 Sabine Richard
Sabine Richard Frédéric Flamant
Frédéric Flamant- Institut de Génomique Fonctionnelle de Lyon, INRA USC 1370, Université de Lyon, Université Lyon 1, CNRS UMR 5242, Ecole Normale Supérieure de Lyon, Lyon, France
Alterations in maternal thyroid physiology may have deleterious consequences on the development of the fetal brain, but the underlying mechanisms remain elusive, hampering the development of appropriate therapeutic strategies. The present review sums up the contribution of genetically modified mouse models to this field. In particular, knocking out genes involved in thyroid hormone (TH) deiodination, transport, and storage has significantly improved the picture that we have of the economy of TH in the fetal brain and the underlying genetic program. These data pave the way for future studies to bridge the gap in knowledge between thyroid physiology and brain development.
Introduction
The major role of thyroid hormone (TH) in brain development has been recognized for a long time, but, in spite of considerable progress in the understanding of TH mode of action, the underlying mechanisms remain elusive. The thyroid gland produces mainly thyroxine (T4), the major circulating form of TH. Deiodination ensures the conversion of T4 into 3,3′,5-triiodothyronine (T3), the active form of TH, in several organs. This enzymatic reaction can be catalyzed either by type 1 deiodinase (DIO1) in the liver or by type 2 deiodinase (DIO2) in several other organs including the brain. TH catabolism is ensured by DIO1 and type 3 deiodinase (DIO3). T3 exerts a pleiotropic influence on development and cell differentiation. it does so by binding to nuclear receptors (TRs, including the TRα1, TRβ1, and TRβ2 isotypes) present in most, if not all, cell types in all vertebrate species. The consequences on neurodevelopment of a deficit in TH, either during pregnancy or during early childhood, are mostly irreversible (1). However, our basic knowledge of TH function during fetal brain development remains limited. Meanwhile, the possibility that genetic alterations (2) or environmental contaminations (3) compromise TH signaling in the fetal brain, without necessarily modifying the circulating levels of TH, is a matter of concern. Moreover, when circulating TH levels are impacted, neither the quantitative relationships nor the timing between serum alterations and brain effects have been well characterized. This review focuses on the contribution of experimental genetics in mice to our basic understanding of the complex relationship that links maternal thyroid physiology to fetal neurodevelopment. Notably, the delivery of T3 to the fetal brain seems to be a highly controlled and complex mechanism, which recent genetic investigations have only begun to unravel.
Time Course of TH Action During Human Brain Neurodevelopment
Thyroid hormone has for a long time been known to exert a major influence on human neurodevelopment, during prenatal and post-natal life. Three prenatal stages are to be considered (4):
(1) The first trimester of pregnancy is essentially dedicated to neurulation, brain patterning, and active neural cell proliferation. By the end of the second month, the rudimentary structures of the brain and central nervous system are established. In the earliest neurodevelopmental stages, TH is present at very low levels in the celomic and amniotic fluids, while TH levels are high in the maternal serum. TH levels increase rapidly in the celomic and amniotic fluids after the 4th week of pregnancy (5). The nuclear receptors of T3 are already present in the embryonic brain at this stage, and partially occupied by T3 (6). As a consequence, it is likely that gene expression can already be activated by T3 at an early stage of development. Indeed, TRs are seen very early in brain development, from 10 weeks of gestation in the human fetal brain (6) well before the fetal thyroid gland is functional. Thus, even minimally reduced maternal TH levels in early pregnancy may result in adverse outcome in the offspring (7). The possibility remains for T3 to be absent in some brain areas where TRs are expressed. In such cases, unliganded TRs would be bound to DNA and recruit corepressors, repressing the expression of the TH-inducible genes.
(2) The second trimester is marked by an acceleration in brain expansion and by massive neuronal and glial cell differentiation. The amniotic cavity grows rapidly and the matured placental barrier controls the passage of biological material from maternal to fetal circulation. While the development of the thyroid gland starts within the first weeks of pregnancy, the capacity of thyrocytes to accumulate iodine appears only at the end of the 4th month, and the secretion of TH by the fetal thyroid gland starts even later. Therefore, maternal TH remains, at mid-gestation, the main supply of TH for the fetus. Although TH concentration remains low in the fetal serum, this concentration increases rapidly in the cortex from the beginning of the second trimester. Thus, the concentration of T3 in the fetal cortex eventually reaches or even exceeds adult values (8). Concomitantly, TR levels in the fetal brain peak around the 16th week, in coincidence with the period of neuroblast multiplication.
(3) During the third trimester, neurodevelopment is clearly asynchronous, and rapid expansion is restricted to a few brain areas, notably the cerebellum and the olfactory bulbs. In other brain areas, terminal maturation processes, axon myelination, and synapse formation predominate. The fetal thyroid gland is functional but the complete maturation of the hypothalamus–pituitary–thyroid gland axis will only be achieved within the last month of pregnancy. Thus, a significant fraction of TH in the fetus is still of maternal origin (9). The levels of T4 and T3 in the fetal serum progressively reach levels found in newborns.
Consequences of TH Deficiency on Human Neurodevelopment
Overall, the risk of presenting neurodevelopmental and psychiatric disorders due to maternal thyroid dysfunction, as well as the nature and severity of the symptoms, depend on two parameters: the time window and the severity of TH deficiency (10, 11). While overt maternal hypothyroidism during pregnancy causes a condition known as “neurological cretinism”—with most dramatic effects if hypothyroidism occurs during the first trimester (12–14), only large epidemiological studies can provide evidence for cognitive defects caused by mild alterations in maternal TH levels taking place during late pregnancy (15). Importantly, therapeutic interventions are risky, because prolonged exposure to excess of TH during pregnancy increases the risk of miscarriage and may also probably cause later cognitive and psychiatric disorders (15, 16).
The Mouse as a Model of Human Pregnancy
Although classical animal studies of TH signaling during gestation have been mainly done in the rat, the mouse is the favorite model for experimental genetics, due to the possibility of generating knock-out (KO) and knock-in mutations. Most of the effects of hypothyroidism that were described in the rat are also found in the mouse. Nevertheless, the differences between the two rodent models require minor adjustments (Figure 1). For example, gestation lasts for 22 days in rats and 19 days in mice, but the stage at which the thyroid gland is functional is similar in both species (around gestational day 17; GD17). As a consequence, the mouse fetus remains dependent on maternal TH up to later stages of in utero development than does the rat fetus (about 89% of the duration of gestation in mice vs about 77% of the duration of gestation in rats) (17). Accordingly, differences between the two species as regards the consequences of hypothyroidism on neurodevelopment have been reported. For example, a malformation of the corpus callosum, with abnormal presence of neurons in the white matter, has been observed in hypothyroid rats but not yet been reported in mice (18).
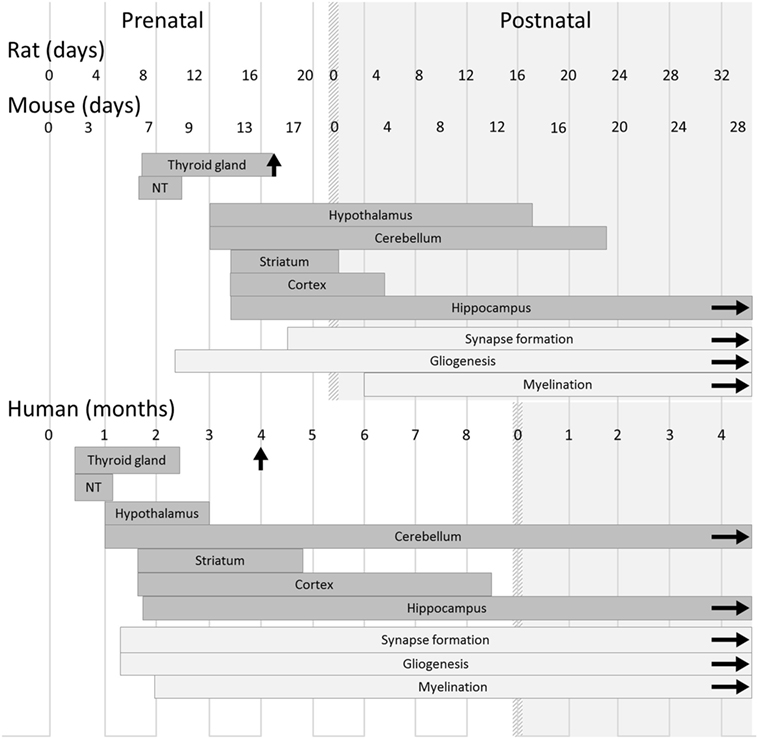
Figure 1. Timeline of thyroid and brain development in humans and rodents. In both rodents and humans, the development of the main brain areas starts before the onset of the fetal thyroid gland function and relies on maternal thyroid hormone supply. However, differences between species must be kept in mind when translating results from rodent models to humans, and even between mouse and rat models. Gestation lasts 22 days in rats, 19 days in mice, and 42 weeks in humans. Gliogenesis, myelination, and synaptogenesis are asynchronous in different brain areas and continue after birth. For each brain region, the corresponding rectangle symbolizes the phase of neurogenesis. Vertical arrows indicate the initiation of iodide uptake by thyrocytes, which marks the end of thyroid gland maturation. The light gray area corresponds to postnatal life. Abbreviation: NT, neural tube [adapted from Ref. (19)].
By contrast, notable differences in the program of neurodevelopment should be carefully taken into account when translating rodent studies to humans (Figure 1) (19). The onset of thyroid gland function in rodents occurs at a late stage of gestation (GD17), and the fetus largely relies on TH of maternal origin, whereas in humans, the thyroid gland becomes functional during the 4th month of gestation. In rodents as in humans, the first third of gestation appears to be of critical importance for the action of TH on neurodevelopment (20). However, at birth, neurodevelopment is less advanced in mice than in humans. Markedly, cerebellum expansion, axon myelination, and synapse formation, which take place during the last trimester of human pregnancy (21), are mainly post-natal events in mice as in rats. Interestingly, this post-natal phase in rodents is marked by a peak in circulating levels of TH (22). It is unclear whether an equivalent peak in TH levels occurs at any point during brain development in humans. As a whole, the mouse post-natal development is, to some extent, relevant to human late pregnancy. Moreover, a recent study has provided evidence that in mice, as in rats and in humans, mild hypothyroidism in the perinatal period is sufficient to cause long-lasting cognitive impairments that persist into adulthood (23).
Brain cells of rodent fetuses are sensitive to TH and already express Thra, the gene encoding TRα1, the most ubiquitous nuclear receptor of TH. Early indirect evidence, based on binding affinity measurements, suggested that TRβ1, rather than TRα1, was the first receptor to be present in human fetal brain (6). However, recent transcriptome data (http://www.brainspan.org) indicate that, like in rodents (24, 25), Thra expression in the human fetal brain is ubiquitous and precocious, while Thrb expression is restricted to specific brain areas and appears at a later stage. This conclusion is also consistent with the fact that neurodevelopmental defects seem to be more frequent in patients with Thra mutations (26), than in patients with Thrb mutations (27). Like TRβ1, TRα1 can exert both ligand-dependent and ligand-independent influence on gene expression. While Thra KO mice lose both activities, knock-in mutations maintain ligand-independent repression. Therefore, Thra knock-in models display severe neurodevelopmental defects, mimicking early TH deficiency, which are not observed in KOs (28, 29). The contribution of mouse genetics to the understanding of Thra and Thrb neurodevelopmental functions has been recently reviewed elsewhere (30).
Neurodevelopmental Consequences of Congenital Hypothyroidism in Rodents
Congenital hypothyroidism is experimentally induced in rodents by reducing thyroid signaling, by exposure to a toxic substance, surgical thyroidectomy, or genetic mutation. Congenital hypothyroidism seems to affect all the main neural cell types in all brain areas in rodents. Many studies have been focused on the cortex and cerebellum, pointing to similarities between the two brain areas, even though they develop during different time windows (1). These similarities suggest the existence of common mechanisms. Studying the two models, cortex and cerebellum, should highlight unifying principles regarding the role of TH in neurodevelopment.
One major defect observed in the hypothyroid brain is that axon myelination is retarded, due to impaired differentiation of oligodendrocytes (31). Another typical feature is delayed maturation of radial glia that act as a scaffold for neuron migration during corticogenesis. Accordingly, the radial migration of glutamatergic neuron progenitors is impaired (32, 33). The migration of GABAergic interneuron progenitors follows different routes but is also disrupted (34, 35). In addition, the terminal differentiation of these inhibitory neurons is deeply altered (34, 36). Cell type-specific blockade of TRα1 function in the cerebellum has led to the conclusion that, during neurodevelopment, the primary targets of T3 are the GABAergic neurons and radial glia (37). These cell types interact with the other neighboring cell types, either by direct contact or by exchanging growth factors and neurotrophins (38). Therefore, while the initial cell-autonomous action of T3 appears to be restricted to few cell types, it is subsequently amplified by these T3-induced secretions. As a consequence, T3 indirectly influences the differentiation of all neural cell types (39). Although the molecular mechanisms underlying all these cellular defects remain largely unknown, genome-wide expression analyses have identified a number of TH responsive genes (40). In particular, Hr (encoding Hairless) and Klf9 (encoding Kruppel-like factor 9 protein) are highly responsive to T3 in several brain areas and cell types and are routinely used as intracerebral sensors of T3 signaling (41, 42).
The Origin of T3 in the Fetal Rodent Brain
Even after the onset of the fetal thyroid gland function, 60% of the total TH content in fetal peripheral tissues remains of maternal origin at late gestation stages in mice (43). In addition, there are good indications supporting the view that the main source of T3 in the fetal brain is maternal T4, not maternal T3. Indeed, when hypothyroid dams are given either T4 or T3 during gestation, T4 is more efficient at restoring T3 level in the fetal cortex (44). Moreover, maternal hypothyroxinemia (i.e., the selective reduction of T4, but not T3, in the maternal serum) causes neurodevelopmental defects in rats similar to maternal hypothyroidism. Therefore, maternal T3 is not sufficient to ensure correct fetal neurodevelopment (45, 46). Such a predominance of maternal T4 over maternal T3 in supplying THs to the fetal brain results from the existence of two barriers, first at the placenta level and second at the blood–brain barrier level, which display some selectivity for T4 or T3.
Maternal T4 and T3 are catabolized in the placenta by DIO3 (47), which has a preference for T3 over T4 as a substrate (48). As the concentration of T4 in the maternal serum exceeds that of T3, the net result is a preferential transfer of maternal T4 to the fetal serum (49). Furthermore, during fetal development, the contribution of fetal circulating T3 to brain T3 appears to be very low, suggesting that T3 is unable to cross the blood–brain barrier at this early stage (43). The collective term “blood–brain barrier” encompasses three putative pathways for TH entry in the brain: the blood–brain barrier proper, the blood–cerebrospinal fluid barrier, and the cerebrospinal fluid–brain barrier. Most of T3 in the fetal brain comes from T4 transport across the blood–cerebrospinal fluid barrier (i.e., the choroid plexus). T4 is locally converted into T3 by DIO2, which is expressed in the brain ventricles as early as GD15 (50). By contrast, after birth, the fraction of brain T3 directly coming from the circulation reaches 50% (43).
Finally, specific mechanisms come into play to ensure an accumulation of T3 in the fetal brain. Indeed, at mid-gestation, the level of T3 in the fetal brain is higher than in the fetal serum or liver (51). Under low levels of maternal T4, fetal T4 levels are decreased in peripheral organs and in the brain, but brain T3 level remains unchanged (43).
Heterogeneity of the Intracerebral Distribution of T3
Quantitative analysis of TH content in small brain areas by biochemical means is technically challenging. To circumvent this problem, transgenic models have been developed to indirectly evaluate T3 signaling. FINDT3 transgenic mice carry a self-inducing reporter construct based on an artificial Gal4-TRα1 receptor (50). In this system, the regulation of lacZ expression allows one to directly visualize TH signaling in the fetal brain. X-gal staining, to visualize lacZ expression, confirms the entry of maternal TH in the fetal mouse brain at mid gestation. Two simpler, but less specific, transgenic devices have also been produced: TRE2x mice carry the lacZ gene, the expression of which is driven by a minimal promoter and two binding sites for endogenous TR (52). THAI mice design followed a similar strategy, but the luciferase gene was preferred to lacZ to facilitate quantification and live imaging (53). The lacZ reporter expression patterns in FINDT3 and TRE2x mice do not always match. Although this is expected, as they rely on very different systems, the presence of experimental biases cannot be completely ruled out, as both systems could be influenced by other factors than T3 local concentration. For example, TRE2x is also regulated by other nuclear receptors recognizing the same response element, notably LXR (53). Meanwhile, the minimal promoter used in FINDT3 mice favors the expression in neurons but is inefficient in glia (50). In spite of these differences, both FINDT3 and TRE2x transgenic models detect the presence of T3 in the fetal brain at mid-gestation (GD12 for TRE2x, GD13 for FINDT3). Both models also converge to suggest a persistent heterogeneity of T3 distribution in the brain. The detailed expression pattern of lacZ in FINDT3 mice during fetal brain development has been reported elsewhere (50). In summary, the reporter expression is high in the midbrain, anterior cortex and hippocampus but low in the cerebellum and olfactory bulbs throughout late fetal and early post-natal development. This expression pattern is maintained in the adult brain and fits with direct measurement of T3 content in different rat brain areas (54). This expression pattern also presents similarities with the situation in humans, where T3 content is high in the fetal cortex and low in the fetal cerebellum (8). While the existence of such heterogeneous distribution of T3 in the brain is confirmed by different approaches, the underlying mechanisms remain unclear.
Deiodinases and Brain T3 Content
As fetal T3 is mainly produced by DIO2-mediated T4 deiodination, the heterogeneous distribution of T3 in the fetal brain has been hypothesized to be a consequence of the heterogeneous distribution of DIO2 in the brain (50, 55). However, there is, to our knowledge, no genetic evidence that DIO2 is necessary to maintain the level of T3 in the fetal brain. Indeed, Dio2 gene KO in mice does not modify the FINDT3 reporter expression pattern throughout gestation (unpublished data). Even the combination of Dio1/Dio2 KO does not induce major changes in the T3 content of different brain areas, as measured at post-natal day 6 (56). In Dio1/Dio2 KO mice, T3 content is not significantly affected until post-natal day 15 (PND15). From that stage onward, T3 levels are significantly reduced in all Dio1/Dio2 KO mouse brain areas (57). However, T3 signaling in neural cells is not immediately affected, as expression of the Hairless gene at PND21 is not changed. As mentioned above, this gene is a well-characterized T3-responsive gene and is often used as an internal sensor of T3 signaling in the brain (58). Taken together, these data seem to indicate that, in mice, T3 levels in the brain do not rely on type 2 deiodination before the second week of post-natal life.
The surprisingly limited consequences of Dio2 KO on fetal neurodevelopment might reflect the intervention of compensatory mechanisms in the brain. In particular, the expression of the Dio3 gene, encoding DIO3, which is responsible for TH catabolism in several tissues, is reduced in the brain of Dio2 KO mice, therefore limiting TH deficiency (17). As a whole, it remains unlikely that DIO2 plays a major role in generating a heterogeneous distribution of T3 in the brain. An alternative hypothesis to the local conversion of T4 into T3 would be a selective catabolism in different brain areas. This possibility was tested by crossing FINDT3 reporter mice with Dio3 KO mice. In FINDT3/Dio3 KO mice, the reporter gene senses an increase in TH signaling in several brain areas and lacZ expression is higher than in FINDT3 mice. However, this difference occurs only weeks after birth, and the expression pattern of the reporter is not markedly altered (59). Therefore, local metabolism by deiodinases has the capacity to buffer variations in TH availability but cannot alone account for the heterogeneous distribution of T3 in fetal and post-natal mouse brains.
TH Transporters
TH Transport: Many Players
If the accumulation and heterogeneous distribution of T3 in the fetal brain are not only the result of local TH metabolism, what could be the underlying mechanisms? An alternative could be that TH transport has a major influence.
It is now recognized that specific transporters are needed for TH to cross the placenta, the blood–brain barrier or the blood–cerebrospinal fluid barrier (43, 60). Intracytoplasmic transporters are also needed for T3 to reach cell nuclei (61). While solute carrier proteins are supposed to be key elements of TH transfer to the fetal brain, progress of genetic investigation in this field has been slow for several reasons. First, THs can be transported with variable efficiency by at least 20 members of the solute carrier family (62). Many of these are present in the brain, which suggests the existence of several entry routes with redundant functions. Therefore, a single gene KO may not be sufficient to alter TH transfer. The second difficulty is that some transporters are expressed in both the placenta and the fetal brain, which might introduce a confounding effect: neurodevelopmental defects due to placental defect might be mistakenly attributed to a direct influence of the KO in the brain (63). Finally, these transporters are often able to transport other low molecular-weight molecules that are also required for proper neurodevelopment. One such example is that of the L-type amino-acid transporter type 1 (LAT1), which is known to facilitate TH cellular uptake (64). The KO of Slc7a5, which encodes LAT1, has clear neurological consequences, even when the KO is restricted to the blood–brain barrier. However, the effects are not attributed to TH deficiency, but to a defect in neutral amino-acid transport, since intra-cerebro-ventricular injection of amino-acids provides a significant improvement in the neurobehavioral abnormalities caused by this mutation (65).
The Intriguing Case of Mct8
Among all solute carrier proteins, MCT8 deserves special attention, for the following reasons: (a) T4, T3, and reverse T3 are the only known substrates of this transporter (66). (b) Mct8 gene expression in the mouse brain starts at embryonic day 15 (67). Its expression is notably high at post-natal stages in the cortex, striatum, hippocampus, and in the tanycytes lining the hypothalamic third ventricle (68). (c) Human Mct8 gene mutations cause a dramatic neurodevelopmental disorder known as the Allan–Herndon–Dudley syndrome (69). Surprisingly, Mct8 KO in mice has only minor neurodevelopmental consequences (70, 71).
The expression pattern of the TH signaling reporter is not visibly changed in the brain of FINDT3/Mct8 KO mice, arguing against a predominant influence of this transporter in mice (unpublished data). Unexpectedly, a transient increase in cortical T3 content is observed between PND0 to PND3 in Mct8 KO mice (72), while the serum content of T4 is high and of T3 is low. The situation is reversed at PND21, when T3 is reduced in the cortex and high in the plasma of Mct8 KO mice. Despite a chronic excess of T4 and T3 in the serum, the adult brain remains hypothyroid (73). In vivo transport of T3, but not T4, is markedly reduced in the brain (71). Taken together, these data suggest that, in mice, the predominance of MCT8 function as a TH transporter in the brain only appears at a late post-natal stage. At this point, Mct8 KO mice start to display a clear deficit in intracerebral T3 content. Interestingly, gene expression analyses also suggest that Mct8 KO selectively affects some brain areas, notably the hypothalamus, but not others, like the striatum (71).
There are two possible explanations for the discrepancy between the severity of the Allan–Herndon–Dudley syndrome and the modest consequences of Mct8 KO in mice: one is that human MCT8, but not mouse MCT8, is necessary to transport some unknown molecule required for neurodevelopment. This might explain why the consequences of the Allan–Herndon–Dudley syndrome are usually much more severe than the symptoms of congenital hypothyroidism. Alternatively, due to their different spatial and temporal patterns of gene expression, compensatory mechanisms might exist in mice which cannot take place in humans (74).
Combining Gene KOs to Reveal Mct8 Function
A number of combinations of KOs have been tested to identify another transporter able to compensate for the absence of MCT8 in mice, but not in humans (Table 1). MCT10 transporter is absent at the blood–brain barrier and, accordingly, the combination of Mct10 and Mct8 KOs does not aggravate the Mct8 KO brain phenotype (68). Combining Mct8 and Lat2 KOs has also failed to demonstrate a major function of LAT2 in TH transport in the brain (72). OATP1C1 is a better candidate, as it is encoded by a gene that is apparently expressed in the mouse fetal brain at higher level than in the human fetal brain. As expected, the combination of both Oatp1c1 and Mct8 KOs markedly increase the post-natal neurodevelopmental defects of single KO, despite a compensatory increase in Dio2 expression (75). Notably, while the reduction of Hairless expression in the cortex (PND 21) is moderate after a single KO, the reduction in gene expression achieved when the two mutations are combined is comparable to the one observed in TH-deficient pups (75). A similar aggravation of the phenotype is observed for axon myelination in the corpus callosum and for GABAergic neuron differentiation in the cortex (75). All these data point to a state of severe hypothyroidism in the brain of Mct8/Oatp1c1 mice at PND21.
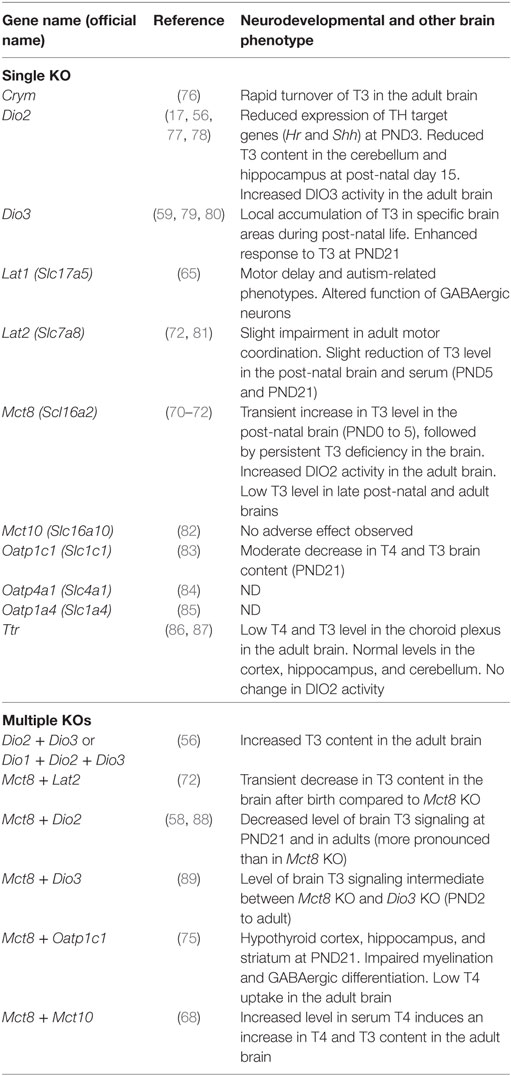
Table 1. Neurodevelopmental consequences of knock-out (KO) mutations in thyroid hormone (TH) transporters and storage proteins.
Interestingly, Hairless gene expression can be restored in the brain of hypothyroid wild-type mice by either T4 or T3 treatment. By contrast, when Mct8 KO mice are made hypothyroid by pharmacological means, only T4 treatment is able to restore Hairless gene expression in the brain (90), suggesting that in the absence of MCT8, deiodination of T4 into T3 becomes mandatory for T3 accumulation in the brain. In agreement with this hypothesis, the combination of Dio2 and Mct8 KOs also alters post-natal neurodevelopment more visibly than either single KO, as judged by gene expression analysis at PND21 (58). The synergy of these two KOs indicates the existence of two independent pathways for T3 entry in the brain. Dio2 and Mct8 are thus key elements of these two pathways ensuring the presence of T3 in neurons (91).
Intracerebral Storage of T3
At least some of the solute carriers that transport TH can act both on cellular influx and efflux (92). Therefore, the maintenance of sustained TH levels in the brain, including when circulating levels are low, might not rely exclusively on transporters. The accumulation of TH in the fetal brain implies the presence of other TH binding proteins for storage, either in neural cells or in the cerebrospinal fluid. One of the main TH storage proteins is transthyretin, which is encoded by the Ttr gene. Transthyretin is present in the serum, placenta and is abundant in the cerebrospinal fluid. While Ttr gene expression is already high at mid-gestation in the choroid plexus, Ttr KO has no visible neurodevelopmental consequences (86). Indirect evidence also indicates that brain TH levels are not altered in Ttr KO mice (93).
Another molecule contributing to TH accumulation in the fetal brain is CRYM (alias μ-crystallin and CTBP), which is a cytosol protein that binds TH in a NADPH-dependent manner. According to the Allen Brain Atlas, Crym gene is already expressed at mid-gestation in the fetal mouse brain. Crym expression is high in some brain areas, notably in the cortex, and its expression pattern changes rapidly during development. Once more, Crym KO has no obvious adverse effect on neurodevelopment. However, the clearance of radiolabeled T3 from the brain is accelerated in adult Crym KO, compared to wild-type, mice (76). Originally considered exclusively as a cytoplasmic storage protein, CRYM was also later identified as an enzyme that reduces the sulfur-containing cyclic ketimines (94), which are potential neurotransmitters. Structural data suggest that T3 is an allosteric regulator that inhibits CRYM enzymatic activity (95). Thus, the regulation of CRYM enzymatic activity by T3 might constitute a way by which T3 influences brain function without binding to its nuclear receptors.
What is the Function of T3 During Fetal Brain Development?
While a number of transcriptome analyses have been performed at post-natal stages in order to identify T3-responsive genes in the post-natal brain (40), little information is available for the fetal brain. Microarray analyses have identified a large number of genes, the expression of which is sensitive to maternal hypothyroidism in rat cortex and hippocampus (96, 97). However, the changes in gene expression observed in these studies might be very indirect consequences of hypothyroidism. Indeed, some of these changes in gene expression may reflect metabolic changes, defects in placenta development, or a modification in the composition of the heterogeneous population of neural cells. Understanding the mechanisms of action of T3 in the fetal brain requires separating T3 direct target genes from genes that are positioned downstream in the cascade of effects induced by hypothyroidism.
A more direct approach to recognize T3-responsive genes was to first submit pregnant mice to a short methimazole treatment to make them hypothyroid and then treat them with THs for 12 h (98). Such a short-term activation of TH signaling pathways reduces the chances of observing indirect effects because it allows little time for activation of secondary target genes, i.e., genes that are activated by proteins encoded by direct T3 target genes. This approach confirmed the existence of a large number of TH-responsive genes in the fetal cortex, although discrepancies between studies were also noticed. These genes produce both messenger RNAs and microRNAs (98). Primary cell cultures from mouse fetal cortices, enriched in either neurons or astrocytes, were also used. When analyzed at a genome-wide scale, the in vitro response to T3 confirmed the existence of many T3-responsive genes in both cell populations. However, none of the above-mentioned protocols allows eliminating completely all secondary effects of TH on gene expression. Thus, the observed changes in gene expression can still either be directly mediated by TRα1 and TRβ1 or be secondary to the initial response of the cells, which consists in overexpressing a number of transcription factors and growth factors.
To distinguish between direct and indirect T3 target genes, cycloheximide was used to inhibit protein synthesis and to prevent secondary responses. Among the previously identified T3-responsive genes, a subset of 371 genes was still responsive in the presence of cycloheximide. This smaller set of genes was likely to contain those that are directly regulated by TRα1 (99, 100). In this in vitro system, T3 response was, at least for a large part, similar to the one observed in more physiological settings (40). Interestingly, in-depth bioinformatics analysis led to the hypothesis that some unidentified cortical cell types displayed a higher sensitivity to T3 than the others (99, 100). However, in that study, it was difficult to ascertain a direct transcriptional response, since there was no knowledge about the genomic sites occupied by TRα1 and TRβ1 in the cells used.
The number of genes that are regulated in neural cells, the variety of their function, and some discrepancies between published studies do not allow for a unified interpretation of these transcriptome data. It is nevertheless striking that T3 activates the expression of genes involved in several signaling pathways known to be important for neurodevelopment, such as the Sonic hedgehog, glucocorticoid, and retinoic acid signaling pathways (99). These regulations suggest that, in the fetal cortex, as in the post-natal cerebellum, the initial response to T3 of a subset of cell types is greatly amplified by the activation of other signaling pathways (37, 101).
Trans-Generational Maternal Transmission of Hypothalamus–Pituitary–Thyroid Axis Set Points?
Only a few studies have to date addressed the capacity of TH to promote hypothalamus development (102). However, one important consequence of a prenatal change in TH signaling is a permanent change in the set points of the hypothalamo–pituitary–thyroid gland axis in the progeny (103). This might reflect, at least in part, a defect in hypothalamus development, perhaps in the development of hypophysiotropic neurons which produce thyrotropin-releasing hormone. For example, mice born from Thrb KO dams are exposed to an excess of TH during fetal life. When they reach adulthood, their circulating level of thyroid-stimulating hormone is lower than in control mice born from euthyroid dams. Despite nearly normal levels of T4 and T3, Dio2 gene expression in the hypothalamus is reduced and Dio3 gene expression in the pituitary is augmented (104, 105). Thus, there is a theoretical possibility for intergenerational and transgenerational maternal transmission of changes in TH signaling acquired during fetal life.
Future Directions and Clinical Relevance
Taken together, these genetic data pave the way for promising studies. One clear gap in our knowledge is the development and physiology of the barriers that TH must cross to reach neural cells and the possible involvement of TH itself in these processes (11). Currently, mouse genetics do not bring much information to our understanding of either the action of TH on placenta development (106) or the mechanisms of trans-placental transfer of TH (107). However, there is a possibility that a placental defect, of maternal or fetal origin, may indirectly compromise neurodevelopment, as observed in other contexts (108, 109). The placental barrier displays some similarities with the blood–brain barrier and also expresses a number of solute carrier family members (92, 110). In fact, placental development in Mct8 KO fetuses is slightly altered (111). Another limitation of current studies is that TH entry in the fetal brain has been mainly considered in a global manner. However, the existence of different pathways for TH entry in the brain is an indication that T3 can be delivered to specific brain areas at different times during neurodevelopment. This may allow the local concentration of T3 to act as a positional and temporal cue for neural cell differentiation.
Our understanding of the molecular mechanisms underlying the influence of T3 in neuronal migration and differentiation also remains very limited and should be integrated in the general model of neurodevelopment that has emerged from a large body of mouse genetic studies (112). The recent advent of “omics” has not yet provided an unbiased and genome-wide view of T3 influence on gene expression, within each of the many cell types that are present in the developing brain.
Improving our understanding of these mechanisms should be beneficial in several respects. First, it may help to understand the pathology of rare genetic diseases altering cognitive function. Also, it should help to better address the neurotoxicity of environmental chemicals acting as TH disruptors. Finally, it could lead to the development of new TH analogs suitable to treat neurodevelopmental and psychiatric diseases.
Author Contributions
FF conceived and wrote the article. SR wrote the article.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Funding
This work was supported by the French National Research Agency [grant number ANR-15-CE14-0011-01].
References
1. Berbel P, Navarro D, Roman GC. An evo-devo approach to thyroid hormones in cerebral and cerebellar cortical development: etiological implications for autism. Front Endocrinol (2014) 5:146. doi:10.3389/fendo.2014.00146
2. Refetoff S, Bassett JH, Beck-Peccoz P, Bernal J, Brent G, Chatterjee K, et al. Classification and proposed nomenclature for inherited defects of thyroid hormone action, cell transport, and metabolism. J Clin Endocrinol Metab (2014) 99:768–70. doi:10.1210/jc.2013-3393
3. Miller MD, Crofton KM, Rice DC, Zoeller RT. Thyroid-disrupting chemicals: interpreting upstream biomarkers of adverse outcomes. Environ Health Perspect (2009) 117:1033–41. doi:10.1289/ehp.0800247
4. de Escobar GM, Ares S, Berbel P, Obregon MJ, del Rey FE. The changing role of maternal thyroid hormone in fetal brain development. Semin Perinatol (2008) 32:380–6. doi:10.1053/j.semperi.2008.09.002
5. Obregon MJ, Calvo RM, Del Rey FE, de Escobar GM. Ontogenesis of thyroid function and interactions with maternal function. Endocr Dev (2007) 10:86–98. doi:10.1159/000106821
6. Bernal J, Pekonen F. Ontogenesis of the nuclear 3,5,3′-triiodothyronine receptor in the human fetal brain. Endocrinology (1984) 114:677–9. doi:10.1210/endo-114-2-677
7. Patel J, Landers K, Li H, Mortimer RH, Richard K. Thyroid hormones and fetal neurological development. J Endocrinol (2011) 209:1–8. doi:10.1530/JOE-10-0444
8. Kester MH, Martinez de Mena R, Obregon MJ, Marinkovic D, Howatson A, Visser TJ, et al. Iodothyronine levels in the human developing brain: major regulatory roles of iodothyronine deiodinases in different areas. J Clin Endocrinol Metab (2004) 89:3117–28. doi:10.1210/jc.2003-031832
9. Santisteban P, Bernal J. Thyroid development and effect on the nervous system. Rev Endocr Metab Disord (2005) 6:217–28. doi:10.1007/s11154-005-3053-9
10. Korevaar TIM, Medici M, Visser TJ, Peeters RP. Thyroid disease in pregnancy: new insights in diagnosis and clinical management. Nat Rev Endocrinol (2017) 13:610–22. doi:10.1038/nrendo.2017.93
11. Moog NK, Entringer S, Heim C, Wadhwa PD, Kathmann N, Buss C. Influence of maternal thyroid hormones during gestation on fetal brain development. Neuroscience (2017) 342:68–100. doi:10.1016/j.neuroscience.2015.09.070
12. Henrichs J, Bongers-Schokking JJ, Schenk JJ, Ghassabian A, Schmidt HG, Visser TJ, et al. Maternal thyroid function during early pregnancy and cognitive functioning in early childhood: the generation R study. J Clin Endocrinol Metab (2010) 95:4227–34. doi:10.1210/jc.2010-0415
13. Chevrier J, Harley KG, Kogut K, Holland N, Johnson C, Eskenazi B. Maternal thyroid function during the second half of pregnancy and child neurodevelopment at 6, 12, 24, and 60 months of age. J Thyroid Res (2011) 2011:426427. doi:10.4061/2011/426427
14. Craig WY, Allan WC, Kloza EM, Pulkkinen AJ, Waisbren S, Spratt DI, et al. Mid-gestational maternal free thyroxine concentration and offspring neurocognitive development at age two years. J Clin Endocrinol Metab (2012) 97:E22–8. doi:10.1210/jc.2011-1772
15. Andersen SL, Laurberg P, Wu CS, Olsen J. Attention deficit hyperactivity disorder and autism spectrum disorder in children born to mothers with thyroid dysfunction: a Danish nationwide cohort study. BJOG (2014) 121:1365–74. doi:10.1111/1471-0528.12681
16. Korevaar TI, Muetzel R, Medici M, Chaker L, Jaddoe VW, de Rijke YB, et al. Association of maternal thyroid function during early pregnancy with offspring IQ and brain morphology in childhood: a population-based prospective cohort study. Lancet Diabetes Endocrinol (2016) 4:35–43. doi:10.1016/S2213-8587(15)00327-7
17. Barez-Lopez S, Obregon MJ, Bernal J, Guadano-Ferraz A. Thyroid hormone economy in the perinatal mouse brain: implications for cerebral cortex development. Cereb Cortex (2018) 28:1783–93. doi:10.1093/cercor/bhx088
18. Goodman JH, Gilbert ME. Modest thyroid hormone insufficiency during development induces a cellular malformation in the corpus callosum: a model of cortical dysplasia. Endocrinology (2007) 148:2593–7. doi:10.1210/en.2006-1276
19. Howdeshell KL. A model of the development of the brain as a construct of the thyroid system. Environ Health Perspect (2002) 110:337–48. doi:10.1289/ehp.02110s3337
20. Morreale de Escobar G, Obregon MJ, Escobar del Rey F. Role of thyroid hormone during early brain development. Eur J Endocrinol (2004) 151:U25–37. doi:10.1530/eje.0.151U025
21. Huttenlocher PR, Dabholkar AS. Regional differences in synaptogenesis in human cerebral cortex. J Comp Neurol (1997) 387:167–78. doi:10.1002/(SICI)1096-9861(19971020)387:2<167::AID-CNE1>3.0.CO;2-Z
22. Hadj-Sahraoui N, Seugnet I, Ghorbel MT, Demeneix B. Hypothyroidism prolongs mitotic activity in the post-natal mouse brain. Neurosci Lett (2000) 280:79–82. doi:10.1016/S0304-3940(00)00768-0
23. Amano I, Takatsuru Y, Khairinisa MA, Kokubo M, Haijima A, Koibuchi N. Effects of mild perinatal hypothyroidism on cognitive function of adult male offspring. Endocrinology (2018) 159:1910–21. doi:10.1210/en.2017-03125
24. Mellstrom B, Naranjo JR, Santos A, Gonzalez AM, Bernal J. Independent expression of the alpha and beta c-erbA genes in developing rat brain. Mol Endocrinol (1991) 5:1339–50. doi:10.1210/mend-5-9-1339
25. Bradley DJ, Young WS III, Weinberger C. Differential expression of alpha and beta thyroid hormone receptor genes in rat brain and pituitary. Proc Natl Acad Sci U S A (1989) 86:7250–4. doi:10.1073/pnas.86.18.7250
26. Moran C, Chatterjee K. Resistance to thyroid hormone alpha-emerging definition of a disorder of thyroid hormone action. J Clin Endocrinol Metab (2016) 101:2636–9. doi:10.1210/jc.2016-2317
27. Weiss RE, Refetoff S. Resistance to thyroid hormone. Rev Endocr Metab Disord (2000) 1:97–108. doi:10.1023/A:1010072605757
28. Flamant F, Poguet AL, Plateroti M, Chassande O, Gauthier K, Streichenberger N, et al. Congenital hypothyroid Pax8(-/-) mutant mice can be rescued by inactivating the TRalpha gene. Mol Endocrinol (2002) 16:24–32. doi:10.1210/me.16.1.24
29. Morte B, Manzano J, Scanlan T, Vennstrom B, Bernal J. Deletion of the thyroid hormone receptor alpha 1 prevents the structural alterations of the cerebellum induced by hypothyroidism. Proc Natl Acad Sci U S A (2002) 99:3985–9. doi:10.1073/pnas.062413299
30. Flamant F, Gauthier K, Richard S. Genetic investigation of thyroid hormone receptor function in the developing and adult brain. Curr Top Dev Biol (2017) 125:303–35. doi:10.1016/bs.ctdb.2017.01.001
31. Billon N, Tokumoto Y, Forrest D, Raff M. Role of thyroid hormone receptors in timing oligodendrocyte differentiation. Dev Biol (2001) 235:110–20. doi:10.1006/dbio.2001.0293
32. Martinez-Galan JR, Escobar del Rey F, Morreale de Escobar G, Santacana M, Ruiz-Marcos A. Hypothyroidism alters the development of radial glial cells in the term fetal and postnatal neocortex of the rat. Brain Res Dev Brain Res (2004) 153:109–14. doi:10.1016/j.devbrainres.2004.08.002
33. Martinez R, Eller C, Viana NB, Gomes FC. Thyroid hormone induces cerebellar neuronal migration and Bergmann glia differentiation through epidermal growth factor/mitogen-activated protein kinase pathway. Eur J Neurosci (2011) 33:26–35. doi:10.1111/j.1460-9568.2010.07490.x
34. Manzano J, Cuadrado M, Morte B, Bernal J. Influence of thyroid hormone and thyroid hormone receptors in the generation of cerebellar gamma-aminobutyric acid-ergic interneurons from precursor cells. Endocrinology (2007) 148:5746–51. doi:10.1210/en.2007-0567
35. Cuevas E, Auso E, Telefont M, Morreale de Escobar G, Sotelo C, Berbel P. Transient maternal hypothyroxinemia at onset of corticogenesis alters tangential migration of medial ganglionic eminence-derived neurons. Eur J Neurosci (2005) 22:541–51. doi:10.1111/j.1460-9568.2005.04243.x
36. Gilbert ME, Sui L, Walker MJ, Anderson W, Thomas S, Smoller SN, et al. Thyroid hormone insufficiency during brain development reduces parvalbumin immunoreactivity and inhibitory function in the hippocampus. Endocrinology (2007) 148:92–102. doi:10.1210/en.2006-0164
37. Fauquier T, Chatonnet F, Picou F, Richard S, Fossat N, Aguilera N, et al. Purkinje cells and Bergmann glia are primary targets of the TRalpha1 thyroid hormone receptor during mouse cerebellum postnatal development. Development (2014) 141:166–75. doi:10.1242/dev.103226
38. Lindholm D, Hamner S, Zirrgiebel U. Neurotrophins and cerebellar development. Perspect Dev Neurobiol (1997) 5:83–94.
39. Picou F, Fauquier T, Chatonnet F, Richard S, Flamant F. Deciphering direct and indirect influence of thyroid hormone with mouse genetics. Mol Endocrinol (2014) 28:429–41. doi:10.1210/me.2013-1414
40. Chatonnet F, Flamant F, Morte B. A temporary compendium of thyroid hormone target genes in brain. Biochim Biophys Acta (2015) 1849:122–9. doi:10.1016/j.bbagrm.2014.05.023
41. Denver RJ, Ouellet L, Furling D, Kobayashi A, Fujii-Kuriyama Y, Puymirat J. Basic transcription element-binding protein (BTEB) is a thyroid hormone-regulated gene in the developing central nervous system. Evidence for a role in neurite outgrowth. J Biol Chem (1999) 274:23128–34. doi:10.1074/jbc.274.33.23128
42. Thompson CC. Thyroid hormone-responsive genes in developing cerebellum include a novel synaptotagmin and a hairless homolog. J Neurosci (1996) 16:7832–40. doi:10.1523/JNEUROSCI.16-24-07832.1996
43. Barez-Lopez S, Guadano-Ferraz A. Thyroid hormone availability and action during brain development in rodents. Front Cell Neurosci (2017) 11:240. doi:10.3389/fncel.2017.00240
44. Grijota-Martinez C, Samarut E, Scanlan TS, Morte B, Bernal J. In vivo activity of the thyroid hormone receptor beta- and alpha-selective agonists GC-24 and CO23 on rat liver, heart, and brain. Endocrinology (2011) 152:1136–42. doi:10.1210/en.2010-0813
45. Auso E, Lavado-Autric R, Cuevas E, Del Rey FE, Morreale De Escobar G, Berbel P. A moderate and transient deficiency of maternal thyroid function at the beginning of fetal neocorticogenesis alters neuronal migration. Endocrinology (2004) 145:4037–47. doi:10.1210/en.2004-0274
46. Lavado-Autric R, Auso E, Garcia-Velasco JV, Arufe Mdel C, Escobar del Rey F, Berbel P, et al. Early maternal hypothyroxinemia alters histogenesis and cerebral cortex cytoarchitecture of the progeny. J Clin Invest (2003) 111:1073–82. doi:10.1172/JCI200316262
47. Emerson CH, Bambini G, Alex S, Castro MI, Roti E, Braverman LE. The effect of thyroid dysfunction and fasting on placenta inner ring deiodinase activity in the rat. Endocrinology (1988) 122:809–16. doi:10.1210/endo-122-3-809
48. Bianco AC, Salvatore D, Gereben B, Berry MJ, Larsen PR. Biochemistry, cellular and molecular biology, and physiological roles of the iodothyronine selenodeiodinases. Endocr Rev (2002) 23:38–89. doi:10.1210/edrv.23.1.0455
49. Calvo RM, Jauniaux E, Gulbis B, Asuncion M, Gervy C, Contempre B, et al. Fetal tissues are exposed to biologically relevant free thyroxine concentrations during early phases of development. J Clin Endocrinol Metab (2002) 87:1768–77. doi:10.1210/jcem.87.4.8434
50. Quignodon L, Legrand C, Allioli N, Guadano-Ferraz A, Bernal J, Samarut J, et al. Thyroid hormone signaling is highly heterogeneous during pre- and postnatal brain development. J Mol Endocrinol (2004) 33:467–76. doi:10.1677/jme.1.01570
51. Grijota-Martinez C, Diez D, Morreale de Escobar G, Bernal J, Morte B. Lack of action of exogenously administered T3 on the fetal rat brain despite expression of the monocarboxylate transporter 8. Endocrinology (2011) 152:1713–21. doi:10.1210/en.2010-1014
52. Nucera C, Muzzi P, Tiveron C, Farsetti A, La Regina F, Foglio B, et al. Maternal thyroid hormones are transcriptionally active during embryo-foetal development: results from a novel transgenic mouse model. J Cell Mol Med (2010) 14:2417–35. doi:10.1111/j.1582-4934.2009.00947.x
53. Mohacsik P, Erdelyi F, Baranyi M, Botz B, Szabo G, Toth M, et al. A transgenic mouse model for detection of tissue-specific thyroid hormone action. Endocrinology (2018) 159:1159–71. doi:10.1210/en.2017-00582
54. Pinna G, Brodel O, Visser T, Jeitner A, Grau H, Eravci M, et al. Concentrations of seven iodothyronine metabolites in brain regions and the liver of the adult rat. Endocrinology (2002) 143:1789–800. doi:10.1210/endo.143.5.8770
55. Guadano-Ferraz A, Obregon MJ, St Germain DL, Bernal J. The type 2 iodothyronine deiodinase is expressed primarily in glial cells in the neonatal rat brain. Proc Natl Acad Sci U S A (1997) 94:10391–6. doi:10.1073/pnas.94.19.10391
56. Galton VA, Schneider MJ, Clark AS, St Germain DL. Life without thyroxine to 3,5,3’-triiodothyronine conversion: studies in mice devoid of the 5’-deiodinases. Endocrinology (2009) 150:2957–63. doi:10.1210/en.2008-1572
57. Galton VA, Wood ET, St Germain EA, Withrow CA, Aldrich G, St Germain GM, et al. Thyroid hormone homeostasis and action in the type 2 deiodinase-deficient rodent brain during development. Endocrinology (2007) 148:3080–8. doi:10.1210/en.2006-1727
58. Morte B, Ceballos A, Diez D, Grijota-Martinez C, Dumitrescu AM, Di Cosmo C, et al. Thyroid hormone-regulated mouse cerebral cortex genes are differentially dependent on the source of the hormone: a study in monocarboxylate transporter-8- and deiodinase-2-deficient mice. Endocrinology (2010) 151:2381–7. doi:10.1210/en.2009-0944
59. Hernandez A, Quignodon L, Martinez ME, Flamant F, St Germain DL. Type 3 deiodinase deficiency causes spatial and temporal alterations in brain T3 signaling that are dissociated from serum thyroid hormone levels. Endocrinology (2010) 151:5550–8. doi:10.1210/en.2010-0450
60. Landers K, Richard K. Traversing barriers – how thyroid hormones pass placental, blood-brain and blood-cerebrospinal fluid barriers. Mol Cell Endocrinol (2017) 458:22–8. doi:10.1016/j.mce.2017.01.041
61. van der Deure WM, Peeters RP, Visser TJ. Molecular aspects of thyroid hormone transporters, including MCT8, MCT10, and OATPs, and the effects of genetic variation in these transporters. J Mol Endocrinol (2010) 44:1–11. doi:10.1677/JME-09-0042
62. Visser WE, Friesema EC, Visser TJ. Minireview: thyroid hormone transporters: the knowns and the unknowns. Mol Endocrinol (2011) 25:1–14. doi:10.1210/me.2010-0095
63. Ferguson KL, Vanderluit JL, Hebert JM, McIntosh WC, Tibbo E, MacLaurin JG, et al. Telencephalon-specific Rb knockouts reveal enhanced neurogenesis, survival and abnormal cortical development. EMBO J (2002) 21:3337–46. doi:10.1093/emboj/cdf338
64. Zevenbergen C, Meima ME, Lima de Souza EC, Peeters RP, Kinne A, Krause G, et al. Transport of iodothyronines by human L-type amino acid transporters. Endocrinology (2015) 156:4345–55. doi:10.1210/en.2015-1140
65. Tarlungeanu DC, Deliu E, Dotter CP, Kara M, Janiesch PC, Scalise M, et al. Impaired amino acid transport at the blood brain barrier is a cause of autism spectrum disorder. Cell (2016) 167:1481–94.e18. doi:10.1016/j.cell.2016.11.013
66. Friesema EC, Ganguly S, Abdalla A, Manning Fox JE, Halestrap AP, Visser TJ. Identification of monocarboxylate transporter 8 as a specific thyroid hormone transporter. J Biol Chem (2003) 278:40128–35. doi:10.1074/jbc.M300909200
67. Braun D, Kinne A, Brauer AU, Sapin R, Klein MO, Kohrle J, et al. Developmental and cell type-specific expression of thyroid hormone transporters in the mouse brain and in primary brain cells. Glia (2011) 59:463–71. doi:10.1002/glia.21116
68. Muller J, Mayerl S, Visser TJ, Darras VM, Boelen A, Frappart L, et al. Tissue-specific alterations in thyroid hormone homeostasis in combined Mct10 and Mct8 deficiency. Endocrinology (2014) 155:315–25. doi:10.1210/en.2013-1800
69. Schwartz CE, May MM, Carpenter NJ, Rogers RC, Martin J, Bialer MG, et al. Allan-Herndon-Dudley syndrome and the monocarboxylate transporter 8 (MCT8) gene. Am J Hum Genet (2005) 77:41–53. doi:10.1086/431313
70. Dumitrescu AM, Liao XH, Weiss RE, Millen K, Refetoff S. Tissue-specific thyroid hormone deprivation and excess in monocarboxylate transporter (mct) 8-deficient mice. Endocrinology (2006) 147:4036–43. doi:10.1210/en.2006-0390
71. Trajkovic M, Visser TJ, Mittag J, Horn S, Lukas J, Darras VM, et al. Abnormal thyroid hormone metabolism in mice lacking the monocarboxylate transporter 8. J Clin Invest (2007) 117:627–35. doi:10.1172/JCI28253
72. Nunez B, Martinez de Mena R, Obregon MJ, Font-Llitjos M, Nunes V, Palacin M, et al. Cerebral cortex hyperthyroidism of newborn mct8-deficient mice transiently suppressed by lat2 inactivation. PLoS One (2014) 9:e96915. doi:10.1371/journal.pone.0096915
73. Iwayama H, Liao XH, Braun L, Barez-Lopez S, Kaspar B, Weiss RE, et al. Adeno associated virus 9-based gene therapy delivers a functional monocarboxylate transporter 8, improving thyroid hormone availability to the brain of Mct8-deficient mice. Thyroid (2016) 26(9):1311–9. doi:10.1089/thy.2016.0060
74. Bernal J, Guadano-Ferraz A, Morte B. Thyroid hormone transporters-functions and clinical implications. Nat Rev Endocrinol (2015) 11:690. doi:10.1038/nrendo.2015.113
75. Mayerl S, Muller J, Bauer R, Richert S, Kassmann CM, Darras VM, et al. Transporters MCT8 and OATP1C1 maintain murine brain thyroid hormone homeostasis. J Clin Invest (2014) 124:1987–99. doi:10.1172/JCI70324
76. Suzuki S, Suzuki N, Mori J, Oshima A, Usami S, Hashizume K. Micro-crystallin as an intracellular 3,5,3’-triiodothyronine holder in vivo. Mol Endocrinol (2007) 21:885–94. doi:10.1210/me.2006-0403
77. Schneider MJ, Fiering SN, Pallud SE, Parlow AF, St Germain DL, Galton VA. Targeted disruption of the type 2 selenodeiodinase gene (DIO2) results in a phenotype of pituitary resistance to T4. Mol Endocrinol (2001) 15:2137–48. doi:10.1210/mend.15.12.0740
78. Fonseca TL, Werneck-De-Castro JP, Castillo M, Bocco BM, Fernandes GW, McAninch EA, et al. Tissue-specific inactivation of type 2 deiodinase reveals multi-level control of fatty acid oxidation by thyroid hormone in the mouse. Diabetes (2014) 63:1594–604. doi:10.2337/db13-1768
79. Hernandez A, Fiering S, Martinez E, Galton VA, St Germain D. The gene locus encoding iodothyronine deiodinase type 3 (dio3) is imprinted in the fetus and expresses antisense transcripts. Endocrinology (2002) 143:4483–6. doi:10.1210/en.2002-220800
80. Hernandez A, Morte B, Belinchon MM, Ceballos A, Bernal J. Critical role of types 2 and 3 deiodinases in the negative regulation of gene expression by T3 in the mouse cerebral cortex. Endocrinology (2012) 153:2919–28. doi:10.1210/en.2011-1905
81. Braun D, Wirth EK, Wohlgemuth F, Reix N, Klein MO, Gruters A, et al. Aminoaciduria, but normal thyroid hormone levels and signalling, in mice lacking the amino acid and thyroid hormone transporter Slc7a8. Biochem J (2011) 439:249–55. doi:10.1042/BJ20110759
82. Mariotta L, Ramadan T, Singer D, Guetg A, Herzog B, Stoeger C, et al. T-type amino acid transporter TAT1 (Slc16a10) is essential for extracellular aromatic amino acid homeostasis control. J Physiol (2012) 590:6413–24. doi:10.1113/jphysiol.2012.239574
83. Mayerl S, Visser TJ, Darras VM, Horn S, Heuer H. Impact of Oatp1c1 deficiency on thyroid hormone metabolism and action in the mouse brain. Endocrinology (2012) 153:1528–37. doi:10.1210/en.2011-1633
84. Alvarez BV, Kieller DM, Quon AL, Robertson M, Casey JR. Cardiac hypertrophy in anion exchanger 1-null mutant mice with severe hemolytic anemia. Am J Physiol Heart Circ Physiol (2007) 292:H1301–12. doi:10.1152/ajpheart.00449.2006
85. Gong L, Aranibar N, Han YH, Zhang Y, Lecureux L, Bhaskaran V, et al. Characterization of organic anion-transporting polypeptide (Oatp) 1a1 and 1a4 null mice reveals altered transport function and urinary metabolomic profiles. Toxicol Sci (2011) 122:587–97. doi:10.1093/toxsci/kfr114
86. Episkopou V, Maeda S, Nishiguchi S, Shimada K, Gaitanaris GA, Gottesman ME, et al. Disruption of the transthyretin gene results in mice with depressed levels of plasma retinol and thyroid hormone. Proc Natl Acad Sci U S A (1993) 90:2375–9. doi:10.1073/pnas.90.6.2375
87. Palha JA, Episkopou V, Maeda S, Shimada K, Gottesman ME, Saraiva MJ. Thyroid hormone metabolism in a transthyretin-null mouse strain. J Biol Chem (1994) 269:33135–9.
88. Liao XH, Di Cosmo C, Dumitrescu AM, Hernandez A, Van Sande J, St Germain DL, et al. Distinct roles of deiodinases on the phenotype of Mct8 defect: a comparison of eight different mouse genotypes. Endocrinology (2011) 152:1180–91. doi:10.1210/en.2010-0900
89. Stohn JP, Martinez ME, Matoin K, Morte B, Bernal J, Galton VA, et al. MCT8 deficiency in male mice mitigates the phenotypic abnormalities associated with the absence of a functional type 3 deiodinase. Endocrinology (2016) 157:3266–77. doi:10.1210/en.2016-1162
90. Ceballos A, Belinchon MM, Sanchez-Mendoza E, Grijota-Martinez C, Dumitrescu AM, Refetoff S, et al. Importance of monocarboxylate transporter 8 for the blood-brain barrier-dependent availability of 3,5,3’-triiodo-L-thyronine. Endocrinology (2009) 150:2491–6. doi:10.1210/en.2008-1616
91. Morte B, Bernal J. Thyroid hormone action: astrocyte-neuron communication. Front Endocrinol (2014) 5:82. doi:10.3389/fendo.2014.00082
92. Friesema EC, Jansen J, Jachtenberg JW, Visser WE, Kester MH, Visser TJ. Effective cellular uptake and efflux of thyroid hormone by human monocarboxylate transporter 10. Mol Endocrinol (2008) 22:1357–69. doi:10.1210/me.2007-0112
93. Palha JA, Fernandes R, de Escobar GM, Episkopou V, Gottesman M, Saraiva MJ. Transthyretin regulates thyroid hormone levels in the choroid plexus, but not in the brain parenchyma: study in a transthyretin-null mouse model. Endocrinology (2000) 141:3267–72. doi:10.1210/endo.141.9.7659
94. Hallen A, Cooper AJ, Jamie JF, Haynes PA, Willows RD. Mammalian forebrain ketimine reductase identified as mu-crystallin; potential regulation by thyroid hormones. J Neurochem (2011) 118:379–87. doi:10.1111/j.1471-4159.2011.07220.x
95. Hallen A, Cooper AJ, Jamie JF, Karuso P. Insights into enzyme catalysis and thyroid hormone regulation of cerebral ketimine reductase/mu-crystallin under physiological conditions. Neurochem Res (2015) 40:1252–66. doi:10.1007/s11064-015-1590-5
96. Morte B, Diez D, Auso E, Belinchon MM, Gil-Ibanez P, Grijota-Martinez C, et al. Thyroid hormone regulation of gene expression in the developing rat fetal cerebral cortex: prominent role of the Ca2+/calmodulin-dependent protein kinase IV pathway. Endocrinology (2010) 151:810–20. doi:10.1210/en.2009-0958
97. Royland JE, Parker JS, Gilbert ME. A genomic analysis of subclinical hypothyroidism in hippocampus and neocortex of the developing rat brain. J Neuroendocrinol (2008) 20:1319–38. doi:10.1111/j.1365-2826.2008.01793.x
98. Dong H, You SH, Williams A, Wade MG, Yauk CL, Thomas Zoeller R. Transient maternal hypothyroxinemia potentiates the transcriptional response to exogenous thyroid hormone in the fetal cerebral cortex before the onset of fetal thyroid function: a messenger and microRNA profiling study. Cereb Cortex (2014) 25:1735–45. doi:10.1093/cercor/bht364
99. Gil-Ibanez P, Bernal J, Morte B. Thyroid hormone regulation of gene expression in primary cerebrocortical cells: role of thyroid hormone receptor subtypes and interactions with retinoic acid and glucocorticoids. PLoS One (2014) 9:e91692. doi:10.1371/journal.pone.0091692
100. Gil-Ibanez P, Garcia-Garcia F, Dopazo J, Bernal J, Morte B. Global transcriptome analysis of primary cerebrocortical cells: identification of genes regulated by triiodothyronine in specific cell types. Cereb Cortex (2017) 27:706–17. doi:10.1093/cercor/bhv273
101. Picou F, Fauquier T, Chatonnet F, Flamant F. A bimodal influence of thyroid hormone on cerebellum oligodendrocyte differentiation. Mol Endocrinol (2012) 26:608–18. doi:10.1210/me.2011-1316
102. Calza L, Forrest D, Vennstrom B, Hokfelt T. Expression of peptides and other neurochemical markers in hypothalamus and olfactory bulb of mice devoid of all known thyroid hormone receptors. Neuroscience (2000) 101:1001–12. doi:10.1016/S0306-4522(00)00420-6
103. Hernandez A, Martinez ME, Fiering S, Galton VA, St Germain D. Type 3 deiodinase is critical for the maturation and function of the thyroid axis. J Clin Invest (2006) 116:476–84. doi:10.1172/JCI26240
104. Alonso M, Goodwin C, Liao X, Page D, Refetoff S, Weiss RE. Effects of maternal levels of thyroid hormone (TH) on the hypothalamus-pituitary-thyroid set point: studies in TH receptor beta knockout mice. Endocrinology (2007) 148:5305–12. doi:10.1210/en.2007-0677
105. Srichomkwun P, Anselmo J, Liao XH, Hones GS, Moeller LC, Alonso-Sampedro M, et al. Fetal exposure to high maternal thyroid hormone levels causes central resistance to thyroid hormone in adult humans and mice. J Clin Endocrinol Metab (2017) 102:3234–40. doi:10.1210/jc.2017-00019
106. Deng WB, Liang XH, Liu JL, Yang ZM. Regulation and function of deiodinases during decidualization in female mice. Endocrinology (2014) 155:2704–17. doi:10.1210/en.2014-1015
107. Patel J, Landers K, Li H, Mortimer RH, Richard K. Delivery of maternal thyroid hormones to the fetus. Trends Endocrinol Metab (2011) 22:164–70. doi:10.1016/j.tem.2011.02.002
108. Lee EY, Chang CY, Hu N, Wang YC, Lai CC, Herrup K, et al. Mice deficient for Rb are nonviable and show defects in neurogenesis and haematopoiesis. Nature (1992) 359:288–94. doi:10.1038/359288a0
109. Ferguson KL, McClellan KA, Vanderluit JL, McIntosh WC, Schuurmans C, Polleux F, et al. A cell-autonomous requirement for the cell cycle regulatory protein, Rb, in neuronal migration. EMBO J (2005) 24:4381–91. doi:10.1038/sj.emboj.7600887
110. Ritchie JW, Taylor PM. Role of the system L permease LAT1 in amino acid and iodothyronine transport in placenta. Biochem J (2001) 356:719–25. doi:10.1042/bj3560719
111. Vasilopoulou E, Loubiere LS, Heuer H, Trajkovic-Arsic M, Darras VM, Visser TJ, et al. Monocarboxylate transporter 8 modulates the viability and invasive capacity of human placental cells and fetoplacental growth in mice. PLoS One (2013) 8:e65402. doi:10.1371/journal.pone.0065402
Keywords: thyroid hormones, neurodevelopment, neurodevelopmental disorder, placenta, brain–blood barrier, transporters
Citation: Richard S and Flamant F (2018) Regulation of T3 Availability in the Developing Brain: The Mouse Genetics Contribution. Front. Endocrinol. 9:265. doi: 10.3389/fendo.2018.00265
Received: 22 December 2017; Accepted: 07 May 2018;
Published: 28 May 2018
Edited by:
Joanne F. Rovet, Hospital for Sick Children, CanadaReviewed by:
Veerle M. Darras, KU Leuven, BelgiumTania M. Ortiga-Carvalho, Universidade Federal do Rio de Janeiro, Brazil
Mary E. Gilbert, Environmental Protection Agency, United States
Copyright: © 2018 Richard and Flamant. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Frédéric Flamant, ZnJlZGVyaWMuZmxhbWFudEBlbnMtbHlvbi5mcg==