 Andrea del Campo1,2,3
Andrea del Campo1,2,3 Catalina Bustos1
Catalina Bustos1 Carolina Mascayano4
Carolina Mascayano4 Claudio Acuña-Castillo1,5
Claudio Acuña-Castillo1,5 Rodrigo Troncoso6
Rodrigo Troncoso6 Leonel E. Rojo1,5*
Leonel E. Rojo1,5*
- 1Departamento de Biología, Facultad de Química y Biología, Universidad de Santiago de Chile, Santiago, Chile
- 2Escuela de Química y Farmacia, Facultad de Ingeniería, Ciencia y Tecnología, Universidad Bernardo O’Higgins, Santiago, Chile
- 3Programa de Biología Celular y Molecular, Facultad de Medicina, Universidad de Chile, Santiago, Chile
- 4Departamento de Ciencias del Ambiente, Facultad de Química y Biología, Universidad de Santiago de Chile, Santiago, Chile
- 5Centro de Biotecnología Acuícola, Universidad de Santiago de Chile, Santiago, Chile
- 6Laboratorio de Investigación en Nutrición y Actividad Física, Instituto de Nutrición y Tecnología de los Alimentos (INTA), Universidad de Chile, Santiago, Chile
Second-generation antipsychotics (SGAs) are known to increase cardiovascular risk through several physiological mechanisms, including insulin resistance, hepatic steatosis, hyperphagia, and accelerated weight gain. There are limited prophylactic interventions to prevent these side effects of SGAs, in part because the molecular mechanisms underlying SGAs toxicity are not yet completely elucidated. In this perspective article, we introduce an innovative approach to study the metabolic side effects of antipsychotics through the alterations of the mitochondrial dynamics, which leads to an imbalance in mitochondrial fusion/fission ratio and to an inefficient mitochondrial phenotype of muscle cells. We believe that this approach may offer a valuable path to explain SGAs-induced alterations in metabolic homeostasis.
Introduction
Second-generation antipsychotics (SGAs) are effective drugs in controlling symptoms of schizophrenia and other psychotic disorders. However, SGAs are also known to induce insulin resistance, hepatic steatosis, and accelerated weight gain, which can lead to morbid obesity in as short as 6 weeks (1). Herein, we introduce an innovative approach to explain metabolic side effects of SGAs through the impairment of the mitochondrial network morphology and insulin signaling, which results in a fusion/fission imbalance of the mitochondrial network. Our hypothesis is that the SGAs-induced disruption of the mitochondrial dynamics, manifested by a highly “fissioned” and inefficient mitochondrial network, results in a reduced capacity to trigger insulin-dependent pathways necessary to preserve adequate energy production and metabolic homeostasis. We believe that this approach may offer a valuable path to further understand the SGAs-induced insulin resistance in different tissues.
This perspective paper briefly reviews the molecular, physiological, and clinical aspects of SGAs-induced metabolic toxicity. Then, we present our explanation and initial findings on an “over-fissioned” phenotype of the mitochondrial network and impairment of the insulin receptor signaling induced by olanzapine, one of the most common drugs of the SGAs pharmacological family. We also propose, based on in silico simulations potential sites for the interactions between olanzapine and the extracellular domain of the insulin receptor. The purpose of this article is not only to provide definite answers on the molecular mechanisms underlying the SGAs-induced metabolic syndrome but also to present a new angle for the study of this problem, which is one of the most clinically relevant, and serious, side effect of SGAs. This perspective article offers a new insight for the development of prophylactic interventions against SGAs-induced metabolic syndrome through the screening of small molecules capable of rescuing SGAs-induced mitochondrial disruption.
Metabolic Syndrome and Antipsychotics
The number of psychiatric patients suffering from SGAs-induced metabolic side effects continues to rise (2, 3), despite all the international guidelines for the clinical use of SGAs, which strongly suggest that this pharmacotherapy should be initiated only after a careful evaluation of basal metabolic parameters to select the appropriate drug (4). Intriguingly, in spite of their metabolic toxicity profile, clozapine, risperidone, olanzapine, quetiapine, and aripiprazole have remain among the world-top selling pharmaceuticals over the past 10 years (5, 6). Preclinical and clinical studies have shown that, among the SGAs, olanzapine is the drug with the strongest metabolic toxicity, due to its effects on weight gain (7–9), plasma glucose levels, and other metabolic parameters (10, 11).
The published evidence regarding the molecular mechanisms underlying the SGAs toxicity is still limited. However, it is known that the metabolic alterations induced by SGAs are partially mediated by hyperphagia linked to alterations in the D1/D2, 5-HT1B, 5-HT2, and 5-HT3 signaling (12), and GABA2 receptor polymorphism (13). On this regard, recent research have demonstrated the participation of serotonin signaling in glucose homeostasis through serotonylation of rab4 proteins (14), moreover other studies have shown that 5HT2 selective antagonism impairs insulin sensitivity. SGAs also induce anomalous cellular differentiation of adipocytes (15), increase lipid accumulation in the liver tissue (16), upregulate the sterol regulatory element-binding protein (17), and inhibit of the glycogen accumulation in skeletal muscle cells (18). In spite of all the current proposed mechanisms, the generation of the secondary effects of SGA is still a matter of controversy. It is important to mention that the literature describes differences of the metabolic problems presented in SGA-induced when compared with type 2 diabetes (3, 19, 20). On this regard, there is also evidence suggesting that metabolic changes due to olanzapine are tissue specific (20–23).
In vivo studies in rodents using the hyperinsulinemic/euglycemic clamp technique have shown that olanzapine impairs insulin sensitivity in the liver (24), skeletal muscle (21), and adipose tissue (21–23). Furthermore, a recent study showed that olanzapine decreases insulin-mediated glucose uptake through a mechanism involving an impaired hypothalamic insulin sensing during pancreatic euglycemic clamps (23). Altogether, these data seemingly confirms the results from the in vitro studies (16, 18) suggesting that olanzapine would induce whole-body insulin resistance. In the context of our hypothesis, it is worth mentioning that olanzapine was shown to impair lipid metabolism by increasing uptake of free fatty acids into peripheral tissues, increasing lipid oxidation in muscle cells, rising levels of long-chain 3-hydroxylated acyl-carnitines, and suppressing the respiratory exchange ratio (20). These events are indicative of an olanzapine-mediated reduced availability of fatty Acyl CoA inside the mitochondrial matrix, which would limit the supply of precursors for the tricarboxylic acids (TCA) cycle. Altogether, these results support the hypothesis that mitochondrial dysfunction plays a major effect of olanzapine-induced metabolic syndrome and the maintenance of mitochondrial homeostasis should be considered as a potential therapeutic target to prevent SGAs-induced metabolic side effects.
In spite of the relevance of skeletal muscle for the insulin-mediated conversion of glucose into ATP (25), the current literature still lacks enough mechanistic studies on the effect of SGAs on energy production and carbohydrates metabolism inside the skeletal muscle (Table 1). In order to explain our perspective, it is important to remember that the intracellular ATP is mainly produced inside the mitochondria, a highly specialized organelle involved in energy production, and that the mitochondrial energy production in skeletal muscle involves lipid metabolism, oxidative phosphorylation, and the cycle of the TCA (26). As it has been largely studied, mitochondrial function is, therefore, a sensitive indicator of the global cellular function.
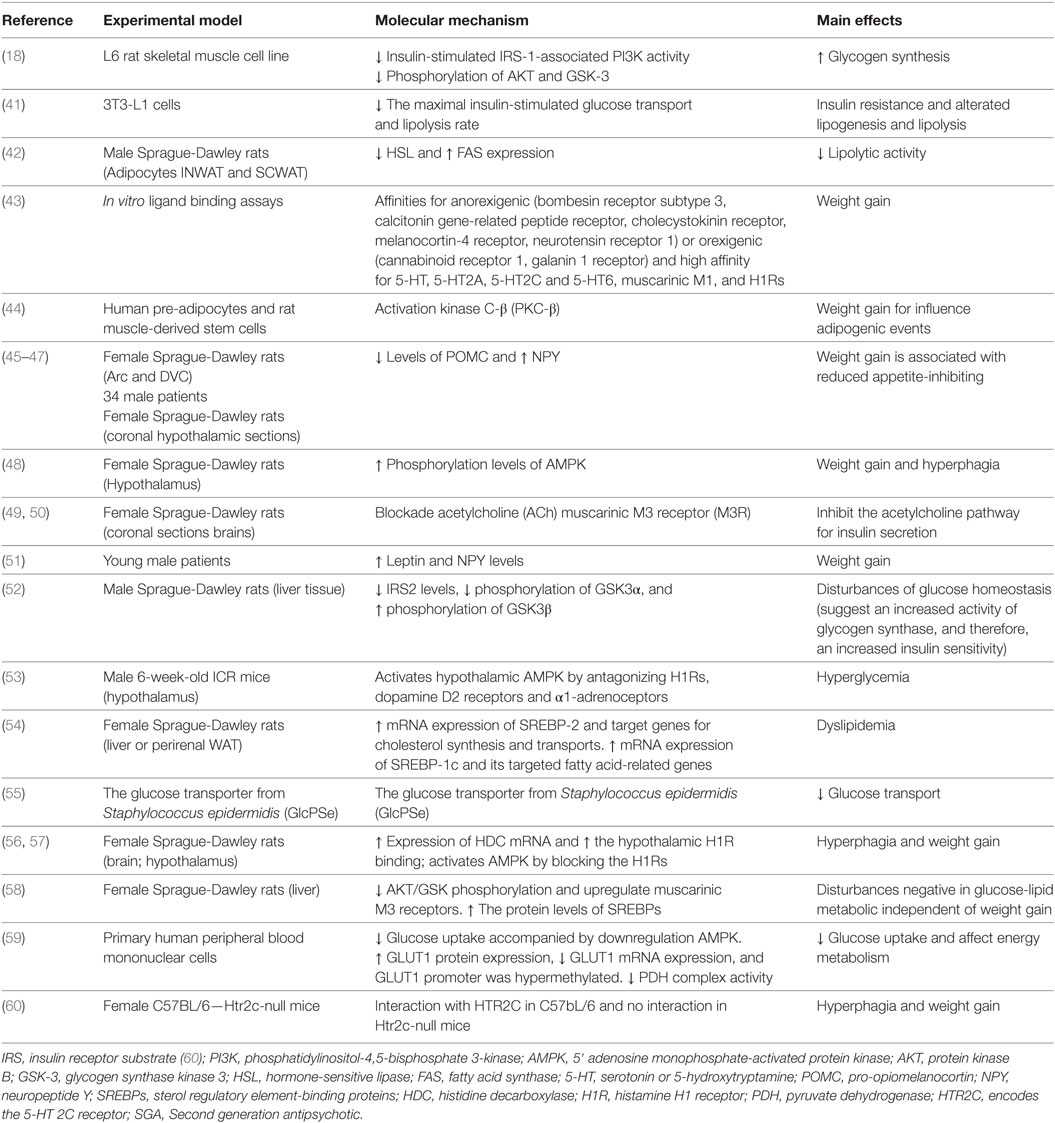
Table 1. Mechanisms of SGA-induced metabolic side effects.
Mitochondrial Network and Metabolic Homeostasis
During the past two decades, several studies have described the functional relationship between the mitochondrial function and mitochondrial dynamics, the latter is defined as the different processes that occur to the mitochondrial network, such as fusion, fission, mitochondrial movements through the cytoskeleton, mitochondrial biogenesis, and mitophagy (27). On this regard, the proper balance between all these processes has been directly linked to a correct mitochondrial function, thus opening new possibilities for regulating the mitochondrial metabolism through the pharmacological interventions of mitochondrial dynamics.
Impaired insulin signaling and mitochondrial dysfunction are two clear signs of abnormal metabolic response in skeletal muscle cells. Given these characteristics, several studies have reported morphological differences of the mitochondrial network in obese and diabetic patients (28), but none of these studies have looked at mitochondrial dynamics in SGAs users. Interestingly, an in vitro study, by Contreras-Shannon et al., showed that Clozapine, a member of the SGA family, alters mitochondrial morphology and ATP levels in cultured insulin-responsive cells in a dose-dependent manner (29).
The relationships between mitochondrial dynamics and insulin physiological responses in skeletal muscle cells are an active field of research, to which we have contributed by describing how insulin promotes mitochondrial fusion after in cardiac myocytes and L6 muscle cells (30). We also reported that the regulation of mitochondrial morphology toward incomplete fusion impairs insulin signaling and glucose uptake in L6 myoblasts (31).
Previous results suggested a direct influence of SGAs on the peripheral insulin resistance. Particularly Engl et al. demonstrated that olanzapine impairs glycogen synthesis by disrupting insulin signaling in a model of L6 skeletal muscle cells (18). Ardizzone and coworkers observed that SGAs inhibit glucose transport in L6 myoblasts (32). These latter in vitro studies suggest that SGAs would induce insulin resistance, although the concentrations of olanzapine used for these studies exceed those observed in human plasma (33). However, these preliminary in vitro studies suggesting insulin resistance induced by SGAs were confirmed and further characterized in rats by Martins and coworkers (34), who demonstrated that olanzapine administered directly to the CNS induces the expression of hypothalamic 5′ adenosine monophosphate-activated protein kinase and hepatic insulin resistance, suggesting a CNS target for the metabolic dysregulation of atypical antipsychotics. Another confirmation came from a clinical study demonstrating that only 9 days of oral olanzapine treatment causes significant elevations in postprandial insulin, glucagon-like peptide 1, and glucagon coincident with insulin resistance (35). According to Teff et al. aripiprazole, another SGAs drug, would also induce insulin resistance.
In view of these findings, we decided to study effects of SGAs through the alterations of the mitochondrial dynamics induced by olanzapine and also to perform an in silico search for potential interactions between the insulin receptor and olanzapine. We believe this model system could help to further explain the multicellular metabolic toxicity of SGAs.
We also examined the question as to whether olanzapine would interact with residues of the extracellular domain of the insulin receptor. It is worth mentioning that the insulin receptor signaling pathways are involved in the peripheral mechanisms of SGAs-induced toxicity (3). We modeled the human insulin receptor (36) using MODELLER 9.14 (37) the crystal structure (PDB ID: 3W14) (38) with an identity of 99%. The best model obtained was refined using Charmm 33b1 with the conjugate gradient.
Dockings of olanzapine (http://zinc.docking.org/; olanzapine code: 52957434) and model of human insulin receptor were performed with the AutoDock4 package (39), using a Lamarckian algorithm and assuming total flexibility of all compounds studied. The grid maps were made up of 40 × 40 × 40 points, with a grid-point spacing of 0.375 Å of the center of the molecule. The AutoTors option was used to define the ligand torsions, and the docking results were then analyzed by a ranked cluster analysis, resulting in conformations with the highest overall binding energy (largest negative −ΔGB value).
We observed that olanzapine displayed a binding energy of −6.89 kcal/mol located at H bond near to Pro309 (2.89 Å). Other interacting residues around the olanzapine were Gln276, Glu287, Cys288, Thr293, and Met294 (Figure 1A). Both, binding energy and interatomic distances suggest there are potential interactions between olanzapine and the extracellular domain of the insulin receptor. This is of course susceptible of experimental verification through binding experiments with radiolabeled ligands, directed mutagenesis of the insulin receptor, or through surface plasmon resonances studies.
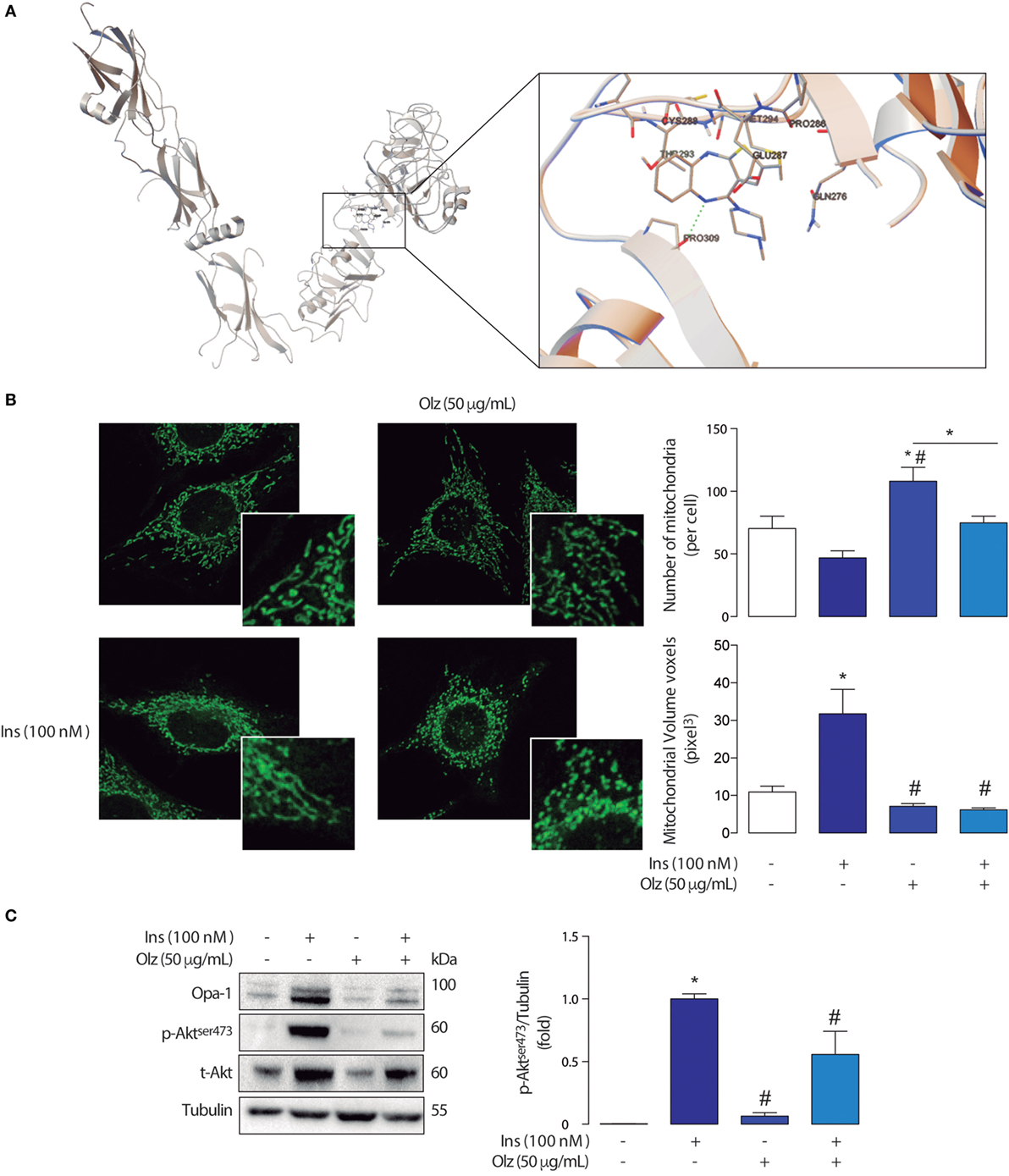
Figure 1. (A) Graphical representation of the interactions sites for olanzapine and the extracellular domain of de insulin receptor according to Docking Studies. The model of human insulin receptor and the structure of olanzapine were obtained using the AutoDock4 package. (B) Mitchondrial morphology of L6 myotubes and densitometry of confocal micrographies. Mitochondrial morphology in control conditions (left) and the olanzapine 160 µM treated cells (right) showing fragmentation of the mitochondrial network. *p < 0.05 vs control #p < 0.05 vs Ins. (C) Detection of insulin signaling proteins from L6 myotubes treated with olanzapine and insulin. The mitochondrial immunoblots for Opa-1, Akt, and p-Akt in L6 myotubes treated with olanzapine (OLZ) and Insulin (Ins). *p < 0.05 vs control #p < 0.05 vs Ins.
The changes in mitochondrial morphology were assessed in L6 myoblasts incubated with 400 nM MitoTracker green in Krebs solution for 25 min and then washed with Krebs solution for 5 min. Confocal images stacks of the mitochondrial network were captured with a Nikon C2 Confocal microscope, using a 60× Plan-Apochromatic λ CFI oil (1,4) objective, as described by del Campo et al. (31). Z-stacks were deconvolved, thresholded, and 3D-reconstructed using ImageJ software (NIH). Number and volume of individual mitochondria were quantified using the 3D Object Counter plug-in.
The analyses of mitochondrial network assessed through variations in mitochondrial number and volume in myoblasts treated with olanzapine showed that this drug induces mitochondrial fragmentation when compared with control (no treatment) myoblasts (Figure 1B). This fragmented phenotype is determined by a significant increase in the number of mitochondria per cell accompanied by a decrease in the volume of each mitochondrion (voxels = pixels3).
Interestingly, olanzapine also disrupted the effects of insulin in mitochondrial dynamics (Figure 1B). As shown in previous studies, insulin 100 nM promotes mitochondrial fusion in L6 myoblasts, our results support the fact that a fused-like phenotype, given by a significant increase in the volume of mitochondria and a reduced number of mitochondria per cell, can be found in myoblasts treated with insulin. On the basis of these previous findings, we investigated whether the pre-incubation of olanzapine impairs the mitochondrial fusion promoted by insulin 100 nM. Our results showed that the pre-incubation with olanzapine actually impairs the action of insulin on the mitochondrial dynamics by significantly decreasing the mitochondrial volume, compared with cells incubated only with insulin 100 nM 3 h only (Figure 1B). Each experiment was repeated at least three times. One-way ANOVA was used as statistic test and a subsequent Tuckey post-test was applied, statistical significance was defined as *p < 0.05.
These results show that olanzapine affects the mitochondrial network probably promoting mitochondrial dysfunction on its own, not only by inducing mitochondrial fragmentation but also by interrupting insulin-mediated changes of the mitochondrial network in skeletal muscle cells. The mitochondrial fragmentation on its own impairs insulin signaling, as proven by the use of antisense adenovirus toward Mfn2 and microRNA toward OPA-1 in L6 skeletal muscle cells, decreases in Akt phosphorylation (31). We observed that olanzapine not only promotes a fragmented phenotype of the mitochondrial network but also inhibits insulin-mediated fusion and decreases Akt phosphorylation (Figure 1C). This is a clear indicative that metabolic alterations induced by olanzapine are related with the fragmentation of the mitochondrial network and mitochondrial metabolic dysfunction. This data is an indicative that olanzapine would limit the utilization of different molecules to produce cellular ATP, which subsequently could lead to maladaptation of the skeletal muscle.
Alterations in the Expression of Mitochondrial Fusion Proteins and Insulin Signaling
Opa-1 is a mitochondrial protein involved in inner mitochondrial membrane fusion and maintenance of mitochondrial cristae (40). As previously reported a 3 h insulin stimulus promotes an increase of Opa-1 protein levels through the Akt-mTOR-NFkB-Opa-1 signaling pathway promoting mitochondrial fusion (30). A mitochondrial fusion-like phenotype has been associated with healthy metabolic homeostasis in eukaryotic cells (27, 30, 31). Interestingly, olanzapine decreases the insulin-induced expression of Opa-1 in L6 cells treated with insulin for 3 h. More specifically, we observed that in L6 skeletal muscle cells insulin (100 nM 3 h) induced an increase of long and short isoforms of Opa-1, which was impaired by olanzapine (Figure 1C). These results suggest that olanzapine disrupts insulin signaling, through a decrease in Akt phosphorylation, and also impairs the subsequent modification of mitochondrial dynamics, contributing to mitochondrial dysfunction.
Concluding Remarks
Based on our data and previous evidence in this field (29, 40), we here propose a new perspective to explore the mechanism of SGAs metabolic toxicity based on the impairment of the mitochondrial dynamics, which could explain the development of accelerated metabolic syndrome manifested by insulin resistance, weight gain, lipid accumulation, and hyperglycemia. In other words, the metabolic disturbances induced by SGAs affect one of the most fundamental functions of living cells, which is ATP production in the mitochondria.
Author Contributions
AC performed experiments and data analysis, CB performed skeletal muscle experiments, CM designed and performed Autodock experiments, CA-C designed experiments and contributed to the manuscript writing, RT summarized SGA studies in Table 1, and LR outlined the manuscript, overviewed the experiments, analyzed data, and wrote the manuscript.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Funding
LR is funded by Proyecto Fondecyt Iniciación No11140915 of CONICYT, Chile.
References
1. Kelly DL, Mcmahon RP, Liu F, Love RC, Wehring HJ, Shim JC, et al. Cardiovascular disease mortality in patients with chronic schizophrenia treated with clozapine: a retrospective cohort study. J Clin Psychiatry (2010) 71:304–11. doi:10.4088/JCP.08m04718yel
2. Gallego JA, Bonetti J, Zhang J, Kane JM, Correll CU. Prevalence and correlates of antipsychotic polypharmacy: a systematic review and meta-regression of global and regional trends from the 1970s to 2009. Schizophr Res (2009) 138:18–28. doi:10.1016/j.schres.2012.03.018
3. Rojo LE, Gaspar PA, Silva H, Risco L, Arena P, Cubillos-Robles K, et al. Metabolic syndrome and obesity among users of second generation antipsychotics: a global challenge for modern psychopharmacology. Pharmacol Res (2015) 101:74–85. doi:10.1016/j.phrs.2015.07.022
4. Cooper SJ, Reynolds GP, Barnes T, England E, Haddad PM, Heald A, et al. BAP guidelines on the management of weight gain, metabolic disturbances and cardiovascular risk associated with psychosis and antipsychotic drug treatment. J Psychopharmacol (2016) 30(8):717–48. doi:10.1177/0269881116645254
5. Geddes J. Generating evidence to inform policy and practice: the example of the second generation “atypical” antipsychotics. Schizophr Bull (2003) 29:105–14. doi:10.1093/oxfordjournals.schbul.a006980
6. Monasterio E, Gleeson D. Pharmaceutical industry behaviour and the Trans Pacific Partnership Agreement. N Z Med J (2014) 127(1389):6–12.
7. Allison DB, Mentore JL, Heo M, Chandler LP, Cappelleri JC, Infante MC, et al. Antipsychotic-induced weight gain: a comprehensive research synthesis. Am J Psychiatry (1999) 156:1686–96.
8. Parsons B, Allison DB, Loebel A, Williams K, Giller E, Romano S, et al. Weight effects associated with antipsychotics: a comprehensive database analysis. Schizophr Res (2009) 110:103–10. doi:10.1016/j.schres.2008.09.025
9. Robinson DG, Woerner MG, Napolitano B, Patel RC, Sevy SM, Gunduz-Bruce H, et al. Randomized comparison of olanzapine versus risperidone for the treatment of first-episode schizophrenia: 4-month outcomes. Am J Psychiatry (2006) 163:2096–102. doi:10.1176/ajp.2006.163.12.2096
10. Briles JJ, Rosenberg DR, Brooks BA, Roberts MW, Diwadkar VA. Review of the safety of second-generation antipsychotics: are they really “atypically” safe for youth and adults? Prim Care Companion CNS Disord (2012) 14(3): PCC.11r01298. doi:10.4088/PCC.11r01298
11. Wampers M, Hanssens L, Van Winkel R, Heald A, Collette J, Peuskens J, et al. Differential effects of olanzapine and risperidone on plasma adiponectin levels over time: results from a 3-month prospective open-label study. Eur Neuropsychopharmacol (2012) 22:17–26. doi:10.1016/j.euroneuro.2011.03.010
12. Kaur G, Kulkarni SK. Studies on modulation of feeding behavior by atypical antipsychotics in female mice. Prog Neuropsychopharmacol Biol Psychiatry (2002) 26:277–85. doi:10.1016/S0278-5846(01)00266-4
13. Zai CCH, Tiwari AK, Chowdhury NI, Brandl EJ, Shaikh SA, Freeman N, et al. Association study of GABAA α2 receptor subunit gene variants in antipsychotic-associated weight gain. J Clin Psychopharmacol (2015) 35:7–12. doi:10.1097/JCP.0000000000000261
14. Al-Zoairy R, Pedrini MT, Khan MI, Engl J, Tschoner A, Ebenbichler C, et al. Serotonin improves glucose metabolism by Serotonylation of the small GTPase Rab4 in L6 skeletal muscle cells. Diabetol Metab Syndr (2017) 9:1. doi:10.1186/s13098-016-0201-1
15. Oh JE, Cho YM, Kwak SN, Kim JH, Lee KW, Jung H, et al. Inhibition of mouse brown adipocyte differentiation by second-generation antipsychotics. Exp Mol Med (2012) 44:545–53. doi:10.3858/emm.2012.44.9.062
16. Sertie AL, Suzuki AM, Sertie RA, Andreotti S, Lima FB, Passos-Bueno MR, et al. Effects of antipsychotics with different weight gain liabilities on human in vitro models of adipose tissue differentiation and metabolism. Prog Neuropsychopharmacol Biol Psychiatry (2011) 35:1884–90. doi:10.1016/j.pnpbp.2011.07.017
17. Hu Y, Kutscher E, Davies GE. Berberine inhibits SREBP-1-related clozapine and risperidone induced adipogenesis in 3T3-L1 cells. Phytother Res (2010) 24:1831–8. doi:10.1002/ptr.3204
18. Engl J, Laimer M, Niederwanger A, Kranebitter M, Starzinger M, Pedrini MT, et al. Olanzapine impairs glycogen synthesis and insulin signaling in L6 skeletal muscle cells. Mol Psychiatry (2005) 10:1089–96. doi:10.1038/sj.mp.4001729
19. Foussias G, Remington G. Antipsychotics and schizophrenia: from efficacy and effectiveness to clinical decision-making. Can J Psychiatry (2010) 55:117–25. doi:10.1177/070674371005500302
20. Albaugh VL, Vary TC, Ilkayeva O, Wenner BR, Maresca KP, Joyal JL, et al. Atypical antipsychotics rapidly and inappropriately switch peripheral fuel utilization to lipids, impairing metabolic flexibility in rodents. Schizophr Bull (2012) 38:153–66. doi:10.1093/schbul/sbq053
21. Albaugh VL, Judson JG, She P, Lang CH, Maresca KP, Joyal JL, et al. Olanzapine promotes fat accumulation in male rats by decreasing physical activity, repartitioning energy and increasing adipose tissue lipogenesis while impairing lipolysis. Mol Psychiatry (2011) 16:569–81. doi:10.1038/mp.2010.33
22. Houseknecht KL, Robertson AS, Zavadoski W, Gibbs EM, Johnson DE, Rollema H. Acute effects of atypical antipsychotics on whole-body insulin resistance in rats: implications for adverse metabolic effects. Neuropsychopharmacology (2007) 32:289–97. doi:10.1038/sj.npp.1301209
23. Kowalchuk C, Teo C, Wilson V, Chintoh A, Lam L, Agarwal SM, et al. In male rats, the ability of central insulin to suppress glucose production is impaired by olanzapine, whereas glucose uptake is left intact. J Psychiatry Neurosci (2017) 42:424–31. doi:10.1503/jpn.170092
24. Chintoh AF, Mann SW, Lam TK, Giacca A, Remington G. Insulin resistance following continuous, chronic olanzapine treatment: an animal model. Schizophr Res (2008) 104:23–30. doi:10.1016/j.schres.2008.06.006
25. Holloszy JO. Regulation by exercise of skeletal muscle content of mitochondria and GLUT4. J Physiol Pharmacol (2008) 59(Suppl 7):5–18.
26. McBride HM, Neuspiel M, Wasiak S. Mitochondria: more than just a powerhouse. Curr Biol (2006) 16:R551–60. doi:10.1016/j.cub.2006.06.054
27. Kuzmicic J, Del Campo A, Lopez-Crisosto C, Morales PE, pennanen C, Bravo-Sagua R, et al. [Mitochondrial dynamics: a potential new therapeutic target for heart failure]. Rev Esp Cardiol (2011) 64:916–23. doi:10.1016/j.recesp.2011.05.018
28. Zorzano A, Liesa M, Palacin M. Role of mitochondrial dynamics proteins in the pathophysiology of obesity and type 2 diabetes. Int J Biochem Cell Biol (2009) 41:1846–54. doi:10.1016/j.biocel.2009.02.004
29. Contreras-Shannon V, Heart DL, Paredes RM, Navaira E, Catano G, Maffi SK, et al. Clozapine-induced mitochondria alterations and inflammation in brain and insulin-responsive cells. PLoS One (2013) 8:e59012. doi:10.1371/journal.pone.0059012
30. Parra V, Verdejo HE, Iglewski M, Del Campo A, Troncoso R, Jones D, et al. Insulin stimulates mitochondrial fusion and function in cardiomyocytes via the Akt-mTOR-NFkappaB-Opa-1 signaling pathway. Diabetes (2014) 63:75–88. doi:10.2337/db13-0340
31. del Campo A, Parra V, Vasquez-Trincado C, Gutierrez T, Morales PE, Lopez-Crisosto C, et al. Mitochondrial fragmentation impairs insulin-dependent glucose uptake by modulating Akt activity through mitochondrial Ca2+ uptake. Am J Physiol Endocrinol Metab (2014) 306:E1–13. doi:10.1152/ajpendo.00146.2013
32. Ardizzone TD, Bradley RJ, Freeman AM III, Dwyer DS. Inhibition of glucose transport in PC12 cells by the atypical antipsychotic drugs risperidone and clozapine, and structural analogs of clozapine. Brain Res (2001) 923:82–90. doi:10.1016/S0006-8993(01)03026-8
33. Nozawa M, Ohnuma T, Matsubara Y, Sakai Y, Hatano T, Hanzawa R, et al. The relationship between the response of clinical symptoms and plasma olanzapine concentration, based on pharmacogenetics: Juntendo University Schizophrenia Projects (JUSP). Ther Drug Monit (2008) 30:35–40. doi:10.1097/FTD.0b013e31816336fd
34. Martins PJ, Haas M, Obici S. Central nervous system delivery of the antipsychotic olanzapine induces hepatic insulin resistance. Diabetes (2010) 59(10):2418–25. doi:10.2337/db10-0449
35. Teff KL, Rickels MR, Grudziak J, Fuller C, Nguyen H-L, Rickels K. Antipsychotic-induced insulin resistance and postprandial hormonal dysregulation independent of weight gain or psychiatric disease. Diabetes (2013) 62:3232–40. doi:10.2337/db13-0430
36. Seino S, Seino M, Bell GI. Human insulin-receptor gene. Partial sequence and amplification of exons by polymerase chain reaction. Diabetes (1990) 39:123–8. doi:10.2337/diabetes.39.1.123
37. Webb B, Sali A. Protein structure modeling with MODELLER. Methods Mol Biol (2014) 1137:1–15. doi:10.1007/978-1-4939-0366-5_1
38. Menting JG, Whittaker J, Margetts MB, Whittaker LJ, Kong GK, Smith BJ, et al. How insulin engages its primary binding site on the insulin receptor. Nature (2013) 493:241–5. doi:10.1038/nature11781
39. Irwin JJ, Shoichet BK. ZINC – a free database of commercially available compounds for virtual screening. J Chem Inf Model (2005) 45:177–82. doi:10.1021/ci049714+
40. Caffin F, Prola A, Piquereau J, Novotova M, David DJ, Garnier A, et al. Altered skeletal muscle mitochondrial biogenesis but improved endurance capacity in trained OPA1-deficient mice. J Physiol (2013) 591:6017–37. doi:10.1113/jphysiol.2013.263079
41. Vestri HS, Maianu L, Moellering DR, Garvey WT. Atypical antipsychotic drugs directly impair insulin action in adipocytes: effects on glucose transport, lipogenesis, and antilipolysis. Neuropsychopharmacology (2007) 32:765. doi:10.1038/sj.npp.1301142
42. Minet-Ringuet J, Even P, Valet P, Carpene C, Visentin V, Prevot D, et al. Alterations of lipid metabolism and gene expression in rat adipocytes during chronic olanzapine treatment. Mol Psychiatry (2007) 12:562. doi:10.1038/sj.mp.4001948
43. Theisen F, Haberhausen M, Firnges M, Gregory P, Reinders J, Remschmidt H, et al. No evidence for binding of clozapine, olanzapine and/or haloperidol to selected receptors involved in body weight regulation. Pharmacogenomics J (2007) 7:275. doi:10.1038/sj.tpj.6500418
44. Pavan C, Vindigni V, Michelotto L, Rimessi A, Abatangelo G, Cortivo R, et al. Weight gain related to treatment with atypical antipsychotics is due to activation of PKC-β. Pharmacogenomics J (2010) 10:408. doi:10.1038/tpj.2009.67
45. Weston-Green K, Huang X-F, Deng C. Alterations to melanocortinergic, GABAergic and cannabinoid neurotransmission associated with olanzapine-induced weight gain. PLoS One (2012) 7:e33548. doi:10.1371/journal.pone.0033548
46. Raposo N, Ferreira A, Gattaz W. Body mass index increase, serum leptin, adiponectin, neuropeptide Y and lipid levels during treatment with olanzapine and haloperidol. Pharmacopsychiatry (2011) 44:169–72. doi:10.1055/s-0031-1280793
47. Fernø J, Varela L, Skrede S, Vázquez MJ, Nogueiras R, Diéguez C, et al. Olanzapine-induced hyperphagia and weight gain associate with orexigenic hypothalamic neuropeptide signaling without concomitant AMPK phosphorylation. PLoS One (2011) 6:e20571. doi:10.1371/journal.pone.0020571
48. Sejima E, Yamauchi A, Nishioku T, Koga M, Nakagama K, Dohgu S, et al. A role for hypothalamic AMP-activated protein kinase in the mediation of hyperphagia and weight gain induced by chronic treatment with olanzapine in female rats. Cell Mol Neurobiol (2011) 31:985–9. doi:10.1007/s10571-011-9663-8
49. Weston-Green K, Huang X-F, Lian J, Deng C. Effects of olanzapine on muscarinic M3 receptor binding density in the brain relates to weight gain, plasma insulin and metabolic hormone levels. Eur Neuropsychopharmacol (2012) 22:364–73. doi:10.1016/j.euroneuro.2011.09.003
50. Weston-Green K, Huang X-F, Deng C. Second generation antipsychotic-induced type 2 diabetes: a role for the muscarinic M3 receptor. CNS Drugs (2013) 27:1069–80. doi:10.1007/s40263-013-0115-5
51. Ak M, Sezlev D, Sutcigil L, Akarsu S, Ozgen F, Yanik T. The investigation of leptin and hypothalamic neuropeptides role in first attack psychotic male patients: olanzapine monotherapy. Psychoneuroendocrinology (2013) 38:341–7. doi:10.1016/j.psyneuen.2012.06.012
52. Mondelli V, Anacker C, Vernon A, Cattaneo A, Natesan S, Modo M, et al. Haloperidol and olanzapine mediate metabolic abnormalities through different molecular pathways. Transl Psychiatry (2013) 3:e208. doi:10.1038/tp.2012.138
53. Ikegami M, Ikeda H, Ohashi T, Kai M, Osada M, Kamei A, et al. Olanzapine-induced hyperglycemia: possible involvement of histaminergic, dopaminergic and adrenergic functions in the central nervous system. Neuroendocrinology (2013) 98:224–32. doi:10.1159/000356119
54. Liu X, Deng C, Cao S, Gong J, Wang B-C, Hu C-H. Acute effects of oral olanzapine treatment on the expression of fatty acid and cholesterol metabolism-related gene in rats. Life Sci (2015) 128:72–8. doi:10.1016/j.lfs.2015.01.033
55. Babkin P, Thompson G, Alayna M, Iancu CV, Walters DE, Choe J-Y. Antipsychotics inhibit glucose transport: determination of olanzapine binding site in Staphylococcus epidermidis glucose/H+ symporter. FEBS Open Bio (2015) 5:335–40. doi:10.1016/j.fob.2015.04.006
56. He M, Zhang Q, Deng C, Jin T, Song X, Wang H, et al. Time-dependent effects of olanzapine treatment on the expression of histidine decarboxylase, H1 and H3 receptor in the rat brain: the roles in olanzapine-induced obesity. Psychoneuroendocrinology (2017) 85:190–9. doi:10.1016/j.psyneuen.2017.08.022
57. He M, Zhang Q, Deng C, Wang H, Lian J, Huang X-F. Hypothalamic histamine H1 receptor-AMPK signaling time-dependently mediates olanzapine-induced hyperphagia and weight gain in female rats. Psychoneuroendocrinology (2014) 42:153–64. doi:10.1016/j.psyneuen.2014.01.018
58. Liu X, Wu Z, Lian J, Hu C-H, Huang X-F, Deng C. Time-dependent changes and potential mechanisms of glucose-lipid metabolic disorders associated with chronic clozapine or olanzapine treatment in rats. Sci Rep (2017) 7:2762. doi:10.1038/s41598-017-02884-w
59. Stapel B, Kotsiari A, Scherr M, Hilfiker-Kleiner D, Bleich S, Frieling H, et al. Olanzapine and aripiprazole differentially affect glucose uptake and energy metabolism in human mononuclear blood cells. J Psychiatr Res (2017) 88:18–27. doi:10.1016/j.jpsychires.2016.12.012
Keywords: second-generation antipsychotic agents, mitochondrial dynamics, insulin resistance, L6 muscle cells, obesity
Citation: del Campo A, Bustos C, Mascayano C, Acuña-Castillo C, Troncoso R and Rojo LE (2018) Metabolic Syndrome and Antipsychotics: The Role of Mitochondrial Fission/Fusion Imbalance. Front. Endocrinol. 9:144. doi: 10.3389/fendo.2018.00144
Received: 01 October 2017; Accepted: 16 March 2018;
Published: 23 April 2018
Edited by:
Chao Deng, University of Wollongong, AustraliaReviewed by:
Pei Jiang, Jining Medical University, ChinaMargaret K. Hahn, University of Toronto, Canada
Copyright: © 2018 del Campo, Bustos, Mascayano, Acuña-Castillo, Troncoso and Rojo. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Leonel E. Rojo, bGVvbmVsLnJvam9AdXNhY2guY2w=