 Joshua P. H. Neale
Joshua P. H. Neale James T. Pearson
James T. Pearson Rajesh Katare
Rajesh Katare Daryl O. Schwenke
Daryl O. Schwenke- 1Department of Physiology-HeartOtago, University of Otago, Dunedin, New Zealand
- 2Department of Cardiac Physiology, National Cerebral and Cardiovascular Center Research Institute, Suita, Japan
- 3Biomedicine Discovery Institute and Department of Physiology, Monash University, Clayton, VIC, Australia
Critical limb ischemia (CLI) is the most severe manifestation of peripheral artery disease. It is characterized by chronic pain at rest, skin ulcerations, and gangrene tissue loss. CLI is a highly morbid condition, resulting in a severely diminished quality of life and a significant risk of mortality. The primary goal of therapy for CLI is to restore blood flow to the affected limb, which is only possible by surgery, but is inadvisable in up to 50% of patients. This subset of patients who are not candidates for revascularisation are referred to as “no-option” patients and are the focus of investigation for novel therapeutic strategies. Angiogenesis, arteriogenesis and vasculogenesis are the processes whereby new blood vessel networks form from the pre-existing vasculature and primordial cells, respectively. In therapeutic angiogenesis, exogenous stimulants are administered to promote angiogenesis and augment limb perfusion, offering a potential treatment option for “no option” patients. However, to date, very few clinical trials of therapeutic angiogenesis in patients with CLI have reported clinically significant results, and it remains a major challenge. Ghrelin, a 28-amino acid peptide, is emerging as a potential novel therapeutic for CLI. In pre-clinical models, exogenous ghrelin has been shown to induce therapeutic angiogenesis, promote muscle regeneration, and reduce oxidative stress via the modulation of microRNAs (miRs). miRs are endogenous, small, non-coding ribonucleic acids of ~20–22 nucleotides which regulate gene expression at the post-transcriptional level by either translational inhibition or by messenger ribonucleic acid cleavage. This review focuses on the mounting evidence for the use of ghrelin as a novel therapeutic for CLI, and highlights the miRs which orchestrate these physiological events.
Introduction
Peripheral artery disease (PAD) is characterized by the narrowing or occlusion of systemic arteries impeding blood supply to the extremities (1). It is rapidly emerging as a significant global socio-economic burden and currently affects over 202 million people (2). PAD is associated with significant cardiovascular morbidity and mortality (3), plus a severely diminished quality of life (4). PAD is most commonly a result of atherosclerosis and, as such, risk factors for the development of the disease include smoking, diabetes mellitus (DM), hypertension (5), and advanced age (6). With the number of people being diagnosed with DM expected to surpass 640 million by 2040 (7) and the acceleration of global population aging (8), the PAD burden is expected to rapidly increase (2).
At the severest end of the PAD spectrum is critical limb ischemia (CLI), which describes patients with chronic pain at rest, skin ulcerations, and gangrene tissue loss (3). CLI accounts for 11% of patients with PAD, increasing to 20% in patients over 70 years of age (9). The prognosis of CLI is very poor; at 6 months post-diagnosis, patients have a 25–40% chance of lower limb amputation and ~20% chance of mortality (10). The quality of life for CLI patients is also severely diminished, similar to those with terminal cancer (11). Management of CLI includes risk factor modification and aggressive pharmacological therapy, accompanied with either an endovascular or surgical intervention (12, 13). However, revascularisation as a primary treatment approach is expensive, labour-intensive, and is often met with poor success (14). Unfortunately, up to 50% of CLI patients are not candidates for surgery due to extreme tissue damage, diffuse atherosclerotic disease, and co-morbidities (15, 16). This subgroup of patients is referred as “no-option” patients and is the focus of research for novel therapies.
Therapeutic angiogenesis is emerging as a potential treatment approach for “no-option” CLI patients by stimulating neovascularization, improving limb perfusion, and aiding tissue regeneration (17). Angiogenic agents, such as gene or cell therapy, have been the focus of investigation, with the aim of inducing a pro-angiogenic milieu in the affected ischemic limb. Regrettably, the results from clinical trials using such agents have shown little clinical benefit regarding primary outcome measures (i.e., patency, amputation-free survival, major limb adverse effects) (18–20). This highlights both the complexity of therapeutic angiogenesis and the need to develop new agents for the management of CLI.
The 28-amino acid peptide hormone ghrelin, first discovered in 1999 as the endogenous ligand for the growth hormone secretagogue receptor (GHS-R) (21), has recently been proposed as a novel therapeutic for CLI (22–24). The GHS-R is a G-protein coupled receptor, ubiquitous throughout the cardiovascular and autonomic nervous systems (25, 26), which plausibly accounts for the diverse range of effects ghrelin has on cardiovascular function (25, 26).
This review presents the concepts of therapeutic angiogenesis and highlights the limitations associated with advancing current treatments for CLI. Furthermore, the emergence of ghrelin as a novel therapeutic for CLI is explored and the molecular mechanisms that underpin ghrelin’s beneficial actions highlighted.
Pathophysiology of CLI
The pathological events which lead to the presentation of CLI are multifactorial, complex, and beyond the scope of this review [reviewed in Ref. (24)]. The underlying pathological events leading to CLI are macro- and microvascular circulation defects, resulting in diminished arterial perfusion. Consequently, the metabolic requirements of the distal tissue outweigh the oxygen and nutrient supply. Although the aetiology of CLI can be vasculitis, thromboembolic disease, trauma, popliteal entrapment, cystic adventitial disease, thromboangiitis, and Buerges disease, it is most commonly associated with diffuse atherosclerosis (27, 28). The compensatory mechanisms against initial ischemia involve angiogenesis and arteriogenesis to increase blood flow to the affected tissue. However, these compensatory mechanisms are ineffective in CLI. Distal arterioles respond to ischemia by decreasing wall thickness, maximally vasodilating, subsequently becoming unresponsive to provasodilatory stimuli; a term referred to as vasomotory paralysis (29). Moreover, arterioles exhibit an inability to control vascular resistance as a result of blunted myogenic autoregulation (29). Combined, these factors lead to an orthostatic-dependent increase in hydrostatic pressure in the distal part of the limb, resulting in the development of oedema (30). Furthermore, chronic inflammation and the production of free radicals further exacerbate endothelial dysfunction. Endothelial damage, inappropriate platelet activation, and leukocyte adhesion contribute to a microthrombi formation, distal oedema, and tissue damage.
Therapeutic Angiogenesis
Angiogenesis is a precisely orchestrated process of events which is essential for optimizing or restoring organ perfusion. Artificially cultivating this complex process of events is the ultimate goal of therapeutic angiogenesis (31). Therapeutic angiogenesis is essentially the promotion of new blood vessel growth in an ischemic tissue bed to supply a local demand for oxygen and aid tissue recovery. This new vasculature can be induced by three distinct means: angiogenesis, arteriogenesis, and vasculogenesis. Angiogenesis is defined as the formation of new capillaries from the pre-existing vasculature (32). It can occur by two distinct mechanisms: intussusception and sprouting (33–35). Intussusception defines the process in which transluminal tissue pillars develop in the capillaries resulting in the vessel splitting, thus creating new vasculature entries and increasing the vascular network (35). Sprouting angiogenesis is the growth of new capillary vessels from a pre-existing vessel and occurs in the following stages: proteolytic degradation of extracellular matrix, endothelial cell migration and proliferation, tubulogenesis, vessel fusion, vessel pruning, and functional maturation (32). Arteriogenesis is characterized by the growth of collateral arteries, in addition to the remodeling and enlargement of the pre-existing arterioles (36, 37). Finally, vasculogenesis is defined as the de novo formation of a primitive vasculature from endothelial precursor cells, which is traditionally considered only to take place during embryogenesis (38). These three events, which to some extent occur naturally in response to ischemia, are influenced by genetic factors but are also the targets of therapeutic agents.
Therapeutic Potential of Angiogenic Factors
In the Western nations, it is estimated that over 300 million people may benefit from a pro-angiogenic therapy (39). Despite considerable research to develop a pro-angiogenic agent for the effective treatment of CLI, no therapeutic agents have been approved by the Medicines and Healthcare Products Regulatory Agency of the United Kingdom or the Food and Drug Administration of the United States of America. However, in Russia, a plasmid DNA gene product encoding vascular endothelial growth factor (VEGF) 165 “Neovasculgen” (40) is approved for clinical use in the treatment of PAD. While this agent is approved in Russia, a recent meta-analysis of randomized control trials shows no consistent benefit of any gene therapy for promoting therapeutic angiogenesis in PAD (20).
Extensive research has identified numerous pro-angiogenic agents for CLI (Table 1 contains a partial list of the studies). However, to date, there is a clear disparity between pre-clinical trials and large randomized control trials. Few clinical trials report long-term positive effects or clinically significant results (18, 41). These disappointing findings with current angiogenic agents can be attributed to numerous factors, including suboptimal delivery strategies (42), disease mediated dysfunction (43), methodological differences, and pre-clinical models which lack common CLI risk factors (41). To ensure the future success of therapeutic angiogenesis, it is imperative that clinical methods are standardized (i.e., angiogenic agent selection, delivery method, in vitro tracking) and focus on more applicable pre-clinical models which better mimic human CLI. Despite potentially promising outcomes in the early stages of some pre-clinical studies, current angiogenic agents had limited long-term success. Due to this limited success, hormone therapy is emerging as a novel treatment strategy.
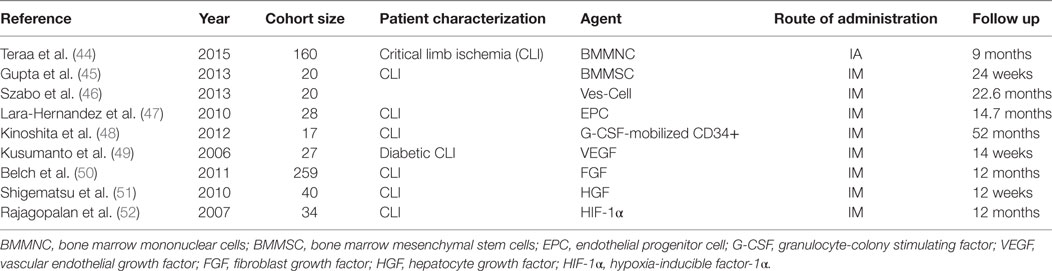
Table 1. Factors with an angiogenic potential.
Ghrelin
The hormone ghrelin was first isolated from rat stomach in 1999 and was subsequently identified as the endogenous ligand for the GHS-R type 1a (21). Ghrelin is predominantly produced by P/D1-like (X/A-like cells in the mouse) cells in the oxyntic glands of the gastric mucosa, but is also expressed in a smaller amount in other tissues including the myocardium, arteries, and veins (25, 26). Ghrelin circulates in two distinct forms: acylated ghrelin (AG) and des-acylated ghrelin (Des-AG). AG possesses a novel octanoylation of serine-3, promoted by ghrelin O-acyltransferase (Figure 1). This is considered essential for facilitating the binding of the peptide to the GHS-R and eliciting its physiological functions (53). Des-AG lacks GH-secretagogue activity and does not bind to the GHS-R type 1a. The mechanisms by which Des-AG exerts its biological functions remain largely unknown, as its target receptor is yet to be identified. Emerging evidence from the literature consistently advocates the therapeutic potential of ghrelin as a novel strategy for the treatment of various metabolic and cardiovascular disorders. These include anorexia, sarcopenia, cardiomyopathy, CLI, inflammatory disorders, metabolic syndrome, epilepsy, and neurodegenerative disorders (54–60).
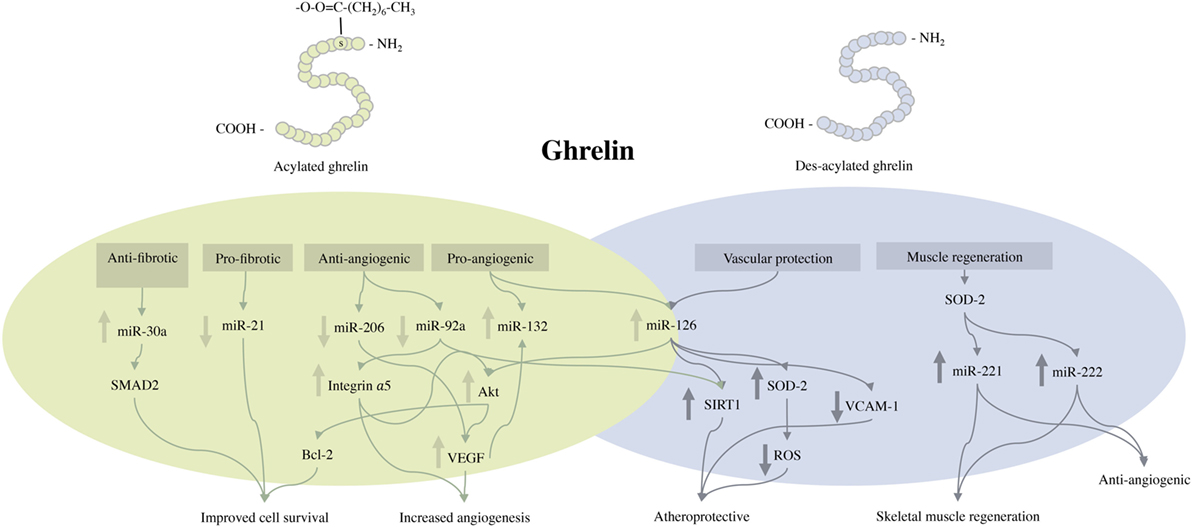
Figure 1. Schematic representation of the structure of acylated and des-acylated ghrelin and the proposed molecular events activated by exogenous ghrelin treatment following critical limb ischemia. miR, indicates microRNA; VEGF, vascular endothelial growth factor; Akt, protein kinase B; Bcl-2, B-cell lymphoma 2; SIRT1, surtuin 1; VCAM-1, vascular cell adhesion molecule 1; SOD-2, superoxide dismutase-2; ROS, reactive oxygen species.
Ghrelin and CLI
Angiogenic Potential
Ghrelin has been shown to have a pro-angiogenic potential in several studies (22, 61–64), but limited potential in others (65–67). Ghrelin’s angiogenic potential was first shown in human microvascular endothelial cells (HMVEC) at a concentration of 0.1 nM. At this concentration, it promoted migration, proliferation, and angiogenesis through phosphorylation and activation of the mitogen activated protein kinases ERK2, which regulates endothelial cell function (63). The same research group has also demonstrated that AG at 1 nM significantly reverses age-related impaired angiogenesis in HMVECs. This is mainly through the activation of the MAPK/ERK2 mitogenic signalling pathway, a central pathway for angiogenesis which promotes endothelial cell motility and survival (62). Using cardiac microvascular endothelial cells, AG at concentrations of 10 nM significantly stimulated proliferation, migration, and angiogenesis in vitro. This was through the GHS-R1a type 1a-mediated kinase MEK and extracellular signal-regulated kinase ERK, in addition to the most commonly studied pro-survival phosphatidylinositol 3-kinase/protein kinase B (PI3K/Akt) pathway, which regulates numerous cellular processes, including cell cycle, angiogenesis, and apoptosis (68). Taken together, these findings indicate exogenous ghrelin administration can activate several independent angiogenesis signalling pathways (61).
In a rat model of chronic myocardial ischemia, following permanent occlusion of the left anterior descending artery, AG (100 µg/kg, twice daily for 4 weeks) increased VEGF expression, inhibited apoptosis, and increased angiogenesis in the myocardial infarct and peri-infarct zones (64). In a murine model of CLI, AG (150 µg/kg, daily for 2 weeks) promoted angiogenesis through the up-regulation of pro-angiogenic microRNAs (miRs)-126 and-132, while preventing the activation of antiangiogenic miRs-92a and -206 (22).
Interestingly, Des-AG (100 µg/kg daily for 2 weeks) also exerted a protective vascular effect in a murine model of CLI, evident from a comparatively similar blood vessel density in the ischemic versus contralateral (non-ischemic) limbs (69). However, Des-AG did not change large vessel perfusion or induce neovascularization, possibly due to its up-regulation of antiangiogenic miRs-221 and -222 (70). This suggests Des-AG may limit adverse changes in the vasculature in response to ischemia, rather than initiate angiogenesis (69, 71).
Although the majority of studies advocate the angiogenic potential of AG, conflicting reports from one research team propose that AG may have antiangiogenic properties. This is based on the observation that AG (10 nM) impeded the in vitro activation of angiogenesis induced by fibroblast growth factor-2 in human umbilical vein endothelial cells (HUVEC), and in rat neuromicrovascular endothelial cells, via inhibition of tyrosine kinase and MAPK pathways (65, 67).
The discrepancy between studies, especially in vitro studies, concerning the angiogenic properties of ghrelin is unquestionably confounded by experimental factors. These include differing cell types, culture conditions, different concentrations, and differing methodological approaches. However, it is clear from in vivo animal models of MI and CLI that ghrelin exerts beneficial effects through vascular protection and neovascularization.
Skeletal Muscle and Peripheral Nerve Regeneration
Impaired perfusion of the lower limbs results in significant atrophy of the calf muscle and increased fat composition, causing severe functional impairment (72). Circulating levels of ghrelin are reduced in diseases of muscle wasting (650 vs. 899 pg/mL) (73). Yet, the therapeutic administration of exogenous ghrelin in a murine model of muscular atrophy induces AktS473 phosphorylation- a direct anti-atrophic signalling pathway in skeletal muscle, blocking skeletal muscle atrophy (74). In murine models of CLI, Des-AG has been shown to induce skeletal muscle regeneration through increased superoxide-dismutase-2 (SOD-2) induced expressions of miRs-221 and-222 (69). AG has been demonstrated to significantly up-regulate anti-fibrotic miR-30a, inhibit pro-fibrotic miR-21, and inhibit miR-206 resulting in increased proliferation of myocytes and aiding in tissue repair (22).
In patients who have DM as a comorbidity with CLI, peripheral neuropathy commonly leads to a delayed clinical presentation of the disease, resulting in severe muscular and vascular damage (75). Several studies have reported that AG administration (300 nmol/kg) ameliorated polyneuropathy in rodent models of diabetic polyneuropathy, evident by increased nerve conduction velocities and temperature sensation (76, 77). Although the mechanisms by which ghrelin alleviates polyneuropathy remain to be identified, these findings further support ghrelin as a potential treatment option for an otherwise intractable disorder. Recently, these findings have also been confirmed in a human study (78), with ghrelin treated patients (1.0 µg/kg for 14 days) having increased motor nerve conduction velocity of the posterior tibial nerve compared with controls. Total symptom score also significantly improved in the treated group compared to controls, suggesting that ghrelin may be a novel therapeutic option for diabetic polyneuropathy. However, a double-blind, placebo-controlled trial is needed in the future.
The results from these studies demonstrate that ghrelin acts to protect against skeletal muscle atrophy, neuropathy, and importantly, aids in muscle regeneration.
Anti-inflammatory and Antioxidant Actions
The diminished supply of blood to the periphery in CLI and the highly oxidative environment result in severe skeletal muscle damage. SOD-2 is the main antioxidant defence against reactive oxygen species (ROS) production and has been shown to be inhibited in skeletal muscle affected by CLI (79). The two isoforms of ghrelin, AG and Des-AG, have been shown to exert potent anti-inflammatory and antioxidant effects throughout the vasculature.
AG has been shown to exhibit anti-apoptotic effects through the strong activation of Akt and ERK, mediated by the PI3K and MAPK pathways (80), and to inhibit pro-inflammatory cytokine production in vitro through the inhibition of nuclear factor-kappa B (81). AG has been shown to inhibit apoptosis, and blunt ROS production in HUVECs treated with high glucose, replicating diabetic vascular complications, via the PI3K/Akt pathway (82). AG (106 M) inhibits advanced glycation end products-mediated cell apoptosis via the ERK1/2 and PI3K/Akt pathways (83). Moreover, ghrelin has been shown to inhibit vascular oxidative stress through inhibition of vascular NAD(P)H oxidases (84).
Des-acylated ghrelin has been shown to protect against oxidative stress-induced apoptosis through the class III histone deacetylase sirtuin (SIRT1), a nicotinamide adenine dinucleotide dependent histone/protein deacetylase signalling pathway (85). Activation of this pathway has been shown to reduce apoptosis and protects endothelial progenitor cells from ROS-mediated damage under conditions of diabetes (86, 87). In a murine model of CLI and glucose intolerance, Des-AG was also demonstrated to rescue miR-126 expression, a regulator SIRT1 and SOD-2, leading to improved oxidative stress levels and improved recovery (71).
Dysregulation of Ghrelin in Atherosclerosis
Atherosclerosis underlies most of CLI. Circulating ghrelin concentrations have been shown to be reduced in several atherosclerotic diseases (88–92). To date, the circulating ghrelin levels in patients with PAD or CLI remain unknown. However, in diseases with a similar aetiology, ghrelin levels have been shown to be dysregulated. These include patients with unstable angina (1.04 ± 0.08 ng/mL), acute myocardial infarction (1.07 ± 0.11 ng/mL), and asymptomatic coronary artery disease (2.1 ± 0.8 ng/mL), which have all been shown to have lower total ghrelin concentrations compared to controls (6.2 ± 4.1 ng/mL) (88). In addition to total ghrelin, low circulating levels of Des-AG (~78.2 fmol/mL) have been shown to be associated with increased risk of cardiovascular events in older hypertensive individuals (89). However, further studies are warranted to elucidate the pathogenic mechanisms underlying this association, giving clarity to the clinical value of the measure. In contrast, high plasma concentration of ghrelin has been shown to protect against coronary heart disease (90), and AG is significantly higher in patients with ischemic heart disease (coronary artery disease) compared to controls (valvular heart disease; 32 ± 3 vs. 16 ± 2 pg/mL) (91), potentially demonstrating a role for ghrelin in auto- and paracrine self-protective mechanisms of the ischemic heart. In addition, total circulating ghrelin concentrations are decreased in patients with acute ischemic stroke compared to age- and sex-matched controls (3.47 ± 1.44 vs. 5.93 ± 2.78 ng/mL) (92).
In a murine model of atherosclerosis, AG (10−9 mol/kg/day for 4 weeks) has also been shown to reduce the formation of atherosclerotic lesions, increase plaque stability, ameliorate activation of endoplasmic reticulum stress, and decrease intima-media thickness (93, 94).
Collectively, this evidence from the literature suggests that plasma ghrelin concentration may be a novel prognostic marker of the extent and severity of various forms of atherosclerotic disease. Additionally, ghrelin may be a promising adjunct therapy for the treatment of CLI and the underlying mechanisms of CLI, atherosclerosis (95).
Vascular Calcification Reduction
Vascular calcification is characterized by the progressive enlargement of calcium deposits in the major arteries, and is an independent risk factor for CLI (96). Serum ghrelin level decreases with the severity of the tibial artery stenosis in diabetic patients (mild stenosis, 167.71 ± 16.73; moderate stenosis, 105.72 ± 10.51; severe stenosis/occlusion, 53.11 ± 5.65 ng/mL) (97). In vivo and in vitro models of vascular calcification consistently demonstrate that ghrelin peptide levels and mRNA expression are significantly reduced during calcification. However, treatment with exogenous AG (10−8–10−5 mol/L) effectively attenuates the severity of this calcification (97–99). This attenuation is likely mediated through the osteoprotegerin, receptor activator of nuclear factor-kappa B ligand, and the receptor activator of NF-κB axis which regulates vascular calcification.
Combined, it is shown that serum ghrelin may be a novel predictor of vascular calcification in diabetic patients and that exogenous administration of ghrelin may be an effective agent in attenuating calcification.
The Influence of Ghrelin on MicroRNAs (miRs)
MicroRNAs are endogenous, small, non-coding RNAs of ~20–22 nucleotides which regulate gene expression at the post-transcriptional level, by either translational inhibition or by messenger ribonucleic acid cleavage (100, 101). So far, 2,588 mature human miRs have been registered at miRBase in release 21,1 which are believed to regulate the expression of 30~50% of human genes (102). Since their discovery in 1993 (103, 104), miRs have gained considerable interest as key modulators in a range of pathological and physiological events (105, 106). A single miR can regulate a plethora of targets and, in doing so, evoke a complex multifactorial physiological process (106). miRs are incredibly stable in circulation due to their transport in small membrane vesicles (exosomes and microvesicles) (107, 108) and, therefore, show promise as clinical biomarkers of disease (105, 109–112). Numerous miRs have been shown to be differentially expressed between PAD/CLI patients and control subjects (113–116). However, the functional significance of these miRs is yet to be truly elucidated. In addition to being biomarkers of disease, miRs have also been shown to be modulated by therapeutic agents.
In murine models of CLI, ghrelin administration has been shown to regulate a variety of miRs (Table 2), leading to a multitude of physiological responses (22, 69). Figure one summarizes the beneficial effects ghrelin has on the expression of several miRs, with the preceding section describing each miR in detail.
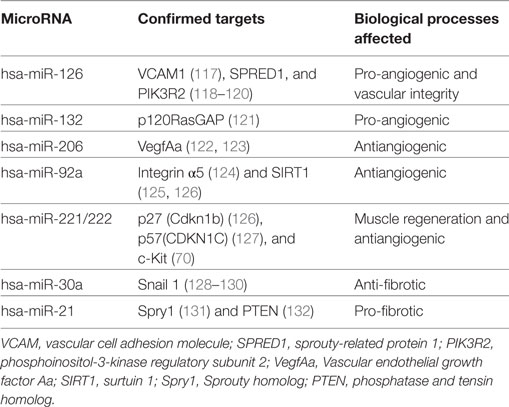
Table 2. Overview of the confirmed targets for micoRNAs regulated by ghrelin.
MicroRNA-126
AG has been shown to induce post-ischemic angiogenesis through the up-regulation of miR-126 (22), the most characterized and abundant pro-angiogenic miR in endothelial cells [reviewed in Ref. (117)]. miR-126 resides within intron seven of the epidermal growth factor-like domain, multiple 7 gene on chromosome 9q34.3, and gives rise to two mature miRs; miR-126-3p and miR-126-5p. miR-126-3p has been well studied for its role in vascular remodeling [reviewed in Ref. (133)]. miR-126 is essential for blood vessel growth in zebrafish (118) and mice (119). Specific deletion of miR-126 is 50% embryonic lethal, with surviving mutants expressing fragile, leaky vessels, and severe haemorrhage (120, 134). These findings highlight the fundamental role of miR-126 in regulation of vascular integrity and angiogenesis. Consequently, this fundamental role of miR-126 in the vasculature has led to research investigating its regenerative potential, which was confirmed in several animal models of ischemic vascular injury (135, 136, 137).
Mechanistically, miR-126 targets sprouty-related protein 1 (SPRED1) and phosphoinositol-3-kinase regulatory subunit 2 (PIK3R2/p85β), both of which suppress VEGF, via PI3K and Akt signalling pathways, respectively. Thus, miR-126 promotes VEGF expression by targeting several pathways, which promotes an angiogenic response (118).
Des-acylated ghrelin has been shown to exhibit an atheroprotective effect by restoring miR-126 expression. In a murine model of CLI, Des-AG administration increased miR-126, which increased SIRT1 expression and prevented cell senescence via reduced p53 and H3K56 acetylation, protecting deoxyribonucleic acids from damage (71, 138). Furthermore, Des-AG induced an anti-adhesive and anti-inflammatory endothelial phenotype, via miR-126 regulated VCAM-1 post-transcriptional regulation (71). Finally, Des-AG administration also restored SOD-2 expression by diminishing ROS production (71).
MicroRNA-132
MicroRNA-132 Is a Highly Conserved miR, Encoded in an Intergenic Region on Chromosome
17p13.3 by the transcription factor cAMP response element-binding protein (CREB) (121). Angiogenic factors such as VEGF and basic fibroblast growth factor lead to phosphorylation of CREB and rapid transcription of miR-132. This transcription suppresses the endothelial GTPase-activating protein p120RasGAP, resulting in Ras activation, and subsequent endothelial cell proliferation and angiogenesis (139). In a murine model of CLI, miR-132/221cluster knockout has been shown to delay perfusion recovery, attributable to the modulation of the Ras-MAPK signalling pathway, a key pathway in neovascularization following ischemia. Katare et al. (22), demonstrated exogenous AG treatment significantly up-regulated miR-132, resulting in angiogenesis in a murine model of CLI, while in vitro inhibition of miR-132 reduced the angiogenic potential of AG, evident from decreased proliferation, tube formation, and survival of HUVECs.
MicroRNA-206
MicroRNA-206 is an evolutionally conserved miR, sharing common expression in muscle from Caenorhabditis elegans to human (122). miR-206 is also similar to miR-1, only differing by four nucleotides as mature miRs. miR-206 is found in the intergenic region on chromosome 6p12.2 and has been proposed to be a negative regulator of angiogenesis. This is shown to be through the modulation of the potent angiogenic factor VEGF (122, 123). Further, miR-206 has been shown to specifically suppress VEGF expression in several types of cancer and smooth muscle cells (140), clearly demonstrating the role of miR-206 as a negative regulator of angiogenesis. AG has been shown to suppress miR-206 expression, leading to increased VEGF expression and angiogenesis (22). This indicates that AG up-regulates several pro-angiogenic miRs and down-regulates antiangiogenic miRs leading to a pro-angiogenic environment.
MicroRNA-92a
MicroRNA-92a is a conserved endothelial cell-specific miR, in the miR-17~92 cluster located at chromosome 13q31.3 (124). miR-92a inhibits endothelial cell sprouting and neovascularization following ischemia, through targeting of the protective endothelial genes integrin α5 and SIRT1 (124, 141). Inhibition of miR-92a has been shown to improve angiogenesis and recovery in murine models of chronic ligation of the anterior descending coronary artery (124), CLI (124), vascular injury (142), and in a porcine model of ischemia/reperfusion injury (143).
Exogenous AG treatment has also shown to decrease miR-92a, which negatively modulates integrin α5, which is crucial for the activation of Akt (144). This leads to improved cell survival through the anti-apoptotic factor B cell lymphoma 2 (145) and improved angiogenesis. Combined, ghrelin’s ability to reduce miR-92a expression leads to further regulation of integrin α5 and SIRT1, both of which critically influence endothelial cell proliferation and migration (146).
MicroRNA-221/-222 Cluster
The miR-221/-222 cluster is encoded by a highly conserved gene cluster on the human Xp11.3 chromosome (147). miR-221/-222 have been shown to control the differentiation and maturation of skeletal muscle cells, by modulating the protein levels of the cell-cycle inhibitors p27 and p57 (126). Des-AG has been shown to induce skeletal muscle regeneration via SOD-2 expression and miR-221/-222 driven post-transcriptional regulation of the Cip/Kip family members P27KIP1 (also known as CDKN1B) and P57KIP2 (also known as CDKN1C) (127), leading to satellite cell differentiation and myofiber regeneration (69). This indicates Des-AG may benefit CLI in that it can up-regulate miR-221/-222, leading to skeletal muscle regeneration. However, miR-221/-222 inhibits endothelial cell proliferation and motility, thus blocking angiogenesis by targeting the pro-angiogenic c-kit (70, 147), suggesting that Des-AG may improve the skeletal muscle recovery following CLI, but at the same time may have detrimental effects on the neovasculature.
MicroRNA-30a
MicroRNA-30a is a member of the miR-30 family, located on chromosome 6q13 (148). miR-30a has recently been shown to play a fundamental role in myocardial (129), peritoneal (130), and hepatic fibrosis (149) through the negative regulation of its target protein Snail 1. AG has been shown to increase miR-30a in a model of CLI, leading to diminished skeletal muscle fibrosis and improved recovery (22).
MicroRNA-21
MicroRNA-21 is a well characterized pro-fibrotic miR [reviewed in Ref. (150)], located on chromosome 17q23.2, within an intron of the transmembrane protein 49 gene (151). miR-21 promotes fibrosis in several tissues including the heart (132, 151), kidneys (150), and skeletal muscle (152) through fibroblast proliferation by activation of ERK–MAPK pathway (131). Sprouty homolog 1 (SPRY1), a potent inhibitor of the Ras/MEK/ERK pathway, has been shown to be a direct target of miR-21 (131, 153). Thus, miR-21 mediates ERK–MAP kinase activity by means of an effect on SPRY1, leading to fibrosis. Moreover, phosphatase and tensin homolog (PTEN) a well-established target of miR-21, is involved in fibrosis by stimulation of the epithelial-to-mesenchymal transition (132) and interrupts the down- stream activation of Akt, a regulator in fibrotic diseases. Pro-fibrotic TGF-β signalling, which contributes to the progression of fibrosis by promoting fibroblasts activation, has been shown to be regulated by miR-21 via a PTEN/Akt-dependent pathway (154). AG has been demonstrated to reduce miR-21 in a model of CLI, thus reducing the TGF-β induced fibrotic signalling cascade (22), leading to decreased fibrosis and improved recovery.
AG and Des-AG exhibit pleiotropic beneficial effects, which all may contribute to improved recovery following CLI. The influence of ghrelin on miRs sheds light on the molecular mechanisms that underpin ghrelin’s therapeutic actions.
Ghrelin as a Novel Therapeutic Agent
Mounting evidence advocates ghrelin as a novel therapeutic candidate for the treatment of CLI, given ghrelin’ potent ability to repair, restore, and regenerate both the vasculature and skeletal muscle. Despite the vast majority of data presented in this review being from in vitro and pre-clinical models, ghrelin has been routinely administered to humans for over a decade with no adverse effects (155). To date, over 400 clinical trials2 are registered under the term “ghrelin” in a broad range of conditions. Whether ghrelin can produce the beneficial effects for CLI in humans as presented in this review remains uncertain but is a promising area of clinical research.
To ease to the transition from mouse to man, further investigation of ghrelin biology in humans with PAD will give valuable insight into the ghrelin systems role in the disease. Future pre-clinical studies will also aim to examine ghrelin administration in an overt diabetic model of CLI, adding to the evidence base for the use of ghrelin mimetics for treating CLI and similar conditions. Ghrelin has also recently been implicated as a therapy for numerous other diseases [see Ref. (59) for a review]. However, few human studies have truly explored its therapeutic potential, leaving an apparent gap in the literature into its efficacy as a treatment option for human disease.
Concluding Remarks
In this review, we have described the multifactorial roles of ghrelin in CLI, as demonstrated by its role in multiple regenerative and regulatory processes. Post-ischemic vascular and skeletal muscle remodeling is stimulated by ghrelin, while it inhibits antiangiogenic and pro-fibrotic signalling pathways. With the incidence of CLI continuing to increase, driven by the rapid increase in DM and population aging, the resulting global socio-economic burden of CLI is significant. Current treatment options for CLI patients are costly and often met with poor success, while emerging novel angiogenic agents have limited success in large clinical trials. If the global burden of CLI is to be tackled, novel therapeutic strategies are urgently required. Collectively, the identification and characterization of novel biological properties of ghrelin opens the door for its administration to be a potential therapeutic agent in the treatment of CLI (Summarised in Figure 2).
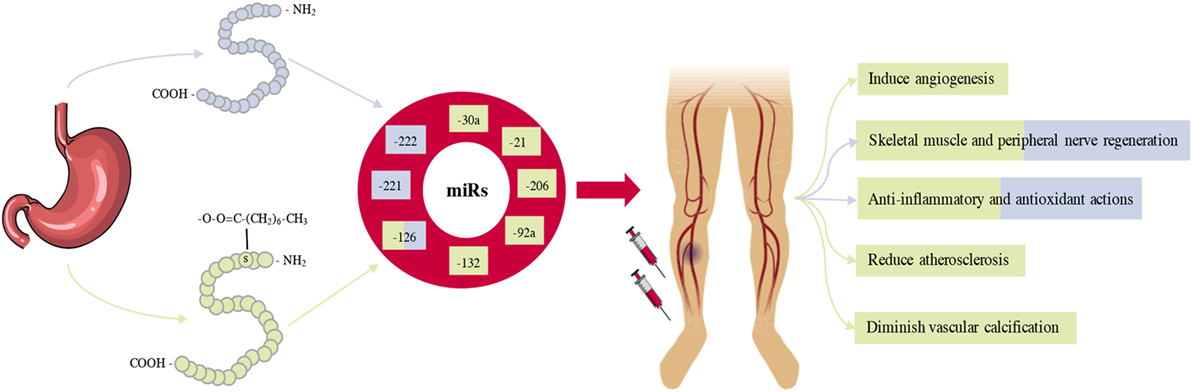
Figure 2. A schematic overview summarizing the role of ghrelin administration in the treatment and management of critical limb ischemia (CLI). Ghrelin is predominantly produced in the stomach and circulates in two forms, Acylated ghrelin (AG) and des-acylated ghrelin (Des-AG). Most of the biological effects of AG and Des-AG in pre-clinical models of CLI appear to be closely related to alterations in several pro-survival microRNAs (miRs). Given ghrelin administration can regulate several miRs, which in turn control a significant proportion of genes, ghrelin administration can result in improved limb perfusion, muscle quality, and ultimately, survival.
The pleiotropic actions of ghrelin as presented in this review will hopefully stimulate further clinical studies to explore the potential of ghrelin as a novel therapeutic agent for the treatment of this highly morbid disease.
Author Contributions
JN performed extensive literature search and wrote the first draft of the review; JP critically reviewed the manuscript; DS and RK equally contributed to conception and design of the manuscript and critically reviewed the drafts of the manuscript. All authors read and approved the final manuscript.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The reviewer HK declared a shared affiliation, with no collaboration, with one of the authors, JP, to the handling Editor.
Funding
This study was supported by the funding from Royal Society of New Zealand Catalyst Fund (CSG-U001601), Heart Foundation New Zealand (1600 and 1698), Lottery Health Board (LHR-2017-46886), and Deans Bequest from School of Biomedical Sciences, University of Otago, New Zealand. JN is a Ph.D. student supported by the scholarship from the University of Otago.
Footnotes
References
1. Annex BH. Therapeutic angiogenesis for critical limb ischaemia. Nat Rev Cardiol (2013) 7:387–96. doi:10.1038/nrcardio.2013.70
2. Fowkes FGR, Rudan D, Rudan I, Aboyans V, Denenberg JO, McDermott MM, et al. Comparison of global estimates of prevalence and risk factors for peripheral artery disease in 2000 and 2010: a systematic review and analysis. Lancet (2013) 382:1329–40. doi:10.1016/S0140-6736(13)61249-0
3. Suzuki J, Shimamura M, Suda H, Wakayama K, Kumagai H, Ikeda Y, et al. Current therapies and investigational drugs for peripheral arterial disease. Hypertens Res (2016) 39:183–91. doi:10.1038/hr.2015.134
4. Cauley JA, Kassem AM, Lane NE, Thorson S. Prevalent peripheral arterial disease and inflammatory burden. BMC Geriatr (2016) 16:213. doi:10.1186/s12877-016-0389-9
5. Lambert MA, Belch JJF. Medical management of critical limb ischaemia: where do we stand today? J Intern Med (2013) 4:295–307. doi:10.1111/joim.12102
6. Criqui MH, Aboyans V. Epidemiology of peripheral artery disease. Circ Res (2015) 9:1509–26. doi:10.1161/CIRCRESAHA.116.303849
7. Gao HX, Regier EE, Close KL. International diabetes federation world diabetes congress 2015. J Diabetes (2016) 3:300–2. doi:10.1111/1753-0407.12328
8. Lutz W, Sanderson WC, Scherbov S. The coming acceleration of global population ageing. Nature (2008) 7179:716–9. doi:10.1038/nature06516
9. Nehler MR, Duval S, Diao L, Annex BH, Hiatt WR, Rogers K, et al. Epidemiology of peripheral arterial disease and critical limb ischemia in an insured national population. J Vasc Surg (2014) 3:686–95. doi:10.1016/j.jvs.2014.03.290
10. Norgren L, Hiatt WR, Dormandy JA, Nehler MR, Harris KA, Fowkes FGR, et al. Inter-Society Consensus for the management of peripheral arterial disease (TASC II). Eur J Vasc Endovasc Surg (2007) 1:S1–75. doi:10.1016/j.jvs.2006.12.037
11. Albers M, Fratezi AC, De Luccia N. Assessment of quality of life of patients with severe ischemia as a result of infrainguinal arterial occlusive disease. J Vasc Surg (1992) 1:54–9. doi:10.1016/0741-5214(92)90417-7
12. Armstrong EJ, Chen DC, Westin GG, Singh S, McCoach CE, Bang H, et al. Adherence to guideline-recommended therapy is associated with decreased major adverse cardiovascular events and major adverse limb events among patients with peripheral arterial disease. J Am Heart Assoc (2014) 3:e000697. doi:10.1161/jaha.113.000697
13. Slovut DP, Sullivan TM. Critical limb ischemia: medical and surgical management. Vasc Med (2008) 3:281–91. doi:10.1177/1358863X08091485
14. Barshes NR, Chambers JD, Cohen J, Belkin M. Cost-effectiveness in the contemporary management of critical limb ischemia with tissue loss. J Vasc Surg (2012) 4:1015–24. doi:10.1016/j.jvs.2012.02.069
15. Marston WA, Davies SW, Armstrong B, Farber MA, Mendes RC, Fulton JJ, et al. Natural history of limbs with arterial insufficiency and chronic ulceration treated without revascularization. J Vasc Surg (2006) 1:108–15. doi:10.1016/j.jvs.2006.03.026
16. Reinecke H, Unrath M, Freisinger E, Bunzemeier H, Meyborg M, Luders F, et al. Peripheral arterial disease and critical limb ischaemia: still poor outcomes and lack of guideline adherence. Eur Heart J (2015) 15:932–8. doi:10.1093/eurheartj/ehv006
17. Ylä-Herttuala S, Bridges C, Katz MG, Korpisalo P. Angiogenic gene therapy in cardiovascular diseases: dream or vision? Eur Heart J (2017) 18:1365–71. doi:10.1093/eurheartj/ehw547
18. Samura M, Hosoyama T, Takeuchi Y, Ueno K, Morikage N, Hamano K. Therapeutic strategies for cell-based neovascularization in critical limb ischemia. J Transl Med (2017) 1:49. doi:10.1186/s12967-017-1153-4
19. Weem SP, Teraa M, de Borst GJ, Verhaar MC, Moll FL. Bone marrow derived cell therapy in critical limb ischemia: a meta-analysis of randomized placebo controlled trials. Eur J Vasc Endovasc Surg (2015) 6:775–83. doi:10.1016/j.ejvs.2015.08.018
20. Hammer A, Steiner S. Gene therapy for therapeutic angiogenesis in peripheral arterial disease – a systematic review and meta-analysis of randomized, controlled trials. Vasa (2013) 5:331–9. doi:10.1024/0301-1526/a000298
21. Kojima M, Hosoda H, Date Y, Nakazato M, Matsuo H, Kangawa K. Ghrelin is a growth-hormone-releasing acylated peptide from stomach. Nature (1999) 6762:656–60. doi:10.1038/45230
22. Katare R, Rawal S, Munasinghe PE, Tsuchimochi H, Inagaki T, Fujii Y, et al. Ghrelin promotes functional angiogenesis in a mouse model of critical limb ischemia through activation of proangiogenic microRNAs. Endocrinology (2015) 2:432–5. doi:10.1210/en.2015-1799
23. Togliatto G, Traversa M, Orsello A, Brizzi MF. Unacylated ghrelin (UnAG): a new treatment option for peripheral arterial disease? J Mol Genet Med (2014) 8:8–10. doi:10.4172/1747-0862.1000108
24. Chakrabarti S. Unacylated ghrelin: a gut-limb connection. Diabetes (2015) 4:1097–8. doi:10.2337/db14-1825
25. Kleinz MJ, Maguire JJ, Skepper JN, Davenport AP. Functional and immunocytochemical evidence for a role of ghrelin and des-octanoyl ghrelin in the regulation of vascular tone in man. Cardiovasc Res (2006) 1:227–35. doi:10.1016/j.cardiores.2005.09.001
26. Gnanapavan S, Kola B, Bustin SA, Morris DG, McGee P, Fairclough P, et al. The tissue distribution of the mRNA of ghrelin and subtypes of its receptor, GHS-R, in humans. J Clin Endocrinol Metab (2002) 6:2988–91. doi:10.1210/jcem.87.6.8739
27. Davies MG. Critical limb ischemia: epidemiology. Methodist DeBakey Cardiovasc J (2012) 4:10–4. doi:10.14797/mdcj-8-4-10
28. Hirsch AT, Haskal ZJ, Hertzer NR, Bakal CW, Creager MA, Halperin JL, et al. ACC/AHA 2005 practice guidelines for the management of patients with peripheral arterial disease (lower extremity, renal, mesenteric, and abdominal aortic). Circulation (2006) 11:e463–654. doi:10.1161/CIRCULATIONAHA.106.174526
29. Coats P, Wadsworth R. Marriage of resistance and conduit arteries breeds critical limb ischemia. Am J Physiol Heart Circ Physiol (2005) 3:H1044–50. doi:10.1152/ajpheart.00773.2004
30. Farber A, Eberhardt RT. The current state of critical limb ischemia. JAMA Surg (2016) 11:1070–7. doi:10.1001/jamasurg.2016.2018
33. Carmeliet P, Jain RK. Molecular mechanisms and clinical applications of angiogenesis. Nature (2011) 7347:298–307. doi:10.1038/nature10144
34. Rissanen TT, Korpisalo P, Markkanen JE, Liimatainen T, Ordén MR, Kholová I, et al. Blood flow remodels growing vasculature during vascular endothelial growth factor gene therapy and determines between capillary arterialization and sprouting angiogenesis. Circulation (2005) 25:3937–46. doi:10.1161/CIRCULATIONAHA.105.543124
35. Makanya AN, Hlushchuk R, Djonov VG. Intussusceptive angiogenesis and its role in vascular morphogenesis, patterning, and remodeling. Angiogenesis (2009) 2:113–23. doi:10.1007/s10456-009-9129-5
36. Simons M. Angiogenesis: where do we stand now? Circulation (2005) 12:1556–66. doi:10.1161/01.CIR.0000159345.00591.8F
37. Schaper W. Collateral circulation. Past and present. Basic Res Cardiol (2009) 1:5–21. doi:10.1007/s00395-008-0760-x
38. Vailhé B, Vittet D, Feige JJ. In vitro models of vasculogenesis and angiogenesis. Lab Invest (2001) 4:439–52. doi:10.1038/labinvest.3780252
39. Briquez PS, Clegg LE, Martino MM, Mac Gabhann F, Hubbell JA. Design principles for therapeutic angiogenic materials. Nat Rev Mater (2016) 1:15006. doi:10.1038/natrevmats.2015.6
40. Deev RV, Bozo IY, Mzhavanadze ND, Voronov DA, Gavrilenko AV, Chervyakov Y, et al. pCMV-vegf165 intramuscular gene transfer is an effective method of treatment for patients with chronic lower limb ischemia. J Cardiovasc Pharmacol Ther (2015) 5:473–82. doi:10.1177/1074248415574336
41. Dragneva G, Korpisalo P, Ylä-Herttuala S. Promoting blood vessel growth in ischemic diseases: challenges in translating preclinical potential into clinical success. Dis Model Mech (2013) 2:312–22. doi:10.1242/dmm.010413
42. Carmeliet P. Angiogenesis in life, disease and medicine. Nature (2005) 7070:932–6. doi:10.1038/nature04478
43. Teraa M, Conte MS, Moll FL, Verhaar MC. Critical limb ischemia: current trends and future directions. J Am Heart Assoc (2016) 2:e002938. doi:10.1161/JAHA.115.002938
44. Teraa M, Sprengers RW, Schutgens REG, Slaper-Cortenbach ICM, van der Graaf Y, Algra A, et al. Effect of repetitive intra-arterial infusion of bone marrow mononuclear cells in patients with no-option limb ischemia: the randomized, double-blind, placebo-controlled JUVENTAS trial. Circulation (2015) 10:851–60. doi:10.1161/CIRCULATIONAHA.114.012913
45. Gupta PK, Chullikana A, Parakh R, Desai S, Das A, Gottipamula S, et al. A double blind randomized placebo controlled phase I/II study assessing the safety and efficacy of allogeneic bone marrow derived mesenchymal stem cell in critical limb ischemia. J Transl Med (2013) 1:143. doi:10.1186/1479-5876-11-143
46. Szabo GV, Kovesd Z, Cserepes J, Daroczy J, Belkin M, Acsady G. Peripheral blood-derived autologous stem cell therapy for the treatment of patients with late-stage peripheral artery disease-results of the short- and long-term follow-up. Cytotherapy (2013) 10:1245–52. doi:10.1016/j.jcyt.2013.05.017
47. Lara-Hernandez R, Lozano-Vilardell P, Blanes P, Torreguitart-Mirada N, Galmés A, Besalduch J. Safety and efficacy of therapeutic angiogenesis as a novel treatment in patients with critical limb ischemia. Ann Vasc Surg (2010) 2:287–94. doi:10.1016/j.avsg.2009.10.012
48. Kinoshita M, Fujita Y, Katayama M, Baba R, Shibakawa M, Yoshikawa K, et al. Long-term clinical outcome after intramuscular transplantation of granulocyte colony stimulating factor-mobilized CD34 positive cells in patients with critical limb ischemia. Atherosclerosis (2012) 2:440–5. doi:10.1016/j.atherosclerosis.2012.07.031
49. Kusumanto YH, van Weel V, Mulder NH, Smit AJ, Van de Dungen JJAM, Hooymans JMM, et al. Treatment with intramuscular vascular endothelial growth factor gene compared with placebo for patients with diabetes mellitus and critical limb ischemia: a double-blind randomized trial. Hum Gene Ther (2006) 6:683–91. doi:10.1089/hum.2006.17.683
50. Belch J, Hiatt WR, Baumgartner I, Driver IV, Nikol S, Norgren L, et al. Effect of fibroblast growth factor NV1FGF on amputation and death: a randomised placebo-controlled trial of gene therapy in critical limb ischaemia. Lancet (2011) 9781:1929–37. doi:10.1016/S0140-6736(11)60394-2
51. Shigematsu H, Yasuda K, Iwai T, Sasajima T, Ishimaru S, Ohashi Y, et al. Randomized, double-blind, placebo-controlled clinical trial of hepatocyte growth factor plasmid for critical limb ischemia. Gene Ther (2010) 9:1152–61. doi:10.1038/gt.2010.51
52. Rajagopalan S, Olin J, Deitcher S, Pieczek A, Laird J, Grossman PM, et al. Use of a constitutively active hypoxia-inducible factor-1α transgene as a therapeutic strategy in no-option critical limb ischemia patients: phase I dose-escalation experience. Circulation (2007) 10:1234–43. doi:10.1161/CIRCULATIONAHA.106.607994
53. Yang J, Brown MS, Liang G, Grishin NV, Goldstein JL. Identification of the acyltransferase that octanoylates ghrelin, an appetite-stimulating peptide hormone. Cell (2008) 3:387–96. doi:10.1016/j.cell.2008.01.017
54. Mao Y, Tokudome T, Kishimoto I. The cardiovascular action of hexarelin. J Geriatr Cardiol (2014) 3:253–8. doi:10.11909/j.issn.1671-5411.2014.03.007
55. Lilleness BM, Frishman WH. Ghrelin and the cardiovascular system. Cardiol Rev (2016) 6:288–97. doi:10.1097/CRD.0000000000000113
56. Zhang G, Yin X, Qi Y, Pendyala L, Chen J, Hou D, et al. Ghrelin and cardiovascular diseases. Curr Cardiol Rev (2010) 1:62–70. doi:10.2174/157340310790231662
57. Virdis A, Lerman LO, Regoli F, Ghiadoni L, Lerman A, Taddei S. Human ghrelin: a gastric hormone with cardiovascular properties ghrelin. Curr Pharm Des (2016) 1:52–8. doi:10.2174/1381612822666151119144458
58. Kishimoto I, Tokudome T, Hosoda H, Miyazato M, Kangawa K. Ghrelin and cardiovascular diseases. J Cardiol (2012) 1:8–13. doi:10.1016/j.jjcc.2011.11.002
59. Colldén G, Tschöp M, Müller T. Therapeutic potential of targeting the ghrelin pathway. Int J Mol Sci (2017) 4:798. doi:10.3390/ijms18040798
60. Ge T, Yang W, Fan J, Li B. Preclinical evidence of ghrelin as a therapeutic target in epilepsy. Oncotarget (2017) 35:59929–39. doi:10.18632/oncotarget.18349
61. Wang L, Chen Q, Li G, Ke D. Ghrelin stimulates angiogenesis via GHSR1a-dependent MEK/ERK and PI3K/Akt signal pathways in rat cardiac microvascular endothelial cells. Peptides (2012) 1:92–100. doi:10.1016/j.peptides.2011.11.001
62. Ahluwalia A, Li A, Cheng G, Deng X, Tarnawski AS. Reduced ghrelin in endothelial cells plays important mechanistic role in aging-related impairment of angiogenesis. J Physiol Pharmacol (2009) 2:29–34.
63. Li A, Cheng G, Zhu GH, Tarnawski AS. Ghrelin stimulates angiogenesis in human microvascular endothelial cells: implications beyond GH release. Biochem Biophys Res Commun (2007) 2:238–43. doi:10.1016/j.bbrc.2006.11.144
64. Yuan MJ, He-Huang , Hu HY, Li-Quan , Hong-Jiang , Huang CX. Myocardial angiogenesis after chronic ghrelin treatment in a rat myocardial infarction model. Regul Pept (2012) 1-3:39–42. doi:10.1016/j.regpep.2012.08.013
65. Conconi MT, Nico B, Guidolin D, Baiguera S, Spinazzi R, Rebuffat P, et al. Ghrelin inhibits FGF-2-mediated angiogenesis in vitro and in vivo. Peptides (2004) 12:2179–85. doi:10.1016/j.peptides.2004.08.011
66. Tahergorabi Z, Khazaei M, Rashidi B. Systemic administration of ghrelin did not restore angiogenesis in hindlimb ischemia in control and diet-induced obese mice. Bratislavsk Lek Listy (2015) 1:35–40. doi:10.4149/BLL_2015_007
67. Baiguera S, Conconi MT, Guidolin D, Mazzocchi G, Malendowicz LK, Parnigotto PP, et al. Ghrelin inhibits in vitro angiogenic activity of rat brain microvascular endothelial cells. Int J Mol Med (2004) 5:849–54. doi:10.3892/ijmm.14.5.849
68. Brazil DP, Park J, Hemmings BA. PKB binding proteins: getting in on the Akt. Cell (2002) 3:293–303. doi:10.1016/S0092-8674(02)01083-8
69. Togliatto G, Trombetta A, Dentelli P, Cotogni P, Rosso A, Tschop MH, et al. Unacylated ghrelin promotes skeletal muscle regeneration following hindlimb ischemia via SOD-2-mediated miR-221/222 expression. J Am Heart Assoc (2013) 6:e000376. doi:10.1161/JAHA.113.000376
70. Poliseno L, Tuccoli A, Mariani L, Evangelista M, Citti L, Woods K, et al. MicroRNAs modulate the angiogenic properties of HUVECs. Blood (2006) 9:3068–71. doi:10.1182/blood-2006-01-012369
71. Togliatto G, Trombetta A, Dentelli P, Gallo S, Rosso A, Cotogni P, et al. Unacylated ghrelin induces oxidative stress resistance in a glucose intolerance and peripheral artery disease mouse model by restoring endothelial cell miR-126 expression. Diabetes (2015) 4:1370–82. doi:10.2337/db14-0991
72. McDermott MM, Hoff F, Ferrucci L, Pearce WH, Guralnik JM, Tian L, et al. Lower extremity ischemia, calf skeletal muscle characteristics, and functional impairment in peripheral arterial disease. J Am Geriatr Soc (2007) 3:400–6. doi:10.1111/j.1532-5415.2007.01092.x
73. Serra-Prat M, Papiol M, Monteis R, Palomera E, Cabre M. Relationship between plasma ghrelin levels and sarcopenia in elderly subjects: a cross-sectional study. J Nutr Health Aging (2015) 6:669–72. doi:10.1007/s12603-015-0550-8
74. Porporato PE, Filigheddu N, Reano S, Ferrara M, Angelino E, Gnocchi VF, et al. Acylated and unacylated ghrelin impair skeletal muscle atrophy in mice. J Clin Invest (2013) 2:611–22. doi:10.1172/JCI39920
75. Jude EB. Intermittent claudication in the patient with diabetes. Br J Diabetes Vasc Dis (2004) 4:238–42. doi:10.1177/14746514040040040401
76. Tsuchimochi W, Kyoraku I, Yamaguchi H, Toshinai K, Shiomi K, Kangawa K, et al. Ghrelin prevents the development of experimental diabetic neuropathy in rodents. Eur J Pharmacol (2013) 1-3:187–93. doi:10.1016/j.ejphar.2013.01.035
77. Kyoraku I, Shiomi K, Kangawa K, Nakazato M. Ghrelin reverses experimental diabetic neuropathy in mice. Biochem Biophys Res Commun (2009) 3:405–8. doi:10.1016/j.bbrc.2009.08.171
78. Ueno H, Shiiya T, Nagamine K, Tsuchimochi W, Sakoda H, Shiomi K, et al. Clinical application of ghrelin for diabetic peripheral neuropathy. Endocr J (2017) 64:53–7. doi:10.1507/endocrj.64.S53
79. Pipinos II, Judge AR, Selsby JT, Zhu Z, Swanson SA, Nella AA, et al. The myopathy of peripheral arterial occlusive disease: part 2. Oxidative stress, neuropathy, and shift in muscle fiber type. Vasc Endovascular Surg (2009) 2:101–12. doi:10.1177/1538574408315995
80. Chung H, Seo S, Moon M, Park S. Phosphatidylinositol-3-kinase/Akt/glycogen synthase kinase-3β and ERK1/2 pathways mediate protective effects of acylated and unacylated ghrelin against oxygen-glucose deprivation-induced apoptosis in primary rat cortical neuronal cells. J Endocrinol (2008) 3:511–21. doi:10.1677/JOE-08-0160
81. Li WG, Gavrila D, Liu X, Wang L, Gunnlaugsson S, Stoll LL, et al. Ghrelin inhibits proinflammatory responses and nuclear factor-kappa B activation in human endothelial cells. Circulation (2004) 18:2221–6. doi:10.1161/01.CIR.0000127956.43874.F2
82. Zhao H, Liu G, Wang Q, Ding L, Cai H, Jiang H, et al. Effect of ghrelin on human endothelial cells apoptosis induced by high glucose. Biochem Biophys Res Commun (2007) 3:677–81. doi:10.1016/j.bbrc.2007.08.021
83. Xiang Y, Li Q, Li M, Wang W, Cui C, Zhang J. Ghrelin inhibits AGEs-induced apoptosis in human endothelial cells involving ERK1/2 and PI3K/Akt pathways. Cell Biochem Funct (2011) 2:149–55. doi:10.1002/cbf.1736
84. Kawczynska-Drozdz A, Olszanecki R, Jawien J, Brzozowski T, Pawlik WW, Korbut R, et al. Ghrelin inhibits vascular superoxide production in spontaneously hypertensive rats. Am J Hypertens (2006) 7:764–7. doi:10.1016/j.amjhyper.2006.01.022
85. Shimada T, Furuta H, Doi A, Ariyasu H, Kawashima H, Wakasaki H, et al. Des-acyl ghrelin protects microvascular endothelial cells from oxidative stress-induced apoptosis through sirtuin 1 signaling pathway. Metabolism (2014) 4:469–74. doi:10.1016/j.metabol.2013.12.011
86. Togliatto G, Trombetta A, Dentelli P, Baragli A, Rosso A, Granata R, et al. Unacylated ghrelin rescues endothelial progenitor cell function in individuals with type 2 diabetes. Diabetes (2010) 4:1016–25. doi:10.2337/db09-0858
87. Granata R, Settanni F, Julien M, Nano R, Togliatto G, Trombetta A, et al. Des-acyl ghrelin fragments and analogues promote survival of pancreatic B-cells and human pancreatic islets and prevent diabetes in streptozotocin-treated rats. J Med Chem (2012) 6:2585–96. doi:10.1021/jm201223m
88. Kadoglou NPE, Lampropoulos S, Kapelouzou A, Gkontopoulos A, Theofilogiannakos EK, Fotiadis G, et al. Serum levels of apelin and ghrelin in patients with acute coronary syndromes and established coronary artery disease-KOZANI STUDY. Transl Res (2010) 5:238–46. doi:10.1016/j.trsl.2010.01.004
89. Yano Y, Nakazato M, Toshinai K, Inokuchi T, Matsuda S, Hidaka T, et al. Circulating des-acyl ghrelin improves cardiovascular risk prediction in older hypertensive patients. Am J Hypertens (2014) 5:727–33. doi:10.1093/ajh/hpt232
90. Laurila M, Santaniemi M, Kesäniemi YA, Ukkola O. High plasma ghrelin protects from coronary heart disease and Leu72Leu polymorphism of ghrelin gene from cancer in healthy adults during the 19 years follow-up study. Peptides (2014) 61:122–9. doi:10.1016/j.peptides.2014.09.012
91. Sax B, Merkely B, Túri K, Nagy A, Ahres A, Hartyánszky I, et al. Regulatory peptides characterization of pericardial and plasma ghrelin levels in patients with ischemic and non-ischemic heart disease. Regul Pept (2013) 186:131–6. doi:10.1016/j.regpep.2013.08.003
92. Kadoglou NPE, Fotiadis G, Lambadiari V, Maratou E, Dimitriadis G, Liapis CD. Serum levels of novel adipokines in patients with acute ischemic stroke: potential contribution to diagnosis and prognosis. Peptides (2014) 57:12–6. doi:10.1016/j.peptides.2014.04.008
93. Ai W, Wu M, Chen L, Jiang B, Mu M, Liu L, et al. Ghrelin ameliorates atherosclerosis by inhibiting endoplasmic reticulum stress. Fundam Clin Pharmacol (2016) 2:147–54. doi:10.1111/fcp.12251
94. Wang L, Chen Q, Ke D, Li G. Ghrelin inhibits atherosclerotic plaque angiogenesis and promotes plaque stability in a rabbit atherosclerotic model. Peptides (2017) 90:17–26. doi:10.1016/j.peptides.2017.01.013
95. Ukkola O. Ghrelin and atherosclerosis. Curr Opin Lipidol (2015) 4:288–91. doi:10.1097/MOL.0000000000000183
96. Levitzky YS, Cupples LA, Murabito JM, Kannel WB, Kiel DP, Wilson PWF, et al. Prediction of intermittent claudication, ischemic stroke, and other cardiovascular disease by detection of abdominal aortic calcific deposits by plain lumbar radiographs. Am J Cardiol (2008) 3:326–31. doi:10.1016/j.amjcard.2007.08.032
97. Xu S, Ye F, Li L, Yan J, Bao Z, Sun Z, et al. Ghrelin attenuates vascular calcification in diabetic patients with amputation. Biomed Pharmacother (2017) 91:1053–64. doi:10.1016/j.biopha.2017.05.031
98. Li GZ, Jiang W, Zhao J, Pan CS, Cao J, Tang CS, et al. Ghrelin blunted vascular calcification in vivo and in vitro in rats. Regul Pept (2005) 129(1–3):167–76. doi:10.1016/j.regpep.2005.02.015
99. Xu M, Liu L, Song C, Chen W, Gui S. Ghrelin improves vascular autophagy in rats with vascular calcification. Life Sci (2016) 179:23–9. doi:10.1016/j.lfs.2016.11.025
100. Bartel DP. MicroRNAs: genomics, biogenesis, mechanism, and function. Cell (2004) 2:281–97. doi:10.1016/S0092-8674(04)00045-5
102. Bartel DP. MicroRNA target recognition and regulatory functions. Cell (2009) 2:215–33. doi:10.1016/j.cell.2009.01.002
103. Lee RC, Feinbaum RL, Ambros V. The C. elegans heterochronic gene lin-4 encodes small RNAs with antisense complementarity to lin-14. Cell (1993) 5:843–54. doi:10.1016/0092-8674(93)90529-Y
104. Wightman B, Ha I, Ruvkun G. Posttranscriptional regulation of the heterochronic gene lin-14 by lin-4 mediates temporal pattern formation in C. elegans. Cell (1993) 5:855–62. doi:10.1016/0092-8674(93)90530-4
105. Rawal S, Manning P, Katare R. Cardiovascular microRNAs: as modulators and diagnostic biomarkers of diabetic heart disease. Cardiovasc Diabetol (2014) 1:44. doi:10.1186/1475-2840-13-44
106. Caporali A, Emanueli C. MicroRNA regulation in angiogenesis. Vascul Pharmacol (2011) 4:79–86. doi:10.1016/j.vph.2011.06.006
107. Valadi H, Ekström K, Bossios A, Sjöstrand M, Lee JJ, Lötvall JO. Exosome-mediated transfer of mRNAs and microRNAs is a novel mechanism of genetic exchange between cells. Nat Cell Biol (2007) 6:654–9. doi:10.1038/ncb1596
108. Collino F, Deregibus MC, Bruno S, Sterpone L, Aghemo G, Viltono L, et al. Microvesicles derived from adult human bone marrow and tissue specific mesenchymal stem cells shuttle selected pattern of miRNAs. PLoS One (2010) 7:e11803. doi:10.1371/journal.pone.0011803
109. Zhang B, Yao Y, Sun QF, Liu S, Jing B, Yuan C, et al. Circulating mircoRNA-21 as a predictor for vascular restenosis after interventional therapy in patients with lower extremity arterial occlusive disease. Biosci Rep (2017) 2:BSR20160502. doi:10.1042/BSR20160502
110. Faccini J, Ruidavets J-B, Cordelier P, Martins F, Maoret J-J, Bongard V. Circulating miR-155, miR-145 and let-7c as diagnostic biomarkers of the coronary artery disease. Sci Rep (2017) 7:42916. doi:10.1038/srep42916
111. Kloos W, Vogel B, Blessing E. miRNAs in peripheral artery disease – something gripping this way comes. Vasa (2014) 3:163–70. doi:10.1024/0301-1526/a000345
112. Creemers EE, Tijsen AJ, Pinto YM. Circulating microRNAs: novel biomarkers and extracellular communicators in cardiovascular disease? Circ Res (2012) 3:483–95. doi:10.1161/CIRCRESAHA.111.247452
113. Stather PW, Sylvius N, Wild JB, Choke E, Sayers RD, Bown MJ. Differential microRNA expression profiles in peripheral arterial disease. Circulation (2013) 5:490–7. doi:10.1161/CIRCGENETICS.113.000053
114. Signorelli SS, Volsi GL, Pitruzzella A, Fiore V, Mangiafico M, Vanella L, et al. Circulating miR-130a, miR-27b, and miR-210 in patients with peripheral artery disease and their potential relationship with oxidative stress. Angiology (2016) 10:945–50. doi:10.1177/0003319716638242
115. Spinetti G, Fortunato O, Caporali A, Shantikumar S, Marchetti M, Meloni M, et al. MicroRNA-15a and MicroRNA-16 impair human circulating proangiogenic cell functions and are increased in the proangiogenic cells and serum of patients with critical limb ischemia. Circ Res (2013) 2:335–46. doi:10.1161/CIRCRESAHA.111.300418
116. Welten S, Tao M, Longchamp A, Kielbasa S, Quax P, Ozaki K, et al. Upregulation of 14q32 microRNAs in human subcutaneous adipose tissue of patients with critical limb ischemia at risk of major amputation. Eur J Vasc Endovasc Surg (2016) 3:409. doi:10.1016/j.ejvs.2016.07.061
117. Harris TA, Yamakuchi M, Ferlito M, Mendell JT, Lowenstein CJ. MicroRNA-126 regulates endothelial expression of vascular cell adhesion molecule 1. Proc Natl Acad Sci U S A (2008) 5:1516–21. doi:10.1073/pnas.0707493105
118. Fish JE, Santoro MM, Morton SU, Yu S, Yeh RF, Wythe JD, et al. miR-126 regulates angiogenic signaling and vascular integrity. Dev Cell (2008) 2:272–84. doi:10.1016/j.devcel.2008.07.008
119. Van Solingen C, Seghers L, Bijkerk R, Duijs JMGJ, Roeten MK, Van Oeveren-Rietdijk AM, et al. Antagomir-mediated silencing of endothelial cell specific microRNA-126 impairs ischemia-induced angiogenesis. J Cell Mol Med (2009) 8a:1577–85. doi:10.1111/j.1582-4934.2008.00613.x
120. Wang S, Aurora AB, Johnson BA, Qi X, McAnally J, Hill JA, et al. The endothelial-specific microRNA miR-126 governs vascular integrity and angiogenesis. Dev Cell (2008) 2:261–71. doi:10.1016/j.devcel.2008.07.002
121. Anand S, Majeti BK, Acevedo LM, Murphy EA, Mukthavaram R, Scheppke L, et al. MicroRNA-132-mediated loss of p120RasGAP activates the endothelium to facilitate pathological angiogenesis. Nat Med (2010) 8:909–14. doi:10.1038/nm.2186
122. Lin C-Y, Lee H-C, Fu C-Y, Ding Y-Y, Chen J-S, Lee M-H, et al. miR-1 and miR-206 target different genes to have opposing roles during angiogenesis in zebrafish embryos. Nat Commun (2013) 4:2829. doi:10.1038/ncomms3829
123. Stahlhut C, Suarez Y, Lu J, Mishima Y, Giraldez AJ. miR-1 and miR-206 regulate angiogenesis by modulating VegfA expression in zebrafish. Development (2012) 23:4356–65. doi:10.1242/dev.083774
124. Bonauer A, Carmona G, Iwasaki M, Mione M, Koyanagi M, Fischer A, et al. MicroRNA-92a controls angiogenesis and functional recovery of ischemic tissues in mice. Science (2009) 5935:1710–3. doi:10.1126/science.1174381
125. Potente M, Ghaeni L, Baldessari D, Mostoslavsky R, Rossig L, Dequiedt F, et al. SIRT1 controls endothelial angiogenic functions during vascular growth. Genes Dev (2007) 20:2644–58. doi:10.1101/gad.435107
126. Cardinalli B, Castellani L, Fasanaro P, Basso A, Alemà S, Martelli F, et al. MicroRNA-221 and microrna-222 modulate differentiation and maturation of skeletal muscle cells. PLoS One (2009) 4:e7607. doi:10.1371/journal.pone.0007607
127. Togliatto G, Trombetta A, Dentelli P, Rosso A, Brizzi MF. MIR221/MIR222-driven post-transcriptional regulation of P27KIP1 and P57KIP2 is crucial for high-glucose- and AGE-mediated vascular cell damage. Diabetologia (2011) 7:1930–40. doi:10.1007/s00125-011-2125-5
128. Zhang L, Wang Y, Li W, Tsonis PA, Li Z, Xie L, et al. MicroRNA-30a regulation of epithelial-mesenchymal transition in diabetic cataracts through targeting SNAI1. Sci Rep (2017) 1:1117. doi:10.1038/s41598-017-01320-3
129. Yuan CT, Li XX, Cheng QJ, Wang YH, Wang JH, Liu CL. MiR-30a regulates the atrial fibrillation-induced myocardial fibrosis by targeting snail 1. Int J Clin Exp Pathol (2015) 8:15527–15136.
130. Zhou Q, Yang M, Lan H, Yu X. miR-30a negatively regulates TGF-β1-induced epithelial-mesenchymal transition and peritoneal fibrosis by targeting snai1. Am J Pathol (2013) 3:808–19. doi:10.1016/j.ajpath.2013.05.019
131. Thum T, Gross C, Fiedler J, Fischer T, Kissler S, Bussen M, et al. MicroRNA-21 contributes to myocardial disease by stimulating MAP kinase signalling in fibroblasts. Nature (2008) 7224:980–4. doi:10.1038/nature07511
132. Roy S, Khanna S, Hussain SRA, Biswas S, Azad A, Rink C, et al. MicroRNA expression in response to murine myocardial infarction: MiR-21 regulates fibroblast metalloprotease-2 via phosphatase and tensin homologue. Cardiovasc Res (2009) 1:21–9. doi:10.1093/cvr/cvp015
133. Welten SM, Goossens EA, Quax PH, Nossent AY. The multifactorial nature of microRNAs in vascular remodelling. Cardiovasc Res (2016) 1:6–22. doi:10.1093/cvr/cvw039
134. Kuhnert F, Mancuso MR, Hampton J, Stankunas K, Asano T, Chen C-Z, et al. Attribution of vascular phenotypes of the murine Egfl7 locus to the microRNA miR-126. Development (2008) 24:3989–93. doi:10.1242/dev.029736
135. Endo-Takahashi Y, Negishi Y, Nakamura A, Ukai S, Ooaku K, Oda Y, et al. Systemic delivery of miR-126 by miRNA-loaded Bubble liposomes for the treatment of hindlimb ischemia. Sci Rep (2014) 1:3883. doi:10.1038/srep03883
136. Bijkerk R, van Solingen C, de Boer HC, van der Pol P, Khairoun M, de Bruin RG, et al. Hematopoietic microRNA-126 protects against renal ischemia/reperfusion injury by promoting vascular integrity. J Am Soc Nephrol (2014) 8:1710–22. doi:10.1681/ASN.2013060640
137. Chistiakov DA, Orekhov AN, Bobryshev YV. The role of miR-126 in embryonic angiogenesis, adult vascular homeostasis, and vascular repair and its alterations in atherosclerotic disease. J Mol Cell Cardiol (2016) 97:47–55. doi:10.1016/j.yjmcc.2016.05.007
138. Zhou S, Chen HZ, Wan YZ, Zhang QJ, Wei YS, Huang S, et al. Repression of P66Shc expression by SIRT1 contributes to the prevention of hyperglycemia-induced endothelial dysfunction. Circ Res (2011) 6:639–48. doi:10.1161/CIRCRESAHA.111.243592
139. Katare R, Riu F, Mitchell K, Gubernator M, Campagnolo P, Cui Y, et al. Transplantation of human pericyte progenitor cells improves the repair of infarcted heart through activation of an angiogenic program involving micro-RNA-132. Circ Res (2011) 8:894–906. doi:10.1161/CIRCRESAHA.111.251546
140. Jalali S, Ramanathan GK, Parthasarathy PT, Aljubran S, Galam L, Yunus A, et al. miR-206 regulates pulmonary artery smooth muscle cell proliferation and differentiation. PLoS One (2012) 10:e46808. doi:10.1371/journal.pone.0046808
141. Doebele C, Bonauer A, Fischer A, Scholz A, Reiss Y, Urbich C, et al. Members of the microRNA-17-92 cluster exhibit a cell-intrinsic antiangiogenic function in endothelial cells. Blood (2010) 23:4944–50. doi:10.1182/blood-2010-01-264812
142. Daniel JM, Penzkofer D, Teske R, Dutzmann J, Koch A, Bielenberg W, et al. Inhibition of miR-92a improves re-endothelialization and prevents neointima formation following vascular injury. Cardiovasc Res (2014) 4:564–72. doi:10.1093/cvr/cvu162
143. Hinkel R, Penzkofer D, Zühlke S, Fischer A, Husada W, Xu QF, et al. Inhibition of microRNA-92a protects against ischemia/reperfusion injury in a large-animal model. Circulation (2013) 10:1066–75. doi:10.1161/CIRCULATIONAHA.113.001904
144. Stenzel D, Lundkvist A, Sauvaget D, Busse M, Graupera M, van der Flier A, et al. Integrin-dependent and -independent functions of astrocytic fibronectin in retinal angiogenesis. Development (2011) 20:4451–63. doi:10.1242/dev.071381
145. Ashkenazi A, Fairbrother WJ, Leverson JD, Souers AJ. From basic apoptosis discoveries to advanced selective BCL-2 family inhibitors. Nat Rev Drug Discov (2017) 4:273–84. doi:10.1038/nrd.2016.253
146. Guarani V, Deflorian G, Franco CA, Krüger M, Phng L-K, Bentley K, et al. Acetylation-dependent regulation of endothelial Notch signalling by the SIRT1 deacetylase. Nature (2011) 7346:234–8. doi:10.1038/nature09917
147. Celic T, Metzinger-Le Meuth V, Six I, Massy ZA, Metzinger L. The mir-221/222 cluster is a key player in vascular biology via the fine-tuning of endothelial cell physiology. Curr Vasc Pharmacol (2016) 1:40–6. doi:10.2174/1570161114666160914175149
148. Rodriguez A, Griffiths-jones S, Ashurst JL, Bradley A. Identification of mammalian microRNA host genes and transcription units. Genome Res (2004) 10a:1902–10. doi:10.1101/gr.2722704
149. Chen Y, Yu S, Li Y, Liu S, Zhou S, Cao J, et al. MicroRNA-30a ameliorates hepatic fibrosis by inhibiting Beclin1-mediated autophagy. J Cell Mol Med (2017) 21:3679–92. doi:10.1111/jcmm.13278
150. Chau BN, Xin C, Hartner J, Ren S, Castano AP, Linn G, et al. MicroRNA-21 promotes fibrosis of the kidney by silencing metabolic pathways. Sci Transl Med (2012) 121:121ra18. doi:10.1126/scitranslmed.3003205
151. Kumarswamy R, Volkmann I, Thum T. Regulation and function of miRNA-21 in health and disease. RNA Biol (2011) 5:706–13. doi:10.4161/rna.8.5.16154
152. Alexander MS, Kunkel LM. Skeletal muscle microRNAs: their diagnostic and therapeutic potentiadl in human muscle disease. J Neuromuscul Dis (2015) 1:1–11. doi:10.3233/JND-140058
153. Huang Y, He Y, Li J. MicroRNA-21: a central regulator of fibrotic diseases via various targets. Curr Pharm Des (2015) 17:2236–42. doi:10.2174/1381612820666141226095701
154. Kumarswamy R, Volkmann I, Jazbutyte V, Dangwal S, Park DH, Thum T. Transforming growth factor-β-induced endothelial-to-mesenchymal transition is partly mediated by microRNA-21. Arterioscler Thromb Vasc Biol (2011) 2:361–9. doi:10.1161/ATVBAHA.111.234286
Keywords: critical limb ischemia, peripheral artery disease, ghrelin, microRNAs, angiogenesis, regeneration, vascular disease, no-option critical limb ischemia patients
Citation: Neale JPH, Pearson JT, Katare R and Schwenke DO (2017) Ghrelin, MicroRNAs, and Critical Limb Ischemia: Hungering for a Novel Treatment Option. Front. Endocrinol. 8:350. doi: 10.3389/fendo.2017.00350
Received: 04 October 2017; Accepted: 29 November 2017;
Published: 13 December 2017
Edited by:
Chun Peng, York University, CanadaReviewed by:
Hiroyuki Kaiya, National Cerebral and Cardiovascular Center, JapanTom Ole Nilsen, University of Bergen, Norway
Copyright: © 2017 Neale, Pearson, Katare and Schwenke. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Rajesh Katare, cmFqZXNoLmthdGFyZUBvdGFnby5hYy5ueg==;
Daryl O. Schwenke, ZGFyeWwuc2Nod2Vua2VAb3RhZ28uYWMubno=
†These authors have contributed equally to this work.