 Terry W. Moody
Terry W. Moody Irene Ramos-Alvarez2
Irene Ramos-Alvarez2 Robert T. Jensen
Robert T. Jensen- 1Department of Health and Human Services, National Cancer Institute, Center for Cancer Research, National Institute of Diabetes, Digestive, and Kidney Disease (NIDDK), Bethesda, MD, United States
- 2Digestive Diseases Branch, National Institute of Diabetes, Digestive, and Kidney Disease (NIDDK), Bethesda, MD, United States
Neuropeptide G protein-coupled receptors (GPCRs) are overexpressed on numerous cancer cells. In a number of tumors, such as small cell lung cancer (SCLC), bombesin (BB) like peptides and neurotensin (NTS) function as autocrine growth factors whereby they are secreted from tumor cells, bind to cell surface receptors and stimulate growth. BB-drug conjugates and BB receptor antagonists inhibit the growth of a number of cancers. Vasoactive intestinal peptide (VIP) increases the secretion rate of BB-like peptide and NTS from SCLC leading to increased proliferation. In contrast, somatostatin (SST) inhibits the secretion of autocrine growth factors from neuroendocrine tumors (NETs) and decreases proliferation. SST analogs such as radiolabeled octreotide can be used to localize tumors, is therapeutic for certain cancer patients and has been approved for four different indications in the diagnosis/treatment of NETs. The review will focus on how BB, NTS, VIP, and SST receptors can facilitate the early detection and treatment of cancer.
Introduction
G protein-coupled receptors (GPCRs) have 7 transmembrane (T M) domains and they interact with G proteins comprised of α, β, and γ subunits (1). The activated GPCRs undergoes a conformation change dissociating the G-protein into a GTP-bound α subunit and β, γ dimer. GPCRs for bombesin (BB) and neurotensin (NTS) interact with Gq/11, whereas receptors for vasoactive intestinal peptide (VIP) interact with Gs and somatostatin (SST) receptors interact with Gi/0 (2). BB and NT receptors cause phosphatidylinositol (PI) turnover resulting in the elevation of cytosolic Ca2+ and activation of protein kinase (PK) C. VIP receptors activate adenylyl cyclase resulting in elevated cAMP whereas SST receptors reduce the elevation of cAMP stimulation caused by VIP.
Neuropeptides modulate neural activity in the brain in a paracrine manner, however, they function as autocrine growth factors in cancer (3). BB, NTS, and VIP stimulate the growth of small cell lung cancer (SCLC) cells whereas SST inhibits growth (3). BB and the structurally related gastrin-releasing peptide (GRP) bind with high affinity to the GRP receptor or BB2R; NTS binds high affinity to NTSR1; VIP binds with high affinity to VPAC1/VPAC2 and SST as well as octreotide/lanreotide bind with high affinity to SSTR2/SSTR5 but reduced affinity to SSTR1, SSTR3, and SSTR4. The agonist-GPCR complex is internalized and the GPCR recycle to the membrane but the peptide is metabolized in lysosomes. Because cancers frequently over-express GPCRs, the cancer GPCRs can be used to deliver neuropeptide-drug conjugates into the cancer cell (4). In contrast, GPCR antagonists bind to the GPCR at the cell surface but are not internalized. PD176252 (GRPR antagonist) and SR48692 (NTSR1 antagonist) inhibit cancer growth (5–7). This review will focus on how neuropeptide GPCRs are oncotargets for the early detection and treatment of cancer.
GRPR, Neuromedin B Receptor and BBR Subtype 3
SCLC has high levels of the BB-like peptide GRP (8, 9). GRP is derived from a 148 amino acid prepropeptide (10). After removal of the N-terminal 23 amino acid signal sequence, pro-GRP1−125 is metabolized by a prohormone convertase to GRP, which contains 27 amino acids and has an amidated C-terminal. GRP (proGRP1−27) is readily metabolized in the blood but SCLC patients have elevated pro-GRP (11). Antibodies to proGRP31−98 have been used to detect high concentrations of proGRP (>100 pg/ml) in the serum of patients with SCLC. Because proGRP is elevated in the serum of 71% of the SCLC patients it may be a biomarker for SCLC (12). BB or GRP, but not proGRP bind with high affinity to the GRPR. The C-terminal octapeptide of BB or GRP can be neutralized by mAb 2A11. mAb2A11 inhibits the growth of SCLC in vitro and in mouse models in vivo (13). In a clinical trial, 2A11 was well tolerated and one patient had SCLC remission whereas four patients had stable disease out of 13 patients treated (14). The results indicate that the GRP precursor may be a biomarker for SCLC.
Table 1 shows that the GRPR, which is localized to chromosome xp22, contains 384 amino acids and is a member of the class A/Rhodopsin-like GPCR (15, 16). The neuromedin B (NMB) R or BB1R, which is localized to chromosome 6q24, contains 390 amino acids whereas BB receptor subtype-3 (BRS-3), which is localized to chromosome xq26, contains 399 amino acids. The NMBR and BRS-3 have about 50% sequence homology with the GRPR (17, 18). The GRPR binds GRP and NMB with high and low affinity, respectively. The NMBR binds GRP and NMB with low and high affinity, respectively. The orphan receptor BRS-3 binds both GRP and NMB with low affinity but MK5046 binds with high affinity (7). The universal agonist BA1, (D-Tyr6, β-Ala11, Phe 13, Nle14) BB6−14, binds with high affinity to the GRPR, NMBR, and BRS-3. Numerous amino acids in TM domains 6 and 7 as well in extracellular loops (EL) 1, 2, and 3 of the GRPR are essential for high affinity binding of GRP (4). While the GPCRs of each family have a similar sequence, the pharmacological profile is different.
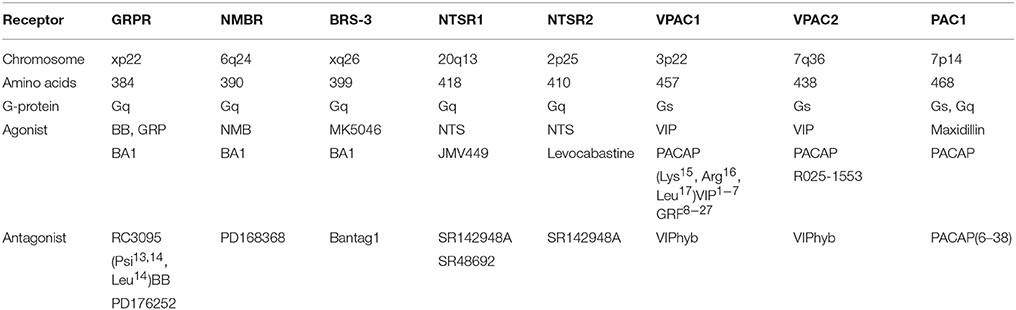
Table 1. Peptide GPCRs (human).
BB-drug conjugates were synthesized which are cytotoxic for lung cancer cells. The topoisomerase-1 inhibitor camptothecin (CPT) was coupled with a linker to the N-terminal of BA1. Surprisingly, the resulting CPT-L2-BA1 bound with higher affinity to the GRPR, NMBR, and BRS-3 than did BA1 (19). CPT-L2-BA1 was an agonist which increased PI turnover and was internalized. The CPT-L2-BA1 was metabolized in the lysosome leading to the release of CPT (20). Also, BB agonists have been coupled to paclitaxel (21), doxorubicin (22), marine toxins (23), magainin II (24), and siRNA to the GRPR (25) resulting in decreased cancer cellular proliferation. Doxorubicin was coupled to a GRPR antagonist and the resulting AN-215 was cytotoxic for gastric, colon, lung, ovarian, endometrial, breast, and pancreatic cancer (26). RC-3095, a GRPR antagonist, was tested in 25 patients with solid tumors. RC-3095 had minimal toxicity but a partial response was only seen in 1 patient (27). Unfortunately, these BB-drug conjugates will not only kill cancer cells, but normal cells with excessive BBR.
BBR antagonists were developed which inhibit the growth of cancer cells. Peptide antagonists such as RC-3095 or (Psi13, 14, Leu14)BB blocked the GRPR, and they inhibited the growth of cancer cells (27, 28). Small molecule antagonists such as PD168368 were synthesized which inhibit the growth of cancer cells which have NMBR (6). Also, bantag-1 is a peptide antagonist for BRS-3 (29). The BB receptor antagonists inhibited the growth of lung cancer cells in vitro and in vivo using nude mice bearing lung cancer xenografts. GRPR, NMBR, and BRS-3 mRNA was detected in 11/13 lung cancer cell lines (7). All lung cancer cell lines tested had at least 1 type of BBR and many cell lines had all 3 receptors.
In contrast, a high density of GRPR but not NMBR or BRS-3 were detected in most prostate and breast cancer cells (30). GRPR agonists were labeled with 111In, 64Cu, 99mTc, 68Ga, 18F for imaging studies. Using a 99mTc-BB2−14 analog 14 prostatic lesions were visualized in patients (31). Using a 99mTc-RGD-BB analog, tumors were visualized in 6/6 breast cancer patients (32). Using a 64Cu-BB6−14 analog, tumors were visualized in 3 of 4 prostate cancer patients (33). It remains to be determined if the imaging of GRPR will be useful in the early detection of breast and/or prostate cancer.
Many of the growth effects of BB-like peptides on non-SCLC (NSCLC) cells may result from transactivation of receptor tyrosine kinases (RTK) such as the epidermal growth factor receptor (EGFR). Activation of the NMBR in NSCLC cells causes PI turnover leading to increased phosphorylation of the EGFR (Figure 1). Addition of NMB to NSCLC cells increases the tyrosine phosphorylation of the EGFR after 1 min leading to the tyrosine phosphorylation of ERK after 2 min (34). The transactivation of the EGFR that is regulated by the NMBR is inhibited by the tyrosine kinase inhibitor (TKI) gefitinib or the NMBR antagonist PD168368. The transactivation process in NSCLC cells is mediated by the EGFR ligand transforming growth factor (TGF)α (Figure 1). The inactive precursor pro-TGFα is metabolized by matrix metalloprotease (MMP) enzymes in the membrane to biologically active TGFα which is secreted and binds to the EGFR. The transactivation of the EGFR caused by addition of NMB to NSCLC cells is inhibited by GM6001 (MMP inhibitor) or anti-TGFα Ab. The transactivation process requires reactive oxygen species (ROS). Addition of N-acetyl cysteine (antioxidant) or tiron (superoxide scavenger) impaired the ability of NMB to increase EGFR tyrosine phosphorylation. The ROS may oxidize Cys773 of the EGFR increasing its tyrosine kinase activity and/ or oxidize protein tyrosine phosphatases (PTP) impairing their ability to metabolize phosphotyrosine (35, 36). The results indicate that GPCRs regulate the transactivation of receptor tyrosin kinases (RTKs) in NSCLC cells.
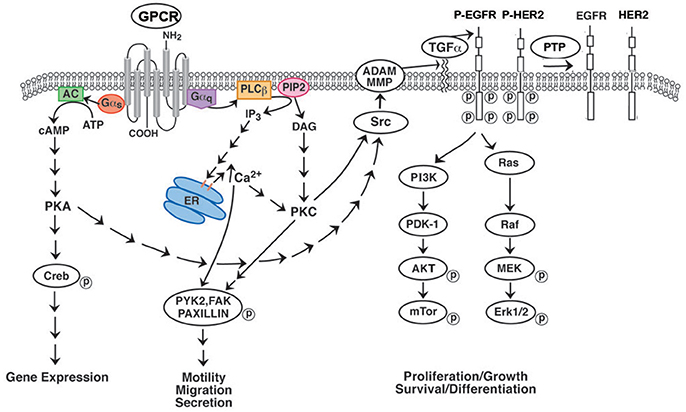
Figure 1. Effect of GPCR's on RTK transactivation. GPCRs for BB and NTS couple to Gq and causes metabolism of PIP2 to DAG (activates PKC) and IP3 (elevates cytosolic Ca2+). Addition of NTS or BB to NSCLC cells increases phosphorylation of PYK2, FAK or paxillin leading to increased cellular migration. GPCR for VIP interact with Gs activating adenylyl cyclase and increasing cAMP. The cAMP activates PKA leading to CREB phosphorylation and altered gene expression. GPCR for PACAP interact with both Gq and Gs. GPCR activate Src and MMP resulting in the production of EGFR ligands such as TGFα. When TGFα binds to the EGFR, tyrosine kinase activity is increased leading to phosphorylated EGFR homodimers or EGFR-HER2 heterodimers. The RTK activates the Ras-Raf-MEK-ERK pathway leading to increased cellular proliferation. The RTK activates the PI3K-PKD-AKT-mTOR pathway leading to increased cellular survival. The phosphorylated EGFR/HER2 is dephosphorylated by protein tyrosine phosphatase (PTP).
The EGFR contains 1,186 amino acids with a 621 and extracellular domains I and III bind EGF or TGFα with high affinity (37). The EGFR has a 23 amino acid TM domain and a 542 intracellular domain with tyrosine kinase activity. Lys721 is essential for binding ATP and the phosphorylation of protein substrates. Upon binding of ligand, the EGFR can undergo homodimerization resulting in the phosphorylation of Tyr1068, Tyr1086, Tyr1148, and Tyr1174. Alternatively, the EGFR can form heterodimers with HER2. The MAPK and PI3K/Akt pathways are downstream of the EGFR and are important for EGFR mediated proliferation and cancer cellular survival, respectively. Currently, we are investigating if GPCRs transactivate additional RTK such as HER2, HER3 or HER4 in cancer cells.
NMB increases the proliferation of NSCLC cells. In contrast, PD168368 and gefitinb inhibit the growth of NSCLC cells (34). Surprisingly, combinations of the NMBR antagonist with the EGFR TKI reduced the proliferation of NSCLC cells in a synergistic manner. The results indicate that GPCR antagonists potentiate the action of TKI in NSCLC. Traditionally NSCLC which kills 130,000 U.S. citizens annually is treated with combination chemotherapy, however, the 5 year survival rate is only 15%. The EGFR is mutated is approximately 13% of the NSCLC patients and those with the L858R mutation have increased tyrosine kinase activity and sensitivity to TKI such as gefitinib or erlotinib (38, 39). Traditionally NSCLC patients are treated with combination chemotherapy, however, the 5 year survival rate is only 15% (40).
Neurotensin Receptors
NTS is present in numerous SCLC cell lines (9, 41). NTS is derived from a 170 amino acid precursor and metabolized to a biologically active peptide which contains 13 amino acids (42). NTS and its C-terminal fragment NTS8−13 bind with high affinity to the NTSR1, which is localized to chromosome 20q13, contains 418 amino acids, and is a class A/Rhodopsin-like GPCR. Table 1 shows that the NTSR2 which is on chromosome 2p25 contains 410 amino acids and binds NTS and levocabastine with high affinity. The NTS−-NTSR1 complex has been crystallized and NTS8−13 sits on top of the NTS binding pocket and interacts with TM domains 6 as well as 7 and EL 2 as well as 3 (43). Both NT and BB receptors have short N-terminals which have little effect on ligand binding (44). The nonpeptide NTSR1 antagonist SR48692 binds deep into the NTSR1 binding pocket and blocks the effects of NTS agonists. Also, SR142948A blocks both the NTSR1 and NTSR2. The NTSR3 is not a GPCR but is sortilin.
NTS binds with high affinity to SCLC cells (45). Addition of NTS to cancer cells causes PI turnover leading to increases PKC activity and elevation of cytosolic Ca2+ (46–48). In contrast, the NTSR2 agonist levocabastin has little effect on lung cancer cells. The effects of NTS on second messenger production and proliferation was antagonized by SR48692 (5). NTS addition to cancer cells causes phosphorylation of various proteins such as focal adhesion kinase (FAK) or ERK (49, 50). The phosphorylated ERK increases the expression of c-fos and c-jun leading to cellular proliferation (51). NTS stimulates proliferation whereas SR48692 inhibits the proliferation of lung cancer cells (5). NT addition to NSCLC cells increased EGFR tyrosine phosphorylation 5-fold (52). NT (5 nM) half-maximally increased EGFR transactivation after 2 min. NTS or NTS8−13 but not NT1−8 or levocabastine increase EGFR tyrosine phosphorylation. The NTSR1 regulation of EGFR transactivation is inhibited by SR48692, gefitinib, PP1, GM6001, TGFα antibodies and antioxidants. SR48692 and gefitinib inhibit the proliferation of NSCLC cells in a synergistic manner. Previously, JMV449, a NT8−13 analog, was found to increase expression of the EGFR, HER2, and HER3 after 24 h (53). JMV449 addition to cells increase MMP activity resulting in HB-EGF and neuregulin 1 release, which activates the EGFR and HER3, respectively.
NTSR1 regulates the EGFR transactivation in numerous cancers including colon, foregut neuroendocrine, lung, and prostate cancer (47, 52, 54, 55). Lung cancer and gastric cancer patients whose tumors had high densities of NTSR1 had decreased survival (53, 56). Addition of NTS to NSCLC cells caused tyrosine phosphorylation of the EGFR in a PLC-dependent manner (52). Phosphorylated β-catenin dissociates from E-cadherin and increases the expression of NTSR1. Wnt/β-catenin signaling increases the expression of E-cadherin leading to epithelial to mesenchymal transitions and cancer metastasis (57). Recently, 3BP-227, a SR142948A analog, was radiolabeled and used to image tumors containing NTSR1. In nude mice containing HT29 colon cancer tumors 177Lu-3BP-227 localized to the tumors with high tumor-to-kidney or tumor-to-liver ratios using whole-body SPECT/CT techniques (58). In 5 out of 6 patients with ductal pancreatic adenocarcinoma tumor uptake of 177Lu-3BP-227 was observed (59). It remains to be determined if 177Lu-3BP-227 will improve survival of patients whose tumors are enriched in NTSR1.
VIPRs and Pituitary Adenylate Cyclase Activating Polypeptide Receptor
The biological activities of the VIP and PACAP family of peptides are mediated by 3 GPCR (VPAC1, VPAC2, and PAC1), which are members of the classB/secretin-like receptors (60). Table 1 shows that VPAC1, which is localized to chromosome 3p22, contains 457 amino acids with a 112 amino acid N-terminal. VPAC2, which is localized to chromosome 7q36, contains 438 amino acids with a 103 amino acid N-terminal. PAC1, which is localized to chromosome 7p14, contains 468 amino acids with a 125 amino acid N-terminal. PAC1 has about 50% sequence homology with VPAC1 or VPAC2 (60). The large N-terminal extracellular domain of PAC1 has antiparallel β-sheets and binds to the C-terminal of PACAP (61, 62). The PAC1 receptor has 3 closed transitional states (G1-G3) and one open state named G4 (63). The N-terminal of PACAP, which activates PAC1 binds to EL and TM domains (64). VPAC1, VPAC2, and PAC1 interact with Gs resulting in elevated cAMP, however, PAC1 interacts with Gq as well resulting in PI turnover (65). VIP, which contains 28 amino acids, is derived from a 170 amino acid precursor protein. PACAP-27 as well as PACAP-38 is derived from a 176 amino acid precursor protein and 67% of the amino acids in PACAP-27 and VIP are identical (60). VPAC1 and VPAC2 binds VIP and PACAP-27 or PACAP-38 with high affinity, whereas PAC1 binds PACAP-27 or PACAP-38 with high affinity but VIP with low affinity. Maxidillin, a 61 amino acid peptide isolated from sand fly, binds with high affinity to PAC1 but not VPAC1 or VPAC2 (66). Recently, a number of PACAP-38 analogs were synthesized which prefer PAC1 relative to VPAC1 or VPAC2 by over an order of magnitude (67). VIPhybrid is a peptide antagonist which binds with moderate affinity to VPAC1 or VPAC2, whereas, PACAP(6–38) is a peptide antagonist for PAC1 (68). Selective non-peptide antagonists for VPAC1, VPAC2 or PAC1 remain unknown.
VPAC1 is present in numerous cancers including breast, colon, liver, lung, neuroblastoma, pancreatic, and prostate cancers in high densities (69). VPAC2 is present in moderate densities in gastric pancreatic adenocarcinomas, gastric leiomyomas, thyroid cancer, and sarcomas (70). PAC1 is present in brain, breast, colon lung, neuroendocrine, pancreatic, pituitary, and prostate cancer as well as neuroblastoma/pheochromocytoma (71). In SH1SY5Y neuroblastoma cells, numerous PAC1 splice variants (SV) were detected in the N-terminal and intracellular loop (IL) 3 (72). PAC1 has 18 exons and deletion of exons 5,6 or 4-6 reduce the N-terminal by 7, 21 (short) or 57 amino acids (very short) (73). The short PAC1 but not the very short PAC1 bind PACAP-38 with high affinity and elevate cAMP (74). Alternative splice variants (SV) of IL3 result in the addition of an additional 28 amino acid segment (hip) to PAC1null (75). Addition of a different set of 28 amino acids to IL3 of the PAC1 results in the hop SV. Finally, both SVs can be added resulting in PAC1hiphop. The order of potency to increase PI turnover was PAC1hop > PAC1null = PAC1hiphop > PAC1hip (76). Thus binding of PACAP and second messenger production can be altered by PAC1 deletions and SVs.
VPAC1 can be utilized to image cancer tumors. 18F(Arg15, 21)VIP localized to T47D breast cancer cells in nude mice (77) and 64Cu-TP3982 localized to mammary tumors in MMTVneu transgenic mice (78). 99mTc-TP3982 was used to image breast tumors in 5 patients (79). The VPAC1-agonist complex internalizes in cancer cells and the ligand is metabolized in lysosomes. VPAC1 has been used to deliver VIP analogs containing cytotoxic CPT, paclitaxel, ellipticin or geldanomycin to cancer cells (80–83). The actions of VIP are antagonized by peptides such as VIP10−28 or VIPhybrid (84). Addition of VIP to cancer cells results in elevated cAMP which activates PKA. Activation of PKA results in CREB phosphorylation which increases nuclear oncogene expression of c-Myc leading to increased proliferation (Figure 1). VIP increases the proliferation of lung cancer cells whereas VIPhybrid inhibits proliferation (84). Addition of PACAP-27 or PACAP-38 to lung cancer cells containing VPAC1, VPAC2 or PAC1 increases cAMP, however, it causes PI turnover in cells containing PAC1. When PI is metabolized, ERK becomes phosphorylated. Phosphorylated ERK increased the expression of the nuclear oncogenes c-fos and c-jun leading to increased cancer cellular proliferation. PACAP(6–38) inhibits the proliferation of lung cancer cells in vitro and in vivo (85).
VIP may be a promoter of carcinogenesis. VPAC1 density is higher in mammary cancer than adjacent normal tissue using rat and mouse models (86). Specific binding of 125I-VIP to mouse mammary tumors was inhibited with high affinity by (Lys15, Arg16, Leu17) VIP1−7GRF8−27 (VPAC1 peptide agonist) but not Ro25-1553 (VPAC2 peptide agonist). Retinoic acid, a chemopreventive agent, down-regulates VPAC1 expression in breast and lung cancer cells (87, 88). Finally VIPhybrid inhibits mammary carcinogenesis in C3(1)SV40T antigen mice (89).
Addition of PACAP-27 or PACAP-38 but not VIP causes transactivation of the EGFR in NSCLC cells (90). The PAC1 regulation of EGFR tyrosine phosphorylation is inhibited by PACAP(6–38), gefitinib, PP2, GM6001, and ROS inhibitors. Diphenyleneiodonium (DPI), a NADPH oxidase (NOX) inhibitor impaired the ability of PACAP to increase EGFR tyrosine phosphorylation. NOX-4, which produces ROS, is present in NSCLC cells (91). PACAP-27 addition to NSCLC cells increased ROS which was inhibited by DPI. VIP addition to breast cancer cells increased EGFR and HER2 phosphorylation (92). The EGFR which dimerizes, may form homodimers with itself or heterodimers with HER2. VPAC1 regulation of EGFR transactivation was blocked by JV-1-53 (VPAC1 antagonist), PP2 or H89 (PKA inhibitor). In contrast, the PAC1 regulation of EGFR transactivation in NSCLC cells was inhibited by U73122 (phospholipase C inhibitor) but not H89. The results indicate that the EGFR can be transactivated by GPCR which interact with Gq or Gs.
SST Receptors
SST occurs endogenously in two principal forms (SST-14, SST-28) and their action is mediated by 5 related subtypes of GPCRs (SSTR1-5) (93, 94). SST receptors are not only widely expressed on normal tissues, but also are frequently overexpressed by many neoplasms, particularly NETs [i.e., carcinoids/pancreatic neuroendocrine tumors (panNETs)] (93–95). SST has a wide range of physiological actions and they are primarily inhibitory (93, 94).
SST and its receptors represent the prototype for a clinically successful peptide/peptide receptor oncotarget. It is the only peptide-GPCR system which has multiple approved indications (four different indications) for the treatment of a class of human neoplasms, NETs. Furthermore, its results in NETs have potential applicability for its clinical utility in a number of other neoplasms.
The initial approved indication for SST analogs was its use in hormone-excess states. Depot long-acting formulations of synthetic SST analogs (octreotide-LAR, lanreotide autogel) (Figure 2) are the drugs of choice to control various functional NET syndromes due to the ectopic release of a biologically active peptides by the NETs (93, 96–99). This includes the control of such widely different functional NET syndromes as the carcinoid syndrome (diarrhea, flushing) due to metastatic carcinoid tumor; severe diarrhea due to VIPomas; rash due to glucagonomas; acromegaly due to excessive growth hormone release primarily by pituitary adenomas, and a number of others (93, 96–100). Almost all (>90%) of the well-differentiated forms of these NETs (>95%) overexpress somatostatin receptor subtype 2 which has high affinity for octreotide and therefore it is generally effective in controlling the hormone-excess state and in many, their growth. Some NETs such as pituitary adenomas do not overexpress SSTR2 with the result that octreotide/lanreotide are only effective in 20–70% of patients (101). One solution to this has been the development of next generation somatostatin analogs such as pasireotide, which has high affinity for multiple subtypes (SSTR5 > SSTR2 > SSTR3 > SSTR1), which has been shown to be effective in these patients and is approved for use in the treatment of a acromegaly.
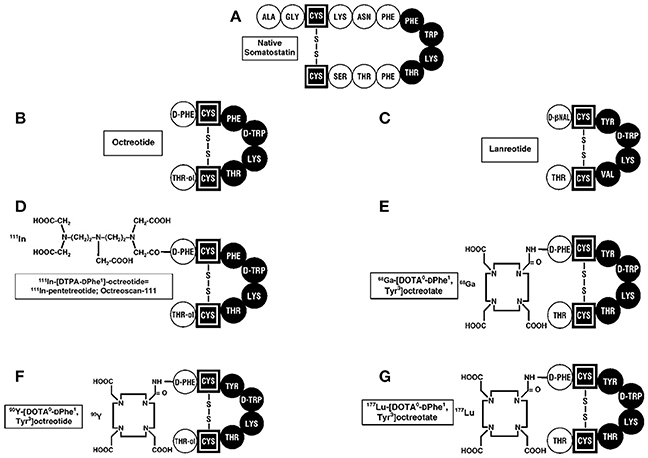
Figure 2. Structures of SST and synthetic analogs used clinically. (A) The 14 amino acid SST is shown and essential amino acids are in black. The SST synthetic analogs, octreotide (B), and lanreotide (C), which have 8 amino acids, are approved to treat patients with neuroendocrine tumors (NETs) producing hormone-excess states (VIPoma, carcinoid syndrome) and for their anti-proliferative activity in patients with advanced, aggressive NETs. 111InDTPA pentetreotide (D) as well as 68GaDOTATATE (E) are used for SSTR imaging in patients with NETs. 90Y-DOTA (F) and 177Lu-DOTA (G) -labeled SST analogs are used for their antitumor activity during PRRT by targeting the cytotoxic radiolabel to the tumor.
The second approved indication for SST analogs is for its anti-proliferative activity on NETs. In numerous pre-clinical studies and animal studies, it was shown that SST analogs have anti-proliferative effects on NETs, as well as number of other human tumors (102–107) Two double-blind Phase 3 studies (108, 109) in patients with advanced NETs treated with lanreotide/octreotide increased the patient's progressive free survival (PFS), which lead to FDA approval. Recent meta-analyses (106, 110) of SST analogs anti-proliferative effects in all publications (106) or the above two studies (110), in patients with advanced NETs, demonstrate good anti-proliferative activity, significant benefit from their use resulting in disease control (HR 0.51, p < 0.01), with the response rates vs. placebo being 58 vs. 32% and a good safety profile. In general, these studies demonstrate that SST analog treatment in patients with advanced NETs result in a tumoristatic effect primarily, rather than a decrease in the tumor size. SST analogs are now recommended in recent guidelines as well as expert reviews as one of the initial treatments for controlling tumor growth in patients with advanced NETs, especially those with well-differentiated NETs, and slower growth rates (99, 111–115). Numerous in vitro and animal studies report that SST receptors are expressed on a number of other non-endocrine tumors, and that SST analogs have anti-proliferative activity in these tumors (93, 94, 97, 103, 104, 107, 116, 117). No controlled trials have established the use of SST analogs for anti-proliferative effects in patients with non-endocrine tumors.
The third approved indication for SST analogs in patients with NETs is for imaging of the tumor. SST receptor imaging (SRI) was originally approved for the use of 111In-labeled pentetreotide with SPECT/CT scanning (Octreoscan), which is now replaced by the use of 68Ga-DOTATATE PET/CT scanning, which has greater resolution, sensitivity and high specificity (118–122). Almost all well differentiated NETs overexpress the somatostatin receptor subtypes (SSTR2 > SSTR5 > SSTR3) that bind this radiolabeled agonist with high affinity (118, 119, 123). A systematic review (122) demonstrated that the use of 68Ga-DOTATATE PET/CT scanning changed the management of the patient in a mean of 44% (range, 16–71%). SRI is now essential for the staging and management of NET patients and is the most sensitive method to allow whole body scanning rapidly to present a complete assessment of the extent of the tumor (112, 118, 121).
The fourth approved indication for SST analogs in patients with NETs is their use to target cytotoxic radiolabeled SST analogs to the tumor in patients with advanced NETs, as an anti-tumor therapy (called PRRT for peptide receptor radionuclide therapy) (124–127). Numerous animal studies as well as uncontrolled studies on patients with advanced NETs, demonstrated this approach resulted in tumor stabilization in progressive tumors as well tumor shrinkage in a significant number of patients with acceptable side-effects (124–127). Various SST analogs were coupled to linkers (DOTATATE, DOTATOC, DOTANOC) (Figure 2) and to different radiolabels including 111Indium,90 Yttrium, and 177Lutetium (127, 128). A recent double-blind controlled trial (129) demonstrated that 177Lutetium-DOTATATE treatment in patients with advanced midgut carcinoids, resulted in marked increase in progression-free survival and a preliminary result demonstrating increased overall survival, with acceptable safely profile. This has led to FDA approval for this treatment in patients with advanced NETs. Almost all of the early studies performed with SRI and PRRT used SST analogs that were agonists because of the belief the peptide should be internalized to provide the best imaging and radionuclide delivery to the tumor. Recent studies demonstrate that SST receptor antagonists recognize more binding sites on the tumor, provide superior tumor targeting to agonists (118, 130, 131) and also demonstrate greater membrane binding suggesting it will be superior for PRRT.
Unfortunately, the over-expression of SST receptors is limited to a subset of tumors and is not seen in many of the more frequent adenocarcinomas, such as breast, colon, lung or prostate and therefore the specific ligands developed for NETs will not be useful in these tumors. However, many of these other more common tumors over-express a number of other GPCRs including receptors for the BB, NTS, VIP/PACAP family (68, 132–134). Furthermore, in many cases selective ligands that are radiolabeled have been developed and studies in animal models and some cases in small numbers of humans with different diseases, support this approach (68, 132–134). Whether in the future they will become established for the imaging of these tumors or for the delivery of cytotoxic substances is unknown.
Conclusions
Neuropeptide GPCR may play an important role in cancer proliferation, angiogenesis, and metastasis. Most of the research conducted on NTS, BB, and VIP has been at the preclinical level. NTSR1 and BBR are class A/Rhodopsin-like receptors which interact with Gq and cause PI turnover. Nonpeptide antagonists are available for the BBR and NTSR which inhibit the proliferation of cancer cells. NTS, BB, and VIP conjugates, which kill cancer cells, have been developed. BB, NTS or PACAP stimulate the proliferation of NSCLC cells in an EGFR-dependent manner. The transactivation of the EGFR is blocked by SR48692 (NTSR1 antagonist), PD176252 (GRPR antagonist) or PACAP(6–38) (PAC1 antagonist) as well as gefitinib (EGFR TKI). The GPCR antagonists potentiate the ability of TKI to reduce NSCLC growth in vitro. It remains to be determined if GPCR antagonists will potentiate the action of TKI in vivo. GPCR antagonists potentiate the effects of chemotherapeutic drugs (135, 136). VIPhybrid, a VPAC1 antagonist, potentiates the effects cytotoxicity of taxol in breast cancer in vitro and in vivo. Also, VIPhybrid potentiated the cytotoxicity of cisplatin, doxorubicin, gemcitabine, irinotecan or vinorelbine on colon cancer in vitro.
VIP and PACAP are class B/Secretin-like receptors which interact with Gs and stimulate adenylyl cyclase. PAC1 has numerous splice variants, which alter second messenger production. VIP has been coupled to radioisotopes, e.g., 18F, 65Cu, and 99mTc to image tumors in animal models. High affinity non-peptide antagonists need to be developed for PAC1 VPAC1 and VPAC2. The use of peptide coated nanoparticles which contain chemotherapeutics is being investigated (137). The GPCR can be used to direct neuropeptide coated nanoparticle to the tumor. Recently, cholecystokinin antagonists were found to potentiate the effects of immune checkpoint inhibitors at impairing the growth of pancreatic tumors in vivo (138). Thus GPCR antagonists can potentiate the effects of various drugs in cancer treatment.
The use of SST analogs and their receptors are now an established part of clinical practice and provide the basis for 4 different FDA approved indications in patients with NETs. Long acting formulations of octreotide or lanreotide are the predominant therapeutic agents used to control excess secretion of peptides or growth hormone causing clinical syndromes in NET patients. Large clinical trials have recently led to the FDA approval of lanreotide/octreotide analogs to reduce NET growth in patients with advanced disease resulting in increased progression-free survival. SST receptor imaging using initially 111In-pentetreotide, and more recently, 68Ga-DOTATATE, which takes advance of the over-expression of SSTRs by NETs is the most sensitive method to image tumor location/extent in these patients. Lastly, numerous animal studies and non- prospective clinical studies, demonstrated that patients with advanced NETs expressing SSTRs could be treated with radiolabeled SST analogs with good antitumor effects. Recently, a large double -blind study in patients with advanced ileal carcinoid NETs confirmed this result leading to FDA approval for this approach using 177Lu labeled octreotate. The SST research shows that neuropeptide GPCRs can be used as oncotargets to detect and treat a human cancer.
A goal is to advance the cancer research on BB, NTS, and VIP. SST is inhibitory in nature and was initially used to inhibit secretions from NETs. Few effective therapies were available and octreotide effectively controlled the symptoms of patients with VIPomas, glucagonomas, GRFomas, insulinomas or gastrinomas. In contrast, BB, NTS, and VIP are stimulatory in nature and a substantial effort has been made to improve the half-life of peptide agonists in the blood and develop specific high affinity antagonists which are stable. SSTRs are present in over 90% of the NETs. BB, NTS, and VIPRs are not so universal in epithelial cancers. By precision medicine, the overexpression of GRPCRs for BB, NTS or VIP in the biopsy specimen will dictate which GPCR should be targeted. To illustrate if the tumor overexpresses NTSR1, the patient may be treated with SR48692 alone or in combination with another drug. Additional clinical trials are needed in BB, NT, and VIP research so that these peptide GPCR's can be used as oncotargets to treat epithelial cancers of the breast, colon, lung, and prostate.
Author Contributions
TM, IR-A and RJ are responsible for the writing of the manuscript and the figure preparation.
Funding
This research is supported by the intramural program of the NCI and NIDDK of the NIH.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
The authors thank Drs. D. Wink and T. Leto for helpful discussions.
References
1. Almendro V, Garcia-Recio S, Gascon P. Tyrosine kinase receptor transactivation associated to Gprotein- coupled receptors. Curr Drug Targets (2010) 11:1160–80. doi: 10.2174/138945010792006807
2. Lappano R, Maggiolinin M. Pharmacotherpeutic targeting of G protein-coupled receptors in oncology: examples of approved therapies and emerging concepts. Drugs (2017) 77:951–65. doi: 10.1007/s40265-017-0738-9
3. Moody TW, Moreno P, Jensen RT. Neuropeptides as lung cancer growth factors. Peptides (2015) 72:106–11. doi: 10.1016/j.peptides.2015.03.018
4. Jensen RT, Battey JF, Spindel ER, Benya RV. Mammalian bombesin receptors: nomenclature, distribution, pharmacology, signaling and functions in normal and disease states. Pharmacol Rev. (2008) 60:1–42. doi: 10.1124/pr.107.07108
5. Moody TW, Chiles J, Casibang M, Moody E, Chan D, Davis TP. SR48692 is a neurotensin receptor antagonist which inhibits the growth of small cell lung cancer cells. Peptides (2001) 22:109–15. doi: 10.1016/S0196-9781(00)00362-4
6. Moody TW, Leyton J, Garcia L, Jensen RT. Nonpeptide gastrin releasing peptide receptor antagonists inhibit the proliferation of lung cancer cells. Eur J Pharmacol. (2003) 474:21–9. doi: 10.1016/S0014-2999(03)01996-4
7. Moreno P, Mantey SA, Lee SH, Ramos-Alvarez I, Moody TW, Jensen RT. A possible new target in lung cancer cells:The orphan receptor, bombesin receptor subtype-3. Peptides (2018) 101:213–26. doi: 10.1016/j.peptides.2018.01.016
8. Moody TW, Pert CB, Gazdar AF, Carney DN, Minna JD. High levels of intracellular bombesin characterize human small cell lung carcinoma. Science (1981) 214:1246–48. doi: 10.1126/science.6272398
9. Wood SM, Wood JR, Ghatei MA, Lee YC, O'Shaughnessy D, Bloom SR. Bombesin somatostatin and neurotensin-like immunoreactivity in bronchial carcinoma. J Clin Endocrinol Metab. (1981) 53:1310–14.
10. Yang HJ, Gu Y, Chen C, Xu C, Bao YX. Diagnostic value of pro-gastrin-releasing peptide for small cell lung cancer:A meta-analysis. Clin Chem Lab Med. (2011) 49:1039–48. doi: 10.1515/CCLM.2011.161
11. Korse CM, Taal BG, Bonfrer JM, Vincent A, val Velthuysen M, Baas P. An elevated progastrin releasing peptide level in patients with well-differentiated neuroendocrine tumours indicates a primary tumour in the lung and predicts a shorter survival. Ann Oncol. (2011) 22:2625–30. doi: 10.1093/annonc/mdr007
12. Wojcik E, Rychik U, Skotnicki P, Jakubowicz J, Kulpa JK. Utility of proGRP determinations in cancer patients. Clin Lab. (2010) 56:527–34.
13. Cuttitta F, Carney DN, Mulshine J, Moody TW, Fedorko J, Fischler A, et al. Bombesin-like peptides can function as autocrine growth factors in human small cell lung cancer. Nature (1985) 316:823–26. doi: 10.1038/316823a0
14. Kelley MJ, Linnoila RI, Avis IL, Georgiakis MS, Cuttitta F, Mulshine JL, et al. Antitumor activity of a monoclonal antibody directed against GRP in patients with small cell lung cancer. Chest (1997) 112:255–61. doi: 10.1378/chest.112.1.256
15. Battey JF, Way J, Corjay MH, Shipira H, Kasano K, Hawkins R, et al. Molecular cloning of the BB/GRP receptor from Swiss 3T3 cells. Proc Natl Acad Sci USA. (1991) 88:395–9. doi: 10.1073/pnas.88.2.395
16. Spindel ER, Giladi E, Brehm P, Goodman RA, Segerson TP. Cloning and functional characterization of a complementary DNA encoding the murine fibroblast BB/GRP receptor. Mol Endocrin. (1990) 4:1956–63. doi: 10.1210/mend-4-12-1956
17. Wada E, Way J, Shapira H, Kusano K, Lebacq-Verheyden AM, Coy D, et al. cDNA cloning, characterization and brain region-specific expression of a neuromedin-B preferring bombesin receptor. Neuron (1991) 6:421–30. doi: 10.1016/0896-6273(91)90250-4
18. Fathi Z, Corjay MH, Shapira H, Wada E, Benya R, Jensen R et al. BRS-3:a novel bombesin receptor subtype selectively expressed in testis and lung carcinoma cells. J Biol Chem. (1993) 268:5979–84.
19. Moody TW, Mantey SA, Pradhan TK, Schumann M, Nakagawa T, Martinez A, et al. Development of high affinity camptothecin-bombesin conjugates that have targeted cytotoxicity for bombesin receptor-containing tumor cells. J Biol Chem. (2004) 279:23580–89. doi: 10.1074/jbc.M401938200
20. Moody TW, Sun LC, Mantey SA, Pradhan T, Mackey LV, Gonzalez N, et al. In vitro and in vivo antitumor effects of cytotoxic camptothecin bombesin conjugates are mediated by specific interaction with cellular bombesin receptors. J Pharmacol Exp Ther. (2006) 318:1265–72. doi: 10.1124/jpet.106.104141
21. Safavy A, Raisch KP, Khazaeli MB, Buchsbaum DJ, Bonner JA. Paclitaxel derivatives for targeted therapy of cancer:Toward the development of smart taxanes. J Med Chem. (1999) 42:419–24. doi: 10.1021/jm990355x
22. Wang K, Sun X, Wang Y, Yang F, Wang H. Breast cancer targeted chemotherapy based on doxorubicin-loaded bombesin peptide modified nanocarriers. Drug Deliv. (2015) 23:2697–702. doi: 10.3109/10717544.2015.1049721
23. Moody TW, Pradhan T, Manety SA, Jensen RT, Dyba M, Moody D, et al. Bombesin marine toxin conjugates inhibit the growth of lung cancer cells. Life Sci. (2008) 82:855–865. doi: 10.1016/j.lfs.2008.01.019
24. Liu S, Yang H, Wan L, Cai HW, Li SF, Li YP, et al. Enhancement of cytotoxicity of anti-microbial peptide magainin II in tumor cells by bombesin-targeted delivery. Acta Pharmacol Sin. (2011) 32:79–88. doi: 10.1038/aps.2010.162
25. Sioud M, Mobergslien A. Efficient siRNA targeted delivery into cancer cells by gastrin-releasing peptides. Bioconjug Chem. (2012) 23:1040–49. doi: 10.1021/bc300050j
26. Hohla F, Schally AV. Targeting gastrin releasing peptide receptors: new options for therapy and diagnosis of cancer. Cell Cycle (2010) 9:1738–41. doi: 10.4161/cc.9.9.11347
27. Schwartzmann G, DiLeone LP, Horowitz M, Schunemann D, Cancella A, Pereira AS et al. A phase I trial of the bombesin/gastrin-releasing peptide (BN/GRP) antagonist RC3095 in patients with advanced solid malignancies. Invest New Drugs (2006) 24:403–12. doi: 10.1007/s10637-006-6886-5
28. Mahmoud S, Staley J, Taylor J, Bodgen A, Moreau JP, Coy D, et al. (Psi13, 14) Bombesin analogues inhibit the growth of small cell lung cancer in vitro and in vivo. Cancer Res. (1991) 51:1798–802.
29. Nakamura T, Ramos-Alvarez I, Iordanskaia T, Moreno P, Mantey SA, Jensen RT. Molecular basis for high affinity and selectivity of the peptide antagonist Bantag-1 for the orphan BB3 receptor. Biochem Pharmacol. (2016) 115:64–7. doi: 10.1016/j.bcp.2016.06.013
30. Reubi JC, Wenger S, Schumuckli-Mauer J, Schaer JC, Gugger M. Bombesin receptor subtypes in human cancers: detection with the universal radioligand (125)I-[D-TYR(6), beta-ALA(11), PHE (13), NLE (14)] bombesin (6-14). Clin Cancer Res. (2002) 8:1139–46.
31. DeVincentis G, Remediani S, Varvarigou AD, DiSanto G, Lori F, Laurenti C, et al. Role of 99mTc-bombesin scan in diagnosis and staging of prostate cancer. Cancer Biother Radiopharm. (2004) 19:81–4. doi: 10.1089/108497804773391711
32. Chen Q, Ma Q, Chen M, Chen B Wen Q, Jia B, et al. An exploratory study of 99mTc-RGD-BBN peptide scintimamography in the assessment of breast malignant lesions compared to 99mTc-3P4-RGD2. PLoS ONE (2015) 10:e0123401. doi: 10.1371/journal.pone.0123401
33. Kahkonen E, Jambor I, Kemppainen J, Lehtio K, Gronroos TJ, Kulsma A, et al. In vivo imaging of prostate cancer using [68Ga]-labeled bombesin analog BAY86-7548. Clin Cancer Res. (2013) 19:5434–43. doi: 10.1158/1078-0432.CCR-12-3490
34. Moody TW, Berna MJ, Mantey S, Sancho V, Ridnour L, Wink DA, et al. Neuromedin B receptors regulate EGF receptor tyrosine phosphorylation in lung cancer cells. Eur J Pharmacol. (2010) 637:38–45. doi: 10.1016/j.ejphar.2010.03.057
35. Truong TH, Carroll KS. Redox regulation of the EGFR signaling through cysteine oxidation. Biochemistry (2012) 51:9954–65. doi: 10.1021/bi301441e
36. Heppner DE, van der Vliet A. Redox-dependent regulation of the epidermal growth factor receptor signaling. Redox Biol. (2016) 8:24–7. doi: 10.1016/j.redox.2015.12.002
37. Lemmon MA, Schlessinger J. Cell signaling by receptor tyrosine kinases. Cell (2010) 141:1117–34. doi: 10.1016/j.cell.2010.06.011
38. Lynch TJ, Bell DW, Sordella R, Gurubhagavatula S, Okimoto RA, Brannigan BW, et al. Activating mutations in the epidermal growth factor receptor underlying responsiveness of non-small-cell lung cancer to gefitinib. N Eng J Med. (2004) 350:2129–39. doi: 10.1056/NEJMoa040938
39. Paez JB, Janne PA, Lee JC, Tracy S, Greuich H, Gabriel S et al. EGFR mutations in lung cancer: Correlation with clinical response to gefitinib. Science (2004) 304:1497–1500. doi: 10.1126/science.1099314
40. Schrump DS, Carter D, Kelsey CR, Marks LB, Giaccone G. Non-small cell lung cancer. In: DeVita Jr V, Lawrence TS, Rosenberg SA, editors. Cancer: Principles and Practice of Oncology. Philadelphia, PA: Lippincott, Williams and Wilkins (2011) p. 799–847.
41. Moody TW, Carney DN, Korman LY, Gazdar A, Minna JD. Neurotensin is produced by and secreted from classic small cell lung cancer cells. Life Sci. (1985) 36:1727–32. doi: 10.1016/0024-3205(85)90555-7
42. Kitabgi P. Neurotensin and neuromedin N are differentially processed from a common precursor by prohormone convertases in tissues and cell lines. Results Probl Cell Differ. (2010) 50:85–96. doi: 10.1007/400_2009_27
43. White JE, Noinaj N, Shibata Y, Love J, Kloss B, Xu F, et al. Structure of the agonist-bound neurotensin receptor. Nature (2012) 490:508–13. doi: 10.1038/nature11558
44. Kitabgi P. Functional domains of the subtype 1 neurotensin receptor (NTS1). Peptides (2006) 27:2461–68. doi: 10.1016/j.peptides.2006.02.013
45. Allen AE, Carney DN, Moody TW. Neurotensin binds with high affinity to small cell lung cancer cells. Peptides (1988) 9:57–61. doi: 10.1016/0196-9781(88)90224-0
46. Staley J, Fiskum G, Davis TP, Moody TW. Neurotensin elevates cyutosolic calcium in small cell lung cancer cells. Peptides (1989) 10:121–7. doi: 10.1016/0196-9781(89)90015-6
47. Muller KM, Tveteraas IH, Aasrum M, Odegard J, Dawood M, Dajani O, et al. Role of protein kinase C and epidermal growth factor receptor signaling in growth stimulation by neurotensin in colon carcinoma cells. BMC Cancer (2011) 11:421. doi: 10.1186/1471-2407-11-421
48. Guha S, Rey O, Rozengurt E. Neurotensin induces protein kinase C-dependent protein kinase D activation and DNA synthesis in human pancreatic carcinoma cell lune PANC-1. Cancer Res. (2002) 62:1632–40.
49. Leyton J, Garcia-Marin L, Jensen RT, Moody TW. Neurotensin causes tyrosine phosphorylation of focal adhesion kinase in lung cancer cells. Eur J Pharmacol. (2002) 442:179–86. doi: 10.1016/S0014-2999(02)01539-X
50. Kisfalvi K, Guha S, Rozengurt E. Neurotensin and EGF induce synergistic stimulation of DNA synthesis by increasing the duration of ERK signaling to ductal pancreatic cancer cells. J Cell Physiol. (2012) 202:880–90. doi: 10.1002/jcp.20187
51. Harrison RJ, McNeil GP, Dobner PR. Synergistic activation of neurotensin/neuromedin N gene expression by c-Jun and glucocorticoids: Novel effects of Fos family proteins. Mol Endocrinol. (1995) 9:981–93.
52. Moody TW, Chan DC, Mantey SA, Moreno P, Jensen RT. SR48692 inhibits non-small cell lung cancer proliferation in an EGF receptor-dependent manner. Life Sci. (2014) 100:25–34. doi: 10.1016/j.lfs.2014.01.072
53. Younes M, Wu Z, Dupouy S, Lupo AM, Mourra N, Takahashi T, et al. Neurotensin (NTS) and its receptor (NTSR1) causes EGFR, HER2, and HER3 over-expression and their autocrine/paracrine activation in lung tumors, confirming responsiveness to erlotinib. Oncotarget (2014) 5:8252–59. doi: 10.18632/oncotarget.1633
54. Hassan S, Dobner PR, Carraway RE. Involvement of MAP kinase, PI-3 kinase and EGF-receptor in the stimulatory effect of neurotensin on DNA synthesis in PC3 cells. Regul Pept. (2004) 120:155–66. doi: 10.1016/j.regpep.2004.03.004
55. DiFlorio A, Sancho V, Moreno P, Della Fave G, Jensen RT. Gastrointestinal hormones stimulate growth of foregut neuroendocrine tumors by transactivating the EGF receptor. Biochem Biophys Acta (2013) 1833:573–82. doi: 10.1016/j.bbamcr.2012.11.021
56. Zhou Z, Xie J, Cai Y, Yang S, Chen Y, Wu H. The significance of NTR1 expression and its correlation with β-catenin and EGFR in gastric cancer. Diagn Pathol. (2015) 100:128. doi: 10.1186/s13000-015-0356-3
57. Ye Y, Long X, Zhang L, Chen J, Liu P, Li H, et al. NTS/NTR1 co-expression enhances epithelial-to-mesenchymal transition and promotes tumor metastasis by activating the Wnt/β-catenin signaling pathway in hepatocellular carcinoma. Oncotarget (2016) 7:70303–22. doi: 10.18632/oncotarget.11854
58. Schulz J, Rohracker M, Stiebler M, Goldschmidt J, Stober F, Noriega M, et al. Proof of therapeutic efficacy of a 177Lu-labeled neurotensin receptor 1 antagonists in a colon carcinoma xenograft model. J Nucl Med. (2017) 58:936–41. doi: 10.2967/jnumed.116.185140
59. Baum RP, Singh A, Schuchardt C, Kulkarni HR, Klette I, Wiessalla S, et al. 177Lu-3BP-227 for neurotensin receptor 1-targeted therapy of metastatic pancreatic adenocarcinoma: first clinical results. J Nucl Med. (2018) 59:809–14. doi: 10.2967/jnumed.117.193847
60. Harmar AJ, Fahrenkrug J, Gozes I, Laburthe M, May V, Pisegna JR, et al. Pharmacology and functions of receptors for vasoactive intestinal peptide and pituitary adenylate cyclase-activating polypeptide:IUPHAR review 1. Br J Pharmacol. (2012) 166:4–17. doi: 10.1111/j.1476-5381.2012.01871.x
61. Sun C, Song D, Davis-Taber R, Barrett LW, Scott V, Richardson PL, et al. Solution structure and mutational analysis of pituitary adenylate cyclase-activating polypeptide binding to the extracellular domain of PAC1-Rs. Proc Natl Acad Sci USA. (2007) 104:7875–7880. doi: 10.1073/pnas.0611397104
62. Kumar S, Ploszak A, Zhang C, Swaminathan K, Xu HE. Crystal structure of the PAC1R extracellular domain unifies a consensus fold for hormone recognition by class B G-protein coupled receptors. PLoS ONE (2011) 6:e19682. doi: 10.1371/journal.pone.0019682
63. Liao C, Zhao X, Brewer M, May V, Li J. Conformational transitions of the pituitary adenylate cyclase-activating polypeptide receptor, a human class B GPCR. Sci Rep. (2017) 7:5427. doi: 10.1038/s41598-017-05815-x
64. Furness SGB, Wootten D, Christopoulos A, Sexton PM. Consequences of splice variation on secretin family G protein-coupled receptor function. Br J Pharmacol. (2012) 166:98–109. doi: 10.1111/j.1476-5381.2011.01571.x
65. Vaudry D, Falluel-Morel A, Vourgault S, Basille M, Burel D, Wurtz O, et al. Pituitary adenylate cyclase-activating polypeptide and its receptor:20 years after discovery. Pharmacol Rev. (2009) 61:283–357. doi: 10.1124/pr.109.001370
66. Lerner EA, Ribeiro JM, Nelson RJ, Lerner MR. Isolation of maxadilan, a potent vasodilatory peptide from the salivary glands of the sand fly Lutzomyia longipalpis. J Biol Chem. (1991) 266:11234–236.
67. Ramos-Alvarez I, Mantey SA, Nakamura T, Nuche-Berenguer B, Moreno P, Moody TW, et al. A structure-function study of PACAP using conformationally restricted analogs: identification of PAC1 receptor-selective PACAP agonists. Peptides (2015) 66:26–42. doi: 10.1016/j.peptides.2015.01.009
68. Moody TW, Nuche-Berenguer B, Jensen RT. VIP/PACAP, and their receptors in cancer. Curr Opin Endocrinol Diabetes (2016) 23:38–47. doi: 10.1097/MED.0000000000000218
69. Reubi JC, Laderach U, Waser B, Gebbers JO, Robberecht P, Laissue JA. Vasoactive intestinal peptide/pituitary adenylate cyclase activating peptide receptor subtypes in human tumors and their tissues of origin. Cancer Res. (2000) 60:3105–12.
70. Tang B, Yong X, Xie R, Li QW, Yang SM. Vasoactive intestinal peptide receptor-based imaging and treatment of tumors. Int J Oncol. (2014) 44:1023–31. doi: 10.3892/ijo.2014.2276
71. Moody TW, Jensen RT. VIP/PACAP receptors. In: Kastin AJ, editor. Handbook of Biologically Active Peptides, Second Edition. San Diego, CA: Academic Press. (2013), p. 556–561.
72. Lutz EM Ronaldson E, Shaw P, Johnson MS, Holland PJ, Mitchell R. Characterization of novel splice variants of the PAC1 receptor in human neuroblastoma cells: consequences for signaling by VIP and PACAP. Mol Cell Neurosci. (2006) 31:193–209. doi: 10.1016/j.mcn.2005.09.008
73. Blechman J, Levkowitz G. Alternative splicing of the pituitary adenylate cyclase activating polypeptide receptor PAC1: mechanisms of fine tuning of brain activity. Front Endocrinol. (2013) 4:55. doi: 10.3389/fendo.2013.00055
74. Pisegna J, Wank S. Cloning and characterization of the signal transduction of four splice variants of the human pituitary adenylate cyclase activating polypeptide receptor: evidence for dual coupling to adenylate cyclase and phospholipase C. J Biol Chem. (1996) 271:17267–74. doi: 10.1074/jbc.271.29.17267
75. Usiyama M, Ikelda R, Sugawara H, Yoshida M, Mori K, Kangawa K, et al. Differential intracellular signaling through PAC1 isoforms as a result of alternative splicing in the first extracellular domain and the third intracellular loop. Mol Pharmacol. (2007) 72:103–11. doi: 10.1124/mol.107.035477
76. Moody TW, Jensen RT. PACAP and Cancer. In: Reglodi D, Tamas A, Editors. Pituitary Adenylate Cyclase Activating Polypeptide-PACAP. Basel: Springerx International Publishing AG. (2016) p. 795–814.
77. Moody TW, Leyton J, Unsworth E, John C, Lang L, Eckelman WC. (Arg15, Arg21) VIP: Evaluation of biological activity and localization to breast cancer tumors. Peptides (1998) 19:585–92. doi: 10.1016/S0196-9781(97)00459-2
78. Thakur ML, Devadhas D, Zhang K, Pestell RG, Wang C, McCue P, et al. Imaging spontaneous MMTV neu transgencic mammary tumors: targeting metabolic activity versus genetic products. J Nucl Med. (2010) 51:106–11. doi: 10.2967/jnumed.109.069542
79. Thakur ML, Marcus CS, Saeed S, Pallela V, Minami C, Diggles L, et al. Imaging tumors in humans with Tc-99m-VIP. Ann NY Acad Sci. (2000) 921:37–44. doi: 10.1111/j.1749-6632.2000.tb06949.x
80. Moody TW, Mantey SA, Fuselier JA, Coy D, Jensen RT. Vasoactive intestinal peptide-camptothecin conjugates inhibit the proliferation of breast cancer cells. Peptides (2007) 28:1883–90. doi: 10.1016/j.peptides.2007.04.017
81. Moody TW, Czerwindki G, Tarasova NI, Moody DL, Michejda CJ. The development of VIP ellipticin conjugates. Regul Pept. (2004) 123:187–92. doi: 10.1016/j.regpep.2004.03.021
82. Oryuksel H, Mohanty PS, Rubinstein J. VIP-grafted sterically stabilized phospholipid nanomicellar 17-allylamino-17-dimethoxy geldanomycin: a novel targeted nanomedicine for breast cancer. Int J Pharm. (2009) 365:157–61. doi: 10.1016/j.ijpharm.2008.08.024
83. Onyuksel H, Jeon E, Rubinstein I. Nanomicellar paclitaxel increases cytotoxicity of multidrug resistant breast cancer cells. Cancer Lett. (2009) 274:327–30. doi: 10.1016/j.canlet.2008.09.041
84. Moody TW, Zia F, Draoui M, Brenneman DE, Fridkin M, Davidson A, et al. A vasoactive intestinal peptide antagonist inhibits non-small cell lung cancer growth. Proc Natl Acad Sci USA. (1993) 90:4345–49. doi: 10.1073/pnas.90.10.4345
85. Zia F, Fagarasan M, Bitar K, Coy DH, Pisegna JR, Wank SA, et al. Pituitary adenylate cyclase activating peptide receptors regulate the growth of non-small cell lung cancer cells. Cancer Res. (1995) 55:4886–91.
86. Dagar S, Sekosan M, Rubinstein I, Onyeksel H. Detection of VIP receptors in MNU-induced breast cancer in rats: implications for breast cancer targeting. Breast Cancer Res Treat. (2001) 65:49–54. doi: 10.1023/A:1006406617497
87. Madsen B, Georg B, Vissing H, Fahrenkrug J. Retinoic acid down-regulates the expression of vasoactive intestinal polypeptide receptor type-1 in human breast carcinoma cell lines. Cancer Res. (1998) 58:4845–50.
88. Jakowlew SB, Zakowicz H, Moody TW. Retinoic acid down-regulates VPAC(1) receptors and TGF-beta-3 but up-regulates TGF-beta 2 in lung cancer cells. Peptides (2000) 2:1831–7. doi: 10.1016/S0196-9781(00)00344-2
89. Moody TW, Dudek J, Zakowicz H, Walters J, Jensen RT, Petricoin E, et al. VIP receptor antagonists inhibit mammary carcinogenesis in C3(1)SV40T antigen mice. Life Sci. (2004) 74:345–57. doi: 10.1016/j.lfs.2003.07.043
90. Moody TW, Osefo N, Nuche-Berenguer B, Ridnour L, Wink D, Jensen RT. Pituitary adenylate cyclase-activating polypeptide causes tyrosine phosphorylation of the epidermal growth factor receptor in lung cancer cells. J Pharm Exp Ther. (2012) 341:873–81. doi: 10.1124/jpet.111.190033
91. Beaudreau HE, Casterline BW, Burke DJ, Leto TL. Wild-type and mutant p53 differentially regulate NADPH oxidase-4 in TGF-β-mediated migration of human lung cancer epithelial cells. Br J Cancer (2014) 110:2569–82. doi: 10.1038/bjc.2014.165
92. Valdehita A, Bajo AM, Schally AV, Varga JL, Carmena MJ, Prieto JC. VIP induces transactivation of the EGFR and HER2 in human breast cancer cells. Mol Cell Endocrinol. (2009) 302:41–8. doi: 10.1016/j.mce.2008.11.024
93. Rai U, Thrimawithana TR, Valery C, Young SA. Therapeutic uses of somatostatin and its analogues: current view and potential applications. Pharmacol Ther. (2015) 152:98–110. doi: 10.1016/j.pharmthera.2015.05.007
94. Sun L, Coy DH. Somatostatin and its analogs. Curr Drug Targets (2016) 17:529–37. doi: 10.2174/1389450116666141205163548
95. Reubi JC, Krenning E, Lamberts SW, Kvols L. In vitro detection of somatostatin receptors in human tumors. Metabolism (1992) 41:104–10. doi: 10.1016/0026-0495(92)90042-9
96. Ito T, Lee L, Jensen RT. Carcinoid-syndrome:recent advances, current status and controversies. Curr Opin Endocrinol Diabetes Obes. (2018) 25:22–35. doi: 10.1097/MED.0000000000000376.
97. Pokuri VK, Fong MK, Iyer R. Octreotide and lanreotide in gastroenteropancreatic neuroendocrine tumors. Curr Oncol Rep. (2016) 18:7. doi: 10.1007/s11912-015-0492-7
98. Narayanan S, Kunz PL. Role of somatostatin analogues in the treatment of neuroendocrine tumors. Hematol Oncol Clin North Am. (2016) 30:163–77. doi: 10.1016/j.hoc.2015.09.008
99. Ito T, Lee L, Jensen RT. Treatment of symptomatic neuroendocrine tumor syndromes: recent advances and controversies. Expert Opin Pharmacother. (2016) 17:2191–205. doi: 10.1080/14656566.2016.1236916
100. Oberg K, Lamberts SW. Somatostatin analogues in acromegaly and gastroenteropancreatic neuroendocrine tumors: past, present and future. Endocr Relat Cancer (2016) 23:R551–66. doi: 10.1530/ERC-16-0151
101. Gadelha MR, Wildemberg LE, Bronstein MD, Gatto F, Ferone D. Somatostatin receptor ligands in the treatment of acromegaly. Pituitary (2017) 20:100–8. doi: 10.1007/s11102-017-0791-0
102. Susini C, Buscail L. Rationale for the use of somatostatin analogs as antitumor agents. Ann. Oncol. (2006) 17:1733–42. doi: 10.1093/annonc/mdl105
103. Schally AV, Szepeshazi K, Nagy A, Comaru-Schally AM, Halmos G. New approaches to therapy of cancers of the stomach, colon and pancreas based on peptide analogs. Cell Mol Life Sci. (2004) 61:1042–68. doi: 10.1007/s00018-004-3434-3
104. Msaouel P, Galanis E, Koutsilieris M. Somatostatin and somatostatin receptors:implications for neoplastic growth and cancer biology. Expert Opin Investig Drugs (2009) 18:1297–316. doi: 10.1517/13543780903176399
105. Verslype C, Carton S, Borbath I, Delaunoit T, Demetter P, Demolin G, et al. The anti-proliferative effect of somatostatin analogs:clinical relevance in patients with neuroendocrine gastro-entero-pancreatic tumours. Acta Gastroenterologica Belgica (2009) 72:54–8.
106. Michael M, Garcia-Carbonero R, Weber MM, Lombard-Bohas C, Toumpanakis C, Hicks RJ. The anti-proliferative role of lanreotide in controlling growth of neuroendocrine tumors: a systematic review. Oncologist (2017) 22:272–85. doi: 10.1634/theoncologist.2016-0305
107. Keskin O, Yalcin S. A review of the use of somatostatin analogs in oncology. Onco Targets Ther. (2013) 6:471–83. doi: 10.2147/OTT.S39987
108. Caplin ME, Pavel M, Cwikla JB, Phan AT, Raderer M, Sedlackova E, et al. Lanreotide in metastatic enteropancreatic neuroendocrine tumors. N Engl J Med. (2014) 371:224–33. doi: 10.1056/NEJMoa1316158
109. Rinke A, Muller HH, Schade-Brittinger C, Klose KJ, Barth P, Wied M, et al. Placebo-controlled, double-blind, prospective, randomized study on the effect of octreotide LAR in the control of tumor growth in patients with metastatic neuroendocrine midgut tumors:a report from the PROMID study group. J Clin Oncol. (2009) 27:4656–63. doi: 10.1200/JCO.2009.22.8510
110. Merola E, Panzuto F, Delle Fave G. Antiproliferative effect of somatostatin analogs in advanced gastro-entero-pancreatic neuroendocrine tumors:a systematic review and meta-analysis. Oncotarget (2017) 8:46624–34. doi: 10.18632/oncotarget.16686
111. Ito T, Igarashi H, Jensen RT. Therapy of metastatic pancreatic neuroendocrine tumors (pNETs): recent insights and advances. J Gastroenterol. (2012) 47:941–60. doi: 10.1007/s00535-012-0642-8
112. Pavel M, O'Toole D, Costa F, Capdevila J, Gross D, Kianmanesh R, et al. ENETS consensus guidelines update for the management of distant metastatic disease of intestinal, pancreatic, bronchial neuroendocrine neoplasms (NEN) and NEN of unknown primary site. Neuroendocrinology (2016) 103:172–85. doi: 10.1159/000443167
113. Kunz PL, Reidy-Lagunes D, Anthony LB, Bertino EM, Brendtro K, Chan JA, et al. Consensus guidelines for the management and treatment of neuroendocrine tumors. Pancreas (2013) 42:557–77. doi: 10.1097/MPA.0b013e31828e34a4
114. Massironi S, Conte D, Rossi RE. Somatostatin analogues in functioning gastroenteropancreatic neuroendocrine tumors:literature review, clinical recommendations and schedules. Scand J Gastroenterol. (2016) 51:513–23. doi: 10.3109/00365521.2015.1115117
115. Enzler T, Fojo T. Long-acting somatostatin analogues in the treatment of unresectable/metastatic neuroendocrine tumors. Seminars in Oncol. (2017) 44:141–56. doi: 10.1053/j.seminoncol.2017.07.001
116. Hasskarl J, Kaufmann M, Schmid HA. Somatostatin receptors in non-neuroendocrine malignancies:the potential role of somatostatin analogs in solid tumors. Future Oncol. (2011) 7:895–913. doi: 10.2217/fon.11.66
117. Smith M C, Maggi M, Orlando C. Somatostatin receptors in non-endocrine tumors. Dig Liver Dis. (2004) 36:S78–85. doi: 10.1016/j.dld.2003.11.019
118. Ito T, Jensen RT. Molecular imaging in neuroendocrine tumors:recent advances, controversies, unresolved issues, and roles in management. Curr Opin Endocrinol Diabetes Obes. (2017) 24:15–24. doi: 10.1097/MED.0000000000000300
119. Dromain C, Deandreis D, Scoazec JY, Goere D, Ducreux M, Baudin E, et al. Imaging of neuroendocrine tumors of the pancreas. Diagn Interv Imaging (2016) 97:1241–57. doi: 10.1016/j.diii.2016.07.012
120. Deppen SA, Blume J, Bobbey AJ, Shah C, Graham MM, Lee P, et al. 68Ga-DOTATATE compared with 111In-DTPA-Octreotide and conventional imaging for pulmonary and gastroenteropancreatic neuroendocrine tumors: a systematic review and meta-analysis. J Nucl Med. (2016) 57:872–78. doi: 10.2967/jnumed.115.165803
121. Ambrosini V, Nanni C, Fanti S. The use of gallium-68 labeled somatostatin receptors in PET/CT imaging. PET Clin. (2014) 9:323–29. doi: 10.1016/j.cpet.2014.03.008
122. Barrio M, Czernin J, Fanti S, Ambrosini V, Binse I, Du L, et al. The impact of somatostatin receptor-directed PET/CT on the management of patients with neuroendocrine tumor: a systematic review and meta-analysis. J Nucl Med. (2017) 58:756–761. doi: 10.2967/jnumed.116.185587
123. Mizutani G, Nakanishi Y, Watanabe N, Honma T, Obana Y, Seki T, et al. Expression of somatostatin receptor (SSTR) subtypes (SSTR-1, 2A, 3, 4 and 5) in neuroendocrine tumors using real-time RT-PCR method and immunohistochemistry. Acta Histochem Cytochem. (2012) 45:167–76. doi: 10.1267/ahc.12006
124. Bodei L, Kwekkeboom DJ, Kidd M, Modlin IM, Krenning EP. Radiolabeled somatostatin analogue therapy of gastroenteropancreatic cancer. Semin Nucl Med. (2016) 46:225–38. doi: 10.1053/j.semnuclmed.2015.12.003
125. Brabander T, Teunissen JJ, van Eijck CH, Franssen GJ, Feelders RA, de Herder WW, et al. Peptide receptor radionuclide therapy of neuroendocrine tumors. Best Pract Res Clin Endocrinol Metab. (2016) 30:103–14. doi: 10.1016/j.beem.2015.10.005
126. Bodei L, Cwikla JB, Kidd M, Modlin IM. The role of peptide receptor radionuclide therapy in advanced/metastatic thoracic neuroendocrine tumors. J Thorac Dis. (2017) 9S1511–23. doi: 10.21037/jtd.2017.09.82
127. Kwekkeboom DJ, Krenning EP. Peptide receptor radionuclide therapy in the treatment of neuroendocrine tumors. Hematol Oncol Clin North Am. (2016) 30:179–91. doi: 10.1016/j.hoc.2015.09.009
128. Kwekkeboom DJ, de Herder WW, Kam BL, van Eijck CH, Van Essen M, Kooij PP, et al. Treatment with the radiolabeled somatostatin analog [177 Lu-DOTA 0, Tyr3]octreotate:toxicity, efficacy, and survival. J Clin Oncol (2008) 26:2124–30. doi: 10.1200/JCO.2007.15.2553
129. Strosberg JR, Wolin EM, Chasen B, Kulke M, Bushnell DL, Caplin ME, et al. NETTER-1 phase III: progression-free survival, radiographic response, and preliminary overall survival results in patients with midgut neuroendocrine tumors treated with 177-Lu-Dotatate. J Clin Oncol. (2016) 34:194. doi: 10.1200/jco.2016.34.4_suppl.194
130. Dalm SU, Nonnekens J, Doeswijk GN, de Blois E, van Gent DC, Konijnenberg M W, et al. Comparison of the therapeutic response to treatment with a 177-lutetium labeled somatostatin receptor agonist and antagonist in preclinical models. J Nucl Med. (2016) 57:260–5. doi: 10.2967/jnumed.115.167007
131. Fani M, Nicolas GP, Wild D. Somatostatin receptor antagonists for imaging and therapy. J Nucl Med. (2017) 58:61S−66S. doi: 10.2967/jnumed.116.186783
132. Reubi JC. Peptide receptors as molecular targets for cancer diagnosis and therapy. Endocr Rev. (2003) 24:389–427. doi: 10.1210/er.2002-0007
133. Reubi JC, Macke HR, Krenning EP. Candidates for peptide receptor radiotherapy today and in the future. J Nucl Med. (2005) 46:67S−75S.
134. Sancho V, Di Florio A, Moody TW, Jensen RT. Bombesin receptor-mediated imaging and cytotoxicity: review and current status. Curr Drug Deliv. (2011) 8:79–134. doi: 10.2174/156720111793663624
135. Moody TW, Leyton J, Chan D, Brenneman DC, Fridkin M, Gelber E, et al. VIP receptor antagonists and chemotherapeutic drugs inhibit the growth of breast cancer cells. Breast Cancer Res Treat. (2001) 68:55–64. doi: 10.1023/A:1017994722130
136. Gelber E, Granoth R, Fridkin M, Dreznik Z, Brenneman DE, Moody TW, et al. A lipophilic vasoactive intestinal peptide analog enhances the anti-proliferative effect of chemotherapeutic agents on cancer cell lines. Cancer (2001) 92:2172–2180. doi: 10.1002/1097-0142(20011015)92:8<2172::AID-CNCR1560>3.0.CO;2-4
137. Khan M, Huang T, Lin CY, Wu J, Fan BM, Blan ZX. Exploiting cancer's phenotypic guise against itself: targeting ectopically expressed peptide G-protein coupled receptors for lung cancer therapy. Oncotarget (2017) 8:104615–637. doi: 10.18632/oncotarget18403
Keywords: cancer GPCR, cancer RTK, bombesin, neurotensin, vasoactive intestinal peptide, pituitary adenylate cyclase activating polypeptide (PACAP), somatostatin, cancer signal transduction
Citation: Moody TW, Ramos-Alvarez I and Jensen RT (2018) Neuropeptide G Protein-Coupled Receptors as Oncotargets. Front. Endocrinol. 9:345. doi: 10.3389/fendo.2018.00345
Received: 15 May 2018; Accepted: 11 June 2018;
Published: 29 June 2018.
Edited by:
Derek LeRoith, Icahn School of Medicine at Mount Sinai, United StatesReviewed by:
Erika Peverelli, Università degli Studi di Milano, ItalyCarsten Grötzinger, Charité Universitätsmedizin Berlin, Germany
Copyright © 2018 Moody, Ramos-Alvarez and Jensen. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Terry W. Moody, bW9vZHl0QG1haWwubmloLmdvdg==