomenon is still not fully understood (70). It has been shown that E2F1 can promote this metabolic switch by both enhancing glycolysis and by repressing glucose oxidation in the mitochondria (Figure 2). During the development of HCC, increased E2F1 levels progressively recruit Pontin and Reptin (two putative DNA helicases) to promote the expression of genes involved in glycolysis and in lactate export, which contributes to the Warburg effect (15). During cell division, E2F1 also promotes the expression of the F-type isoform of the enzyme 6-phosphofructo-2-kinase/fructose-2,6-bisphosphatase, which results in the synthesis of fructose-2,6-bisphosphate, a potent stimulator of glycolysis (71, 72). Moreover, E2F1 also enhances glycolysis in bladder and prostate cancer cell lines through the suppression of the expression of Sirtuin 6, a NAD(+)-dependent deacetylase that inhibits the transcription of several key glycolytic genes (73, 74). Besides enhancing glycolytic gene expression, as previously mentioned, E2F1 also blocks glucose oxidation in the mitochondria by promoting the expression of the PDK enzymes. While in the heart E2F1 regulates PDK4 (57), in pancreatic cancer cells E2F1 enhances the expression of PDK1 and PDK3 isoforms, which results in increased aerobic glycolysis and proliferation (75).
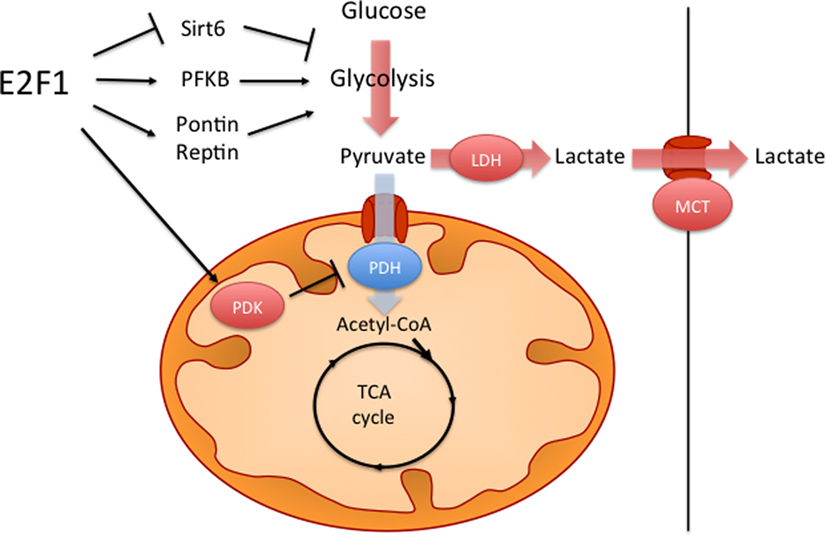
Figure 2. E2F1 contributes to the Warburg effect. E2F1 participates in the characteristic aerobic glycolysis observed in many tumors by different mechanisms. E2F1 promotes glycolysis by repressing the expression of Sirtuin 6 (Sirt6), a negative regulator of glycolytic gene expression and by promoting the expression of the F-type isoform of 6-phosphofructo-2-kinase/fructose-2,6-bissphosphatase (PFKB). E2F1 also recruits a Pontin/Reptin complex to promote the expression of genes involved in glycolysis and lactate export. Additionally, E2F1 blocks glucose oxidation in the mitochondria by promoting the expression of pyruvate dehydrogenase kinase (PDK) enzymes, which inhibit the pyruvate dehydrogenase complex (PDH).
E2F1 and Oxidative Metabolism
In addition to regulating oxidative metabolism in non-proliferative conditions (34), E2F1 also repress mitochondrial biogenesis during proliferation. Like in the muscle, knocking down E2F1 in HeLa cells led to increased expression of several genes involved in mitochondrial biogenesis and oxidative phosphorylation, which resulted in increased ATP production (76). E2F1 depletion in Mesenchymal Stem Cells also increased mitochondrial biogenesis and oxygen consumption (77). Additionally, it has been shown that E2F1-mediated repression of oxidative metabolism results in a self-renewal of tumor-initiating stem-like cells that contributes to the progression of HCC (78). Some evidences show that mitochondrial function, in turn, also impacts E2F1 activity. For instance, inhibition of ATP synthase or of the electron transport chain leads to the downregulation of E2F1 activity and to cell cycle arrest (79, 80). On the other hand, mitochondrial ROS production can promote E2F1-mediated apoptosis (81, 82). For a more detailed perspective of the complex interplay between E2F transcription factors and the mitochondrial function, we address you to recent specific reviews about the topic (83, 84).
E2F1 and Anabolic Metabolism
Cancer cells undergo different anabolic processes to fulfill the high demand of macromolecules required for proliferation. E2F1 participates in DNA synthesis by regulating the expression of several genes involved in nucleotide metabolism such as Thymidine kinase and Dihydrofolate reductase (85, 86). Tumors also normally present high rates of lipid synthesis, which are used both for membrane production and as signaling molecules (87). Lipogenesis is not only important during proliferation; it also contributes to the metastatic capacity of cancer cells (88). Besides promoting lipogenesis in the liver (16), in medulloblastoma E2F1 enhances fatty acid synthase expression in response to Sonic hedgehog signaling (89).
mTORC1 is a master regulator of cell growth and survival, and it is involved in the progression of many cancers (90). It was recently shown that E2F1 promotes mTORC1 activity by enhancing the expression of lysosomal v-ATPase. This in turn, blocked autophagy, one of the main metabolic processes regulated by mTORC1 (91). Conversely, it was shown that E2F1 can also stimulate upregulation of genes involved in autophagy in response to DNA damage (92). Hence, the contribution of E2F1 to autophagy is still a matter of debate. Additionally, numerous studies have highlighted the crosstalk between E2F1 activity and other signaling pathways involved in cancer metabolism, such as the AKT or the HIF pathways (93–95). Whether E2F1 promotes anabolic reprogramming in cancer cells through the interaction with these signaling pathways remain to be explored.
Overall, these studies show that the transcription factor E2F1 plays a pivotal role integrating the cell cycle regulatory machinery with metabolic pathways essential for cell growth and survival. This, in turn, determines cell fate by affecting cell stemness, proliferation rate, or apoptosis. Therefore, inhibiting E2F1 activity could potentially impact tumor development at different levels simultaneously by blocking cell cycle progression and by impairing metabolic flexibility in cancer cells. In this regard, CDK4/6 inhibitors that block pRB phosphorylation and that are currently used for treating hormone-positive breast tumors have been reported to block proliferation, in part, by inducing a metabolic reprogramming in cancer cells (96, 97).
Conclusion and Perspectives
Here, we have collected the current and emerging evidence showing that E2F1 regulates metabolism in non-proliferating conditions and, more importantly, that dysregulation of E2F1 activity leads to complications associated with obesity. Many studies have focused on the mitogenic signals that drive E2F1 activation in cancer cells, but how E2F1 is activated in other pathological conditions such as obesity is just beginning to be understood. The CDK4-pRB-E2F1 pathway can be stimulated both by glucose and by insulin in different tissues involved in global metabolic homeostasis (16, 24, 29, 95, 98). One possibility is that during obesity, hyperglycemia and/or hyperinsulinemia render pRB hyperphosphorylated (50, 61, 65). This in turn, would increase E2F1 activity and, in a positive feedback loop, E2F1 could promote its own expression (99). Other possible candidates for exacerbated E2F1 activation during obesity could be chronic inflammation or increased ROS production due to mitochondrial stress, two factors that promote E2F1 activity in other contexts (52, 82). Despite the specific mechanisms that lead to E2F1 hyperactivation during obesity, targeting E2F1 could potentially be used to ameliorate some of the deleterious effects of this condition. Notably, E2f1−/− mice present increased insulin sensitivity and are resistant to HFD-induced obesity (25, 34). However, it should be considered that systemically inhibiting E2F1 activity would likely impair insulin secretion (100), which could be detrimental in the initial phases of insulin resistance, when insulin production is enhanced to maintain normoglycemia.
Given its dual role in proliferation and metabolism, it is tempting to speculate that E2F1 might be a central actor in the interplay between obesity and some types of cancer. One of those cases could be HCC, for which there is an increased risk in obese patients (101). We have recently shown that hepatic E2F1 expression is augmented during obesity (16), while numerous studies have demonstrated that increased E2F1 activity promotes the development of HCC (15, 78, 102, 103). Notably, it was also recently reported that E2F1 mediates the proliferative effects of insulin in hepatocytes (95). Indeed, obesity-associated hyperinsulinemia is one mechanism proposed to explain the epidemiological observations of increased HCC in obese patients (104). Therefore, under obesity conditions, enhanced hepatic E2F1 activity—maybe in response to hyperinsulinemia—may first lead to enhanced de novo lipogenesis, NAFLD development and fibrosis (16, 62). Subsequently, E2F1 may contribute to HCC progression by promoting the expression of genes involved in cell cycle machinery and cancer metabolism (15).
In conclusion, research over the past 15 years has given an increasingly complex picture of the multiple roles of E2F1. Beyond being a mere cell cycle regulator, this transcription factor has emerged as a novel player in the control of metabolism not only in normal physiology but also under pathological conditions such as obesity and cancer.
Author Contributions
PDD, LF, and AG conceived and wrote the manuscript.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
The authors thank Isabel C. Lopez-Mejía and Jenny Sandström for their critical reading of the manuscript.
Funding
This work was supported by grants from the Swiss Ligue Contre le Cancer, the Swiss National Science Foundation, and the Fondation de France.
References
1. Kovesdi I, Reichel R, Nevins JR. Identification of a cellular transcription factor involved in E1A trans-activation. Cell (1986) 45(2):219–28. doi:10.1016/0092-8674(86)90386-7
2. Black EP, Hallstrom T, Dressman HK, West M, Nevins JR. Distinctions in the specificity of E2F function revealed by gene expression signatures. Proc Natl Acad Sci U S A (2005) 102(44):15948–53. doi:10.1073/pnas.0504300102
3. Bagchi S, Weinmann R, Raychaudhuri P. The retinoblastoma protein copurifies with E2F-I, an E1A-regulated inhibitor of the transcription factor E2F. Cell (1991) 65(6):1063–72. doi:10.1016/0092-8674(91)90558-G
4. Dyson NJ. RB1: a prototype tumor suppressor and an enigma. Genes Dev (2016) 30(13):1492–502. doi:10.1101/gad.282145.116
5. Dyson N. The regulation of E2F by pRB-family proteins. Genes Dev (1998) 12(15):2245–62. doi:10.1101/gad.12.15.2245
6. Frolov MV, Dyson NJ. Molecular mechanisms of E2F-dependent activation and pRB-mediated repression. J Cell Sci (2004) 117(Pt 11):2173–81. doi:10.1242/jcs.01227
7. Polager S, Kalma Y, Berkovich E, Ginsberg D. E2Fs up-regulate expression of genes involved in DNA replication, DNA repair and mitosis. Oncogene (2002) 21(3):437–46. doi:10.1038/sj.onc.1205102
8. La Thangue NB. DP and E2F proteins: components of a heterodimeric transcription factor implicated in cell cycle control. Curr Opin Cell Biol (1994) 6(3):443–50. doi:10.1016/0955-0674(94)90038-8
9. Wu L, Timmers C, Maiti B, Saavedra HI, Sang L, Chong GT, et al. The E2F1-3 transcription factors are essential for cellular proliferation. Nature (2001) 414(6862):457–62. doi:10.1038/35106593
10. Ginsberg D. E2F1 pathways to apoptosis. FEBS Lett (2002) 529(1):122–5. doi:10.1016/S0014-5793(02)03270-2
11. Dimri GP, Itahana K, Acosta M, Campisi J. Regulation of a senescence checkpoint response by the E2F1 transcription factor and p14(ARF) tumor suppressor. Mol Cell Biol (2000) 20(1):273–85. doi:10.1128/MCB.20.1.273-285.2000
12. Stevens C, La Thangue NB. The emerging role of E2F-1 in the DNA damage response and checkpoint control. DNA Repair (Amst) (2004) 3(8–9):1071–9. doi:10.1016/j.dnarep.2004.03.034
13. Bieda M, Xu X, Singer MA, Green R, Farnham PJ. Unbiased location analysis of E2F1-binding sites suggests a widespread role for E2F1 in the human genome. Genome Res (2006) 16(5):595–605. doi:10.1101/gr.4887606
14. Ouyang Z, Zhou Q, Wong WH. ChIP-Seq of transcription factors predicts absolute and differential gene expression in embryonic stem cells. Proc Natl Acad Sci U S A (2009) 106(51):21521–6. doi:10.1073/pnas.0904863106
15. Tarangelo A, Lo N, Teng R, Kim E, Le L, Watson D, et al. Recruitment of Pontin/Reptin by E2f1 amplifies E2f transcriptional response during cancer progression. Nat Commun (2015) 6:10028. doi:10.1038/ncomms10028
16. Denechaud PD, Lopez-Mejia IC, Giralt A, Lai Q, Blanchet E, Delacuisine B, et al. E2F1 mediates sustained lipogenesis and contributes to hepatic steatosis. J Clin Invest (2016) 126(1):137–50. doi:10.1172/JCI81542
17. Poppy Roworth A, Ghari F, La Thangue NB. To live or let die – complexity within the E2F1 pathway. Mol Cell Oncol (2015) 2(1):e970480. doi:10.4161/23723548.2014.970480
18. Shats I, Deng M, Davidovich A, Zhang C, Kwon JS, Manandhar D, et al. Expression level is a key determinant of E2F1-mediated cell fate. Cell Death Differ (2017) 24(4):626–37. doi:10.1038/cdd.2017.12
19. Field SJ, Tsai FY, Kuo F, Zubiaga AM, Kaelin WG Jr, Livingston DM, et al. E2F-1 functions in mice to promote apoptosis and suppress proliferation. Cell (1996) 85(4):549–61. doi:10.1016/S0092-8674(00)81255-6
20. Yamasaki L, Jacks T, Bronson R, Goillot E, Harlow E, Dyson NJ. Tumor induction and tissue atrophy in mice lacking E2F-1. Cell (1996) 85(4):537–48. doi:10.1016/S0092-8674(00)81254-4
21. Li FX, Zhu JW, Tessem JS, Beilke J, Varella-Garcia M, Jensen J, et al. The development of diabetes in E2f1/E2f2 mutant mice reveals important roles for bone marrow-derived cells in preventing islet cell loss. Proc Natl Acad Sci U S A (2003) 100(22):12935–40. doi:10.1073/pnas.2231861100
22. Fajas L, Annicotte JS, Miard S, Sarruf D, Watanabe M, Auwerx J. Impaired pancreatic growth, beta cell mass, and beta cell function in E2F1 (-/-) mice. J Clin Invest (2004) 113(9):1288–95. doi:10.1172/JCI18555
23. Kim SY, Rane SG. The Cdk4-E2f1 pathway regulates early pancreas development by targeting Pdx1+ progenitors and Ngn3+ endocrine precursors. Development (2011) 138(10):1903–12. doi:10.1242/dev.061481
24. Annicotte JS, Blanchet E, Chavey C, Iankova I, Costes S, Assou S, et al. The CDK4-pRB-E2F1 pathway controls insulin secretion. Nat Cell Biol (2009) 11(8):1017–23. doi:10.1038/ncb1915
25. Fajas L, Landsberg RL, Huss-Garcia Y, Sardet C, Lees JA, Auwerx J. E2Fs regulate adipocyte differentiation. Dev Cell (2002) 3(1):39–49. doi:10.1016/S1534-5807(02)00190-9
26. Docquier A, Augereau P, Lapierre M, Harmand PO, Badia E, Annicotte JS, et al. The RIP140 gene is a transcriptional target of E2F1. PLoS One (2012) 7(5):e35839. doi:10.1371/journal.pone.0035839
27. Liew CW, Boucher J, Cheong JK, Vernochet C, Koh HJ, Mallol C, et al. Ablation of TRIP-Br2, a regulator of fat lipolysis, thermogenesis and oxidative metabolism, prevents diet-induced obesity and insulin resistance. Nat Med (2013) 19(2):217–26. doi:10.1038/nm.3056
28. Abella A, Dubus P, Malumbres M, Rane SG, Kiyokawa H, Sicard A, et al. Cdk4 promotes adipogenesis through PPARgamma activation. Cell Metab (2005) 2(4):239–49. doi:10.1016/j.cmet.2005.09.003
29. Lagarrigue S, Lopez-Mejia IC, Denechaud PD, Escoté X, Castillo-Armengol J, Jimenez V, et al. CDK4 is an essential insulin effector in adipocytes. J Clin Invest (2016) 126(1):335–48. doi:10.1172/JCI81480
30. Wang J, Helin K, Jin P, Nadal-Ginard B. Inhibition of in vitro myogenic differentiation by cellular transcription factor E2F1. Cell Growth Differ (1995) 6(10):1299–306.
31. Wang J, Huang Q, Tang W, Nadal-Ginard B. E2F1 inhibition of transcription activation by myogenic basic helix-loop-helix regulators. J Cell Biochem (1996) 62(3):405–10. doi:10.1002/(SICI)1097-4644(199609)62:3<405::AID-JCB10>3.0.CO;2-H
32. Gong W, Gohla RM, Bowlin KM, Koyano-Nakagawa N, Garry DJ, Shi X. Kelch repeat and BTB domain containing protein 5 (Kbtbd5) regulates skeletal muscle myogenesis through the E2F1-DP1 complex. J Biol Chem (2015) 290(24):15350–61. doi:10.1074/jbc.M114.629956
33. Zappia MP, Frolov MV. E2F function in muscle growth is necessary and sufficient for viability in Drosophila. Nat Commun (2016) 7:10509. doi:10.1038/ncomms10509
34. Blanchet E, Annicotte JS, Lagarrigue S, Aguilar V, Clapé C, Chavey C, et al. E2F transcription factor-1 regulates oxidative metabolism. Nat Cell Biol (2011) 13(9):1146–52. doi:10.1038/ncb2309
35. Hansen JB, Jørgensen C, Petersen RK, Hallenborg P, De Matteis R, Bøye HA, et al. Retinoblastoma protein functions as a molecular switch determining white versus brown adipocyte differentiation. Proc Natl Acad Sci U S A (2004) 101(12):4112–7. doi:10.1073/pnas.0301964101
36. Blanchet E, Annicotte JS, Pradelli LA, Hugon G, Matecki S, Mornet D, et al. E2F transcription factor-1 deficiency reduces pathophysiology in the mouse model of Duchenne muscular dystrophy through increased muscle oxidative metabolism. Hum Mol Genet (2012) 21(17):3910–7. doi:10.1093/hmg/dds219
37. Dali-Youcef N, Mataki C, Coste A, Messaddeq N, Giroud S, Blanc S, et al. Adipose tissue-specific inactivation of the retinoblastoma protein protects against diabesity because of increased energy expenditure. Proc Natl Acad Sci U S A (2007) 104(25):10703–8. doi:10.1073/pnas.0611568104
38. Mercader J, Ribot J, Murano I, Feddersen S, Cinti S, Madsen L, et al. Haploinsufficiency of the retinoblastoma protein gene reduces diet-induced obesity, insulin resistance, and hepatosteatosis in mice. Am J Physiol Endocrinol Metab (2009) 297(1):E184–93. doi:10.1152/ajpendo.00163.2009
39. Scimè A, Grenier G, Huh MS, Gillespie MA, Bevilacqua L, Harper ME, et al. Rb and p107 regulate preadipocyte differentiation into white versus brown fat through repression of PGC-1alpha. Cell Metab (2005) 2(5):283–95. doi:10.1016/j.cmet.2005.10.002
40. Petrov PD, Ribot J, López-Mejía IC, Fajas L, Palou A, Bonet ML. Retinoblastoma protein knockdown favors oxidative metabolism and glucose and fatty acid disposal in muscle cells. J Cell Physiol (2016) 231(3):708–18. doi:10.1002/jcp.25121
41. Scimè A, Soleimani VD, Bentzinger CF, Gillespie MA, Le Grand F, Grenier G, et al. Oxidative status of muscle is determined by p107 regulation of PGC-1alpha. J Cell Biol (2010) 190(4):651–62. doi:10.1083/jcb.201005076
42. Ciavarra G, Zacksenhaus E. Rescue of myogenic defects in Rb-deficient cells by inhibition of autophagy or by hypoxia-induced glycolytic shift. J Cell Biol (2010) 191(2):291–301. doi:10.1083/jcb.201005067
43. Sankaran VG, Orkin SH, Walkley CR. Rb intrinsically promotes erythropoiesis by coupling cell cycle exit with mitochondrial biogenesis. Genes Dev (2008) 22(4):463–75. doi:10.1101/gad.1627208
44. Nicolay BN, Danielian PS, Kottakis F, Lapek JD Jr, Sanidas I, Miles WO, et al. Proteomic analysis of pRb loss highlights a signature of decreased mitochondrial oxidative phosphorylation. Genes Dev (2015) 29(17):1875–89. doi:10.1101/gad.264127.115
45. Váraljai R, Islam AB, Beshiri ML, Rehman J, Lopez-Bigas N, Benevolenskaya EV. Increased mitochondrial function downstream from KDM5A histone demethylase rescues differentiation in pRB-deficient cells. Genes Dev (2015) 29(17):1817–34. doi:10.1101/gad.264036.115
46. Font-Burgada J, Sun B, Karin M. Obesity and cancer: the oil that feeds the flame. Cell Metab (2016) 23(1):48–62. doi:10.1016/j.cmet.2015.12.015
47. Haim Y, Blüher M, Slutsky N, Goldstein N, Klöting N, Harman-Boehm I, et al. Elevated autophagy gene expression in adipose tissue of obese humans: a potential non-cell-cycle-dependent function of E2F1. Autophagy (2015) 11(11):2074–88. doi:10.1080/15548627.2015.1094597
48. Choi Y, Jang S, Choi MS, Ryoo ZY, Park T. Increased expression of FGF1-mediated signaling molecules in adipose tissue of obese mice. J Physiol Biochem (2016) 72(2):157–67. doi:10.1007/s13105-016-0468-6
49. Kim S, Jin Y, Choi Y, Park T. Resveratrol exerts anti-obesity effects via mechanisms involving down-regulation of adipogenic and inflammatory processes in mice. Biochem Pharmacol (2011) 81(11):1343–51. doi:10.1016/j.bcp.2011.03.012
50. Moreno-Navarrete JM, Petrov P, Serrano M, Ortega F, García-Ruiz E, Oliver P, et al. Decreased RB1 mRNA, protein, and activity reflect obesity-induced altered adipogenic capacity in human adipose tissue. Diabetes (2013) 62(6):1923–31. doi:10.2337/db12-0977
51. Haim Y, Blüher M, Konrad D, Goldstein N, Klöting N, Harman-Boehm I, et al. ASK1 (MAP3K5) is transcriptionally upregulated by E2F1 in adipose tissue in obesity, molecularly defining a human dys-metabolic obese phenotype. Mol Metab (2017) 6(7):725–36. doi:10.1016/j.molmet.2017.05.003
52. Lim CA, Yao F, Wong JJ, George J, Xu H, Chiu KP, et al. Genome-wide mapping of RELA(p65) binding identifies E2F1 as a transcriptional activator recruited by NF-kappaB upon TLR4 activation. Mol Cell (2007) 27(4):622–35. doi:10.1016/j.molcel.2007.06.038
53. Warg LA, Oakes JL, Burton R, Neidermyer AJ, Rutledge HR, Groshong S, et al. The role of the E2F1 transcription factor in the innate immune response to systemic LPS. Am J Physiol Lung Cell Mol Physiol (2012) 303(5):L391–400. doi:10.1152/ajplung.00369.2011
54. Kenchaiah S, Evans JC, Levy D, Wilson PW, Benjamin EJ, Larson MG, et al. Obesity and the risk of heart failure. N Engl J Med (2002) 347(5):305–13. doi:10.1056/NEJMoa020245
55. Vara D, Bicknell KA, Coxon CH, Brooks G. Inhibition of E2F abrogates the development of cardiac myocyte hypertrophy. J Biol Chem (2003) 278(24):21388–94. doi:10.1074/jbc.M212612200
56. Zhang L, Mori J, Wagg C, Lopaschuk GD. Activating cardiac E2F1 induces up-regulation of pyruvate dehydrogenase kinase 4 in mice on a short term of high fat feeding. FEBS Lett (2012) 586(7):996–1003. doi:10.1016/j.febslet.2012.02.027
57. Hsieh MC, Das D, Sambandam N, Zhang MQ, Nahlé Z. Regulation of the PDK4 isozyme by the Rb-E2F1 complex. J Biol Chem (2008) 283(41):27410–7. doi:10.1074/jbc.M802418200
58. Wang K, Zhou LY, Wang JX, Wang Y, Sun T, Zhao B, et al. E2F1-dependent miR-421 regulates mitochondrial fragmentation and myocardial infarction by targeting Pink1. Nat Commun (2015) 6:7619. doi:10.1038/ncomms8619
59. Wu M, Zhou J, Cheng M, Boriboun C, Biyashev D, Wang H, et al. E2F1 suppresses cardiac neovascularization by down-regulating VEGF and PlGF expression. Cardiovasc Res (2014) 104(3):412–22. doi:10.1093/cvr/cvu222
60. Musso G, Gambino R, Cassader M. Cholesterol metabolism and the pathogenesis of non-alcoholic steatohepatitis. Prog Lipid Res (2013) 52(1):175–91. doi:10.1016/j.plipres.2012.11.002
61. Lee Y, Dominy JE, Choi YJ, Jurczak M, Tolliday N, Camporez JP, et al. Cyclin D1-Cdk4 controls glucose metabolism independently of cell cycle progression. Nature (2014) 510(7506):547–51. doi:10.1038/nature13267
62. Zhang Y, Xu N, Xu J, Kong B, Copple B, Guo GL, et al. E2F1 is a novel fibrogenic gene that regulates cholestatic liver fibrosis through the Egr-1/SHP/EID1 network. Hepatology (2014) 60(3):919–30. doi:10.1002/hep.27121
63. Lai Q, Giralt A, Le May C, Zhang L, Cariou B, Denechaud PD, et al. E2F1 inhibits circulating cholesterol clearance by regulating Pcsk9 expression in the liver. JCI Insight (2017) 2(10). doi:10.1172/jci.insight.89729
64. Descamps OS, Fraass U, Dent R, Marz W, Gouni-Berthold I. Anti-PCSK9 antibodies for hypercholesterolaemia: Overview of clinical data and implications for primary care. Int J Clin Pract (2017) 71:e12979.
65. Lu Z, Marcelin G, Bauzon F, Wang H, Fu H, Dun SL, et al. pRb is an obesity suppressor in hypothalamus and high-fat diet inhibits pRb in this location. EMBO J (2013) 32(6):844–57. doi:10.1038/emboj.2013.25
66. Pavlova NN, Thompson CB. The emerging hallmarks of cancer metabolism. Cell Metab (2016) 23(1):27–47. doi:10.1016/j.cmet.2015.12.006
67. Fendt SM. Is there a therapeutic window for metabolism-based cancer therapies? Front Endocrinol (2017) 8:150. doi:10.3389/fendo.2017.00150
68. Floter J, Kaymak I, Schulze A. Regulation of metabolic activity by p53. Metabolites (2017) 7(2). doi:10.3390/metabo7020021
69. Stine ZE, Walton ZE, Altman BJ, Hsieh AL, Dang CV. MYC, metabolism, and cancer. Cancer Discov (2015) 5(10):1024–39. doi:10.1158/2159-8290.CD-15-0507
70. Liberti MV, Locasale JW. The Warburg Effect: how does it benefit cancer cells? Trends Biochem Sci (2016) 41(3):211–8. doi:10.1016/j.tibs.2016.01.004
71. Darville MI, Antoine IV, Mertens-Strijthagen JR, Dupriez VJ, Rousseau GG. An E2F-dependent late-serum-response promoter in a gene that controls glycolysis. Oncogene (1995) 11(8):1509–17.
72. Fernandez de Mattos S, Lam EW, Tauler A. An E2F-binding site mediates the activation of the proliferative isoform of 6-phosphofructo-2-kinase/fructose-2,6-bisphosphatase by phosphatidylinositol 3-kinase. Biochem J (2002) 368(Pt 1):283–91. doi:10.1042/bj20020622
73. Wu M, Seto E, Zhang J. E2F1 enhances glycolysis through suppressing Sirt6 transcription in cancer cells. Oncotarget (2015) 6(13):11252–63. doi:10.18632/oncotarget.3594
74. Zhong L, D’Urso A, Toiber D, Sebastian C, Henry RE, Vadysirisack DD, et al. The histone deacetylase Sirt6 regulates glucose homeostasis via Hif1alpha. Cell (2010) 140(2):280–93. doi:10.1016/j.cell.2009.12.041
75. Wang LY, Hung CL, Chen YR, Yang JC, Wang J, Campbell M, et al. KDM4A coactivates E2F1 to regulate the PDK-dependent metabolic switch between mitochondrial oxidation and glycolysis. Cell Rep (2016) 16(11):3016–27. doi:10.1016/j.celrep.2016.08.018
76. Goto Y, Hayashi R, Kang D, Yoshida K. Acute loss of transcription factor E2F1 induces mitochondrial biogenesis in HeLa cells. J Cell Physiol (2006) 209(3):923–34. doi:10.1002/jcp.20802
77. Tan PY, Chang CW, Duan K, Poidinger M, Ng KL, Chong YS, et al. E2F1 orchestrates transcriptomics and oxidative metabolism in Wharton’s Jelly-derived mesenchymal stem cells from growth-restricted infants. PLoS One (2016) 11(9):e0163035. doi:10.1371/journal.pone.0163035
78. Chen CL, Uthaya Kumar DB, Punj V, Xu J, Sher L, Tahara SM, et al. NANOG metabolically reprograms tumor-initiating stem-like cells through tumorigenic changes in oxidative phosphorylation and fatty acid metabolism. Cell Metab (2016) 23(1):206–19. doi:10.1016/j.cmet.2015.12.004
79. Mori K, Uchida T, Fukumura M, Tamiya S, Higurashi M, Sakai H, et al. Linkage of E2F1 transcriptional network and cell proliferation with respiratory chain activity in breast cancer cells. Cancer Sci (2016) 107(7):963–71. doi:10.1111/cas.12953
80. Gemin A, Sweet S, Preston TJ, Singh G. Regulation of the cell cycle in response to inhibition of mitochondrial generated energy. Biochem Biophys Res Commun (2005) 332(4):1122–32. doi:10.1016/j.bbrc.2005.05.061
81. Espada L, Meo-Evoli N, Sancho P, Real S, Fabregat I, Ambrosio S, et al. ROS production is essential for the apoptotic function of E2F1 in pheochromocytoma and neuroblastoma cell lines. PLoS One (2012) 7(12):e51544. doi:10.1371/journal.pone.0051544
82. Raimundo N, Song L, Shutt TE, McKay SE, Cotney J, Guan MX, et al. Mitochondrial stress engages E2F1 apoptotic signaling to cause deafness. Cell (2012) 148(4):716–26. doi:10.1016/j.cell.2011.12.027
83. Lopez-Mejia IC, Fajas L. Cell cycle regulation of mitochondrial function. Curr Opin Cell Biol (2015) 33:19–25. doi:10.1016/j.ceb.2014.10.006
84. Benevolenskaya EV, Frolov MV. Emerging links between E2F control and mitochondrial function. Cancer Res (2015) 75(4):619–23. doi:10.1158/0008-5472.CAN-14-2173
85. Li Y, Slansky JE, Myers DJ, Drinkwater NR, Kaelin WG, Farnham PJ. Cloning, chromosomal location, and characterization of mouse E2F1. Mol Cell Biol (1994) 14(3):1861–9. doi:10.1128/MCB.14.3.1861
86. Slansky JE, Li Y, Kaelin WG, Farnham PJ. A protein synthesis-dependent increase in E2F1 mRNA correlates with growth regulation of the dihydrofolate reductase promoter. Mol Cell Biol (1993) 13(3):1610–8. doi:10.1128/MCB.13.3.1610
87. Beloribi-Djefaflia S, Vasseur S, Guillaumond F. Lipid metabolic reprogramming in cancer cells. Oncogenesis (2016) 5:e189. doi:10.1038/oncsis.2015.49
88. Pascual G, Avgustinova A, Mejetta S, Martín M, Castellanos A, Attolini CS, et al. Targeting metastasis-initiating cells through the fatty acid receptor CD36. Nature (2017) 541(7635):41–5. doi:10.1038/nature20791
89. Bhatia B, Hsieh M, Kenney AM, Nahlé Z. Mitogenic Sonic hedgehog signaling drives E2F1-dependent lipogenesis in progenitor cells and medulloblastoma. Oncogene (2011) 30(4):410–22. doi:10.1038/onc.2010.454
90. Saxton RA, Sabatini DM. mTOR signaling in growth, metabolism, and disease. Cell (2017) 169(2):361–71. doi:10.1016/j.cell.2017.03.035
91. Meo-Evoli N, Almacellas E, Massucci FA, Gentilella A, Ambrosio S, Kozma SC, et al. V-ATPase: a master effector of E2F1-mediated lysosomal trafficking, mTORC1 activation and autophagy. Oncotarget (2015) 6(29):28057–70. doi:10.18632/oncotarget.4812
92. Polager S, Ofir M, Ginsberg D. E2F1 regulates autophagy and the transcription of autophagy genes. Oncogene (2008) 27(35):4860–4. doi:10.1038/onc.2008.117
93. Dynlacht BD. Live or let die: E2F1 and PI3K pathways intersect to make life or death decisions. Cancer Cell (2008) 13(1):1–2. doi:10.1016/j.ccr.2007.12.017
94. Moniz S, Bandarra D, Biddlestone J, Campbell KJ, Komander D, Bremm A, et al. Cezanne regulates E2F1-dependent HIF2 alpha expressiond. J Cell Sci (2015) 128(16):3082–93. doi:10.1242/jcs.168864
95. Morzyglod L, Caüzac M, Popineau L, Denechaud PD, Fajas L, Ragazzon B, et al. Growth factor receptor binding protein 14 inhibition triggers insulin-induced mouse hepatocyte proliferation and is associated with hepatocellular carcinoma. Hepatology (2017) 65(4):1352–68. doi:10.1002/hep.28972
96. Franco J, Balaji U, Freinkman E, Witkiewicz AK, Knudsen ES. Metabolic reprogramming of pancreatic cancer mediated by CDK4/6 inhibition elicits unique vulnerabilities. Cell Rep (2016) 14(5):979–90. doi:10.1016/j.celrep.2015.12.094
97. Olmez I, Brenneman B, Xiao A, Serbulea V, Benamar M, Zhang Y, et al. Combined CDK4/6 and mTOR inhibition is synergistic against glioblastoma via multiple mechanisms. Clin Cancer Res (2017). doi:10.1158/1078-0432.CCR-17-0803
98. Chirivella L, Kirstein M, Ferrón SR, Domingo-Muelas A, Durupt FC, Acosta-Umanzor C, et al. Cdk4 regulates adult neural stem cell proliferation and differentiation in response to insulin-IRS2 signals. Stem Cells (2017). doi:10.1002/stem.2694
99. Johnson DG, Ohtani K, Nevins JR. Autoregulatory control of E2F1 expression in response to positive and negative regulators of cell cycle progression. Genes Dev (1994) 8(13):1514–25. doi:10.1101/gad.8.13.1514
100. Fajas L, Blanchet E, Annicotte JS. The CDK4-pRB-E2F1 pathway: a new modulator of insulin secretion. Islets (2010) 2(1):51–3. doi:10.4161/isl.2.1.10338
101. Reeves HL, Zaki MY, Day CP. Hepatocellular carcinoma in obesity, type 2 diabetes, and NAFLD. Dig Dis Sci (2016) 61(5):1234–45. doi:10.1007/s10620-016-4085-6
102. Conner EA, Lemmer ER, Omori M, Wirth PJ, Factor VM, Thorgeirsson SS. Dual functions of E2F-1 in a transgenic mouse model of liver carcinogenesis. Oncogene (2000) 19(44):5054–62. doi:10.1038/sj.onc.1203885
103. Kent LN, Bae S, Tsai SY, Tang X, Srivastava A, Koivisto C, et al. Dosage-dependent copy number gains in E2f1 and E2f3 drive hepatocellular carcinoma. J Clin Invest (2017) 127(3):830–42. doi:10.1172/JCI87583
Keywords: E2F1, cell cycle regulators, cancer metabolism, obesity, metabolic diseases
Citation: Denechaud PD, Fajas L and Giralt A (2017) E2F1, a Novel Regulator of Metabolism. Front. Endocrinol. 8:311. doi: 10.3389/fendo.2017.00311
Received: 04 September 2017; Accepted: 26 October 2017;
Published: 10 November 2017
Edited by:
Timo Dirk Müller, Helmholtz Zentrum München (HZ), GermanyReviewed by:
Daniele Baiz, Plymouth University, United KingdomDario Palmieri, The Ohio State University Columbus, United States
Copyright: © 2017 Denechaud, Fajas and Giralt. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Albert Giralt, YWxiZXJ0LmdpcmFsdGNvbGxAdW5pbC5jaA==