 Fazal Wahab
Fazal Wahab Bibi Atika2
Bibi Atika2 Muhammad Shahab
Muhammad Shahab Rüdiger Behr
Rüdiger Behr- 1Platform Degenerative Diseases, German Primate Center, Leibniz Institute for Primate Research, Göttingen, Germany
- 2Department of Developmental Biology, Faculty of Biology, University of Göttingen, Göttingen, Germany
- 3Department of Zoology, Islamia College University, Peshawar, Pakistan
- 4Laboratory of Reproductive Neuroendocrinology, Department of Animal Sciences, Faculty of Biological Sciences, Quiad-i-Azam University, Islamabad, Pakistan
- 5DZHK (German Center for Cardiovascular Research), Partner Site Göttingen, Göttingen, Germany
A large body of data has established the hypothalamic kisspeptin (KP) and its receptor, KISS1R, as major players in the activation of the neuroendocrine reproductive axis at the time of puberty and maintenance of reproductive capacity in the adult. Due to its strategic location, this ligand-receptor pair acts as an integrator of cues from gonadal steroids as well as of circadian and seasonal variation-related information on the reproductive axis. Besides these cues, the activity of the hypothalamic KP signaling is very sensitive to the current metabolic status of the body. In conditions of energy imbalance, either positive or negative, a number of alterations in the hypothalamic KP signaling pathway have been documented in different mammalian models including nonhuman primates and human. Deficiency of metabolic fuels during fasting causes a marked reduction of Kiss1 gene transcript levels in the hypothalamus and, hence, decreases the output of KP-containing neurons. Food intake or exogenous supply of metabolic cues, such as leptin, reverses metabolic insufficiency-related changes in the hypothalamic KP signaling. Likewise, alterations in Kiss1 expression have also been reported in other situations of energy imbalance like diabetes and obesity. Information related to the body’s current metabolic status reaches to KP neurons both directly as well as indirectly via a complex network of other neurons. In this review article, we have provided an updated summary of the available literature on the regulation of the hypothalamic KP-Kiss1r signaling by metabolic cues. In particular, the potential mechanisms of metabolic impact on the hypothalamic KP-Kiss1r signaling, in light of available evidence, are discussed.
Introduction
Kisspeptin (KP), a hypothalamic neuropeptide, and KISS1R/Kiss1r, the KP receptor, are the main components of an important hypothalamic signaling pathway (1, 2). KP and KISS1R are encoded by KISS1 and KISS1R genes, respectively (3, 4). A large body of data has established an important role for the KP-Kiss1r signaling in the initiation of puberty in both non-primate and primate vertebrates (5–8). Loss of function mutations in human KISS1 or KISS1R genes causes absence of or delayed puberty (8–11), whereas a gain of function mutation in KISS1R gene results in precocious puberty (12). Likewise, administration of KP in immature rats elicits an early onset of puberty, whereas KP antagonist infusion leads to a delay in the achievement of pubertal hallmarks (5, 13, 14).
Kisspeptin signaling also plays an important role in the maintenance of the reproductive capacity in the adult (1, 15–17). Administration of KP, peripherally as well as centrally, has been reported to markedly increase systemic levels of reproductive hormones both in normal as well as subjects with reproductive insufficiency phenotype (7, 15, 18, 19). Due to their strategic position in the hypothalamus, the KP-containing neurons also act as a conduit for transferring information related to a number of different intrinsic and extrinsic cues to the gonadotropin-releasing hormone (GnRH) neurons (Figure 1). These neurons are involved in circadian and seasonal regulation of reproduction (20, 21). Moreover, this ligand-receptor pair acts as an integrator of the action of gonadal steroids and metabolic cues on the reproductive axis (22–26).
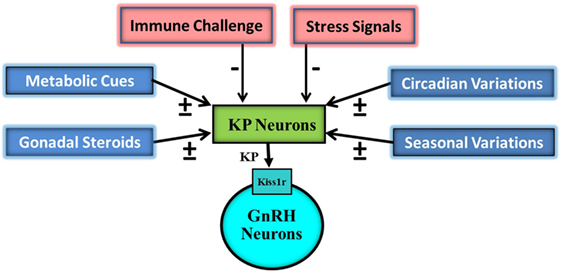
Figure 1. Schematic representation of impact of various external and internal signals on the hypothalamic Kisspeptin (KP) system. KP neurons are targeted by gonadal steroids, metabolic, circadian, seasonal, immune, and stress signals. Some of gonadal steroids, metabolic, circadian, and seasonal signals result in upregulation (+) of KP expressions while others in downregulation. Immune and stress signals cause down regulation (−) of KP expression. KP neurons then on the basis of this information modulate pulsatile discharge of gonadotropin-releasing hormone (GnRH) from GnRH neurons.
Proper functioning of the hypothalamic KP signaling is very sensitive to the current metabolic status of the body (23, 25) (Table 1). Conditions of energy imbalance, either positive or negative, induce a number of alterations in the hypothalamic KP signaling pathway in different mammalian experimental animal models (22, 26, 27). Deficiency of metabolic fuels during fasting causes a clear reduction of the Kiss1 gene transcript levels in the hypothalamus and hence decreases the output of KP-containing neurons (5, 28). Food intake or exogenous supply of metabolic cues, such as leptin, overcomes metabolic insufficiency-related changes in the hypothalamic KP signaling (29, 30). Likewise, alterations in Kiss1 expression have also been reported in other situations of energy imbalance like diabetes and obesity (30, 31). All these findings indicate a high sensitivity of KP signaling to alterations in the body’s energy homeostasis. In this review, we summarize and discuss the presently available pieces of evidence indicating an impact of metabolic status-related cues on the hypothalamic KP-Kiss1r signaling in conditions of energy imbalance. We also discuss potential mechanisms of the transmission of the metabolic information on the hypothalamic KP system and ultimately reproduction.
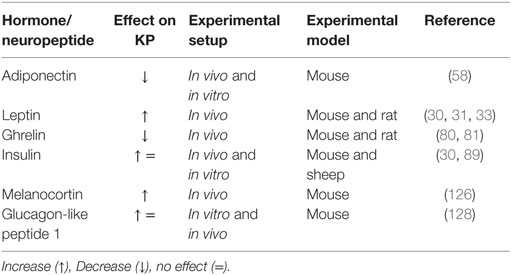
Table 1. Effect of different metabolic hormones and neuropeptides on the hypothalamic Kisspeptin (KP) system under different experimental setup in rodents and primates.
Sensitivity of the Hypothalamic KP-Kiss1r Signaling Pathway to Metabolic Alterations in Conditions of Altered Energy Homeostasis
The hypothalamic KP-Kiss1r system is highly sensitive to alterations in the metabolic cues levels in the systemic circulation. All sorts of metabolic perturbances exert negative impact on the Kiss1 expressing neurons (Figure 2) (22, 23, 26, 27, 32). It is well established that reduction of metabolic fuels in food-deprived conditions causes a decrease in Kiss1 transcript levels in the arcuate nucleus (ARC) (5, 28). In some conditions of energy imbalance, such as diabetes and obesity, very high energy reserves are present in the body, but due to the body’s inability to properly utilize them, an attenuation of Kiss1 mRNA expression was observed (30, 31, 33).
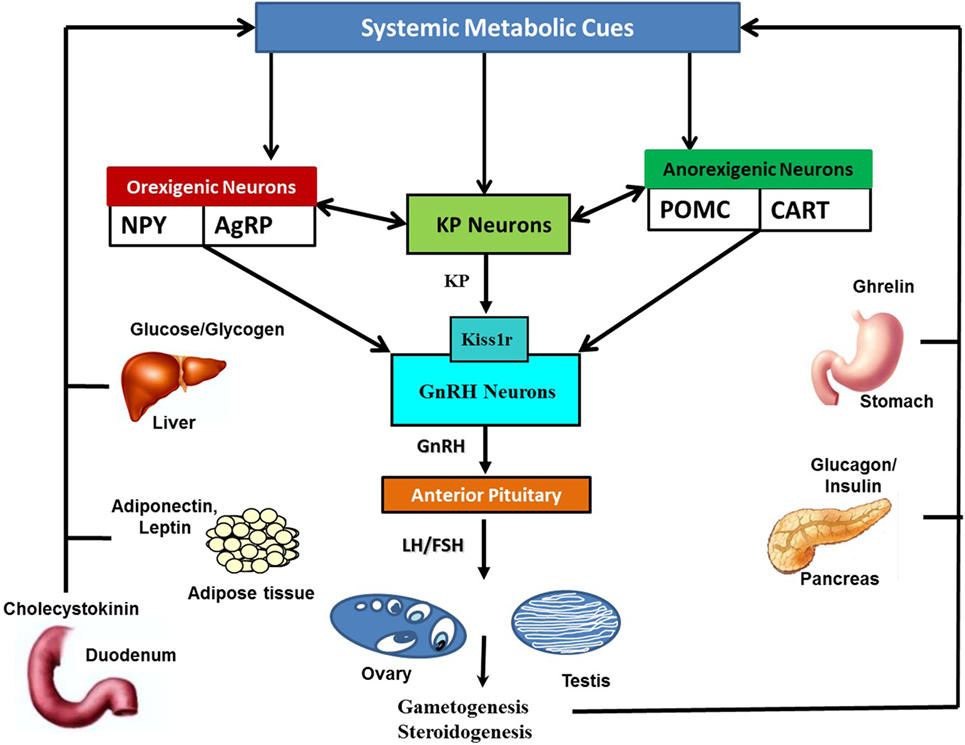
Figure 2. Schematic representation of the interaction of systemic metabolic cues with Kisspeptin (KP), orexigenic, and anorexigenic neurons: metabolic cues are secreted by metabolic organs in responses to alterations in metabolic status. Metabolic cues include insulin and glucagon from pancreas, leptin, adiponectin and leptin from adipose tissues, ghrelin from stomach, glucose, fatty acid, cholecystokinin, glucocorticoids, and thyroid hormones, among many others. Alterations in metabolic cues, either directly or indirectly via anorexigenic and orexigenic neurons, modulate KP neuronal activities. KP neurons in turn transfer this information to the HPG axis via gonadotropin-releasing hormone (GnRH) neuronal network. Likewise, orexigenic and anorexigenic neurons can also directly convey current metabolic status related information to GnRH neurons.
Both direct and indirect evidence suggests that deficiency in metabolic fuels severely affects the KP neuronal network in the hypothalamus. Short-term fasting-associated metabolic cues alterations lead to a marked reduction of hypothalamic Kiss1 expression in prepubertal as well as adult rats (5, 29, 34–36). Castellano et al. (5) carried out a first comprehensive analysis of the short-term fasting impact on the hypothalamic Kiss1 system in prepubertal rats. In fasted rats, delayed puberty, as monitored by vaginal opening, was associated with a reduction of whole hypothalamic Kiss1 gene transcript levels. However, Kiss1r mRNA expression was increased in these rats as compared to normally fed control animals. A possible explanation, as provided by authors (5), for this opposite change in Kiss1 and Kiss1r is that a major reduction in ligand (KP encoding gene) expression might cause a compensatory increase in the expression of its receptor gene, leading to a situation of sensitization to the effects of KP. Importantly, exogenous administration of KP not only rescues the suppression of the reproductive axis in these rats but also overcomes the negative energy balance-induced pubertal delay (5). This finding indicates that a proper reserve of energy is critical for the achievement of reproductive capacity at the time of puberty. The energy reserve related cues, in turn, communicate with the neuroendocrine center for the regulation of reproduction through the hypothalamic neural circuitry of KP neurons (37). Subsequent studies analyzed the impact of food restriction on the distinct hypothalamic KP neuron subpopulations in ARC and anteroventral periventricular nucleus (AVPV). In adult ovariectomized female rats, fasting decreased AVPV Kiss1 mRNA levels, but not Kiss1 mRNA expression in the ARC (34). In the intact adult female rats, food deprivation resulted in a prolongation of the reproductive cycle via a reduction in ARC Kiss1 mRNA expression (34). However, these researchers did not observe any changes in AVPV Kiss1 mRNA expression. Likewise, chronic food deprivation in pubertal female rats diminished expression of Kiss1 in ARC, but not in the AVPV (38). In mice and rhesus macaques, in contrast to rats, the hypothalamic transcript levels of both Kiss1 and Kiss1r are reduced by a 48-h fast (28, 30).
In addition to the aforementioned expression data, KP administration data also indirectly pinpointed a high sensitivity of the hypothalamic KP system to fasting-induced negative energy balance (5, 39). Administration of exogenous KP has been documented to overcome the negative energy balance-induced suppression of the reproductive axis, further advocating the idea that the endogenous KP system is negatively affected by fasting (5, 39).
Besides the condition of fasting, experimental data from other paradigms of energy imbalance such as diabetes, obesity, and lactation also indicate an impact of metabolic perturbations on the KP neurons output (30, 31, 40). The hypothalamic expression of Kiss1 gene is significantly reduced not only in the rat model of diabetes but also in obesity rodent models (30, 31, 40). In both congenital leptin deficiency and high-fat-diet-induced models of obesity, Kiss1-expressing neurons output is greatly reduced (33, 40). Likewise, a reduction in Kiss1 expression has also been reported in lactating female rats (41, 42). Moreover, exogenous administration of KP has been noted to rescue the energy imbalance impact on the reproductive axis (41, 42).
Taken together, the evidence summarized above strongly suggests a very high sensitivity of KP-containing neurons to metabolic alterations in the body.
Mechanism of Metabolic Impact on the Hypothalamic KP System
The exact mechanism by which changes in metabolic cues alter the hypothalamic KP system is still not fully clear. Available data suggests both direct and indirect mechanisms. Hypothalamic KPergic neurons can most likely sense metabolic cues directly because receptors for a number of peripheral metabolic hormones have been shown to be expressed by these neurons (22, 23, 25, 26, 32). Indirect sensing of metabolic status-related information is also possible because KP neurons receive information from various neuronal networks by direct cell-cell-communication, and neurons capable of sensing systemic metabolic cues are part of these networks (23, 25, 32, 43, 44). In this section, we summarize available data on both direct and indirect impact of metabolic cues on the hypothalamic KPergic neurons.
Direct Impact of Peripheral Metabolic Factors on KP Secreting Neurons in the Hypothalamus
Adiponectin
Adiponectin, a white adipocyte-secreted adipocytokine, was first documented in 1995 independently by various groups (45–48). It is a 244 amino acid protein hormone encoded by the APMI gene. It is secreted in very large amount into the systemic circulation. It has been noted to be about 0.01–0.05% of the total systemically circulating proteins (45–49). Systemic concentration of adiponectin is ranged from 3 to 30 µg/mL (45). Adiponectin levels are sexually dimorphic as its concentration is higher in females than in males (45). In various metabolic disorders, such as obesity and diabetes, a marked reduction in plasma adiponectin levels has been reported (49, 50). Nevertheless, its levels are markedly elevated during fasting and are positively associated with severe weight reduction although in these situations the body has a greatly reduced adipose tissue mass (51, 52). This elevation in plasma adiponectin levels during food restriction condition is caused by adipose tissue in bone marrow. In contrast to other parts of the body, a prominent increase in the mass of adipose tissue in bone marrow has been noted in food restriction conditions (53).
Adiponectin exerts its biological action via two 7-transmembrane receptors, AdipoR1 and AdipoR2 (45, 51), which are structurally as well as functionally different from 7-transmembrane G protein-coupled receptors. These receptors constitute a subgroup of 7-transmembrane receptors together with 11 progestin AdipoQ receptors (45). Besides peripheral organs, studies have demonstrated expression of both AdipoR1 and AdipoR2 in various brain regions, including the hypothalamus, although evidence for the transport of adiponectin across the blood-brain barrier is still lacking (54–56).
Binding of adiponectin to its receptor leads to the activation of 5’ AMP-activated protein kinase (AMPK). The activated AMPK acts to regulate energy homeostasis of the cell via fatty acid oxidation and stimulation of glucose uptake (45, 49, 51). Moreover, adiponectin has been shown to modulate the release of reproductive hormones. Adiponectin inhibits LH, GnRH-stimulated LH, and GnRH secretion while no impact on follicle-stimulating hormone (FSH) secretion was noted (54, 56). Recently, Wen et al. (57). analyzed the adiponectin effect on hypothalamic Kiss1 mRNA expression in GT1-7 cells, which are immortalized mouse hypothalamic neuronal cells, and in vivo in rats. They showed that adiponectin, as well as a synthetic activator of AMPK, greatly reduced transcription of Kiss1 mRNA while inhibition of AMPK caused an increase in expression of Kiss1 mRNA in both in vitro and in vivo studies. Taken together, these findings suggest a negative impact of adiponectin on the activities of KP-containing neurons. The negative impact of adiponectin on Kiss1 expression suggests that it might be involved in short-term fasting induced suppression of the reproductive axis. In fasting condition, an increase in systemic levels of adiponectin has been reported.
Leptin
Leptin is another important adipokine of white adipose tissue. In contrast to systemic adiponectin levels, leptin levels in the bloodstream are directly related to the body mass of adipose tissues. Leptin plays a vital role in the maintenance of energy balance in the body (58–60). One of the key functions of leptin is to communicate information on the body’s current metabolic status to brain centers for energy homeostasis (61, 62). Systemic concentrations of leptin are reduced in food restriction conditions while food intake augments leptin concentrations (63). Available experimental data show that leptin is an important regulator of the metabolic deficiency/sufficiency-induced alterations in the neuroendocrine axes. Thereby, it also affects reproductive functions (37, 60, 64).
Besides peripheral reproductive organs, expression of the leptin receptor (LepR) has also been noted in several central neuronal networks in the hypothalamus, including KP-secreting neurons (33, 37). In situations of energy imbalance, low levels of leptin cause a clear reduction in Kiss1 transcripts levels in the hypothalamus (28, 30, 31, 33, 40) while the elevation of systemic leptin concentrations via exogenous administration greatly ameliorates expression of Kiss1 transcripts levels (30, 33). Similarly, ablation of leptin in ob/ob mice and hypoleptinemia in experimental diabetic rats diminish Kiss1 mRNA expression while leptin infusion in both, ob/ob mice and in the rat model augments Kiss1 transcript levels (30, 31, 33). Leptin can also indirectly change activities of KP-secreting neurons because many studies have reported the expression of LepR in numerous discrete regions of the hypothalamus (58). Important neuronal populations that express LepR include the GABAergic, neuropeptide Y (NPY), proopiomelanocortin (POMC), and agouti-related peptide (AgRP) populations (44, 58, 65, 66). These neurons are known to communicate with KP neurons (43, 44, 67). The indirect impact of leptin on KP neurons is supported by the evidence that exogenous leptin injection was unable to induce signal transducer and activator of transcription-3 (STAT3), a leptin action mediating intracellular signaling pathway, expression in KP neurons (65).
However, Donato et al. (68) have recently shown that hypothalamic KP neuronal LepR deletion did not change LH secretion. Likewise, re-expression of LepR on KP cells in LepR null mice also did not improve hypogonadotropic hypogonadism phenotype in these mice (69). These observations, together with above mentioned findings (28, 30, 31, 33, 40) of a pivotal role of leptin in KP secretion, suggest a potential developmental compensation or an indirect effect of leptin in modulating KP secretion in mice. Nevertheless, more studies are required in other species to further clarify the link between leptin and KP.
Ghrelin
Ghrelin, an orexigenic peptide hormone of the upper gastrointestinal track, is a ligand of growth hormone secretagogue receptor (GHSR), which is also a member of the seven transmembrane receptor family (70–72). Ghrelin has been implicated in the short-term regulation of food intake. The systemic concentrations of ghrelin increase at the preprandial time, whereas they decrease postprandially (70, 72, 73). In food restriction conditions, increased ghrelin levels in the circulation are associated with a decrease in reproductive hormones (74). Exogenous ghrelin administration rapidly induces food intake and inhibits the reproductive axis (70, 72, 74, 75). Besides short-term actions on food intake, ghrelin is also involved in the regulation of long-term body weight. Chronic administration of ghrelin increases the body weight through a number of mechanisms, including continuous stimulation of food intake, alterations in energy expenditure, and induction of adiposity (75). In mice, congenital loss of ghrelin or of the GHSR gene causes resistance to high-fat-diet-induced adiposity and weight gain (76, 77). Likewise, ablation of both ligand and receptor resulted in reduced body weight of mice, high energy expenditure, and increased motor activity on a standard chow without exposure to a high-fat diet (78). All in all, the available data pinpoint an important role of ghrelin in monitoring and transferring metabolic information to the brain centers implicated in the regulation of reproduction and intake of food intake.
Ghrelin acts centrally in the brain via GHSR in the hypothalamus to stimulate food intake and to alter reproduction (72, 75). Expression of GHSRs has been observed on a subset of Kiss1-expressing neurons. In 2009, Forbes et al. (79) reported a reduction in the hypothalamic transcript levels of Kiss1 in response to an increase in circulating ghrelin levels either due to food deprivation or exogenous injection of ghrelin. Besides this direct action of ghrelin on the hypothalamic Kiss1 gene expression, an indirect action via interneurons like the AgRP/NPY neurons (75), which will be discussed below, is also possible.
An important role of estradiol has been reported in the modulation of KP neuronal response to ghrelin by Frazao et al. (80). These researchers found that elevated levels of estradiol augment transcript levels of GHSR in the hypothalamic ARC. Moreover, an increase in the number of KPergic neurons responding to ghrelin was noted (80). Very recently, it has been reported that an increase in ghrelin levels during the short-term fasting condition leads to a stimulatory effect of central KP on growth hormone secretion. This effect has not been observed in normal fed condition. Moreover, a ghrelin receptor antagonist or a block of increase in its systemic levels abolishes this effect of KP on growth hormone secretion. On the basis of these findings, it has been proposed that central KP neuronal networks might transfer reproductive and metabolic status related cues onto the somatotropic axis thus causing a change in the release of growth hormone (81).
Insulin
Insulin, a metabolic hormone secreted by the pancreatic β cells, is involved in metabolic regulation of reproduction through actions on both central and peripheral components of the reproductive axis (82, 83). Central injections of insulin cause a dose-dependent attenuation in feeding and body weight (84). Ablation of insulin receptor (IR) from neurons results in hypogonadotropic hypogonadism in mice via central hypothalamic mechanism (85). Moreover, central injection of insulin has been reported to reinstate normal LH secretion in an experimental rat model of diabetes (86).
Besides many other neurons, expression of IRs has been noted on the ARC KP cells (87). It has been found that the specific deletion of the IR gene from KP neurons delayed the onset of puberty in mice but reproductive capacity was normal in adulthood (87). Therefore, these observations indicate that insulin signaling in KP neurons is important for the normal pubertal awakening of the reproductive axis but not an absolutely critical signal for the achievement of ultimate pubertal hallmarks. Additionally, reproductive ability, feeding, glucose regulation, distribution of fat, and body weight were normal in adult mutants. Of note, administration of insulin in the late follicular ovarian phase significantly stimulated expression of the c-fos protein in sheep ARC KP neurons (88), although it is not clear whether this effect is direct or indirect.
Some indirect evidence supports a possible role of insulin in altering the activity of hypothalamic KP-secreting neurons. In rats, experimental chronic diabetes has been noted to cause a marked reduction of Kiss1 transcript levels in the hypothalamus (31, 89). Likewise, during short-term fasting, which is characterized by reduced levels of insulin, a reduction in Kiss1 expression was reported (28, 90). However, exogenous injections of insulin did not reverse the decreased Kiss1 gene expression, which was induced by fasting- and diabetes-associated metabolic perturbations (30, 31). Additionally, in vitro data did not show any effect of insulin on KP expression in the mouse hypothalamic cell line N6 (30). Of note, leptin and NPY applications have stimulated Kiss1 expression in this cell line.
Indirect Impact of Metabolic Cues on the Hypothalamic KPergic Neurons
Besides the per se impact of metabolic cues on the hypothalamic KPergic neurons, a number of other hypothalamic neuronal networks are also sensitive to metabolic status-related cues. The major hypothalamic neuronal systems, which express the LepR, the GHSR, and the IR, include gamma-aminobutyric acid (GABA), glutamate, NPY/AgRP, and POMC/CART neurons. Many of these neurons, in turn, can alter activities of the hypothalamic KPergic neurons either directly or indirectly.
Glutamate and GABA Expressing Neurons’ Input to KP Secreting Neurons in the Hypothalamus
Glutamate and GABA neurons are playing important roles in the regulation of reproduction (91). These neurons have been documented to contain receptors for metabolic hormones, and their activities are modulated by metabolic cues (44, 67, 92–94). In a recent study, we checked changes in the hypothalamic glutamate and GABA systems in fed and 48 hours fasted monkeys via checking transcripts levels of Kiss1, Kiss1r, NR1 (N-methyl-d-aspartate receptor subunit 1) and GAD67 (glutamic acid decarboxylase67) in the mediobasal hypothalamus (MBH) and pre-optic area (POA) of the adult male rhesus macaque (Macaca mulatta) (95). The expression of Kiss1, Kiss1r, and NR1 mRNA was greatly decreased in fasted macaques as compared to ad libitum fed monkeys. A noteworthy reduction was also noted in the expression of KP and the interactions of NR1 with KPergic neurons in the hypothalamus of fasted monkeys. Taken together, these observations indicate that a reduction in inputs of glutamate-containing neurons to KPergic neurons may be responsible for the reduction in the hypothalamic KP signaling in the fasted monkey. However, no obvious change in expression of GAD67 mRNA between fed and fasted monkey was observed, suggesting that the fasting-induced reduction in the hypothalamic KP signaling is not mediated through GABAergic neurons (95).
RFamide-Related Peptide-3 Expressing Neurons Input to KP-Secreting Neurons in the Hypothalamus
The hypothalamic gonadotropin-inhibitory hormone and its mammalian ortholog RFamide-related peptide-3 (RFRP-3) neurons have been implicated as the potent inhibitors of reproduction in a number of vertebrate species (26, 96–98). RFRP-3 binds to a G protein-coupled receptor namely GPR147. GPR147 is expressed in different regions of the hypothalamus including a subset of the hypothalamic KPergic neurons in the ARC (99). Moreover, a direct contact between GnIH fibers and about 35% of ARC KPergic neurons was also noted.
Different studies in animal models and human subjects analyzed of RFRP-3 effect on KP stimulation of GnRH (99, 100). In human subjects, although RFRP-3 exerts an inhibitory effect on LH secretion in postmenopausal women, no noteworthy effect of RFRP-3 was observed on KP-stimulated LH secretion in men during concomitant KP and RFRP-3 administration (100). In mouse hypothalamic explant culture, research from Tsutsui’s group showed that RFRP3 significantly reduced KP-induced GnRH release (99). Of note, no effect of RFRP-3 on KP-induced GnRH release was noted in the mouse hypothalamic GT1-7 cells (101).
Leon et al. performed an analysis of the GPR147 ablation on the hypothalamic Kiss1 mRNA expression (102). They reported that GPR147 null mice showed normal pubertal awakening of the reproductive axis. Of note, an increase in expression of Kiss1 mRNA was noted in the hypothalamic ARC of the adult GPR147 null male mice. Additionally, an increase in systemic levels of FSH and response of LH to GnRH stimulation was observed in GPR147 null mice. However, ablation of GPR147 did not rescue hypogonadotropic hypogonadism in Kiss1r-ablated mice. More importantly, in the GPR147 null mouse energy imbalance conditions induced a lesser degree of disruption in the secretion of LH (102). These findings indicate that a lack of RFRP3 signaling may partly prevent metabolic perturbation induced inhibition of the reproductive axis. However, expression of Kiss1 mRNA was not checked in GPR147 ablated mice in these conditions of metabolic perturbations. Therefore, it will be important to check Kiss1 expression in GPR147 null mice in situations of metabolic insufficiency in order to know whether RFRP-3 signaling mediates nutritional challenge induced suppression of the reproductive axis.
Orexigenic Neuronal Input to the KP Secreting Neurons in Hypothalamus
Hypothalamic orexigenic neurons include NPY and AgRP neurons among others (103–108). AgRP is utterly secreted by a specific neuronal population in the ARC, which also co-expresses NPY. These neurons are playing a crucial role in feeding. They stimulate feeding when they are activated by metabolic deficiency-associated signals (105, 109).
These neurons express receptors for several key metabolic hormones like leptin, insulin, and ghrelin (103, 107, 109). Hence, AgRP/NPY neurons are direct targets of leptin action (109, 110) Exogenous injection of leptin induces a mark activation of STAT3, a prominent leptin action mediating intracellular signaling pathway, in AgRP/NPY neurons (109–111). Insulin has been noted to inhibit the electrophysiological properties of NPY/AgRP neurons. Insulin causes inhibition of NPY/AgRP neurons through activation of ATP-sensitive K+ channels (112). However, ablation of IR from AgRP/NPY neurons does not induce prominent alterations of the reproductive axis, while deletion of both, IR and LepR, adversely affected the reproductive axis (113, 114). Ghrelin stimulates the activity of ARC AgRP/NPY neurons (75, 109, 115) via activation of GHSR present on these neurons (109, 116). More importantly, it has been reported that ghrelin’s orexigenic effects are lost in Agrp and Npy knockout mice, suggesting that intact NPY and AgRP neurons are essential for orexigenic effects of ghrelin (115).
Although a large body of data established the perception of metabolic cues by the AgRP/NPY neurons, there is only very limited information on possible routes via which the effects are transmitted to KP neurons in the ARC. In the ovine brain, Backholer et al. (43) observed the occurrence of reciprocal transsynaptic neural connections between the hypothalamic NPY-containing cells and the perikarya of the KP-expressing neurons. This anatomical evidence indicates that NPY-containing neurons can affect the output of KP neurons. More importantly, a normal NPY neuronal circuitry is essential for proper functioning of hypothalamic KPergic neurons, as mice with NPY deficiency have defective hypothalamic KP expression (30).
Recently, Foradori et al. provided more comprehensive evidence for cross-talk between KP and NPY neurons (81), especially from KP to NPY neurons in presence of the fasting-induced alteration in metabolic cues. KP administration in fasted ewe has been noted to cause a significant increase in growth hormone level via stimulation of NPY neurons and growth hormone releasing-hormone in ARC and an inhibition of somatostatin neurons in the periventricular nucleus.
Anorexigenic Neuronal Input to KP-Containing Neurons in the ARC
The hypothalamic ARC POMC (POMC)/CART (cocaine- and amphetamine-regulated transcript) neurons have been implicated as a pivotal central controller of metabolic homeostasis (109, 117, 118). These neurons have been described to constitute a major part of the hypothalamic satiety center. The anorexigenic role of POMC neurons is pinpointed by the evidence that ablation of the Pomc gene results in a state of severe hyperphagia, which ultimately leads to an enormous amount of weight gain (119). Moreover, food deprivation reduces mRNA levels of Pomc in the hypothalamic ARC, whereas transcript levels of hypothalamic Pomc are augmented in overfed rats (120). Similarly, CART mRNA expression is also at the nadir in fasting, while food intake restores ARC CART mRNA expression (121).
The possible metabolic cues that may be sensed by POMC neurons include leptin and insulin. Presence of both LepR and IR has been noted on POMC neurons (109, 117, 122–124). Recently, researchers documented via the whole-cell recording that both leptin and insulin excite POMC neurons and nearby KP cells via stimulation of TRPC5 (short transient receptor potential channel 5) channels (112), which are abundantly present in these hypothalamic neurons. Moreover, central administration of exogenous insulin greatly suppressed feeding and enhanced expression of the c-fos protein in ARC POMC neurons (112).
Indeed, POMC neurons are strategically located in the hypothalamus. Thereby, they can integrate the information provided by many different metabolic cues and can link these to the KP neurons. A direct action of POMC neurons is supported by the presence of reciprocal connections between POMC and KP neurons in the hypothalamus (43, 125). POMC neuronal projections were observed in close apposition with a number of other neurons which cross-talk with KP neurons. This also suggests an indirect connection.
Very recently, Tena-Sempere’s group uncovered a melanocortin-KP-GnRH regulatory pathway (126). This pathway was reported to be involved in transmitting leptin actions and plays an important role in regulating the onset of puberty. Of important note, KP neurons were noted to play a vital role in relaying the stimulatory effects of melanocortin signaling onto the reproductive centers (126). In this regard, they reported the existence of a close contact between α-MSH fibers and KP-containing neuronal cell bodies in the ARC of pubertal female rats while the chronic block of the melanocortin receptor, MC3/4R, results in a significant reduction of Kiss1 transcript levels. Moreover, the LH responses to the MT-II melanocortin agonist, which stimulates LH release, greatly reduced in Gpr54-ablated mice and also in DREADD-induced inhibition of ARC Kiss1 neurons. Altogether, these findings suggest central role KP in mediating impact of POMC neurons on to GnRH neurons during development and metabolic cues related changes.
Very recently, True et al. have reported several differences in coexpression patterns of various hypothalamic neuropeptides in female nonhuman primates as compared to rodents (127). They did not observe coexpression of CART with POMC but instead with NPY. They also noted co-expression of the CART in a subpopulation of KP cells. These CARP + KP neurons were noted to show close appositions with GnRH neurons. In contrast, the single-labeled KP and CART fibers were in synaptic contacts with GnRH neurons.
Heppner et al. (128) reported that KP neurons in the hypothalamic ARC receive synaptic input from glucagon-like peptide 1 (GLP-1), which is an anorexigenic neuropeptide. Moreover, KP neurons also express Glp1r mRNA. More importantly, they noted an increase in KP neurons action potential firing after application of the GLP-1R agonist. GLP-1R agonist also results in a direct membrane depolarization of ARC KP cells. However, central infusions of the GLP-1R antagonist, exendin (9–39), did not exert any effect on expression of ARC Kiss1mRNA or plasma LH in the normal fed mice (128).
Conclusion and Future Recommendations
In summary, emerging and increasing evidence indicates that metabolic cues exert a profound impact on the hypothalamic Kiss1-expressing neurons, both directly and indirectly. The direct sensing of metabolic cues is indicated by the presence of metabolic hormone receptors on Kiss1-expressing neurons while indirect sensing of metabolic information is suggested by cross-talk of these neurons with other hypothalamic neuronal populations which also respond to metabolic cues.
Most of the current evidence for the metabolic regulation of the hypothalamic KP system is provided by non-primate studies. Therefore, in the future, further studies in nonhuman primates are required to get more insight into the mechanism by which various peripheral metabolic cues (leptin, adiponectin, testosterone, estrogen, cortisol/corticosterone, ghrelin, insulin, glucagon, thyroid hormones, etc.) exert effects on their central neuronal targets (KPergic, AgRP/NPY, POMC/CART, GABA, corticotropin-releasing hormone, etc.). Indeed, deeper understanding of the metabolic impact on the hypothalamic KP signaling in animal models phylogenetically closer to humans and therefore with high clinical significance will more likely put Kiss1-Kiss1r signaling in the focus as a potential drug target. This may include improvement and management of reproductive functions as well as treatment of disorders of energy balance. Notably, an important role of KP has been shown in the restoration of the reproductive axis after its quiescence in metabolic disorders such as diabetes and hypothalamic amenorrhea (129, 130).
In 2014, Tolson et al. (131) have shown that ablation of KP signaling leads to a reduction in the body’s metabolic activities. They also noted that a lack of the KP system leads to glucose intolerance and obesity (131). However, it is not known whether KP exerts an impact on metabolic activities peripherally or centrally or both. Therefore, it will be interesting to check the impact of various organ-specific knockdowns of KP signaling on metabolism. Very recently, De Bond et al. (132) compared the expression of different metabolically important genes, such as Npy, Pomc, lepr, Ghsr (ghrelin receptor), Mc3r (melanocortin receptors 3), and Mc4r (melanocortin receptors 4). Unexpectedly, they observed no clear alterations in gonadectomized kiss1r-ablated mice compared to intact controls. These findings indicate that the etiology of obesity in the lack of KP-Kiss1r signaling may show an impairment in metabolic cues peripherally instead of central metabolic impairments (132).
Author Contributions
FW and BA have written first draft of this review article and have drawn figures. FU, MS, and RB have edited and revised the review article. All authors have read and approved the final version of the manuscript.
Conflict of Interest Statement
Authors declare that there is no conflict of interest that could be perceived as prejudicing the impartiality of the research reported.
Funding
FW was supported by a Georg Forster Fellowship of the Alexander von Humboldt Foundation. Research work in RB laboratory is funded by the German Primate Center, which is a Leibniz Institute financed by the Bundesrepublik Deutschland and the Bundesländer.
Abbreviations
KP, kisspeptin; GnRH, gonadotropin-releasing hormone; ARC, arcuate nucleus; AVPV, anteroventral periventricular nucleus; NPY, neuropeptide Y; AgRP, agouti-related protein; POMC, proopiomelanocortin; CART, cocaine- and amphetamine-related transcript; LepR, leptin receptor; IR, insulin receptor; GHSR, growth hormone secretagogue receptor.
References
1. Oakley AE, Clifton DK, Steiner RA. Kisspeptin signaling in the brain. Endocr Rev (2009) 30:713–43. doi:10.1210/er.2009-0005
2. Pinilla L, Aguilar E, Dieguez C, Millar RP, Tena-Sempere M. Kisspeptins and reproduction: physiological roles and regulatory mechanisms. Physiol Rev (2012) 92:1235–316. doi:10.1152/physrev.00037.2010
3. Kotani M, Detheux M, Vandenbogaerde A, Communi D, Vanderwinden JM, Le Poul E, et al. The metastasis suppressor gene KiSS-1 encodes kisspeptins, the natural ligands of the orphan G protein-coupled receptor GPR54. J Biol Chem (2001) 276:34631–6. doi:10.1074/jbc.M104847200
4. Lee DK, Nguyen T, O’Neill GP, Cheng R, Liu Y, Howard AD, et al. Discovery of a receptor related to the galanin receptors. FEBS Lett (1999) 446:103–7. doi:10.1016/S0014-5793(99)00009-5
5. Castellano JM, Navarro VM, Fernandez-Fernandez R, Nogueiras R, Tovar S, Roa J, et al. Changes in hypothalamic KiSS-1 system and restoration of pubertal activation of the reproductive axis by kisspeptin in undernutrition. Endocrinology (2005) 146:3917–25. doi:10.1210/en.2005-0337
6. Navarro VM, Fernandez-Fernandez R, Castellano JM, Roa J, Mayen A, Barreiro ML, et al. Advanced vaginal opening and precocious activation of the reproductive axis by KiSS-1 peptide, the endogenous ligand of GPR54. J Physiol (2004) 561:379–86. doi:10.1113/jphysiol.2004.072298
7. Shahab M, Mastronardi C, Seminara SB, Crowley WF, Ojeda SR, Plant TM. Increased hypothalamic GPR54 signaling: a potential mechanism for initiation of puberty in primates. Proc Natl Acad Sci U S A (2005) 102:2129–34. doi:10.1073/pnas.0409822102
8. Seminara SB, Messager S, Chatzidaki EE, Thresher RR, Acierno JS Jr, Shagoury JK, et al. The GPR54 gene as a regulator of puberty. N Engl J Med (2003) 349:1614–27. doi:10.1056/NEJMoa035322
9. de Roux N, Genin E, Carel JC, Matsuda F, Chaussain JL, Milgrom E. Hypogonadotropic hypogonadism due to loss of function of the KiSS1-derived peptide receptor GPR54. Proc Natl Acad Sci U S A (2003) 100:10972–6. doi:10.1073/pnas.1834399100
10. Nimri R, Lebenthal Y, Lazar L, Chevrier L, Phillip M, Bar M, et al. A novel loss-of-function mutation in GPR54/KISS1R leads to hypogonadotropic hypogonadism in a highly consanguineous family. J Clin Endocrinol Metab (2011) 96:E536–45. doi:10.1210/jc.2010-1676
11. Chan YM, Broder-Fingert S, Paraschos S, Lapatto R, Au M, Hughes V, et al. GnRH-deficient phenotypes in humans and mice with heterozygous variants in KISS1/Kiss1. J Clin Endocrinol Metab (2011) 96:E1771–81. doi:10.1210/jc.2011-0518
12. Teles MG, Bianco SD, Brito VN, Trarbach EB, Kuohung W, Xu S, et al. A GPR54-activating mutation in a patient with central precocious puberty. N Engl J Med (2008) 358:709–15. doi:10.1056/NEJMoa073443
13. Guerriero KA, Keen KL, Millar RP, Terasawa E. Developmental changes in GnRH release in response to kisspeptin agonist and antagonist in female rhesus monkeys (Macaca mulatta): implication for the mechanism of puberty. Endocrinology (2012) 153:825–36. doi:10.1210/en.2011-1565
14. Pineda R, Aguilar E, Pinilla L, Tena-Sempere M. Physiological roles of the kisspeptin/GPR54 system in the neuroendocrine control of reproduction. Prog Brain Res (2010) 181:55–77. doi:10.1016/S0079-6123(08)81005-9
15. Dhillo WS, Chaudhri OB, Patterson M, Thompson EL, Murphy KG, Badman MK, et al. Kisspeptin-54 stimulates the hypothalamic-pituitary gonadal axis in human males. J Clin Endocrinol Metab (2005) 90:6609–15. doi:10.1210/jc.2005-1468
16. Irwig MS, Fraley GS, Smith JT, Acohido BV, Popa SM, Cunningham MJ, et al. Kisspeptin activation of gonadotropin releasing hormone neurons and regulation of KiSS-1 mRNA in the male rat. Neuroendocrinology (2004) 80:264–72. doi:10.1159/000083140
17. Thompson EL, Patterson M, Murphy KG, Smith KL, Dhillo WS, Todd JF, et al. Central and peripheral administration of kisspeptin-10 stimulates the hypothalamic-pituitary-gonadal axis. J Neuroendocrinol (2004) 16:850–8. doi:10.1111/j.1365-2826.2004.01240.x
18. Ramaswamy S, Seminara SB, Pohl CR, DiPietro MJ, Crowley WF Jr, Plant TM. Effect of continuous intravenous administration of human metastin 45-54 on the neuroendocrine activity of the hypothalamic-pituitary-testicular axis in the adult male rhesus monkey (Macaca mulatta). Endocrinology (2007) 148:3364–70. doi:10.1210/en.2007-0207
19. Corander MP, Challis BG, Thompson EL, Jovanovic Z, Loraine Tung YC, Rimmington D, et al. The effects of neurokinin B upon gonadotrophin release in male rodents. J Neuroendocrinol (2010) 22:181–7. doi:10.1111/j.1365-2826.2009.01951.x
20. Clarke IJ, Caraty A. Kisspeptin and seasonality of reproduction. Adv Exp Med Biol (2013) 784:411–30. doi:10.1007/978-1-4614-6199-9_19
21. Kriegsfeld LJ. Circadian regulation of kisspeptin in female reproductive functioning. Adv Exp Med Biol (2013) 784:385–410. doi:10.1007/978-1-4614-6199-9_18
22. Castellano JM, Bentsen AH, Mikkelsen JD, Tena-Sempere M. Kisspeptins: bridging energy homeostasis and reproduction. Brain Res (2010) 1364:129–38. doi:10.1016/j.brainres.2010.08.057
23. De Bond JA, Smith JT. Kisspeptin and energy balance in reproduction. Reproduction (2014) 147:R53–63. doi:10.1530/REP-13-0509
24. Smith JT. Sex steroid regulation of kisspeptin circuits. Adv Exp Med Biol (2013) 784:275–95. doi:10.1007/978-1-4614-6199-9_13
25. Wahab F, Atika B, Shahab M. Kisspeptin as a link between metabolism and reproduction: evidences from rodent and primate studies. Metabolism (2013) 62:898–910. doi:10.1016/j.metabol.2013.01.015
26. Wahab F, Shahab M, Behr R. The involvement of gonadotropin inhibitory hormone and kisspeptin in the metabolic regulation of reproduction. J Endocrinol (2015) 225:R49–66. doi:10.1530/JOE-14-0688
27. Castellano JM, Tena-Sempere M. Metabolic regulation of kisspeptin. Adv Exp Med Biol (2013) 784:363–83. doi:10.1007/978-1-4614-6199-9_17
28. Wahab F, Ullah F, Chan YM, Seminara SB, Shahab M. Decrease in hypothalamic Kiss1 and Kiss1r expression: a potential mechanism for fasting-induced suppression of the HPG axis in the adult male rhesus monkey (Macaca mulatta). Horm Metab Res (2011) 43:81–5. doi:10.1055/s-0030-1269852
29. Ladyman SR, Woodside B. Food restriction during lactation suppresses Kiss1 mRNA expression and kisspeptin-stimulated LH release in rats. Reproduction (2014) 147:743–51. doi:10.1530/REP-13-0426
30. Luque RM, Kineman RD, Tena-Sempere M. Regulation of hypothalamic expression of KiSS-1 and GPR54 genes by metabolic factors: analyses using mouse models and a cell line. Endocrinology (2007) 148:4601–11. doi:10.1210/en.2007-0500
31. Castellano JM, Navarro VM, Fernandez-Fernandez R, Roa J, Vigo E, Pineda R, et al. Expression of hypothalamic KiSS-1 system and rescue of defective gonadotropic responses by kisspeptin in streptozotocin-induced diabetic male rats. Diabetes (2006) 55:2602–10. doi:10.2337/db05-1584
32. Castellano JM, Tena-Sempere M. Metabolic control of female puberty: potential therapeutic targets. Expert Opin Ther Targets (2016) 20:1181–93. doi:10.1080/14728222.2016.1212015
33. Smith JT, Acohido BV, Clifton DK, Steiner RA. KiSS-1 neurones are direct targets for leptin in the ob/ob mouse. J Neuroendocrinol (2006) 18:298–303. doi:10.1111/j.1365-2826.2006.01417.x
34. Kalamatianos T, Grimshaw SE, Poorun R, Hahn JD, Coen CW. Fasting reduces KiSS-1 expression in the anteroventral periventricular nucleus (AVPV): effects of fasting on the expression of KiSS-1 and neuropeptide Y in the AVPV or arcuate nucleus of female rats. J Neuroendocrinol (2008) 20:1089–97. doi:10.1111/j.1365-2826.2008.01757.x
35. Matsuzaki T, Iwasa T, Kinouchi R, Yoshida S, Murakami M, Gereltsetseg G, et al. Fasting reduces the kiss1 mRNA levels in the caudal hypothalamus of gonadally intact adult female rats. Endocr J (2011) 58:1003–12. doi:10.1507/endocrj.K11E-131
36. True C, Kirigiti MA, Kievit P, Grove KL, Smith MS. Leptin is not the critical signal for kisspeptin or luteinising hormone restoration during exit from negative energy balance. J Neuroendocrinol (2011) 23:1099–112. doi:10.1111/j.1365-2826.2011.02144.x
37. Sanchez-Garrido MA, Tena-Sempere M. Metabolic control of puberty: roles of leptin and kisspeptins. Horm Behav (2013) 64:187–94. doi:10.1016/j.yhbeh.2013.01.014
38. Roa J, Garcia-Galiano D, Varela L, Sanchez-Garrido MA, Pineda R, Castellano JM, et al. The mammalian target of rapamycin as novel central regulator of puberty onset via modulation of hypothalamic Kiss1 system. Endocrinology (2009) 150:5016–26. doi:10.1210/en.2009-0096
39. Wahab F, Aziz F, Irfan S, Zaman WU, Shahab M. Short-term fasting attenuates the response of the HPG axis to kisspeptin challenge in the adult male rhesus monkey (Macaca mulatta). Life Sci (2008) 83:633–7. doi:10.1016/j.lfs.2008.09.001
40. Quennell JH, Howell CS, Roa J, Augustine RA, Grattan DR, Anderson GM. Leptin deficiency and diet-induced obesity reduce hypothalamic kisspeptin expression in mice. Endocrinology (2011) 152:1541–50. doi:10.1210/en.2010-1100
41. Roa J, Vigo E, Castellano JM, Navarro VM, Fernandez-Fernandez R, Casanueva FF, et al. Hypothalamic expression of KiSS-1 system and gonadotropin-releasing effects of kisspeptin in different reproductive states of the female Rat. Endocrinology (2006) 147:2864–78. doi:10.1210/en.2005-1463
42. Yamada S, Uenoyama Y, Kinoshita M, Iwata K, Takase K, Matsui H, et al. Inhibition of metastin (kisspeptin-54)-GPR54 signaling in the arcuate nucleus-median eminence region during lactation in rats. Endocrinology (2007) 148:2226–32. doi:10.1210/en.2006-1529
43. Backholer K, Smith JT, Rao A, Pereira A, Iqbal J, Ogawa S, et al. Kisspeptin cells in the ewe brain respond to leptin and communicate with neuropeptide Y and proopiomelanocortin cells. Endocrinology (2010) 151:2233–43. doi:10.1210/en.2009-1190
44. Martin C, Navarro VM, Simavli S, Vong L, Carroll RS, Lowell BB, et al. Leptin-responsive GABAergic neurons regulate fertility through pathways that result in reduced kisspeptinergic tone. J Neurosci (2014) 34:6047–56. doi:10.1523/JNEUROSCI.3003-13.2014
45. Kadowaki T, Yamauchi T. Adiponectin and adiponectin receptors. Endocr Rev (2005) 26:439–51. doi:10.1210/er.2005-0005
46. Maeda K, Okubo K, Shimomura I, Funahashi T, Matsuzawa Y, Matsubara K. cDNA cloning and expression of a novel adipose specific collagen-like factor, apM1 (AdiPose most abundant gene transcript 1). Biochem Biophys Res Commun (1996) 221:286–9. doi:10.1006/bbrc.1996.0587
47. Nakano Y, Tobe T, Choi-Miura NH, Mazda T, Tomita M. Isolation and characterization of GBP28, a novel gelatin-binding protein purified from human plasma. J Biochem (1996) 120:803–12. doi:10.1093/oxfordjournals.jbchem.a021483
48. Scherer DC, Brockman JA, Chen Z, Maniatis T, Ballard DW. Signal-induced degradation of I kappa B alpha requires site-specific ubiquitination. Proc Natl Acad Sci U S A (1995) 92:11259–63. doi:10.1073/pnas.92.24.11259
49. Nigro E, Scudiero O, Monaco ML, Palmieri A, Mazzarella G, Costagliola C, et al. New insight into adiponectin role in obesity and obesity-related diseases. Biomed Res Int (2014) 2014:658913. doi:10.1155/2014/658913
50. Achari AE, Jain SK. Adiponectin, a therapeutic target for obesity, diabetes, and endothelial dysfunction. Int J Mol Sci (2017) 18:1321. doi:10.3390/ijms18061321
51. Kadowaki T, Yamauchi T, Okada-Iwabu M, Iwabu M. Adiponectin and its receptors: implications for obesity-associated diseases and longevity. Lancet Diabetes Endocrinol (2014) 2:8–9. doi:10.1016/S2213-8587(13)70120-7
52. Wahab F, Bano R, Jabeen S, Irfan S, Shahab M. Effect of peripheral kisspeptin administration on adiponectin, leptin, and resistin secretion under fed and fasting conditions in the adult male rhesus monkey (Macaca mulatta). Horm Metab Res (2010) 42:570–4. doi:10.1055/s-0030-1252016
53. Cawthorn WP, Scheller EL, Learman BS, Parlee SD, Simon BR, Mori H, et al. Bone marrow adipose tissue is an endocrine organ that contributes to increased circulating adiponectin during caloric restriction. Cell Metab (2014) 20:368–75. doi:10.1016/j.cmet.2014.06.003
54. Palin MF, Bordignon VV, Murphy BD. Adiponectin and the control of female reproductive functions. Vitam Horm (2012) 90:239–87. doi:10.1016/B978-0-12-398313-8.00010-5
55. Kaminski T, Smolinska N, Maleszka A, Kiezun M, Dobrzyn K, Czerwinska J, et al. Expression of adiponectin and its receptors in the porcine hypothalamus during the oestrous cycle. Reprod Domest Anim (2014) 49:378–86. doi:10.1111/rda.12282
56. Rak A, Mellouk N, Froment P, Dupont J. Adiponectin and resistin: potential metabolic signals affecting hypothalamo-pituitary gonadal axis in females and males of different species. Reproduction (2017) 153:R215–26. doi:10.1530/REP-17-0002
57. Wen JP, Liu C, Bi WK, Hu YT, Chen Q, Huang H, et al. Adiponectin inhibits KISS1 gene transcription through AMPK and specificity protein-1 in the hypothalamic GT1-7 neurons. J Endocrinol (2012) 214:177–89. doi:10.1530/JOE-12-0054
58. Wada N, Hirako S, Takenoya F, Kageyama H, Okabe M, Shioda S. Leptin and its receptors. J Chem Neuroanat (2014) 61-62:191–9. doi:10.1016/j.jchemneu.2014.09.002
60. Mantzoros CS, Magkos F, Brinkoetter M, Sienkiewicz E, Dardeno TA, Kim SY, et al. Leptin in human physiology and pathophysiology. Am J Physiol Endocrinol Metab (2011) 301:E567–84. doi:10.1152/ajpendo.00315.2011
61. Anubhuti V, Arora S. Leptin and its metabolic interactions: an update. Diabetes Obes Metab (2008) 10:973–93. doi:10.1111/j.1463-1326.2008.00852.x
62. Bjorbaek C, Kahn BB. Leptin signaling in the central nervous system and the periphery. Recent Prog Horm Res (2004) 59:305–31. doi:10.1210/rp.59.1.305
63. Dardeno TA, Chou SH, Moon HS, Chamberland JP, Fiorenza CG, Mantzoros CS. Leptin in human physiology and therapeutics. Front Neuroendocrinol (2010) 31:377–93. doi:10.1016/j.yfrne.2010.06.002
64. Bajari TM, Nimpf J, Schneider WJ. Role of leptin in reproduction. Curr Opin Lipidol (2004) 15:315–9. doi:10.1097/00041433-200406000-00012
65. Louis GW, Greenwald-Yarnell M, Phillips R, Coolen LM, Lehman MN, Myers MG Jr. Molecular mapping of the neural pathways linking leptin to the neuroendocrine reproductive axis. Endocrinology (2011) 152:2302–10. doi:10.1210/en.2011-0096
66. Ratra DV, Elias CF. Chemical identity of hypothalamic neurons engaged by leptin in reproductive control. J Chem Neuroanat (2014) 61-62:233–8. doi:10.1016/j.jchemneu.2014.05.005
67. Zuure WA, Roberts AL, Quennell JH, Anderson GM. Leptin signaling in GABA neurons, but not glutamate neurons, is required for reproductive function. J Neurosci (2013) 33:17874–83. doi:10.1523/JNEUROSCI.2278-13.2013
68. Donato J Jr, Cravo RM, Frazao R, Elias CF. Hypothalamic sites of leptin action linking metabolism and reproduction. Neuroendocrinology (2011) 93:9–18. doi:10.1159/000322472
69. Cravo RM, Frazao R, Perello M, Osborne-Lawrence S, Williams KW, Zigman JM, et al. Leptin signaling in Kiss1 neurons arises after pubertal development. PLoS One (2013) 8:e58698. doi:10.1371/journal.pone.0058698
70. Patterson CM, Leshan RL, Jones JC, Myers MG Jr. Molecular mapping of mouse brain regions innervated by leptin receptor-expressing cells. Brain Res (2011) 1378:18–28. doi:10.1016/j.brainres.2011.01.010
71. Camina JP, Carreira MC, Micic D, Pombo M, Kelestimur F, Dieguez C, et al. Regulation of ghrelin secretion and action. Endocrine (2003) 22:5–12. doi:10.1385/ENDO:22:1:5
72. Pusztai P, Sarman B, Ruzicska E, Toke J, Racz K, Somogyi A, et al. Ghrelin: a new peptide regulating the neurohormonal system, energy homeostasis and glucose metabolism. Diabetes Metab Res Rev (2008) 24:343–52. doi:10.1002/dmrr.830
73. Wren AM, Seal LJ, Cohen MA, Brynes AE, Frost GS, Murphy KG, et al. Ghrelin enhances appetite and increases food intake in humans. J Clin Endocrinol Metab (2001) 86:5992. doi:10.1210/jcem.86.12.8111
74. Tena-Sempere M. Interaction between energy homeostasis and reproduction: central effects of leptin and ghrelin on the reproductive axis. Horm Metab Res (2013) 45:919–27. doi:10.1055/s-0033-1355399
75. De Vriese C, Delporte C. Influence of ghrelin on food intake and energy homeostasis. Curr Opin Clin Nutr Metab Care (2007) 10:615–9. doi:10.1097/MCO.0b013e32829fb37c
76. Zigman JM, Nakano Y, Coppari R, Balthasar N, Marcus JN, Lee CE, et al. Mice lacking ghrelin receptors resist the development of diet-induced obesity. J Clin Invest (2005) 115:3564–72. doi:10.1172/JCI26002
77. Wortley KE, Anderson KD, Garcia K, Murray JD, Malinova L, Liu R, et al. Genetic deletion of ghrelin does not decrease food intake but influences metabolic fuel preference. Proc Natl Acad Sci U S A (2004) 101:8227–32. doi:10.1073/pnas.0402763101
78. Pfluger PT, Kirchner H, Gunnel S, Schrott B, Perez-Tilve D, Fu S, et al. Simultaneous deletion of ghrelin and its receptor increases motor activity and energy expenditure. Am J Physiol Gastrointest Liver Physiol (2008) 294:G610–8. doi:10.1152/ajpgi.00321.2007
79. Forbes S, Li XF, Kinsey-Jones J, O’Byrne K. Effects of ghrelin on kisspeptin mRNA expression in the hypothalamic medial preoptic area and pulsatile luteinising hormone secretion in the female rat. Neurosci Lett (2009) 460:143–7. doi:10.1016/j.neulet.2009.05.060
80. Frazao R, Dungan Lemko HM, da Silva RP, Ratra DV, Lee CE, Williams KW, et al. Estradiol modulates Kiss1 neuronal response to ghrelin. Am J Physiol Endocrinol Metab (2014) 306:E606–14. doi:10.1152/ajpendo.00211.2013
81. Foradori CD, Whitlock BK, Daniel JA, Zimmerman AD, Jones MA, Read CC, et al. Kisspeptin stimulates growth hormone release by utilizing neuropeptide Y pathways and is dependent on the presence of ghrelin in the ewe. Endocrinology (2017) 158:3526–39. doi:10.1210/en.2017-00303
82. Plum L, Schubert M, Bruning JC. The role of insulin receptor signaling in the brain. Trends Endocrinol Metab (2005) 16:59–65. doi:10.1016/j.tem.2005.01.008
83. Sliwowska JH, Fergani C, Gawalek M, Skowronska B, Fichna P, Lehman MN. Insulin: its role in the central control of reproduction. Physiol Behav (2014) 133:197–206. doi:10.1016/j.physbeh.2014.05.021
84. Woods SC, Lotter EC, McKay LD, Porte D Jr. Chronic intracerebroventricular infusion of insulin reduces food intake and body weight of baboons. Nature (1979) 282:503–5. doi:10.1038/282503a0
85. Bruning JC, Gautam D, Burks DJ, Gillette J, Schubert M, Orban PC, et al. Role of brain insulin receptor in control of body weight and reproduction. Science (2000) 289:2122–5. doi:10.1126/science.289.5487.2122
86. Kovacs P, Parlow AF, Karkanias GB. Effect of centrally administered insulin on gonadotropin-releasing hormone neuron activity and luteinizing hormone surge in the diabetic female rat. Neuroendocrinology (2002) 76:357–65. doi:10.1159/000067585
87. Qiu X, Dowling AR, Marino JS, Faulkner LD, Bryant B, Bruning JC, et al. Delayed puberty but normal fertility in mice with selective deletion of insulin receptors from Kiss1 cells. Endocrinology (2013) 154:1337–48. doi:10.1210/en.2012-2056
88. Fergani C, Routly JE, Jones DN, Pickavance LC, Smith RF, Dobson H. Kisspeptin, c-Fos and CRFR type 2 expression in the preoptic area and mediobasal hypothalamus during the follicular phase of intact ewes, and alteration after LPS. Physiol Behav (2013) 110-111:158–68. doi:10.1016/j.physbeh.2012.12.016
89. Castellano JM, Navarro VM, Roa J, Pineda R, Sanchez-Garrido MA, Garcia-Galiano D, et al. Alterations in hypothalamic KiSS-1 system in experimental diabetes: early changes and functional consequences. Endocrinology (2009) 150:784–94. doi:10.1210/en.2008-0849
90. Wahab F, Riaz T, Shahab M. Study on the effect of peripheral kisspeptin administration on basal and glucose-induced insulin secretion under fed and fasting conditions in the adult male rhesus monkey (Macaca mulatta). Horm Metab Res (2011) 43:37–42. doi:10.1055/s-0030-1268458
91. Terasawa E, Fernandez DL. Neurobiological mechanisms of the onset of puberty in primates. Endocr Rev (2001) 22:111–51. doi:10.1210/er.22.1.111
92. Evans MC, Rizwan M, Mayer C, Boehm U, Anderson GM. Evidence that insulin signalling in gonadotrophin-releasing hormone and kisspeptin neurones does not play an essential role in metabolic regulation of fertility in mice. J Neuroendocrinol (2014) 26:468–79. doi:10.1111/jne.12166
93. Xu Y, Kim ER, Zhao R, Myers MG Jr, Munzberg H, Tong Q. Glutamate release mediates leptin action on energy expenditure. Mol Metab (2013) 2:109–15. doi:10.1016/j.molmet.2013.01.004
94. Zuure WA, Quennell JH, Anderson GM. Leptin responsive and GABAergic projections to the rostral preoptic area in mice. J Neuroendocrinol (2016) 28:12357. doi:10.1111/jne.12357
95. Shamas S, Khan SU, Khan MY, Shabbir N, Zubair H, Shafqat S, et al. Fasting induced kisspeptin signaling suppression is regulated by glutamate mediated cues in adult male rhesus macaque (Macaca mulatta). Neuropeptides (2015) 52:39–45. doi:10.1016/j.npep.2015.06.005
96. Clarke IJ, Parkington HC. Gonadotropin inhibitory hormone (GnIH) as a regulator of gonadotropes. Mol Cell Endocrinol (2014) 385:36–44. doi:10.1016/j.mce.2013.08.017
97. Ullah R, Shen Y, Zhou YD, Huang K, Fu JF, Wahab F, et al. Expression and actions of GnIH and its orthologs in vertebrates: current status and advanced knowledge. Neuropeptides (2016) 59:9–20. doi:10.1016/j.npep.2016.05.004
98. Tsutsui K, Osugi T, Son YL, Ubuka T. Review: structure, function and evolution of GnIH. Gen Comp Endocrinol (2017). doi:10.1016/j.ygcen.2017.07.024
99. Poling MC, Quennell JH, Anderson GM, Kauffman AS. Kisspeptin neurones do not directly signal to RFRP-3 neurones but RFRP-3 may directly modulate a subset of hypothalamic kisspeptin cells in mice. J Neuroendocrinol (2013) 25:876–86. doi:10.1111/jne.12084
100. George JT, Hendrikse M, Veldhuis JD, Clarke IJ, Anderson RA, Millar RP. Effect of gonadotropin-inhibitory hormone on luteinizing hormone secretion in humans. Clin Endocrinol (Oxf) (2017) 86:731–8. doi:10.1111/cen.13308
101. Poling MC, Kauffman AS. Organizational and activational effects of sex steroids on kisspeptin neuron development. Front Neuroendocrinol (2013) 34:3–17. doi:10.1016/j.yfrne.2012.06.001
102. Leon S, Garcia-Galiano D, Ruiz-Pino F, Barroso A, Manfredi-Lozano M, Romero-Ruiz A, et al. Physiological roles of gonadotropin-inhibitory hormone signaling in the control of mammalian reproductive axis: studies in the NPFF1 receptor null mouse. Endocrinology (2014) 155:2953–65. doi:10.1210/en.2014-1030
103. Hill JW, Elmquist JK, Elias CF. Hypothalamic pathways linking energy balance and reproduction. Am J Physiol Endocrinol Metab (2008) 294:E827–32. doi:10.1152/ajpendo.00670.2007
104. Hokfelt T, Stanic D, Sanford SD, Gatlin JC, Nilsson I, Paratcha G, et al. NPY and its involvement in axon guidance, neurogenesis, and feeding. Nutrition (2008) 24:860–8. doi:10.1016/j.nut.2008.06.010
105. Alex Thomas M, Xue B. Mechanisms for AgRP neuron-mediated regulation of appetitive behaviors in rodents. Physiol Behav (2017). doi:10.1016/j.physbeh.2017.10.006
106. Bingham NC, Cone RD. Regulation of orexigenic AgRP neurons: a third way? Trends Endocrinol Metab (2015) 26:339–40. doi:10.1016/j.tem.2015.05.008
107. Simpson KA, Martin NM, Bloom SR. Hypothalamic regulation of food intake and clinical therapeutic applications. Arq Bras Endocrinol Metabol (2009) 53:120–8. doi:10.1590/S0004-27302009000200002
108. Chen Y, Lin YC, Zimmerman CA, Essner RA, Knight ZA. Hunger neurons drive feeding through a sustained, positive reinforcement signal. Elife (2016) 5:e18640. doi:10.7554/eLife.18640
109. Warne JP, Xu AW. Metabolic transceivers: in tune with the central melanocortin system. Trends Endocrinol Metab (2013) 24:68–75. doi:10.1016/j.tem.2012.10.005
110. Barsh GS, Schwartz MW. Genetic approaches to studying energy balance: perception and integration. Nat Rev Genet (2002) 3:589–600. doi:10.1038/nrg862
111. Barsh G, Gunn T, He L, Wilson B, Lu X, Gantz I, et al. Neuroendocrine regulation by the agouti/Agrp-melanocortin system. Endocr Res (2000) 26:571. doi:10.3109/07435800009048572
112. Qiu J, Zhang C, Borgquist A, Nestor CC, Smith AW, Bosch MA, et al. Insulin excites anorexigenic proopiomelanocortin neurons via activation of canonical transient receptor potential channels. Cell Metab (2014) 19:682–93. doi:10.1016/j.cmet.2014.03.004
113. Hill JW. Gene expression and the control of food intake by hypothalamic POMC/CART neurons. Open Neuroendocrinol J (2010) 3:21–7. doi:10.2174/1876528901003010021
114. Konner AC, Janoschek R, Plum L, Jordan SD, Rother E, Ma X, et al. Insulin action in AgRP-expressing neurons is required for suppression of hepatic glucose production. Cell Metab (2007) 5:438–49. doi:10.1016/j.cmet.2007.05.004
115. Chen HY, Trumbauer ME, Chen AS, Weingarth DT, Adams JR, Frazier EG, et al. Orexigenic action of peripheral ghrelin is mediated by neuropeptide Y and agouti-related protein. Endocrinology (2004) 145:2607–12. doi:10.1210/en.2003-1596
116. Willesen MG, Kristensen P, Romer J. Co-localization of growth hormone secretagogue receptor and NPY mRNA in the arcuate nucleus of the rat. Neuroendocrinology (1999) 70:306–16. doi:10.1159/000054491
117. Millington GW. The role of proopiomelanocortin (POMC) neurones in feeding behaviour. Nutr Metab (Lond) (2007) 4:18. doi:10.1186/1743-7075-4-18
118. Anderson EJ, Cakir I, Carrington SJ, Cone RD, Ghamari-Langroudi M, Gillyard T, et al. 60 YEARS OF POMC: regulation of feeding and energy homeostasis by alpha-MSH. J Mol Endocrinol (2016) 56:T157–74. doi:10.1530/JME-16-0014
119. Yaswen L, Diehl N, Brennan MB, Hochgeschwender U. Obesity in the mouse model of pro-opiomelanocortin deficiency responds to peripheral melanocortin. Nat Med (1999) 5:1066–70. doi:10.1038/12506
120. Belgardt BF, Okamura T, Bruning JC. Hormone and glucose signalling in POMC and AgRP neurons. J Physiol (2009) 587:5305–14. doi:10.1113/jphysiol.2009.179192
121. Lau J, Herzog H. CART in the regulation of appetite and energy homeostasis. Front Neurosci (2014) 8:313. doi:10.3389/fnins.2014.00313
122. Berglund ED, Vianna CR, Donato J Jr, Kim MH, Chuang JC, Lee CE, et al. Direct leptin action on POMC neurons regulates glucose homeostasis and hepatic insulin sensitivity in mice. J Clin Invest (2012) 122:1000–9. doi:10.1172/JCI59816
123. Castro-Gonzalez D, Fuente-Martin E, Sanchez-Garrido MA, Argente-Arizon P, Tena-Sempere M, Barrios V, et al. Increased prepubertal body weight enhances leptin sensitivity in proopiomelanocortin and neuropeptide y neurons before puberty onset in female rats. Endocrinology (2015) 156:1272–82. doi:10.1210/en.2014-1759
124. Shi H, Sorrell JE, Clegg DJ, Woods SC, Seeley RJ. The roles of leptin receptors on POMC neurons in the regulation of sex-specific energy homeostasis. Physiol Behav (2010) 100:165–72. doi:10.1016/j.physbeh.2010.02.018
125. Backholer K, Smith J, Clarke IJ. Melanocortins may stimulate reproduction by activating orexin neurons in the dorsomedial hypothalamus and kisspeptin neurons in the preoptic area of the ewe. Endocrinology (2009) 150:5488–97. doi:10.1210/en.2009-0604
126. Manfredi-Lozano M, Roa J, Ruiz-Pino F, Piet R, Garcia-Galiano D, Pineda R, et al. Defining a novel leptin-melanocortin-kisspeptin pathway involved in the metabolic control of puberty. Mol Metab (2016) 5:844–57. doi:10.1016/j.molmet.2016.08.003
127. True C, Takahashi D, Kirigiti M, Lindsley SR, Moctezuma C, Arik A, et al. Arcuate nucleus neuropeptide coexpression and connections to gonadotrophin-releasing hormone neurones in the female rhesus macaque. J Neuroendocrinol (2017) 29. doi:10.1111/jne.12491
128. Heppner KM, Baquero AF, Bennett CM, Lindsley SR, Kirigiti MA, Bennett B, et al. GLP-1R signaling directly activates arcuate nucleus kisspeptin action in brain slices but does not rescue luteinizing hormone inhibition in ovariectomized mice during negative energy balance. eNeuro (2017) 4. doi:10.1523/ENEURO.0198-16.2016
129. George JT, Veldhuis JD, Tena-Sempere M, Millar RP, Anderson RA. Exploring the pathophysiology of hypogonadism in men with type 2 diabetes: kisspeptin-10 stimulates serum testosterone and LH secretion in men with type 2 diabetes and mild biochemical hypogonadism. Clin Endocrinol (Oxf) (2013) 79:100–4. doi:10.1111/cen.12103
130. Jayasena CN, Abbara A, Veldhuis JD, Comninos AN, Ratnasabapathy R, De Silva A, et al. Increasing LH pulsatility in women with hypothalamic amenorrhoea using intravenous infusion of kisspeptin-54. J Clin Endocrinol Metab (2014) 99:E953–61. doi:10.1210/jc.2013-1569
131. Tolson KP, Garcia C, Yen S, Simonds S, Stefanidis A, Lawrence A, et al. Impaired kisspeptin signaling decreases metabolism and promotes glucose intolerance and obesity. J Clin Invest (2014) 124:3075–9. doi:10.1172/JCI71075
Keywords: Kiss1, kisspeptin, Kiss1r, metabolism, reproduction, metabolic hormones, proopiomelanocortin, AgRP
Citation: Wahab F, Atika B, Ullah F, Shahab M and Behr R (2018) Metabolic Impact on the Hypothalamic Kisspeptin-Kiss1r Signaling Pathway. Front. Endocrinol. 9:123. doi: 10.3389/fendo.2018.00123
Received: 18 December 2017; Accepted: 12 March 2018;
Published: 28 March 2018
Edited by:
Rosanna Chianese, Università degli Studi della Campania "Luigi Vanvitelli" Caserta, ItalyReviewed by:
Qingchun Tong, University of Texas Health Science Center at Houston, United StatesWei-jiang Zhao, Shantou University, China
Copyright: © 2018 Wahab, Atika, Ullah, Shahab and Behr. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Fazal Wahab, ZndhaGFiQGRwei5ldQ==