Abstract
In thyroid health, the pituitary hormone thyroid-stimulating hormone (TSH) raises glandular thyroid hormone production to a physiological level and enhances formation and conversion of T4 to the biologically more active T3. Overstimulation is limited by negative feedback control. In equilibrium defining the euthyroid state, the relationship between TSH and FT4 expresses clusters of genetically determined, interlocked TSH–FT4 pairs, which invalidates their statistical correlation within the euthyroid range. Appropriate reactions to internal or external challenges are defined by unique solutions and homeostatic equilibria. Permissible variations in an individual are much more closely constrained than over a population. Current diagnostic definitions of subclinical thyroid dysfunction are laboratory based, and do not concur with treatment recommendations. An appropriate TSH level is a homeostatic concept that cannot be reduced to a fixed range consideration. The control mode may shift from feedback to tracking where TSH becomes positively, rather than inversely related with FT4. This is obvious in pituitary disease and severe non-thyroid illness, but extends to other prevalent conditions including aging, obesity, and levothyroxine (LT4) treatment. Treatment targets must both be individualized and respect altered equilibria on LT4. To avoid amalgamation bias, clinically meaningful stratification is required in epidemiological studies. In conclusion, pituitary TSH cannot be readily interpreted as a sensitive mirror image of thyroid function because the negative TSH–FT4 correlation is frequently broken, even inverted, by common conditions. The interrelationships between TSH and thyroid hormones and the interlocking elements of the control system are individual, dynamic, and adaptive. This demands a paradigm shift of its diagnostic use.
Introduction
In the healthy body, multiple interlocking biochemical mechanisms interact homeostatically both to achieve biological equilibrium and to respond suitably to any challenges, internal or external, that might occur. However, the way in which different individuals maintain a steady homeostatic state varies considerably, including their appropriate reaction to any changes that may be demanded of the system. In this regard, the possible biochemical expressions of a healthy framework within which viable variations of response can occur have finite limits (1). Appropriate reactions in individuals are defined by their own unique solutions both to homeostatic equilibrium and to biochemical challenges, which are much more closely constrained than those permissible over the whole population. This has profound implications as to how disequilibria are confronted by individual responses to the challenge or therapeutic interventions administered to obvert it.
The classical definitions of hypothyroidism and hyperthyroidism reflect this concept when referring to inadequate—either reduced or exaggerated—supply and response to thyroid hormones (2). Pathophysiological conditions or diseases may arise at various levels including deficiencies of hormone supply, alterations of control or resistance to cellular responses to the hormones (3–5). Thyroid gland failure represents a more restricted view, termed primary hypothyroidism. Current diagnostic definitions of thyroid disease are primarily laboratory based, and include subclinical dysfunctions that are dissociated from treatment recommendations (6–9). In this review, we revisit underlying principles of thyroid control, aligning or contrasting them with the current use of thyroid-stimulating hormone (TSH) in the diagnosis and treatment of thyroid diseases.
The Current Paradigm of Thyroid Diagnosis
Two events have shaped the current paradigm of thyroid function testing (1) the discovery of negative feedback by thyroid hormones on pituitary TSH and (2) methodological advances in sensitively measuring TSH (6, 10, 11). If TSH serum concentrations provided a more sensitive and accurate mirror image of thyroid hormone status than thyroid hormones themselves, this would be an ideal diagnostic tool. This argument emerged in the 1980s, and was readily accepted by clinicians (12, 13). TSH measurement subsequently gained a dominant role in thyroid function testing, facilitating cost effective disease screening, introducing new definitions of subclinical hypothyroidism or hyperthyroidism, and delivering biochemical treatment targets (14–17). A plethora of epidemiological studies then associated TSH concentrations with clinical outcomes (18–21). This resulted in a questionable paradigm elevating an indirect controlling parameter to such prominence and dominance as expressed in current guidelines for diagnosis and treatment of thyroid diseases (14–16). With the holy grail in the hand of scientists, clinically orientated approaches contradicting the TSH paradigm and noting discrepancies between a biochemically euthyroid and clinically hypothyroid status were readily dismissed (22–26). However, concerns have resurfaced in the light of recent basic and clinical studies revealing fundamental issues with the interpretation of TSH measurements and documenting unsatisfactory improvement in the quality of life of patients treated for hypothyroidism according to the current TSH paradigm (1, 27–29).
Shifting the Paradigm
Is the TSH concentration at all times a reliable mirror image of the thyroid hormone status of the patient?
Firstly, we must note that this question reverses causality of process. While TSH is subject to negative feedback by thyroid hormones predominantly FT4, thereby reflecting on the thyroid hormone status, its primary physiological role is to elevate the hypothyroid to the euthyroid state (1). Without appropriate stimulation by TSH, the basal hormone output by the thyroid gland remains insufficient, as becomes apparent in pituitary deficiency (secondary hypothyroidism) (4). Conversely, in thyroid health TSH raises the glandular hormone production to its physiological level, and it also enhances the formation of the biologically more active T3 and its conversion from T4 (30–32). The integration of the TSH–T3 shunt into the homeostatic control of the thyroid–pituitary axis is illustrated in Figure 1A. Feedback details have been discussed elsewhere (27). Clinical studies and theoretical modeling suggest that the shunt facilitates FT3 stability against variations in the glandular T4 output, and its absence may lead to T3 production instability in athyreotic patients (31, 33). This relational control loop operates still within the euthyroid TSH range (34). It also appears to mediate subtle expressions of central control over peripheral tissues, such as circadian or seasonal variation in FT3, but not FT4, the former lagging the TSH rhythm (35). In thyroid autoimmune disease, the feedforward control increases T4 to T3 conversion, compensating for latent thyroid failure until the disease has progressed beyond tolerance (34).
Figure 1
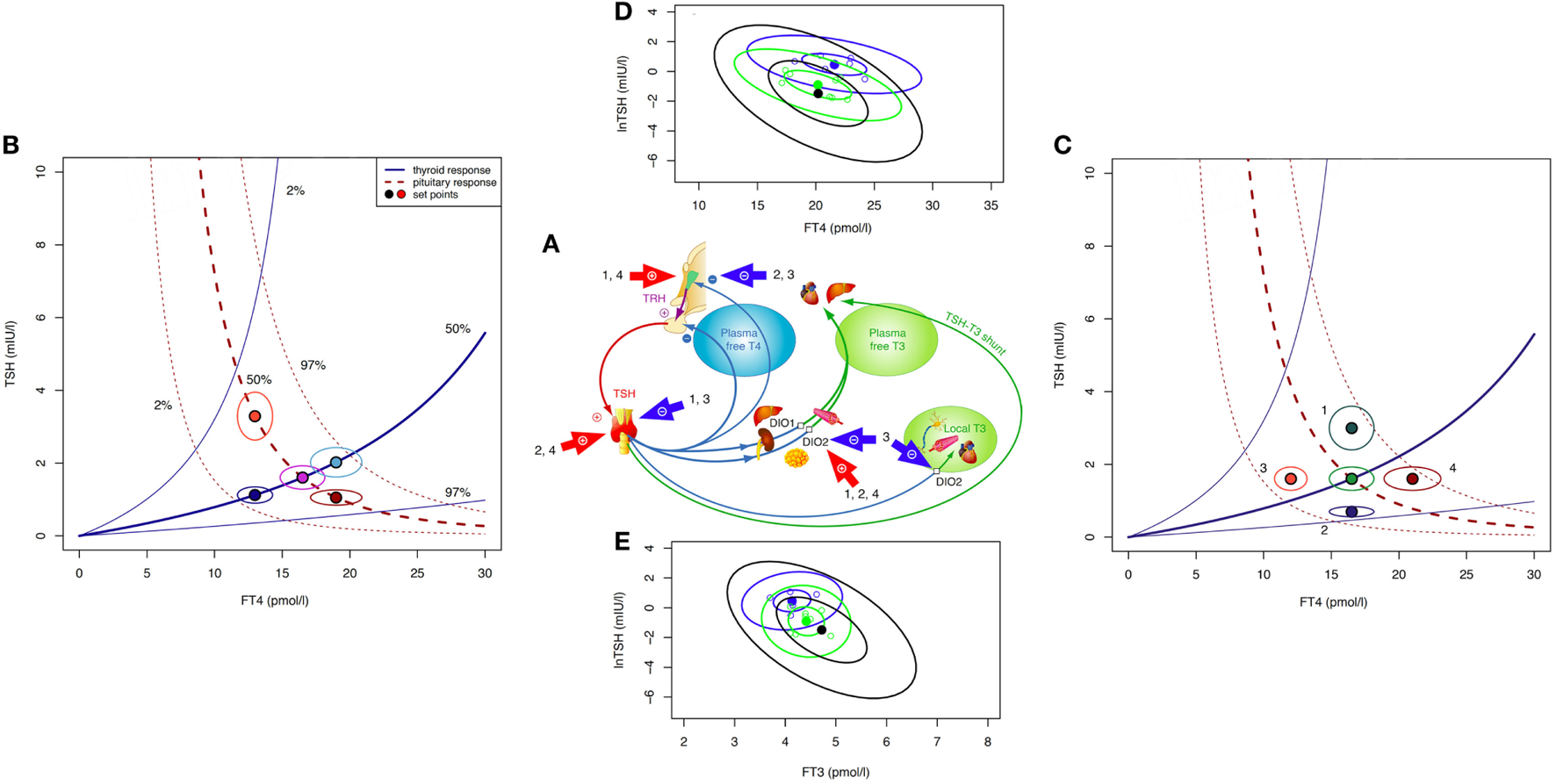
Theoretical and observed relationships between thyroid-stimulating hormone (TSH) and thyroid hormones. The central system provides an integrated solution to the location of the setpoint (settling point) of thyroid homeostasis, as determined by the various genetic, epigenetic, and allostatic parameters (27, 36). This includes monogenic factors such as polymorphic variants of receptors and transporters, changes in deiodinase activity or variations in T4 production efficiency, and environmental impacts, e.g., nutritional factors, body weight, body composition, age, and extends to diseases of the thyroid and other organs where severe disequilibria may arise and a “euthyroid” solution is not achieved (27, 36). (A) Schematic overview of main regulatory pathways and control loops in the hypothalamic–pituitary–thyroid regulation. External factors of influence include (1) obesity (hyperthyrotropinemia), (2) pregnancy (hCG-mediated suppression of TSH release and stimulation of T4 secretion), (3) non-thyroidal illness (NTI) (both pituitary and thyroid function down-regulated), and (4) certain psychiatric diseases (both pituitary and thyroid function stimulated). (B) The response of the thyroid to TSH by hormone release and corresponding feedback on pituitary TSH secretion produces a finite equilibrium solution, thereby defining interlocking TSH–FT4 pairs (setpoints). These are characteristic for each person and show only little variation unless disturbed by internal or external strain. In the event of progressive thyroid capacity stress, irrespective and independent of other influences, setpoints for FT4 and TSH translocate along a homeostatic pituitary response curve (isocline) unique for the individual. Hence, in the case of a diminished or exaggerated pituitary response the translocation moves along the thyroid isocline. The open circles indicate the expected variation (10% for FT4 and 30% for TSH) surrounding the individual setpoint. The percentiles for the isoclines of the response curves were derived from previous data in a healthy sample and the included area between the 2.5 and 97.5 percentiles represents the population’s reference range (37). (C) Influences additional to direct thyroid activity change (e.g., allostatic changes such as obesity, age, pregnancy, and NTI) may have dislocating effects on isoclines and setpoints. (D) Observed TSH–FT4 pairs (setpoints) in two individual patients (blue and green symbols) and the averaged group of 250 patients (black ellipse) followed long term on stable treatment conditions. Data ellipses indicate the 50 and 95% confidence limits for the setpoint. Levothyroxine dose was 100 μg/day, 1.5 µg/kg body weight for each of the individual patients, and 1.5 (SD 0.27) μg/kg for the whole group. Data are from a published longitudinal study (38). (E) Observed TSH–FT3 pairs depicted in the same patients as in (D). FT3 concentrations vary with conversion efficiency and impact on the location of the TSH–FT4 setpoint (38).
Pathophysiological challenges other than thyroid disease may disturb the equilibria between TSH and thyroid hormones, requiring setpoint adjustment of the hypothalamic–pituitary–thyroid axis (Figures 1A–C). Weight gain or loss, alterations in body composition, and aging may cause profound changes (Figure 1C), frequently switching the control mode from negative feedback to tracking FT4 rather than opposing it (36, 39, 40). This inversion of the TSH–FT4 correlation is clinically important to recognize, but it may be missed or misinterpreted as subclinical hypothyroidism when relying on TSH measurement as sole diagnostic test. Several mechanisms provide a physiological interface for shifting the control modes of the hypothalamic–pituitary–thyroid axis (36, 39–46). While detailed examination of the complex subject is beyond the scope of this article and dedicated reviews are available (36, 39, 41, 43, 45, 46), a brief explanation may be helpful. Hypothalamic tancytes strongly express the relatively more T3- than T4-affine thyroid hormone transporter MCT8 and deiodinase type 2 and 3, enabling them to sense FT3 and FT4 levels (41, 44–46). They also respond to alterations in the energetic and metabolic needs of the body and interact with TRH neurons (41, 42, 44–48). Fat cells release adipokines such as leptin into the circulation which directly or indirectly stimulate pituitary TSH secretion (36, 39, 40, 42, 45). In a vicious cycle, TSH may promote leptin release through TSH receptor activation on adipocytes (49). The controlling system stays informed on fat and energy reserves, and may act on adjusting energy expenditure accordingly. While rising with weight gain TSH decreases again after weight loss (36, 39, 50, 51). Central and peripheral deiodinases are also sensitive to nutritional factors and body composition, adjusting T4 to T3 conversion to maintain T3 stability under varying conditions (39, 40). As transporters, deiodinases and thyroid hormone receptors subtypes differ in both their expressions and T3 affinities, central responses may readily diverge from peripheral equilibria (52–54). This may explain why none of these physiological changes is associated with thyroid dysfunction, although the equilibria between thyroid hormones and TSH are profoundly altered. Similar changes in control modes are observed in aging (55). In the Baltimore Longitudinal Study of Aging, some participants experienced a parallel rise/fall of TSH and FT4, whereas others showed a rising TSH with declining FT4 concentrations (55). Interestingly, the two patterns carried different mortality risks (55). Underlying mechanisms are complex and confounded by other pathologies, but pituitary responsiveness is clearly age related, as is deiodinase activity (56–58). Generally, higher TSH ranges are deemed acceptable in the elderly patient (57, 59, 60). More dramatic perturbations occur in severe non-thyroidal illness (NTI) including psychiatric disease (Figure 1C) where the allostatic stress response operates to alter both the setpoint and peripheral transfer parameters of the control loop, as reviewed elsewhere (36).
These lines of evidence suggest that equilibria and correlations between TSH, FT4, and FT3 are by no means invariant; rather being situationally adjusted in response to minor disturbances, and completely deranged in severe pathology (Figure 1B).
The pituitary–thyroid feedback loop must be revisited in the light of these findings (27). The strong log-linear correlation between TSH and FT4, observed in primary hypothyroidism, disintegrates toward the euthyroid range (61–65). The statistical correlation between TSH and FT4 is invalidated by clustering of genetically determined TSH–FT4 pairs (Figure 1B), and influences other than TSH shape the interrelations and equilibria (Figure 1C) (37, 65). This results in a high-individuality index of TSH, which gravely limits the use of population statistics in determining the euthyroid range (66, 67). Hypothalamic–pituitary–thyroid control is more complex and frequently denies the diagnostic simplification of a fixed log-linear TSH–FT4 gradient (Figures 1A–C) (27, 68). Studies in levothyroxine (LT4)-treated patients with thyroid carcinoma favor cascade-type control involving FT4, FT3 and their interaction over simple proportional control (38). Setpoint simulations corroborated these clinical results (37, 38). Together these findings question, the current diagnostic use of the TSH response as a reliable mirror of thyroid status. They demand a conceptional evolution of thyroid regulation to underpin a more differentiated clinical use.
Appropriateness Versus Normality
The appropriateness of a TSH level is a different concept from range consideration. In secondary hypothyroidism or hypopituitarism, TSH measurement may be frequently inappropriate, residing within its reference range, yet abnormally low relative to the low-FT4 level. Deranged setpoints together with normal or elevated FT4 concentrations have been well recognized in TSH or thyroid hormone resistance syndromes and TSH producing pituitary tumors. Similarly, low-FT3 and/or low-FT4 serum concentrations in the NTI syndrome are accompanied by variable TSH values within or outside the reference range (4, 5, 36, 69–72). The isolated interpretation of TSH is not diagnostically useful in these conditions. While these limitations are well known, the dominance of TSH in the diagnosis of euthyroidism and as a treatment target for primary hypothyroidism also warrants closer scrutiny.
The high-individuality index of TSH and its correlative variation with thyroid hormones cause major statistical issues, because the basic concept of statistical hypothesis testing demands that the sample is randomly drawn from a population and thus representative of the population (73). This tenet is, however, violated where a measured TSH value is not an expression of normality surrounded by some margin of error or variation within a given population, rather a heterogeneous group of individuals sharing the same TSH value with different physiological meaning. For some individuals, the same TSH value may indicate a perfectly normal situation, for others subtle thyroid failure with declining FT4 concentrations and negative correlation between the two hormones, and for others a control shift to a positive TSH–FT4 correlation induced by non-thyroidal influences. A known response heterogeneity, however, precludes the use of statistical regression models based on normality assumption, requiring multilevel, hierarchical, or latent class models instead (74). The heterogeneity bias is known as Simpson’s amalgamation paradox (75, 76). Misleading and conflicting results may therefore be expected when mixing different underlying physiologies without proper stratification. Some authors reported a protective effect of TSH in the elderly, others an increased mortality risk of higher TSH within the normal range, and others no association of death with TSH, but with higher FT4 (21, 77–81). A dissociation between TSH and FT4 is also apparent in atrial fibrillation in euthyroid individuals where the risk increases with higher FT4 concentrations, but not lower TSH levels (82). We face a similar paradox in RCTs comparing T3–T4 combinations with T4 monotherapy, where patients invariably expressed their preference for the combination, but statistical analysis finds no evidence of superiority (83, 84).
We conclude that a TSH measurement representing ambiguous and overlapping categories between thyroid health and disease is by itself unreliable as a diagnostic tool. Statistical averaging or arbitrary cut-offs such as tertiles or quartiles should not be based on false assumptions of normality without further clinically meaningful stratification.
Another situation that differs from any naturally occurring condition is LT4 monotherapy for primary hypothyroidism. The homeostatic equilibria between TSH, FT4, and FT3 adequate for the previously healthy state no longer apply equally to the treated state, where dissociations between FT4 and FT3 and TSH and FT3 are apparent (Figures 1D,E) (31, 33, 65, 85–87). The T3/T4 conversion rate is reduced in LT4-treated athyreotic patients, compared with their rate prior to surgery (38, 86). FT3 is displaced from TSH in these patients, being relatively lower at the same TSH level (33, 85). While the consequences of the altered ratios are widely unknown and require further study (88), implications for the interpretation of TSH measurements can be readily derived.
Thyroid-stimulating hormone concentrations in normal health cannot be a therapeutic target for establishing LT4 dose adequacy (31, 89, 90). The treatment situation has to be re-evaluated on the basis of the shifted equilibria rather than pre-existing range considerations.
Can’t we just rely on bringing TSH within an acceptable population range and assume peripheral autoregulation at the tissue level should take care of the adequate tissue supply with T3? There are two major issues with this popular belief in the ability of the patient’s own pituitary gland—except for pituitary deficiency—to be the best judge of dose adequacy. First, this leaves patients frequently dissatisfied, because their quality of life is not generally restored with LT4 treatment to the same level seen in healthy persons despite their TSH concentrations being within the reference range (29). Second, the TSH for a patient on LT4 is not what it is for an untreated patient (65). The clinical and biochemical treatment response to LT4 turns out to be diverse and is influenced by many treatment-related or unrelated factors (Figures 1D,E) (38, 89). As a consequence of low-conversion efficiency, at least in some patients, the equilibrium for TSH may be shifted below the reference range of the healthy population (38, 86, 89). This poses a dilemma to clinicians.
Toward an Optimized Treatment Strategy
Given the fundamental changes in thyroid control related to LT4 medication, we can no longer dismiss patient complaints when discrepancies arise between clinically hypothyroid versus biochemically euthyroid states (23–26). A recent retrospective study in patients with thyroid carcinoma followed long term found that displaced equilibria and resulting lower FT3 concentrations were associated with a lack of symptom relief, independently of known confounders such as gender, age, body weight, and weight-adjusted LT4 dose (38). This study and others contradict the assumption that patients may invariably gain satisfaction from LT4 treatment when TSH levels are kept within or even below reference range (29, 38, 90). Unfortunately, tissue T3 concentrations or T3 receptor saturation cannot be readily determined in humans. This was, however, done in rodents where various tissues remained in the hypothyroid state on LT4 monotherapy despite normal serum TSH levels, and only T3/T4 combination could restore euthyroidism in tissues (91, 92). These experiments uncovered limiting biochemical pathways that affect local sensitivity to T4 such as ubiquitination of hypothalamic type 2 deiodinase (92).
Implications emerging from advances in the understanding of the diverse facets of pituitary control (Figures 1A–E), clinical studies, mathematical simulations, and experimental treatments suggest replacing the current TSH paradigm with a more inclusive interpretation. This should take into account clinical signs and symptoms, all three thyroid parameters, and their relationships. The appropriateness of the respective levels relative to each other, to the previous healthy state and to a specific condition becomes more important than a blanket categorization according to placements within or outside any set range (Table 1).
Table 1
| TSH paradigm | New relational paradigm |
|---|---|
| Normality-based statistics | Homeostatic equilibria |
| Univariate normal distribution | Multivariate distributions |
| Population-based range | Setpoint, joined TSH–FT4 pairs |
| Low degree of individuality | High-individuality index |
| TSH is reflective of thyroid hormone status | TSH is interlocked with FT4 and FT3 |
| The reference range is fixed across individuals and conditions | The setpoint is genetically determined and adjustable to various conditions |
| The parameters are treated as singularities, even when interpreted in combination | The parameters are interpreted in relation to each other |
| Interpreting ranges | Reconstructing setpoints |
| Levels are interpreted as within the reference range or outside its limits | Levels are interpreted as relatively appropriate or inappropriate |
| A TSH within its reference range in a healthy population indicates euthyroidism | The population-based TSH reference range is too wide to reliably define euthyroidism in a person |
| A high TSH indicates overt or subclinical hypothyroidism with rare exceptions | A high TSH originates from diverse physiologies |
| The setting of reference ranges and their interpretation is a simple process | The derivation of conjoined homeostatic equilibria is an intricate process |
| Subclinical thyroid disease entities are solely based on laboratory measurements and do not correspond to treatable clinical entities | The clinical change or challenge is considered primary mounting a defensive reaction that may alter the setpoint or transfer function |
| TSH is frequently interpreted without sufficient consideration of the clinical situation | The interpretation of TSH is tied to the clinical presentation |
| TSH reference range is universally suitable to judge treatment success | TSH level is inadequate as a measure of treatment success and LT4 dose adequacy |
| The suitable TSH range remains unchanged in LT4-treated patients | The suitable TSH range is shifted in LT4-treated patients |
| Exclusive role of TSH in guiding treatment targets | Supportive role of FT3 in guiding treatment targets |
Differences between the thyroid-stimulating hormone (TSH) paradigm and the newly proposed paradigm.
If required for symptom relief, considering wide variations in individual responses, clinicians may find it acceptable to suppress the TSH close to or slightly below its reference range while avoiding overt clinical or biochemical hyperthyroidism. In fact, in a recent study, patients after thyroidectomy with mildly suppressed TSH levels were closest to euthyroid, based on surrogate markers for end organ response, those with normal TSH mildly hypothyroid and those with strongly suppressed levels mildly hyperthyroid (90). T3/T4 combination therapy may be preferable to patients with persistent symptoms or a failure to sufficiently raise their FT3 concentration despite LT4 dose escalation and TSH suppression (93).
This practice should not be discouraged by incorrect statistical analyses of epidemiological studies. Many studies reported increased risks associated with suppressed TSH such as atrial fibrillation and osteoporosis but failed to properly classify the hormone status of patients into euthyroid versus hyperthyroid, and frequently did not even distinguish between treatment-induced TSH suppression and endogenous hyperthyroidism (94). Importantly, thyroid hormones, while suppressing pituitary TSH, have been reported to upregulate the locally produced osteoprotective TSHβv variant (95). Statistical associations with TSH cannot establish causality, as the opposing effects of low-TSH and low-FT3/TSHβv frequently occur together in LT4-treated patients.
However, it is equally appropriate to stress a caveat that not every patient on LT4 may require or tolerate a suppressed TSH. Unfortunately, conventional range considerations for TSH do not apply to the LT4-treated patient. There is no easy fix, but a paradigm shift could be a first step toward a solution. Until more personalized methods such as setpoint reconstruction have been evaluated (37), treatment adequacy must be judged on an individual basis by a combination of clinical and biochemical outcomes. The frequent overlapping and unspecific nature of hypothyroid symptoms presents yet another challenge (96–99). Unfortunately, reliable and readily accessible markers of the tissue effects are lacking.
However, the hypothyroid patient should not opt for under-treatment and forego symptom relief out of an exaggerated fear of over-treatment. On the other hand, long-term risks of LT4 treatment should also be carefully evaluated. Assessing scientific studies is a difficult task because statistical masking, confounding, and response heterogeneity can all be expected in a statistical parameter with a high-individuality index (67, 100, 101). This includes RCTs, regarded as the highest class of empirical evidence, which are not immune from violations of the underlying statistical assumptions.
Simplified concepts such as the negative thyroid pituitary feedback control contributed a fundamental understanding of endocrine principles, but were subsequently not refined for dealing with more complex facets of the various thyroid disorders and the clinical requirements of the diagnostic process. The pituitary TSH response is too diverse to be viewed as a sensitive mirror image of thyroid function. The interlocking elements of the control system are highly individual, dynamic, and adaptive. The assumed negative TSH–FT4 association that underpins the diagnostic use of TSH becomes frequently uncorrelated or even inverted, involving various influences, and mechanisms such as statistically multivariate clustering of TSH–FT4 setpoints, non-proportional cascade-type control, control mode shifts from negative feedback to positive tracking, and prevalent extra-pituitary–thyroid modulators of the relationship. Discrepancies arise between individually appropriate TSH levels and population-based reference ranges, laboratory-based disease definitions and treatment requirements, feedback adjustments and thyroid failure, and non-shared equilibria between thyroid health and LT4 treatment (Table 1). These have to be conceptionally reconciled to meet the needs of patients and clinical practitioners. TSH should be viewed as a controlling element and interpreted within its physiological role as a more narrowly defined and conditionally adaptive setpoint (Figure 1). Novel testable concepts are emerging and await clinical study.
Statements
Author contributions
RH and JM drafted the main part of the manuscript. RL and JD edited the text and contributed additional ideas, material, and text passages. JD also contributed to figures. All authors read and approved the final version.
Conflict of interest
JD received funding and personal fees by Sanofi-Henning, Hexal AG, Bristol-Myers, Squibb, and Pfizer, and is co-owner of the intellectual property rights for the patent “System and Method for Deriving Parameters for Homeostatic Feedback Control of an Individual” (Singapore Institute for Clinical Sciences, Biomedical Sciences Institutes, Application Number 201208940-5, WIPO number WO/2014/088516). All other authors declare that there is no conflict of interest that could be perceived as prejudicing the impartiality of the research reported.
References
1
HoermannRMidgleyJEMLarischRDietrichJW. Relational stability in the expression of normality, variation, and control of thyroid function. Front Endocrinol (2016) 7:57–8.10.3389/fendo.2016.00142
2
BravermanLEUtigerRD. Introduction to thyrotoxicosis. In: BravermanLEUtigerRD, editors. Werner and Ingbar’s The Thyroid. Philadelphia: J.B. Lippincott Company (1995). p. 645–7.
3
ChakerLBiancoACJonklaasJPeetersRP. Hypothyroidism. Lancet (2017) 390:1550–62.10.1016/S0140-6736(17)30703-1
4
Beck-PeccozPRodariGGiavoliCLaniaA. Central hypothyroidism—a neglected thyroid disorder. Nat Rev Endocrinol (2017) 13:588–98.10.1038/nrendo.2017.47
5
GrasbergerHRefetoffS. Resistance to thyrotropin. Best Pract Res Clin Endocrinol Metab (2017) 31:183–94.10.1016/j.beem.2017.03.004
6
BalochZCarayonPConte-DevolxBDemersLMFeldt-RasmussenUHenryJ-Fet alLaboratory medicine practice guidelines. Laboratory support for the diagnosis and monitoring of thyroid disease. Thyroid (2003) 13:3–126.10.1089/105072503321086962
7
SpencerCA. Thyroid function tests: assay of thyroid hormones and related substances. Thyroid Disease Manager. (2017). Available from: http://www.thyroidmanager.org/chapter/assay-of-thyroid-hormones-and-related-substances3/
8
CooperDS. Clinical practice. Subclinical hypothyroidism. N Engl J Med (2001) 345:260–5.10.1056/NEJM200107263450406
9
RedfordCVaidyaB. Subclinical hypothyroidism: should we treat?Post Reprod Health (2017) 23:55–62.10.1177/2053369117705058
10
LarsenPR. Thyroid-pituitary interaction: feedback regulation of thyrotropin secretion by thyroid hormones. N Engl J Med (1982) 306:23–32.10.1056/NEJM198201073060107
11
ThienpontLMVan UytfangheKBeastallGFaixJDIeiriTMillerWGet alReport of the IFCC Working Group for standardization of thyroid function tests; part 1: thyroid-stimulating hormone. Clin Chem (2010) 56:902–11.10.1373/clinchem.2009.140178
12
CaldwellGKellettHAGowSMBeckettGJSweetingVMSethJet alA new strategy for thyroid function testing. Lancet (1985) 1:1117–9.10.1016/S0140-6736(85)92429-8
13
SheehanMT. Biochemical testing of the thyroid: TSH is the best and, oftentimes, only test needed—a review for primary care. Clin Med Res (2016) 14:83–92.10.3121/cmr.2016.1309
14
PearceSHSBrabantGDuntasLHMonzaniFPeetersRPRazviSet al2013 ETA guideline: management of subclinical hypothyroidism. Eur Thyroid J (2013) 2:215–28.10.1159/000356507
15
JonklaasJBiancoACBauerAJBurmanKDCappolaARCeliFSet alGuidelines for the treatment of hypothyroidism: prepared by the American Thyroid Association task force on thyroid hormone replacement. Thyroid (2014) 24:1670–751.10.1089/thy.2014.0028
16
RossDSBurchHBCooperDSGreenleeMCLaurbergPMaiaALet al2016 American Thyroid Association guidelines for diagnosis and management of hyperthyroidism and other causes of thyrotoxicosis. Thyroid (2016) 26:1343–421.10.1089/thy.2016.0229
17
AlexanderEKPearceENBrentGABrownRSChenHDosiouCet al2017 guidelines of the American Thyroid Association for the diagnosis and management of thyroid disease during pregnancy and the postpartum. Thyroid (2017) 27:315–89.10.1089/thy.2016.0457
18
RodondiNElzen denWPJBauerDCCappolaARRazviSWalshJPet alSubclinical hypothyroidism and the risk of coronary heart disease and mortality. JAMA (2010) 304:1365–74.10.1001/jama.2010.1361
19
RiebenCSegnaDda CostaBRColletT-HChakerLAubertCEet alSubclinical thyroid dysfunction and the risk of cognitive decline: a meta-analysis of prospective cohort studies. J Clin Endocrinol Metab (2016) 101:4945–54.10.1210/jc.2016-2129
20
ChakerLKorevaarTIMRizopoulosDColletTHVölzkeHHofmanAet alDefining optimal health range for thyroid function based on the risk of cardiovascular disease. J Clin Endocrinol Metab (2017) 102:2853–61.10.1210/jc.2017-00410
21
InoueKTsujimotoTSaitoJSugiyamaT. Association between serum thyrotropin levels and mortality among euthyroid adults in the United States. Thyroid (2016) 26:1457–65.10.1089/thy.2016.0156
22
ZulewskiHMüllerBExerPMiserezARStaubJJ. Estimation of tissue hypothyroidism by a new clinical score: evaluation of patients with various grades of hypothyroidism and controls. J Clin Endocrinol Metab (1997) 82:771–6.10.1210/jc.82.3.771
23
SaravananPChauW-FRobertsNVedharaKGreenwoodRDayanCM. Psychological well-being in patients on “adequate” doses of l-thyroxine: results of a large, controlled community-based questionnaire study. Clin Endocrinol (Oxf) (2002) 57:577–85.10.1046/j.1365-2265.2002.01654.x
24
WekkingEMAppelhofBCFliersEScheneAHHuyserJTijssenJGPet alCognitive functioning and well-being in euthyroid patients on thyroxine replacement therapy for primary hypothyroidism. Eur J Endocrinol (2005) 153:747–53.10.1530/eje.1.02025
25
SkinnerGRBHolmesDAhmadADaviesJABenitezJ. Clinical response to thyroxine sodium in clinically hypothyroid but biochemically euthyroid patients. J Nutr Environ Med (2009) 10:115–24.10.1080/13590840050043530
26
PanickerVEvansJBjøroTAsvoldBODayanCMBjerkesetO. A paradoxical difference in relationship between anxiety, depression and thyroid function in subjects on and not on T4: findings from the HUNT study. Clin Endocrinol (Oxf) (2009) 71:574–80.10.1111/j.1365-2265.2008.03521.x
27
HoermannRMidgleyJEMLarischRDietrichJW. Homeostatic control of the thyroid–pituitary axis: perspectives for diagnosis and treatment. Front Endocrinol (2015) 6:1–17.10.3389/fendo.2015.00177
28
McAninchEABiancoAC. New insights into the variable effectiveness of levothyroxine monotherapy for hypothyroidism. Lancet Diabetes Endocrinol (2015) 3:756–8.10.1016/S2213-8587(15)00325-3
29
WintherKHCramonPWattTBjornerJBEkholmOFeldt-RasmussenUet alDisease-specific as well as generic quality of life is widely impacted in autoimmune hypothyroidism and improves during the first six months of levothyroxine therapy. PLoS One (2016) 11:e0156925.10.1371/journal.pone.0156925
30
IshiiHInadaMTanakaKMashioYNaitoKNishikawaMet alInduction of outer and inner ring monodeiodinases in human thyroid gland by thyrotropin. J Clin Endocrinol Metab (1983) 57:500–5.10.1210/jcem-57-3-500
31
HoermannRMidgleyJEMLarischRDietrichJW. Integration of peripheral and glandular regulation of triiodothyronine production by thyrotropin in untreated and thyroxine-treated subjects. Horm Metab Res (2015) 47:674–80.10.1055/s-0034-1398616
32
CitterioCEVeluswamyBMorganSJGaltonVABangaJPAtkinsSet alDe novo triiodothyronine formation from thyrocytes activated by thyroid-stimulating hormone. J Biol Chem (2017) 292:15434–44.10.1074/jbc.M117.784447
33
HoermannRMidgleyJEMLarischRDietrichJW. Is pituitary TSH an adequate measure of thyroid hormone-controlled homoeostasis during thyroxine treatment?Eur J Endocrinol (2013) 168:271–80.10.1530/EJE-12-0819
34
HoermannRMidgleyJEMLarischRDietrichJW. Relational stability of thyroid hormones in euthyroid subjects and patients with autoimmune thyroid disease. Eur Thyroid J (2016) 5:171–9.10.1159/000447967
35
RussellWHarrisonRFSmithNDarzyKShaletSWeetmanAPet alFree triiodothyronine has a distinct circadian rhythm that is delayed but parallels thyrotropin levels. J Clin Endocrinol Metab (2008) 93:2300–6.10.1210/jc.2007-2674
36
ChatzitomarisAHoermannRMidgleyJEHeringSUrbanADietrichBet alThyroid allostasis-adaptive responses of thyrotropic feedback control to conditions of strain, stress, and developmental programming. Front Endocrinol (2017) 8:163.10.3389/fendo.2017.00163
37
HoermannRMidgleyJEMLarischRDietrichJW. Advances in applied homeostatic modelling of the relationship between thyrotropin and free thyroxine. PLoS One (2017) 12(11):e0187232.10.1371/journal.pone.0187232
38
HoermannRMidgleyJEMDietrichJWLarischR. Dual control of pituitary thyroid stimulating hormone secretion by thyroxine and triiodothyronine in athyreotic patients. Ther Adv Endocrinol Metab (2017) 8:83–95.10.1177/2042018817716401
39
ReinehrT. Thyroid function in the nutritionally obese child and adolescent. Curr Opin Pediatr (2011) 23:415–20.10.1097/MOP.0b013e328344c393
40
HoermannRCheungASMilneM. Hypothalamic-pituitary-thyroid axis setpoint alterations are associated with body composition in androgen deprived men. J Edocr Soc (2017) 1:874–85.10.1210/js.2017-00057
41
RodríguezEMBlázquezJLPastorFEPeláezBPeñaPPeruzzoBet alHypothalamic tanycytes: a key component of brain–endocrine interaction. Int Rev Cytol (2005) 247:89–164.10.1016/S0074-7696(05)47003-5
42
FeketeCLechanRM. Negative feedback regulation of hypophysiotropic thyrotropin-releasing hormone (TRH) synthesizing neurons: role of neuronal afferents and type 2 deiodinase. Front Neuroendocrinol (2007) 28:97–114.10.1016/j.yfrne.2007.04.002
43
VisserWEFriesemaECHVisserTJ. Minireview: thyroid hormone transporters: the knowns and the unknowns. Mol Endocrinol (2011) 25:1–14.10.1210/me.2010-0095
44
KallóIMohácsikPVidaBZeöldABardócziZZavackiAMet alA novel pathway regulates thyroid hormone availability in rat and human hypothalamic neurosecretory neurons. PLoS One (2012) 7:e37860.10.1371/journal.pone.0037860
45
FeketeCLechanRM. Central regulation of hypothalamic-pituitary-thyroid axis under physiological and pathophysiological conditions. Endocr Rev (2014) 35:159–94.10.1210/er.2013-1087
46
GerebenBMcAninchEARibeiroMOBiancoAC. Scope and limitations of iodothyronine deiodinases in hypothyroidism. Nat Rev Endocrinol (2015) 11:642–52.10.1038/nrendo.2015.155
47
HerwigACampbellGMayerC-DBoelenAAndersonRARossAWet alA thyroid hormone challenge in hypothyroid rats identifies T3 regulated genes in the hypothalamus and in models with altered energy balance and glucose homeostasis. Thyroid (2014) 24:1575–93.10.1089/thy.2014.0169
48
Joseph-BravoPJaimes-HoyLCharliJ-L. Regulation of TRH neurons and energy homeostasis-related signals under stress. J Endocrinol (2015) 224:R139–59.10.1530/JOE-14-0593
49
MenendezCBaldelliRCamiñaJPEscuderoBPeinoRDieguezCet alTSH stimulates leptin secretion by a direct effect on adipocytes. J Endocrinol (2003) 176:7–12.10.1677/joe.0.1760007
50
ReinehrTde SousaGAndlerW. Hyperthyrotropinemia in obese children is reversible after weight loss and is not related to lipids. J Clin Endocrinol Metab (2006) 91:3088–91.10.1210/jc.2006-0095
51
BiondiB. Thyroid and obesity: an intriguing relationship. J Clin Endocrinol Metab (2010) 95:3614–7.10.1210/jc.2010-1245
52
GerebenBZavackiAMRibichSKimBWHuangSASimonidesWSet alCellular and molecular basis of deiodinase-regulated thyroid hormone signaling. Endocr Rev (2008) 29:898–938.10.1210/er.2008-0019
53
BernalJGuadaño-FerrazAMorteB. Thyroid hormone transporters—functions and clinical implications. Nat Rev Endocrinol (2015) 11:406–17.10.1038/nrendo.2015.66
54
WanWFarboudBPrivalskyML. Pituitary resistance to thyroid hormone syndrome is associated with T3 receptor mutants that selectively impair beta2 isoform function. Mol Endocrinol (2005) 19:1529–42.10.1210/me.2005-0014
55
MammenJSMcGreadyJLadensonPWSimonsickEM. Unstable thyroid function in older adults is caused by alterations in both thyroid and pituitary physiology and associated with increased mortality. Thyroid (2017) 27:1370–7.10.1089/thy.2017.0211
56
CarleALaurbergPPedersenIBPerrildHOvesenLRasmussenLBet alAge modifies the pituitary TSH response to thyroid failure. Thyroid (2007) 17:139–44.10.1089/thy.2006.0191
57
PeetersRP. Thyroid hormones and aging. Hormones (2008) 7:28–35.10.14310/horm.2002.1111035
58
StrichDKaravaniGEdriSGillisD. TSH enhancement of FT4 to FT3 conversion is age dependent. Eur J Endocrinol (2016) 175:49–54.10.1530/EJE-16-0007
59
VadivelooTDonnanPTMurphyMJLeeseGP. Age- and gender-specific TSH reference intervals in people with no obvious thyroid disease in Tayside, Scotland: the thyroid epidemiology, audit, and research study (TEARS). J Clin Endocrinol Metab (2013) 98:1147–53.10.1210/jc.2012-3191
60
EhrenkranzJBachPRSnowGLSchneiderALeeJLIlstrupSet alCircadian and circannual rhythms in thyroid hormones: determining the TSH and free T4 reference intervals based upon time of day, age, and sex. Thyroid (2015) 25:954–61.10.1089/thy.2014.0589
61
HoermannREcklWAHoermannCLarischR. Complex relationship between free thyroxine and TSH in the regulation of thyroid function. Eur J Endocrinol (2010) 162:1123–9.10.1530/EJE-10-0106
62
ClarkPMSHolderRLHaqueSMHobbsFDRRobertsLMFranklynJA. The relationship between serum TSH and free T4 in older people. J Clin Pathol (2012) 65:463–5.10.1136/jclinpath-2011-200433
63
MidgleyJEMHoermannRLarischRDietrichJW. Physiological states and functional relation between thyrotropin and free thyroxine in thyroid health and disease: in vivo and in silico data suggest a hierarchical model. J Clin Pathol (2013) 66:335–42.10.1136/jclinpath-2012-201213
64
HadlowNCRothackerKMWardropRBrownSJLimEMWalshJP. The relationship between TSH and free T4 in a large population is complex and nonlinear and differs by age and sex. J Clin Endocrinol Metab (2013) 98:2936–43.10.1210/jc.2012-4223
65
HoermannRMidgleyJEMGiacobinoAEcklWAWahlHGDietrichJWet alHomeostatic equilibria between free thyroid hormones and pituitary thyrotropin are modulated by various influences including age, body mass index and treatment. Clin Endocrinol (Oxf) (2014) 81:907–15.10.1111/cen.12527
66
AndersenSPedersenKMBruunNHLaurbergP. Narrow individual variations in serum T(4) and T(3) in normal subjects: a clue to the understanding of subclinical thyroid disease. J Clin Endocrinol Metab (2002) 87:1068–72.10.1210/jcem.87.3.8165
67
van de VenACNetea-MaierRTMediciMSweepFCGJRossHAHofmanAet alUnderestimation of effect of thyroid function parameters on morbidity and mortality due to intra-individual variation. J Clin Endocrinol Metab (2011) 96:E2014–7.10.1210/jc.2011-0680
68
BrownSJBremnerAPHadlowNCFeddemaPLeedmanPJO’LearyPCet alThe log TSH-free T4 relationship in a community-based cohort is nonlinear and is influenced by age, smoking and thyroid peroxidase antibody status. Clin Endocrinol (Oxf) (2016) 85:789–96.10.1111/cen.13107
69
FliersEKalsbeekABoelenA. Beyond the fixed setpoint of the hypothalamus-pituitary-thyroid axis. Eur J Endocrinol (2014) 171:R197–208.10.1530/EJE-14-0285
70
BoonenEVan den BergheG. Endocrine responses to critical illness: novel insights and therapeutic implications. J Clin Endocrinol Metab (2014) 99:1569–82.10.1210/jc.2013-4115
71
Van den BergheG. Non-thyroidal illness in the ICU: a syndrome with different faces. Thyroid (2014) 24:1456–65.10.1089/thy.2014.0201
72
GoemannIMRomittiMMeyerELSWajnerSMMaiaAL. Role of thyroid hormones in the neoplastic process: an overview. Endocr Relat Cancer (2017) 24:R367–85.10.1530/ERC-17-0192
73
YatesDSMooreDSStarnesDS. The Practice of Statistics. 3rd ed. New York: WH Freeman and Company (2008).
74
MuthénBO. Latent variable modeling in heterogeneous populations. Psychometrika (1989) 54:557–85.10.1007/BF02296397
75
SimpsonEH. The interpretation of interaction in contingency tables. J R Stat Soc (1951) 13:238–41.
76
WagnerCH. Simpson’s paradox in real life. Am Stat (1982) 36:46.10.2307/2684093
77
GusseklooJvan ExelEde CraenAJMMeindersAEFrölichMWestendorpRGJ. Thyroid status, disability and cognitive function, and survival in old age. JAMA (2004) 292:2591–9.10.1001/jama.292.21.2591
78
van den BeldAWVisserTJFeeldersRAGrobbeeDELambertsSWJ. Thyroid hormone concentrations, disease, physical function, and mortality in elderly men. J Clin Endocrinol Metab (2005) 90:6403–9.10.1210/jc.2005-0872
79
WaringACArnoldAMNewmanABBùzkováPHirschCCappolaAR. Longitudinal changes in thyroid function in the oldest old and survival: the cardiovascular health study all-stars study. J Clin Endocrinol Metab (2012) 97:3944–50.10.1210/jc.2012-2481
80
YeapBBAlfonsoHHankeyGJFlickerLGolledgeJNormanPEet alHigher free thyroxine levels are associated with all-cause mortality in euthyroid older men: the health in men study. Eur J Endocrinol (2013) 169:401–8.10.1530/EJE-13-0306
81
SelmerCOlesenJBHansenMLKappelgaard vonLMMadsenJCHansenPRet alSubclinical and overt thyroid dysfunction and risk of all-cause mortality and cardiovascular events: a large population study. J Clin Endocrinol Metab (2014) 99:2372–82.10.1210/jc.2013-4184
82
BaumgartnerCda CostaBRColletT-HFellerMFlorianiCBauerDCet alThyroid function within the normal range, subclinical hypothyroidism and the risk of atrial fibrillation. Circulation (2017) 136:2100–16.10.1161/CIRCULATIONAHA.117.028757
83
Grozinsky-GlasbergSFraserANahshoniEWeizmanALeiboviciL. Thyroxine-triiodothyronine combination therapy versus thyroxine monotherapy for clinical hypothyroidism: meta-analysis of randomized controlled trials. J Clin Endocrinol Metab (2006) 91:2592–9.10.1210/jc.2006-0448
84
WiersingaWM. Therapy of endocrine disease: T4+T3 combination therapy: is there a true effect?Eur J Endocrinol (2017) 177:R287–96.10.1530/EJE-17-0645
85
GulloDLatinaAFrascaFLe MoliRPellegritiGVigneriR. Levothyroxine monotherapy cannot guarantee euthyroidism in all athyreotic patients. PLoS One (2011) 6:e22552.10.1371/journal.pone.0022552
86
ItoMMiyauchiAMoritaSKudoTNishiharaEKiharaMet alTSH-suppressive doses of levothyroxine are required to achieve preoperative native serum triiodothyronine levels in patients who have undergone total thyroidectomy. Eur J Endocrinol (2012) 167:373–8.10.1530/EJE-11-1029
87
ItoMMiyauchiAKangSHisakadoMYoshiokaWIdeAet alEffect of the presence of remnant thyroid tissue on the serum thyroid hormone balance in thyroidectomized patients. Eur J Endocrinol (2015) 173:333–40.10.1530/EJE-15-0138
88
PetersonSJMcAninchEABiancoAC. Is a normal TSH synonymous with “euthyroidism” in levothyroxine monotherapy?J Clin Endocrinol Metab (2016) 101:4964–73.10.1210/jc.2016-2660
89
MidgleyJEMLarischRDietrichJWHoermannR. Variation in the biochemical response to L-thyroxine therapy and relationship with peripheral thyroid hormone conversion. Endocr Connect (2015) 4:196–205.10.1530/EC-15-0056
90
ItoMMiyauchiAHisakadoMYoshiokaWIdeAKudoTet alBiochemical markers reflecting thyroid function in athyreotic patients on levothyroxine monotherapy. Thyroid (2017) 27:484–90.10.1089/thy.2016.0426
91
Escobar-MorrealeHFdel ReyFEObregónMJde EscobarGM. Only the combined treatment with thyroxine and triiodothyronine ensures euthyroidism in all tissues of the thyroidectomized rat. Endocrinology (1996) 137:2490–502.10.1210/endo.137.6.8641203
92
Werneck de CastroJPFonsecaTLUetaCBMcAninchEAAbdallaSMWittmannGet alDifferences in hypothalamic type 2 deiodinase ubiquitination explain localized sensitivity to thyroxine. J Clin Invest (2015) 125:769–81.10.1172/JCI77588
93
WiersingaWMDuntasLFadeyevVNygaardBVanderpumpMP. 2012 ETA guidelines: the use of L-T4 + L-T3 in the treatment of hypothyroidism. Eur Thyroid J (2012) 1:55–71.10.1159/000339444
94
BiondiBWartofskyL. Treatment with thyroid hormone. Endocr Rev (2014) 35:433–512.10.1210/er.2013-1083
95
BaliramRLatifRZaidiMDaviesTF. Expanding the role of thyroid-stimulating hormone in skeletal physiology. Front Endocrinol (2017) 8:758–9.10.3389/fendo.2017.00252
96
OttJPrombergerRKoberFNeuholdNTeaMHuberJCet alHashimoto’s thyroiditis affects symptom load and quality of life unrelated to hypothyroidism: a prospective case-control study in women undergoing thyroidectomy for benign goiter. Thyroid (2011) 21:161–7.10.1089/thy.2010.0191
97
Kelderman-BolkNVisserTJTijssenJPBerghoutA. Quality of life in patients with primary hypothyroidism related to BMI. Eur J Endocrinol (2015) 173:507–15.10.1530/EJE-15-0395
98
ApplewhiteMKJamesBCKaplanSPAngelosPKaplanELGroganRHet alQuality of life in thyroid cancer is similar to that of other cancers with worse survival. World J Surg (2016) 40:551–61.10.1007/s00268-015-3300-5
99
HedmanCDjärvTStrangPLundgrenCI. Effect of thyroid-related symptoms on long-term quality of life in patients with differentiated thyroid carcinoma: a population-based study in Sweden. Thyroid (2017) 27:1034–42.10.1089/thy.2016.0604
100
AndersenSBruunNHPedersenKMLaurbergP. Biologic variation is important for interpretation of thyroid function tests. Thyroid (2003) 13:1069–78.10.1089/105072503770867237
101
HoermannRMidgleyJEM. TSH measurement and its implications for personalised clinical decision-making. J Thyroid Res (2012) 2012:1–9.10.1089/thy.2008.0155
Summary
Keywords
setpoint, thyroid homeostasis, thyroid-stimulating hormone, levothyroxine treatment
Citation
Hoermann R, Midgley JEM, Larisch R and Dietrich JW (2017) Recent Advances in Thyroid Hormone Regulation: Toward a New Paradigm for Optimal Diagnosis and Treatment. Front. Endocrinol. 8:364. doi: 10.3389/fendo.2017.00364
Received
30 October 2017
Accepted
12 December 2017
Published
22 December 2017
Volume
8 - 2017
Edited by
Noriyuki Koibuchi, Gunma University, Japan
Reviewed by
Xuguang Zhu, National Cancer Institute (NIH), United States; Pieter de Lange, Università degli Studi della Campania “Luigi Vanvitelli” Caserta, Italy
Updates
Copyright
© 2017 Hoermann, Midgley, Larisch and Dietrich.
This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Rudolf Hoermann, rudolf.hoermann@gmail.com
Specialty section: This article was submitted to Thyroid Endocrinology, a section of the journal Frontiers in Endocrinology
Homeostasis and allostasis of thyroid function: https://www.frontiersin.org/research-topics/4262/homeostasis-and-allostasis-of-thyroid-function
Disclaimer
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.