 Caifei Liu
Caifei Liu Jing Li*
Jing Li*- Beijing Key Laboratory of DNA Damage Response and College of Life Sciences, Capital Normal University, Beijing, China
The addition and removal of O-linked N-acetylglucosamine (O-GlcNAc) to and from the Ser and Thr residues of proteins is an emerging post-translational modification. Unlike phosphorylation, which requires a legion of kinases and phosphatases, O-GlcNAc is catalyzed by the sole enzyme in mammals, O-GlcNAc transferase (OGT), and reversed by the sole enzyme, O-GlcNAcase (OGA). With the advent of new technologies, identification of O-GlcNAcylated proteins, followed by pinpointing the modified residues and understanding the underlying molecular function of the modification has become the very heart of the O-GlcNAc biology. O-GlcNAc plays a multifaceted role during the unperturbed cell cycle, including regulating DNA replication, mitosis, and cytokinesis. When the cell cycle is challenged by DNA damage stresses, O-GlcNAc also protects genome integrity via modifying an array of histones, kinases as well as scaffold proteins. Here we will focus on both cell cycle progression and the DNA damage response, summarize what we have learned about the role of O-GlcNAc in these processes and envision a sweeter research future.
Introduction
The study of O-linked N-acetylglucosamine (O-GlcNAc), O-GlcNAc transferase (OGT), and O-GlcNAcase (OGA) was pioneer by Dr. Hart in 1984 (1). OGT modifies the substrate protein at Ser/Thr residues with the O-GlcNAc group, while OGA reverses it. Since then, both biologists and chemists have been working hand in hand to solve the sweet mystery. In this review, we will first cover the laboratory routine to study protein O-GlcNAcylation, and then venture onto the recently identified function of O-GlcNAc in regulating cell cycle and the DNA damage response (DDR). For the versatile role of O-GlcNAc in other biological processes, please refer to other comprehensive and exhaustive reviews (2–5).
An Overview of O-GlcNAc
Of all the glucose that we consume every day, ~2−5% enters the hexosamine biosynthetic pathway (HBP), which provides UDP-N-acetyl-D-glucosamine (UDP-GlcNAc) (6), the donor substrate for OGT. UDP-GlcNAc is highly responsive to cellular nutrient variations, as its synthesis relies heavily on the metabolism of glucose, amino acids, fatty acids, and nucleotides (7). Hence, O-GlcNAc may serve as a reporter for the functional status of multiple pathways and is considered an ideal metabolic sensor (8), and defunct O-GlcNAc signaling underscores many metabolic diseases (3, 5).
O-GlcNAc is Implicated in Various Human Diseases
O-GlcNAcylation is most abundant in the pancreas, followed by the brain (3). Thus it has been intimately linked with a plethora of human diseases, especially diabetes, and Alzheimer's disease (AD).
In diabetes, hyperglycemia leads to chronic hyper-O-GlcNAcylation, which brings mayhem to cellular signaling networks. And inhibiting O-GlcNAc was found to blockade arrhythmias in diabetic animals (9). Recent investigations show that OGT in pancreatic β cells regulates the β cell mass. And OGT disruption results in diabetes via the endoplasmic reticulum (ER) stress and the Akt pathway (10). The vital player in AD, Tau, is O-GlcNAcylated in normal brains, but hyperphosphorylated in AD brains (3). Interestingly, increasing O-GlcNAc in mice decreases neuronal losses (11).
O-GlcNAc in Cancer Metabolism
One characteristic of cancer cells is the Warburg effect, namely, elevated glycolytic flux, including glycolysis, the pentose phosphate pathway (PPP), and the HBP. Moreover, enhanced O-GlcNAcylation levels have been pinpointed as a common cancer feature (12). Many studies have impinged O-GlcNAc as a sweet accomplice of cancer.
Upon entering the cell, glucose is phosphorylated by hexokinase to become glucose-6-phosphate (G6P). G6P then either undergoes glycolysis and the tricarboxylic acid (TCA) cycle to generate ATP, NADPH, and pyruvate, or produce ribose-5-phosphate and NADPH through PPP (13). PPP maintains redox homoeostasis in rapidly dividing cells, especially in cancer cells. In the glycolytic pathway, phosphofructokinase 1 (PFK1) is O-GlcNAcylated at S529 upon hypoxia, inhibiting PFK1 activity and directing the glucose metabolism to the PPP (14). Concomitantly, the rate-limiting enzyme of the PPP, G6P dehydrogenase (G6PD), is also O-GlcNAcylated during hypoxia, thus being activated to increase glucose flux via PPP (15). Taken together, O-GlcNAc activates the PPP to promote cancer proliferation.
In the TCA cycle, fumarase both reversibly catalyzes fumarate to malate in the mitochondria, and regulates amino acid and fumarate metabolism in the cytoplasm. Fumarase is O-GlcNAcylated at S75, which antagonizes phosphorylation by AMPK at the exact same residue when glucose is scarce (16). pS75 mediates fumarase-ATF2 interaction, blocks KDM2A activity, stabilizes H3K36me2 and thus redirecting the cell proliferation to growth arrest (16). Hence in cancer cells, where OGT activity is particularly high, pS75 levels are relatively low, conferring growth advantage to cancer cells.
Equally important, OGT also regulates lipid metabolism in cancer. To sustain growth, cancer cells usually utilize de novo lipogenesis, which encompass activation of key enzymes, such as fatty acid synthase (FAS), and master transcription factors, such as the sterol regulatory element binding protein (SREBP-1). OGT suppression leads to lipogenic defects, which could be rescued by SREBP-1 overproduction (17). OGT regulates SREBP-1 protein abundance, probably via AMPK (17). On the other hand, FAS binds with OGA, and the interaction increases during oxidative stress (18). FAS inhibits OGA activity, so the O-GlcNAc levels elevate under oxidative stress in mammalian cells (18).
In conclusion, O-GlcNAc integrates various nutrient signaling with growth signaling and may provide new venues for therapeutic purposes.
Crosstalks Between O-GlcNAc and Other Post-translational Modifications (PTMs)
Then how does O-GlcNAc weave its magic wand? The answer lies largely in crosstalk with other PTMs. Due to the large size of the O-GlcNAc moiety, steric hindrance is imposed upon the O-GlcNAcylated protein, thus impeding other PTMs at the same or adjoining sites. For instance, when a mixture of two OGA inhibitors, PUGNAc and NAG-thiazoline, was utilized to analyze the ~700 phosphopeptides, phosphorylation levels of 131 peptides (18.4%) escalated and 234 (32.9%) peptides dampened (19). In a quantitative phosphoproteomics study using OGT wild-type and null cells, 232 phosphosites increased and 133 decreased out of the 5,529 sites in the null cells (20). Thus, a yin-yang relationship has been proposed between O-GlcNAcylation and phosphorylation (3).
Discordant results against the “yin-yang” model have also been recorded. In a 2008 study, researchers closely monitored phosphorylation sites when O-GlcNAcylation was elevated (19). As a result, 280 phosphorylation sites decreased, while 148 sites increased (19). In a recent quantitative phosphoproteomics study, a great many DDR proteins were identified to be O-GlcNAcylated, among which was checkpoint kinase 1 (Chk1) (20). When OGT is deleted, pT113 of Chk1 increases, consistent with the “yin-yang” model, but pS151 decreases, against the model (20). Another case in hand is the intermediate filament protein, vimentin. Vimentin filament severing is a key step for completion of cytokinesis, and many phosphorylation events intricately mediate this process. In particular, cyclin-dependent kinase 1 (CDK1) phosphorylates vimentin at S55 (21) to prime vimentin for subsequent phosphorylation by polo-like kinase 1 (Plk1) at S82 (22), thus inhibiting vimentin filament assembly. Other kinases, such as Aurora B and the Rho kinase, also phosphorylate vimentin at S72 and S71, respectively, thus localizing vimentin to the cleavage furrow (23–25). When cells were depleted of OGT by siRNA, pS71 levels were hampered; vimentin filament thus could not be severed during cytokinesis, leading to cytokinesis failure (26). In sum, the relationship between phosphorylation and O-GlcNAcylation needs to be analyzed case by case, and there might not be a clear-cut rule.
The relationship between O-GlcNAcylation and ubiquitination and hence protein stability was tested recently. Using a newly developed quantitative time-resolved O-linked GlcNAc proteomics (qTOP), 533 O-GlcNAcylated proteins were examined for stability, and 14% were identified to be hyper-stably O-GlcNAcylated (27). Of this pool of ~75 proteins, O-GlcNAcylation has a significant impact on the protein stability, and O-GlcNAcylation mainly promotes protein stability (27).
Congruent with this study, O-GlcNAcylation has been identified to augment protein abundance in an array of studies. O-GlcNAcylation of the circadian clock proteins, BMAL1 and CLOCK, inhibits their ubiquitination and stabilizes protein levels (28). During gluconeogenesis, the master regulator PGC-1α is O-GlcNAcylated to bind the deubiquitinase BAP1, thus dampening ubiquitination, and enhancing protein abundance (29). The mixed lineage leukemia 5 (MLL5) protein forms a stable complex with OGT and ubiquitin specific protease 7 (USP7), and OGT suppresses MLL5 ubiquitination and increases its stability (30). The histone methyltransferase enhancer of zeste homolog 2 (EZH2) is O-GlcNAcylated at S75, which maintains its protein stability (31). Besides proteasome-mediated degradation, ubiquitination also plays other roles in signal transduction. And crosstalks have also been identified in these scenarios. For instance, O-GlcNAcylation of DNA polymerase Polη promotes its polyubiquitination and subsequent removal from replication forks (32).
With more people joining in the O-GlcNAc venture, communication among O-GlcNAc and more PTMs, such as methylation, SUMOylation, acetylation, ADP-ribosylation is bound to be unveiled.
The Toolbox of O-GlcNAc Research
As the Chinese saying goes, “Nice craftsmanship entails utilization of nice tools.” The toolbox of O-GlcNAc has been limited, compared to other PTMs, e. g., phosphorylation and ubiquitination. The common practice to identify O-GlcNAcylated proteins is by immunoprecipitating (IP) proteins of interest, then immunoblotting (IB) with O-GlcNAc antibodies. Below we will briefly delineate the lab routine to identify and study protein O-GlcNAcylation.
Antibodies
O-GlcNAc antibodies encompass RL2 and CTD110.6, among others (33). CTD110.6, an IgM, recognizes YSPTS(O-GlcNAc)PSK and also non-specifically binds terminal β-linked-GlcNAc (β-GlcNAc) and other N-glycan cores (34). RL2, on the contrary, is an IgG, and its antigen is pore complex-lamina fraction purified from rat liver nuclear envelopes (34). The RL2 and CTD110.6 are considered pan-O-GlcNAc antibodies, and might be promiscuous. They have overlapping, yet somewhat distinct ranges of protein targets. Hence it has been recommended to adopt both antibodies to conclude the O-GlcNAc modification. Alternatively, GlcNAc could be added during antibody blotting to compete against antibody binding, so that signals from O-GlcNAcylated proteins could be validated (35).
Another method is to perform in vitro O-GlcNAcylation assays. Using tritiated UDP-Galactose (UDP-[3H]-Galactose) as a donor, the addition of “hot” O-GlcNAc onto proteins can be traced. However, this is not a trivial experiment. First, the OGT enzyme is sensitive to salt and reducing agents, thus the purified proteins need to be desalted (36). Second, tritium has a long half-life of 12.3 years, so the storage of residue materials is a valid concern. Third, tritium is not as sensitive as 32P, hence autoradiography might take an extended period of time to be detected. Alternatively, RL2 and CTD110.6 could be used in IB experiments on the in vitro reaction products (37), thus avoiding the tritium issue.
Inhibitors
The aforementioned identification of O-GlcNAcylated proteins could be facilitated by using OGA inhibitors, thus enhancing O-GlcNAc signals (36). Two most commonly utilized OGA inhibitors are O-(2-acetamido-2-deoxy-D-glucopyranosylidene) amino N-phenylcarbamate (PUGNAc) and Thiamet-G (TMG). PUGNAc is a 1,5-hydroximolactone. It is a non-selective inhibitor of OGA and inhibits glycosyl hydrolases in general, in particular lysosomal hexosaminidases (38). Treating 3T3-L1 adipocytes with PUGNAc increased globular O-GlcNAc levels and resulted in insulin resistance, the hallmark of diabetes (39), consistent with the current view that elevated O-GlcNAcylation correlates with diabetes. However, such effects could not be repeated by treating cells or animals with TMG (40). Since TMG is more specific for OGA compared to PUGNAc, the results above suggest that the effect of PUGNAc was non-specific. It is more promising in treating AD, as treating AD mice with TMG increased Tau O-GlcNAcylation, attenuated Tau phosphorylation and hence aggregation, thus opening new venues for AD therapy (11).
Unlike OGA inhibitors, OGT inhibitors have been difficult to come along (38). Alloxan is the first reported inhibitor for OGT (41). As a uracil analog, it inhibits OGT, but its toxicity affects many cellular processes (41). Other inhibitors have been used anecdotally. 6-diazo-5-oxo-L-norleucine (DON), which is an inhibitor of glutamine-utilizing enzymes, such as the CTP synthase (CTPS) and NAD synthase, has been shown to inhibit O-GlcNAcylation (35). Two OGT inhibitors have been reported recently. One is a naturally produced OGT inhibitor, L01 (42), whose effects are yet to be tested. The other is uridine diphospho-5-thio-N-acetylglucosamine (UDP-5SGlcNAc), a substrate analog of O-GlcNAc that might be a competitive inhibitor of OGT (43). When the inhibitor per-O-acetylated 2-acetamido-2-deoxy-5-thioglucopyranose (Ac-5SGlcNAc) is added into cells, it is coverted to UDP-5S-GlcNAc, which is the most frequently used OGT inhibitor today (44–46).
Mass Spectrometry (MS)
Last three decades have witnessed significant strides in developing MS instruments, especially electron transfer dissociation (ETD) MS (47, 48) and higher energy collisional dissociation (HCD) MS (49). These two apparatuses differ greatly from each other: HCD cannot specify the amino acid that is modified, while ETD is only efficient toward peptides. For more details about identification of O-GlcNAc sites by MS, please refer to a more in-depth review (33).
Mutagenesis Studies
Following MS, the potential sites will be mutagenized to confirm whether they are indeed the modification sites. O-GlcNAc-deficient mutations are normally Ser/Thr to Ala, but O-GlcNAc-mimicking mutations are not well defined. Wang et al. (50) mutated the O-GlcNAc sites of the Ser/Thr kinase AKT1, T305, and T312, to Tyr, to mimic the bulky steric hindrance imposed by the O-GlcNAc moiety. But it has not been commonly adopted.
The mutant proteins will subsequently be IBed against the O-GlcNAc antibodies, RL2 and CTD110.6. Site-specific O-GlcNAc antibodies are yet to be developed, unlike site-specific phospho-antibodies, which have been a standard practice to study protein phosphorylation.
Most of the time, MS identifies more than one O-GlcNAc site (32, 51, 52). It has been noted that in this scenario, single mutants will sometimes display elevated O-GlcNAcylation, while the double/triple/quadruple mutants will down-regulate O-GlcNAcylation (32, 51, 52). The single residue might be a preferred modification site, the abolishing of which might result in prolonged interaction between OGT and the target protein. But the exact molecular details are lacking.
O-GlcNAc Regulates Cell Cycle Progression
The cell cycle, comprising G1, S, G2, and M phases, has entranced biologists from the earliest times. How is the genomic DNA faithfully replicated? How is the chromosome segregation process synchronized? How do the two daughter cells part from each other? These are just a few questions that have baffled biologists and the answers are still much sought after.
As has long been appreciated, the faithful execution and success completion of cell cycle is governed by a multitude of master kinases, phosphatases, ubiquitin E3 ligases and a network of protein machineries. Recent years have witnessed a surge of reports on the role of OGT in the cell cycle. Henthforth, we will first have a bird's eye view on the role of OGT, and then delve into the substrates of O-GlcNAc in cell cycle (Tables 1, 2). Of note, these tables are by no means comprehensive. Table 2 only contains proteins whose molecular mechanism is well studied. Many other proteins have been identified in many proteomic screens, but their functional significance is not well understood.
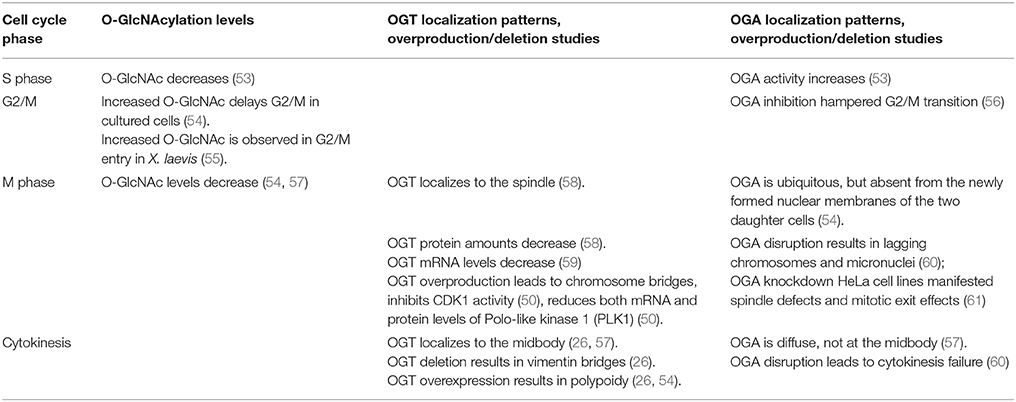
Table 1. Effects of OGT and OGA on cell cycle progression.
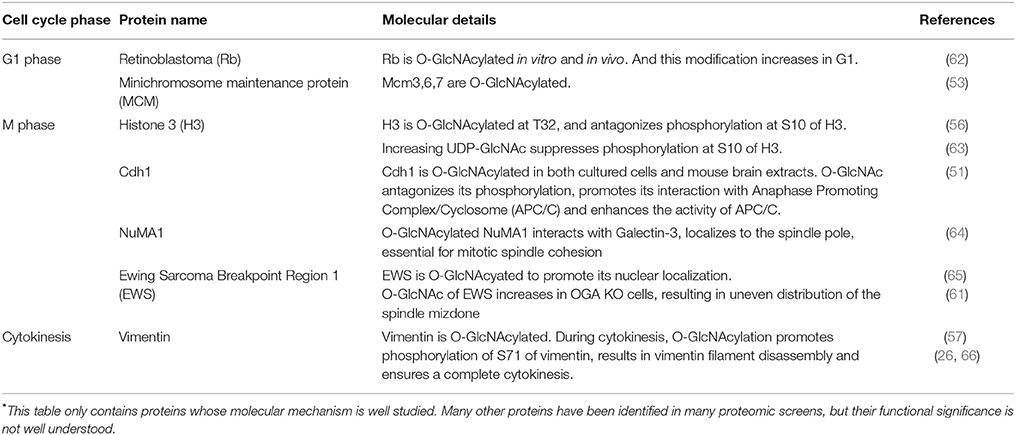
Table 2. O-GlcNAcylated proteins during the unperturbed cell cycle*.
An Overall Role of OGT/OGA in the Cell Cycle
Although a late comer to the cell cycle arena, there has been mounting evidence that OGT underscores cellular proliferation (67).O-GlcNAc levels fluctuate during distinct phases of the cell cycle. Upon entering the S phase, the OGA activity increases and the global O-GlcNAc levels decrease (53). During G2/M, reports have been incongruous. In X. laevis oocytes, global increase of O-GlcNAc is discernable during G2/M transition (55). In cultured mammalian cells, however, increased O-GlcNAc delays G2/M transition (54), and OGA disruption hinders G2/M transition (56). The discrepancy could be partly due to different species.
During mitosis, O-GlcNAc levels decrease (54, 57). OGA disruption results in lagging chromosomes and micronuclei (60), and stable OGA knockdown HeLa cell lines manifest aberrant spindles and mitotic exit effects (61). Meanwhile, OGT overproduction leads to chromosome bridges and delays mitosis (50). The mitotic spindle is an important apparatus to ensure accurate chromosome segregation (68), and its integrity impinges on the appropriate level of O-GlcNAcylation, as either OGT or OGA overproduction leads to aberrant mitotic spindles (69).
During cytokinesis, both OGA disruption and OGT overexpression lead to polyploidy and cytokinesis failure (26, 54, 60). Studies from our lab show that Chk1 phosphorylates OGT at S20 specifically during cytokinesis (26). This modification stabilizes OGT abundance and ensures a sufficient level of pS71 of vimentin (26). Thus OGT knockdown leads to vimentin bridges (26). In sum it is suffice to say that an appropriate O-GlcNAc level is pivotal for all phases. If chemical interrogation or genetic ablation renders O-GlcNAc above or below that level, cellular reproduction will be seriously affected.
Cytologically, OGT localizes to the spindle during M phase (58, 54). And it localizes to the midbody during cytokinesis (26, 54, 57). The localization pattern of OGA is quite different. At mitosis, OGA is ubiquitous, but absent from the newly formed nuclear membranes of the two daughter cells (54). During cytokinesis, OGA is diffused, without a distinct midbody localization (57).
O-GlcNAc and the Mitotic Master Kinases Are Intertwined
A link between O-GlcNAc and the mitotic master kinases have also been identified. The cell cycle is regulated by a concerted choreography of cyclins and CDKs (70). Investigations show that O-GlcNAc modulates cyclin stability. To begin with, cyclin D abundance increases during G1 and declines in S and M phases. OGA overproduction delays the increase of cyclin D (54), while OGT overproduction constitutively decreases cyclin D levels, partly due to delayed mitosis (54). Another example is cyclins A and B. Cyclin B peaks during prophase and decreases during metaphase, and cyclin A peaks in G2. In cells that overproduce OGT or OGA, protein abundance of cyclins A and B fails to decline, probably due to mitotic exit defects (54).
O-GlcNAc regulates two key mitotic kinases, CDK1 and Plk1 (Figure 1). CDK1 regulates Plk1 through Myosin phosphatase targeting protein 1 (MYPT1), which is a targeting subunit of protein phosphatase 1cβ(PP1cβ) (72). During mitosis, Cdk1 phosphorylates MYPT1 at S473, creating a binding pocket between MYPT1 and Plk1. Thus MYPT1 recruits PP1cβ to dephosphorylate Plk1 at pT210, the activation phosphorylation site (71). Intriguingly, MYPT1 is also a targeting subunit of OGT in neuroblastoma cells (73), raising the tantalizing possibilty that MYPT1 targets both PP1β and OGT to the same substrate, so that the substrate protein could be dephosphorylated and O-GlcNAcylated simultaneously. Indeed, Plk1 is identified to be O-GlcNAcylated in an in vitro OGT assay (74), but whether this occurs in vivo, or O-GlcNAcylation interplays with pT210 of Plk1 is elusive. To add yet another layer of complexity, MYPT1 itself is O-GlcNAcylated (73). We are yet to find out the exact function of this PTM.
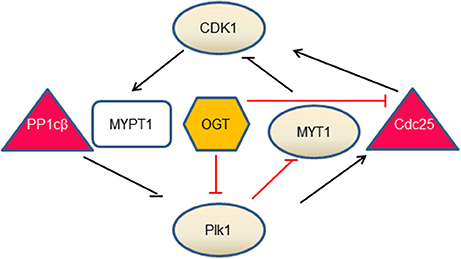
Figure 1. The interlinked network between O-GlcNAc and mitotic kinases. Our current understandings comprise three branches. During mitosis, CDK1 phosphorylates MYPT1 to promote its interaction with Plk1. MYPT1 recruits PP1cβ to dephosphorylate and inactivate Plk1 (71). In the second branch, OGT decreases Plk1 protein levels, which further increases MYT1 levels and decreases MYT1 phosphorylation. As MYT1 inhibits CDK1 by phosphorylation, OGT thus promotes inhibitory phosphorylation of CDK1. In the third branch, OGT inactivates CDK1, via down-regulating the mRNA levels of Cdc25-the activating phosphatase of CDK1 (50). Whether OGT is enmeshed in the MYPT1 branch is not understood. Kinases are in circles, phosphatases are in triangles, MYPT1 is in a square and OGT is in a hexagon. Lines in black denote direct effects (phosphorylation or dephosphorylation), and lines in red demarcate indirect effects (mRNA or protein levels).
Overproduction of OGT decreases mRNA levels of Plk1 and therefore Plk1 protein levels, which further decreases the abundance of Membrane Associated Tyrosine and Threonine cdc2 inhibitory Kinase (MYT1) and increases the activity of MYT1. As MYT1 phosphorylates CDK1 in an inhibitory manner, thus OGT overexpression increases the inhibitory phosphorylation of CDK1 (50). Another branch of the network concerns Cdc25, the activating phosphatase of CDK1. On one hand, Plk1 phosphorylates Cdc25, which in turn dephosphorylates and activates CDK1. On the other hand, OGT overproduction curbs mRNA levels of Cdc25 and subsequent protein levels, thus inactivating CDK1 (50). In sum, O-GlcNAc exerts its role through protein abundance at the transcriptional level. OGT overproduction increases the inhibitory phosphorylation of CDK1, and decreases phosphorylation levels of its substrates (50).
A third key kinase, Aurora B, is also extensively studied (75). Aurora B is a component of the chromosomal passenger complex (CPC) that also comprises INCENP, Survivin, and Borealin. CPC is an essential driver of the late mitotic events, including assembly and disassembly of the mitotic spindle, activation of the spindle assembly checkpoint and cytokinesis (75). Both OGT and OGA are in a complex with Aurora B and protein phosphatase 1 (PP1) during M phase by coIP studies (57). Moreover, chemical inhibition of Aurora B enhances OGT protein abundance and therefore cellular O-GlcNAcylation levels during M phase (57). Importantly, the spindle localization of OGT is derailed by Aurora B inhibition (57). It will be of particular interest to investigate whether Aurora B phosphorylates OGT or OGA in due course.
O-GlcNAcylated Proteins in the S Phase
The retinoblastoma protein (RB) is a classical tumor suppressor that is key for the replication process (76). In the G1 phase, Rb is hypophosphorylated and binds the transcription factor E2F-1, an essential transcription factor for many S phase genes. In the late G1 phase, Rb is hyperphosphorylated by CDKs, thus releasing E2F-1 to transcribe downstream S phase genes (76). Consequently, cells will enter S phase. Both in vitro and in vivo experiments show that Rb is O-GlcNAcylated, with the highest O-GlcNAc levels in the G1 phase, suggesting that O-GlcNAcylation of Rb antagonizes CDK-dependent phosphorylation and inactivates the transcriptional activities of E2F1 (62). It will be of further interest to pinpoint the modification residues and study its effects on cell cycle.
Conversely, the transcription levels of OGT and OGA are also regulated by Rb/E2F1 (77). OGT and OGA expression is reduced by overproduction of E2F1 in HEK293 cells and is increased in E2F1−/− mouse fibroblasts and Rb−/−mouse embryonic fibroblasts (MEFs). E2F1 overproduction does not change OGT or OGA expression levels in Rb−/−MEFs, suggesting that E2F1 negatively regulates OGT and OGA expression in an Rb-dependent manner (77). Mechanistically, consensus E2F binding sequences were identified on OGT and OGA and confirmed by reporter-based assays and in silico modeling (77). Therefore, expression of OGT or OGA is not constant, but subject to multiple layers of regulations.
The minichromosome maintenance (MCM) replicative helicase, comprising Mcm2-7, is loaded onto the DNA in the G1 phase, thus restricting one round of DNA replication per cell cycle (78). The Mcm2-7 is activated in S phase to unwind DNA. Investigations have shown that loading and activation never occur concomitantly, and not all MCMs loaded are activated in one cell cycle (79). Based on the recent cryoelectron microscopy (cryo-EM) data, an elaborate Acrobat Model has been proposed to elucidate how the MCMs are loaded to form a double hexamer (80). Phosphorylation (79), SUMOylation (81), and ubiquitination (82, 83) have been identified to distinct subunits of MCMs to regulate their recruitment, activation and disassembly. Interestingly, MCM3, 6, 7 are O-GlcNAcylated, with the alteration pattern of Mcm7 different from Mcm3 and 6 (53). The significance of this modification is still up in the air.
O-GlcNAcylated Proteins in Mitosis
All four core histones, H2A, H2B, H3, and H4 are found to be O-GlcNAcylated (63). By centrifugal elutriation, histone O-GlcNAcylation is found to be high in the G1 phase, decreases during the S phase and elevates during S/G2 transition (63). Glycosylated H3, specifically, is higher in interphase than mitosis (56). Histone H3 Ser10 phosphorylation (H3pS10), a landmark of mitosis, is interconnected with O-GlcNAcylation. Mechanistically, H3 is O-GlcNAcylated at T32, and O-GlcNAcylated H3 reduces H3pS10 (56). In general, overproduction of OGT increases H3K9Ac and H3K27me3 levels in mitosis, decreases H3pS10 levels in mitosis and decreases H3R17me2 levels in both asynchronous and mitotic cells (58).
Besides histones, our study about Fizzy-related protein homolog (Cdh1) provides another example of the yin-yang paradigm of O-GlcNAc. Cdh1 is the coactivator of the E3 ligase, anaphase promoting complex (APC), and is phosphorylated at multiple sites (84), with four significant phosphorylation residues at S40, T121, S151, and S163 suppressing the activity of APC (85). O-GlcNAc of Cdh1 has been identified in vitro (86). Our assays reveal that O-GlcNAcylation occurs at a peptide harboring S40, and O-GlcNAcylation antagonizes phosphorylation, thus activating APC (51).
The proto-oncoprotein Ewing Sarcoma Breakpoint Region 1 (EWS) binds with both RNA and DNA and localizes to the nucleus, cytosol and cell membranes. Upon adipogenic stimuli EWS is O-GlcNAcylated, which partly promotes its nuclear localization (65). In mitosis, EWS recruits Aurora B and CPC to the spindle midzone (87). In OGA-knockdown cells, O-GlcNAcylation of EWS elevates significantly, resulting in uneven distribution of the spindle mizdone (61).
In an extensive glycoproteomic and phosphoproteomics screen, 141 new O-GlcNAc sites were pinpointed on proteins involved in the spindle apparatus and cytokinesis (50). Many proteins involved in mitosis have also been identified to be O-GlcNAcylated, including the nuclear mitotic apparatus protein 1 (NuMA1), nuclear pore protein 153 (Nup153) and BRCA2-interacting transcriptional repressor EMSY (50). Follow-up investigations reveal that NuMA1 interacts with Galectin-3 in an O-GlcNAcylation-dependent manner (64). Galectin-3 is a small soluble lectin of the Galectin family that localizes to the centrosomes. The newly identified Galectin-3-O-GlcNAc-NuMA1 complex not only localizes NuMA1 to spindle poles, but also is essential for mitotic spindle cohesion (64).
Paradoxically, OGT itself is subject to O-GlcNAcylation (58, 88), which is completely abolished during mitosis, and reappears in G1 (58). It will be an intriguing possibility that O-GlcNAcylation of OGT itself promotes its own stability, so that its abundance declines when the modification drops. Further studies will surely provide more mechanistic insights of O-GlcNAc in mitosis.
O-GlcNAclated Proteins in Cytokinesis
Vimentin is an intermediate filament protein, and there have been a few reports on its O-GlcNAcylation. Vimentin is O-GlcNAcylated during M phase, and is also subject to phosphorylation by Cdk1, Plk1, Aurora B, and Rho kinase (21–25, 57). In vitro studies suggest that vimentin interacts with GlcNAc-bearing polymers, which promotes phosphorylation of vimentin S71 (66). Cellular assays suggest that vimentin pS71 levels increase when either OGT or OGA is overexpressed (57). Our studies show that when cells are synchronized in cytokinesis, O-GlcNAcylation of vimentin promotes pS71 (26). OGT knockdown by siRNA significantly attenuates vimentin pS71, thus forming elongated vimentin bridges during cytokinesis, and impeding daughter cell separation (26). Moreover, Chk1, a kinase pivotal for both DDR and cytokinesis, phosphorylates OGT at S20 specifically during cytokinesis. This phosphorylation is vital for OGT to localize to midbodies and maintain cellular O-GlcNAcylation levels (26). Thus, OGT-S20A mutant cells display vimentin bridge defects due to compromised O-GlcNAcylation levels and consequently quench pS71 of vimentin (26). It will be of keen interest to explore more O-GlcNAcylated proteins in cytokinesis.
O-GlcNAc in the DDR: a Guardian of the Genome
Our discussion on cell cycle events will not be complete without DDR. The genomic DNA is subject to many intrinsic or extrinsic lesions, such as double-strand breaks (DSBs), DNA crosslinking damage or DNA alkylation damage. Failure to repair these damages will lead to not only chromosomal abnormalities on the cellular level, but also fatal diseases such as cancer (89). Facing these genomic insults, organisms develop elaborate surveillance mechanisms-DNA damage checkpoints-to ensure genome integrity (90). The term “checkpoint” was coined by the Nobel Laureate, Dr. Leland Hartwell, almost three decades ago to describe the series of concerted responses to deleterious signals, including cell cycle arrest, transcription activation or suppression, and DNA repair. If the damage is beyond repair, then cells might undergo senescence or apoptosis (91).
In-depth investigations have identified an extensive network of proteins as guardians of our genomes to sense, mediate, and repair the damages (92). And these proteins are subject to phosphorylation, mono- or polyubiquitination, or SUMOylation. The very first step in the DDR pathway is the phosphorylation of H2AX (γH2AX) by a group of phosphatidylinositide-3 (PI-3)-like kinases, including ataxia telangiectasia mutated (ATM), ATM, and Rad3 related (ATR) and DNA-dependent protein kinase (DNA-PK) (92). Then γH2AX is recognized by checkpoint mediator proteins, such as mediator of DNA damage checkpoint protein 1 (MDC1). MDC1, along with other mediators, is phosphorylated by ATM, and the DDR signal is propagated downstream to transducer kinases, such as Chk1 and checkpoint kinase 2 (Chk2) (92), to execute homologous recombination (HR) or non-homologous end joining (93) and other repair pathways (92) (Figure 2).
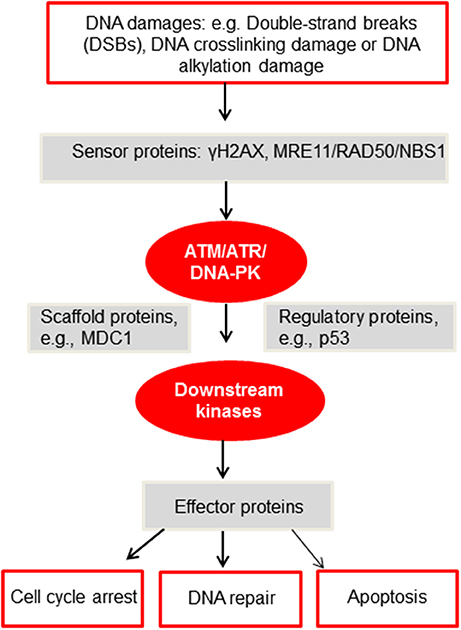
Figure 2. A simplified diagram of the DDR network adapted from Furgason and Bahassi (94). When genomic DNA is faced with various DNA damages, such as but not limited to DSBs, DNA crosslinking damage or alkylation damage, sensor proteins such as γH2AX and the MRE11/RAD50/NBS1 (MRN) complex will then recruit and activate the PIKK kinases, including ATM, ATR and DNA-PK. These PIKK kinases will then phosphorylate downstream targets, including scaffold proteins, regulatory proteins and downstream kinases. Upon activation, these proteins will subsequently phosphorylate effector proteins to undergo cell cycle arrest, DNA repair or apoptosis. For a detailed mechanistic analysis of the DDR, please refer to Ciccia and Elledge (92).
As O-GlcNAcylation is deemed as a stress signal, its effects on DNA damage have been of intense interest. As early as 2004, global O-GlcNAcylation levels were investigated after cells were irradiated by the ultraviolet (UV)-B light for 90 s, then recovered for 8 h. It was discovered that O-GlcNAc increased dose-dependently on the UV radiation durations (95). Then in 2015, a comprehensive proteomic study using OGT-null MEFs demonstrated that 232 phosphosites were upregulated compared with OGT wild-type cells (20), among which were ATM pS1987 and Chk1 pS317. Recently, O-GlcNAc was caught at the scene of crime. DNA damages increase O-GlcNAc levels at the damage sites, where both OGT and OGA are recruited (96). OGT abrogation reduces cell viability during DDR (96).
In spite of the significance of DDR, a search of “O-GlcNAc, DNA damage response” in the PubMed only yielded 12 publications. The 2015 quantitative phosphoproteomics study (20) thus provides us a timely route map to explore this uncharted territory. Herein we will review the O-GlcNAcylated DDR proteins, and more importantly, its underlying molecular underpinnings (Table 3).
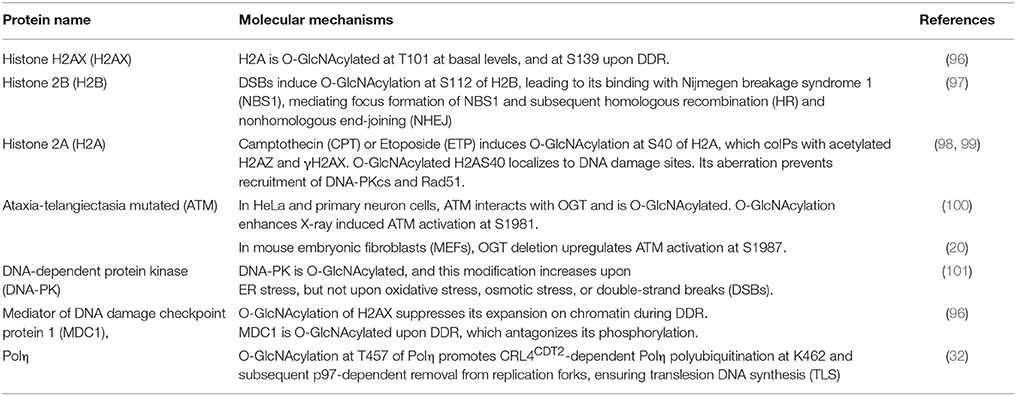
Table 3. O-GlcNAcylated proteins during the DNA damage response.
O-GlcNAcylation of Histones
Histone modification is integral to epigenetics, and OGT has been central to epigenetics (102). Here we will only cover histones involved in DDR, including H2AX, H2AS40, and H2B.
Histone H2A has many variants (103) and γH2AX is vital to DDR. H2AX is O-GlcNAcylated at T101 at basal levels and at S139 upon DDR, which restricts the expansion of damage signals, thus limiting DDR (96).
Viviparous animals have either Ser40 (S40) or Ala40 (A40) types of H2A, while non-mammals only have A40 (98, 99). Specific to viviparous organisms, Camptothecin (CPT) or Etoposide (ETP) (both inhibitors for Topoisomerase) induce O-GlcNAcylation at S40 of H2A (98, 99). This modification interacts with γH2AX, recruits DNA-PK and the recombinase Rad51 to the damage sites, and facilitates DDR (98, 99).
Besides H2AX and H2AS40, H2B is O-GlcNAcylated at S112 upon DSBs (97). Then the O-GlcNAcylated H2B binds with the Nijmegen breakage syndrome 1 (NBS1) protein, promoting its recruitment to DSBs and the following repairing events.
O-GlcNAc of Kinases and Scaffolds
Among the DDR proteins, the ATM, DNA-PK kinases, and the scaffold protein MDC1 have been reported to be O-GlcNAcylated. O-GlcNAc of DNA-PK is elevated upon ER stress, but not during oxidative, osmotic or DSB stresses (101). The mediator MDC1 is also O-GlcNAcylated, which counteracts ATM-dependent phosphorylation (96).
Reports about ATM have been controversial. Miura et al. (100) reported that in HeLa cells and primary cultured neurons ATM is O-GlcNAcylated (with CTD110.6) and endogenous ATM coIPs with OGT. O-GlcNAc of ATM enhances ATM activation at S1981 upon X-ray (100), thus O-GlcNAc plays a positive role. Meanwhile, Zhong et al. (20) failed to detect O-GlcNAc of ATM in MEFs. OGT deletion upregulates the activation phosphorylation of ATM at S1987 (the mouse equivalent of human S1981) (20), thus OGT plays a negative role. The discrepancy might lie in different species. Phosphorylation of S1981 has been a hallmark of ATM activation in humans, but mutation of S1987 to Ala does not impede ATM function in mice (104, 105). More in-depth studies are needed to solve the mystery.
The exact modification sites of these proteins were not investigated, probably because ATM, DNA-PK, and MDC1 are all large proteins. Fragmentation studies and precise site mapping will be needed in the future. And whether there are other distinctions between mice and humans, as far as O-GlcNAc is concerned, is at anyone's wild guess.
OGT in DNA Translesion Synthesis (TLS)
TLS utilizes a flurry of distinct and specialized DNA polymerases to replicate damaged regions of DNA (106). Polη is specifically involved in replicative bypass of cyclobutane pyrimidine dimers (CPDs) induced by UV, as well as damages induced by cisplatin (107). It is error prone, with an error rate of 10−2-10−3, thus it is vital to timely remove Polη after DDR.
A peptide of Polη was O-GlcNAcylated in vitro (86) and T457 was identified to be O-GlcNAcylated upon DDR in cultured cells (32). This modification promotes polyubiquitination at K462 and subsequent removal of Polη from replication forks. Moreover, O-GlcNAcylated Polη is essential for cisplatin-induced DDR. The deficient T457A mutant enhances UV-induced mutagenesis and also increases cellular sensitivity to cisplatin (32).
Collectively, the reports of OGT in DDR are emerging. From the current few investigations, O-GlcNAc seems to play an overall positive role: OGT is recruited to damaged sites, where it helps recruit NBS1; it antagonizes phosphorylated H2AX and MDC1 to restrict damage signal expansion; it promotes error-prone Polη to dissociate from chromatin after TLS to bypass UV-induced CPDs. Considering that DDR utilizes distinct pathways in response to each and every DNA damage type, we need more time and energy to elucidate how O-GlcNAc fine-tunes the DDR process.
Conclusions
In less than two score years, great strides have been made in the field of O-GlcNAcylation. We have witnessed really exciting things in glycobiology, and its utilization in medicine and clinical practice. As we have realized, most bedside applications will depend on our understandings of underlying molecular mechanisms to the ultimate level of resolution. Achieving that will not only enable us to comprehend various biological events, but also bestow upon us rich information as well as rich opportunities to be exploited. As we begin to elucidate the molecular basis of O-GlcNAc and its interaction, we are going to be able to leap profoundly forward into our future.
Author Contributions
All authors listed have made a substantial, direct and intellectual contribution to the work, and approved it for publication.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
This work was supported by the Capacity Building for Sci-Tech Innovation - Fundamental Scientific Research Funds (025185305000) to JL. We apologize to other colleagues whose work we cannot cite due to space limitations.
Abbreviations
(O-GlcNAc), O-linked N-acetylglucosamine; (OGT), O-GlcNAc transferase; OGA, O-GlcNAcase; DDR, DNA damage response; TLS, DNA translesion synthesis; ETD, electron transfer dissociation; MS, mass spectrometry; HCD, higher energy collisional dissociation; PTM, post-translational modification; IP, immunoprecipitate; IB, immunoblot; UV, ultraviolet; MEF, mouse embryonic fibroblast; DSB, double-strand break; ATM, ataxia telangiectasia mutated; ATR, ATM and Rad3 related; DNA-PK, and DNA-dependent protein kinase; MDC1, mediator of DNA damage checkpoint protein 1.
References
1. Torres CR, Hart GW. Topography and polypeptide distribution of terminal N-acetylglucosamine residues on the surfaces of intact lymphocytes. Evidence for O-linked GlcNAc. J Biol Chem. (1984) 259:3308–17.
2. Slawson C, Hart GW. O-GlcNAc signalling: implications for cancer cell biology. Nat Rev Cancer (2011) 11:678–84. doi: 10.1038/nrc3114
3. Hart GW, Slawson C, Ramirez-Correa G, Lagerlof O. Cross talk between O-GlcNAcylation and phosphorylation: roles in signaling, transcription, and chronic disease. Annu Rev Biochem. (2011) 80:825–58. doi: 10.1146/annurev-biochem-060608-102511
4. Levine ZG, Walker S. The Biochemistry of O-GlcNAc transferase: which functions make it essential in mammalian cells? Annu Rev Biochem. (2016) 85:631–57. doi: 10.1146/annurev-biochem-060713-035344
5. Yang X, Qian K. Protein O-GlcNAcylation: emerging mechanisms and functions. Nat Rev Mol Cell Biol. (2017) 18:452–65. doi: 10.1038/nrm.2017.22
6. Marshall S, Bacote V, Traxinger RR. Discovery of a metabolic pathway mediating glucose-induced desensitization of the glucose transport system. Role of hexosamine biosynthesis in the induction of insulin resistance. J Biol Chem. (1991) 266:4706–12.
7. Lucena MC, Carvalho-Cruz P, Donadio JL, Oliveira IA, De Queiroz RM, Marinho-Carvalho MM, et al. Epithelial mesenchymal transition induces aberrant glycosylation through hexosamine biosynthetic pathway activation. J Biol Chem. (2016) 291:12917–29. doi: 10.1074/jbc.M116.729236
8. Vasconcelos-Dos-Santos A, Oliveira IA, Lucena MC, Mantuano NR, Whelan SA, Dias WB, et al. Biosynthetic machinery involved in aberrant glycosylation: promising targets for developing of drugs against cancer. Front Oncol. (2015) 5:138. doi: 10.3389/fonc.2015.00138
9. Erickson JR, Pereira L, Wang L, Han G, Ferguson A, Dao K, et al. Diabetic hyperglycaemia activates CaMKII and arrhythmias by O-linked glycosylation. Nature (2013) 502:372–6. doi: 10.1038/nature12537
10. Alejandro EU, Bozadjieva N, Kumusoglu D, Abdulhamid S, Levine H, Haataja L, et al. Disruption of O-linked N-acetylglucosamine signaling induces ER stress and beta cell failure. Cell Rep. (2015) 13:2527–38. doi: 10.1016/j.celrep.2015.11.020
11. Yuzwa SA, Shan X, Macauley MS, Clark T, Skorobogatko Y, Vosseller K, et al. Increasing O-GlcNAc slows neurodegeneration and stabilizes tau against aggregation. Nat Chem Biol. (2012) 8:393–9. doi: 10.1038/nchembio.797
12. Ma Z, Vosseller K. Cancer metabolism and elevated O-GlcNAc in oncogenic signaling. J Biol Chem. (2014) 289:34457–65. doi: 10.1074/jbc.R114.577718
13. Buchakjian MR, Kornbluth S. The engine driving the ship: metabolic steering of cell proliferation and death. Nat Rev Mol Cell Biol. (2010) 11:715–27. doi: 10.1038/nrm2972
14. Yi W, Clark PM, Mason DE, Keenan MC, Hill C, Goddard WA, et al. Phosphofructokinase 1 glycosylation regulates cell growth and metabolism. Science (2012) 337:975–80. doi: 10.1126/science.1222278
15. Rao X, Duan X, Mao W, Li X, Li Z, Li Q, et al. O-GlcNAcylation of G6PD promotes the pentose phosphate pathway and tumor growth. Nat Commun. (2015) 6:8468. doi: 10.1038/ncomms9468
16. Wang T, Yu Q, Li J, Hu B, Zhao Q, Ma C, et al. O-GlcNAcylation of fumarase maintains tumour growth under glucose deficiency. Nat Cell Biol. (2017) 19:833–43. doi: 10.1038/ncb3562
17. Sodi VL, Bacigalupa ZA, Ferrer CM, Lee JV, Gocal WA, Mukhopadhyay D, et al. Nutrient sensor O-GlcNAc transferase controls cancer lipid metabolism via SREBP-1 regulation. Oncogene (2018) 37:924–34. doi: 10.1038/onc.2017.395
18. Groves JA, Maduka AO, O'meally RN, Cole RN, Zachara NE. Fatty acid synthase inhibits the O-GlcNAcase during oxidative stress. J Biol Chem. (2017). 292:6493–511. doi: 10.1074/jbc.M116.760785
19. Wang Z, Gucek M, Hart GW. Cross-talk between GlcNAcylation and phosphorylation: site-specific phosphorylation dynamics in response to globally elevated O-GlcNAc. Proc Natl Acad Sci USA. (2008) 105:13793–8. doi: 10.1073/pnas.0806216105
20. Zhong J, Martinez M, Sengupta S, Lee A, Wu X, Chaerkady R, et al. Quantitative phosphoproteomics reveals crosstalk between phosphorylation and O-GlcNAc in the DNA damage response pathway. Proteomics (2015) 15:591–607. doi: 10.1002/pmic.201400339
21. Tsujimura K, Ogawara M, Takeuchi Y, Imajoh-Ohmi S, Ha MH, Inagaki M. Visualization and function of vimentin phosphorylation by cdc2 kinase during mitosis. J Biol Chem. (1994) 269:31097–106.
22. Yamaguchi T, Goto H, Yokoyama T, Sillje H, Hanisch A, Uldschmid A, et al. Phosphorylation by Cdk1 induces Plk1-mediated vimentin phosphorylation during mitosis. J Cell Biol. (2005) 171:431–6. doi: 10.1083/jcb.200504091
23. Goto H, Yasui Y, Kawajiri A, Nigg EA, Terada Y, Tatsuka M, et al. Aurora-B regulates the cleavage furrow-specific vimentin phosphorylation in the cytokinetic process. J Biol Chem. (2003) 278:8526–30. doi: 10.1074/jbc.M210892200
24. Goto H, Kosako H, Tanabe K, Yanagida M, Sakurai M, Amano M, et al. Phosphorylation of vimentin by Rho-associated kinase at a unique amino-terminal site that is specifically phosphorylated during cytokinesis. J Biol Chem. (1998) 273:11728–36. doi: 10.1074/jbc.273.19.11728
25. Yasui Y, Goto H, Matsui S, Manser E, Lim L, Nagata K, et al. Protein kinases required for segregation of vimentin filaments in mitotic process. Oncogene (2001) 20:2868–76. doi: 10.1038/sj.onc.1204407
26. Li Z, Li X, Nai S, Geng Q, Liao J, Xu X, et al. Checkpoint kinase 1-induced phosphorylation of O-linked beta-N-acetylglucosamine transferase regulates the intermediate filament network during cytokinesis. J Biol Chem. (2017) 292:19548–55. doi: 10.1074/jbc.M117.811646
27. Qin W, Lv P, Fan X, Quan B, Zhu Y, Qin K, et al. Quantitative time-resolved chemoproteomics reveals that stable O-GlcNAc regulates box C/D snoRNP biogenesis. Proc Natl Acad Sci USA. (2017) 114:E6749–58. doi: 10.1073/pnas.1702688114
28. Li MD, Ruan HB, Hughes ME, Lee JS, Singh JP, Jones SP, et al. O-GlcNAc signaling entrains the circadian clock by inhibiting BMAL1/CLOCK ubiquitination. Cell Metab. (2013) 17:303–10. doi: 10.1016/j.cmet.2012.12.015
29. Ruan HB, Han X, Li MD, Singh JP, Qian K, Azarhoush S, et al. O-GlcNAc transferase/host cell factor C1 complex regulates gluconeogenesis by modulating PGC-1alpha stability. Cell Metab. (2012) 16:226–37. doi: 10.1016/j.cmet.2012.07.006
30. Ding X, Jiang W, Zhou P, Liu L, Wan X, Yuan X, et al. Mixed Lineage Leukemia 5 (MLL5) Protein Stability Is Cooperatively Regulated by O-GlcNac Transferase (OGT) and Ubiquitin Specific Protease 7 (USP7). PLoS ONE (2015) 10:e0145023. doi: 10.1371/journal.pone.0145023
31. Chu CS, Lo PW, Yeh YH, Hsu PH, Peng SH, Teng YC, et al. O-GlcNAcylation regulates EZH2 protein stability and function. Proc Natl Acad Sci USA. (2014) 111:1355–60. doi: 10.1073/pnas.1323226111
32. Ma X, Liu H, Li J, Wang Y, Ding YH, Shen H, et al. Poleta O-GlcNAcylation governs genome integrity during translesion DNA synthesis. Nat Commun. (2017) 8:1941. doi: 10.1038/s41467-017-02164-1
33. Ma J, Hart GW. O-GlcNAc profiling: from proteins to proteomes. Clin Proteomics (2014) 11:8. doi: 10.1186/1559-0275-11-8
34. Tashima Y, Stanley P. Antibodies that detect O-linked beta-D-N-acetylglucosamine on the extracellular domain of cell surface glycoproteins. J Biol Chem. (2014) 289:11132–42. doi: 10.1074/jbc.M113.492512
35. Ramakrishnan P, Clark PM, Mason DE, Peters EC, Hsieh-Wilson LC, Baltimore D. Activation of the transcriptional function of the NF-kappaB protein c-Rel by O-GlcNAc glycosylation. Sci Signal. (2013) 6:ra75. doi: 10.1126/scisignal.2004097
36. Zachara NE, Vosseller K, Hart GW. Detection and analysis of proteins modified by O-linked N-acetylglucosamine. Curr Protoc Mol Biol. (2011). Chapter 17:Unit 17.6 doi: 10.1002/0471142727.mb1706s95
37. Xu Q, Yang C, Du Y, Chen Y, Liu H, Deng M, et al. AMPK regulates histone H2B O-GlcNAcylation. Nucleic Acids Res. (2014) 42:5594–604. doi: 10.1093/nar/gku236
38. Banerjee PS, Hart GW, Cho JW. Chemical approaches to study O-GlcNAcylation. Chem Soc Rev. (2013) 42:4345–57. doi: 10.1039/C2CS35412H
39. Vosseller K, Wells L, Lane MD, Hart GW. Elevated nucleocytoplasmic glycosylation by O-GlcNAc results in insulin resistance associated with defects in Akt activation in 3T3-L1 adipocytes. Proc Natl Acad Sci USA. (2002) 99:5313–8. doi: 10.1073/pnas.072072399
40. Macauley MS, Shan X, Yuzwa SA, Gloster TM, Vocadlo DJ. Elevation of Global O-GlcNAc in rodents using a selective O-GlcNAcase inhibitor does not cause insulin resistance or perturb glucohomeostasis. Chem Biol (2010) 17:949–58. doi: 10.1016/j.chembiol.2010.07.005
41. Trapannone R, Rafie K, Van Aalten DM. O-GlcNAc transferase inhibitors: current tools and future challenges. Biochem Soc Trans. (2016) 44:88–93. doi: 10.1042/BST20150189
42. Liu Y, Ren Y, Cao Y, Huang H, Wu Q, Li W, et al. Discovery of a low toxicity O-GlcNAc transferase (OGT) inhibitor by structure-based virtual screening of natural products. Sci Rep. (2017) 7:12334. doi: 10.1038/s41598-017-12522-0
43. Gloster TM, Zandberg WF, Heinonen JE, Shen DL, Deng L, Vocadlo DJ. Hijacking a biosynthetic pathway yields a glycosyltransferase inhibitor within cells. Nat Chem Biol. (2011) 7:174–81. doi: 10.1038/nchembio.520
44. Sodi VL, Khaku S, Krutilina R, Schwab LP, Vocadlo DJ, Seagroves TN, et al. mTOR/MYC Axis Regulates O-GlcNAc Transferase Expression and O-GlcNAcylation in Breast Cancer. Mol Cancer Res. (2015) 13:923–33. doi: 10.1158/1541-7786.MCR-14-0536
45. Ma Z, Vocadlo DJ, Vosseller K. Hyper-O-GlcNAcylation is anti-apoptotic and maintains constitutive NF-kappaB activity in pancreatic cancer cells. J Biol Chem. (2013) 288:15121–30. doi: 10.1074/jbc.M113.470047
46. Perez-Cervera Y, Dehennaut V, Aquino Gil M, Guedri K, Solorzano Mata CJ, Olivier-Van Stichelen S, et al. Insulin signaling controls the expression of O-GlcNAc transferase and its interaction with lipid microdomains. FASEB J. (2013) 27:3478–86. doi: 10.1096/fj.12-217984
47. Syka JE, Coon JJ, Schroeder MJ, Shabanowitz J, Hunt DF. Peptide and protein sequence analysis by electron transfer dissociation mass spectrometry. Proc Natl Acad Sci USA. (2004) 101:9528–33. doi: 10.1073/pnas.0402700101
48. Schroeder MJ, Webb DJ, Shabanowitz J, Horwitz AF, Hunt DF. Methods for the detection of paxillin post-translational modifications and interacting proteins by mass spectrometry. J Proteome Res. (2005) 4:1832–41. doi: 10.1021/pr0502020
49. Chalkley RJ, Burlingame AL. Identification of GlcNAcylation sites of peptides and alpha-crystallin using Q-TOF mass spectrometry. J Am Soc Mass Spectrom. (2001) 12:1106–13. doi: 10.1016/S1044-0305(01)00295-1
50. Wang Z, Udeshi ND, Slawson C, Compton PD, Sakabe K, Cheung WD, et al. Extensive crosstalk between O-GlcNAcylation and phosphorylation regulates cytokinesis. Sci Signal. (2010) 3:ra2. doi: 10.1126/scisignal.2000526
51. Tian J, Geng Q, Ding Y, Liao J, Dong MQ, Xu X, et al. O-GlcNAcylation Antagonizes Phosphorylation of CDH1 (CDC20 Homologue 1). J Biol Chem. (2016) 291:12136–44. doi: 10.1074/jbc.M116.717850
52. Fardini Y, Perez-Cervera Y, Camoin L, Pagesy P, Lefebvre T, Issad T. Regulatory O-GlcNAcylation sites on FoxO1 are yet to be identified. Biochem Biophys Res Commun. (2015) 462:151–8. doi: 10.1016/j.bbrc.2015.04.114
53. Drougat L, Olivier-Van Stichelen S, Mortuaire M, Foulquier F, Lacoste AS, Michalski JC, et al. Characterization of O-GlcNAc cycling and proteomic identification of differentially O-GlcNAcylated proteins during G1/S transition. Biochim Biophys Acta (2012) 1820:1839–48. doi: 10.1016/j.bbagen.2012.08.024
54. Slawson C, Zachara NE, Vosseller K, Cheung WD, Lane MD, Hart GW. Perturbations in O-linked beta-N-acetylglucosamine protein modification cause severe defects in mitotic progression and cytokinesis. J Biol Chem. (2005) 280:32944–56. doi: 10.1074/jbc.M503396200
55. Lefebvre T, Baert F, Bodart JF, Flament S, Michalski JC, Vilain JP. Modulation of O-GlcNAc glycosylation during Xenopus oocyte maturation. J Cell Biochem. (2004) 93:999–1010. doi: 10.1002/jcb.20242
56. Fong JJ, Nguyen BL, Bridger R, Medrano EE, Wells L, Pan S, et al. beta-N-Acetylglucosamine (O-GlcNAc) is a novel regulator of mitosis-specific phosphorylations on histone H3. J Biol Chem. (2012) 287:12195–203. doi: 10.1074/jbc.M111.315804
57. Slawson C, Lakshmanan T, Knapp S, Hart GW. A mitotic GlcNAcylation/phosphorylation signaling complex alters the posttranslational state of the cytoskeletal protein vimentin. Mol Biol Cell (2008) 19:4130–40. doi: 10.1091/mbc.e07-11-1146
58. Sakabe K, Hart GW. O-GlcNAc transferase regulates mitotic chromatin dynamics. J Biol Chem. (2010) 285:34460–8. doi: 10.1074/jbc.M110.158170
59. Whitfield ML, Sherlock G, Saldanha AJ, Murray JI, Ball CA, Alexander KE, et al. Identification of genes periodically expressed in the human cell cycle and their expression in tumors. Mol Biol Cell (2002) 13:1977–2000. doi: 10.1091/mbc.02-02-0030
60. Yang YR, Song M, Lee H, Jeon Y, Choi EJ, Jang HJ, et al. O-GlcNAcase is essential for embryonic development and maintenance of genomic stability. Aging Cell (2012) 11:439–48. doi: 10.1111/j.1474-9726.2012.00801.x
61. Lanza C, Tan EP, Zhang Z, Machacek M, Brinker AE, Azuma M, et al. Reduced O-GlcNAcase expression promotes mitotic errors and spindle defects. Cell Cycle (2016) 15:1363–75. doi: 10.1080/15384101.2016.1167297
62. Wells L, Slawson C, Hart GW. The E2F-1 associated retinoblastoma-susceptibility gene product is modified by O-GlcNAc. Amino Acids (2011) 40:877–83. doi: 10.1007/s00726-010-0709-x
63. Zhang S, Roche K, Nasheuer HP, Lowndes NF. Modification of histones by sugar beta-N-acetylglucosamine (GlcNAc) occurs on multiple residues, including histone H3 serine 10, and is cell cycle-regulated. J Biol Chem. (2011) 286:37483–95. doi: 10.1074/jbc.M111.284885
64. Magescas J, Sengmanivong L, Viau A, Mayeux A, Dang T, Burtin M, et al. Spindle pole cohesion requires glycosylation-mediated localization of NuMA. Sci Rep. (2017) 7:1474. doi: 10.1038/s41598-017-01614-6
65. Li Q, Kamemura K. Adipogenesis stimulates the nuclear localization of EWS with an increase in its O-GlcNAc glycosylation in 3T3-L1 cells. Biochem Biophys Res Commun. (2014) 450:588–92. doi: 10.1016/j.bbrc.2014.06.013
66. Komura K, Ise H, Akaike T. Dynamic behaviors of vimentin induced by interaction with GlcNAc molecules. Glycobiology (2012) 22:1741–59. doi: 10.1093/glycob/cws118
67. Tan EP, Duncan FE, Slawson C. The sweet side of the cell cycle. Biochem Soc Trans (2017) 45:313–22. doi: 10.1042/BST20160145
68. Prosser SL, Pelletier L. Mitotic spindle assembly in animal cells: a fine balancing act. Nat Rev Mol Cell Biol. (2017) 18:187–201. doi: 10.1038/nrm.2016.162
69. Tan EP, Caro S, Potnis A, Lanza C, Slawson C. O-linked N-acetylglucosamine cycling regulates mitotic spindle organization. J Biol Chem. (2013) 288:27085–99. doi: 10.1074/jbc.M113.470187
70. Guardavaccaro D, Pagano M. Stabilizers and destabilizers controlling cell cycle oscillators. Mol Cell (2006) 22:1–4. doi: 10.1016/j.molcel.2006.03.017
71. Yamashiro S, Yamakita Y, Totsukawa G, Goto H, Kaibuchi K, Ito M, et al. Myosin phosphatase-targeting subunit 1 regulates mitosis by antagonizing polo-like kinase 1. Dev Cell (2008) 14:787–97. doi: 10.1016/j.devcel.2008.02.013
72. Ito M, Nakano T, Erdodi F, Hartshorne DJ. Myosin phosphatase: structure, regulation and function. Mol Cell Biochem. (2004) 259:197–209. doi: 10.1023/B:MCBI.0000021373.14288.00
73. Cheung WD, Sakabe K, Housley MP, Dias WB, Hart GW. O-linked beta-N-acetylglucosaminyltransferase substrate specificity is regulated by myosin phosphatase targeting and other interacting proteins. J Biol Chem. (2008) 283:33935–41. doi: 10.1074/jbc.M806199200
74. Dias WB, Cheung WD, Hart GW. O-GlcNAcylation of kinases. Biochem Biophys Res Commun. (2012) 422:224–8. doi: 10.1016/j.bbrc.2012.04.124
75. Carmena M, Wheelock M, Funabiki H, Earnshaw WC. The chromosomal passenger complex (CPC): from easy rider to the godfather of mitosis. Nat Rev Mol Cell Biol. (2013) 13:789–803. doi: 10.1038/nrm3474
76. Fischer M, Muller GA. Cell cycle transcription control: DREAM/MuvB and RB-E2F complexes. Crit Rev Biochem Mol Biol. (2017) 52:638–62. doi: 10.1080/10409238.2017.1360836
77. Muthusamy S, Hong KU, Dassanayaka S, Hamid T, Jones SP. E2F1 Transcription Factor Regulates O-linked N-acetylglucosamine (O-GlcNAc) Transferase and O-GlcNAcase Expression. J Biol Chem. (2015) 290:31013–24. doi: 10.1074/jbc.M115.677534
78. Bochman ML, Schwacha A. The Mcm complex: unwinding the mechanism of a replicative helicase. Microbiol Mol Biol Rev. (2009) 73:652–83. doi: 10.1128/MMBR.00019-09
79. Deegan TD, Diffley JF. MCM: one ring to rule them all. Curr Opin Struct Biol. (2016) 37:145–51. doi: 10.1016/j.sbi.2016.01.014
80. Zhai Y, Li N, Jiang H, Huang X, Gao N, Tye BK. Unique Roles of the Non-identical MCM Subunits in DNA replication licensing. Mol Cell (2017) 67:168–79. doi: 10.1016/j.molcel.2017.06.016
81. Wei L, Zhao X. A new MCM modification cycle regulates DNA replication initiation. Nat Struct Mol Biol. (2016) 23:209–16. doi: 10.1038/nsmb.3173
82. Moreno SP, Bailey R, Campion N, Herron S, Gambus A. Polyubiquitylation drives replisome disassembly at the termination of DNA replication. Science (2014) 346:477–81. doi: 10.1126/science.1253585
83. Maric M, Maculins T, De Piccoli G, Labib K. Cdc48 and a ubiquitin ligase drive disassembly of the CMG helicase at the end of DNA replication. Science (2014) 346:1253596. doi: 10.1126/science.1253596
84. Kramer ER, Scheuringer N, Podtelejnikov AV, Mann M, Peters JM. Mitotic regulation of the APC activator proteins CDC20 and CDH1. Mol Biol Cell (2000) 11:1555–69. doi: 10.1091/mbc.11.5.1555
85. Lukas C, Sorensen CS, Kramer E, Santoni-Rugiu E, Lindeneg C, Peters JM, et al. Accumulation of cyclin B1 requires E2F and cyclin-A-dependent rearrangement of the anaphase-promoting complex. Nature (1999) 401:815–8. doi: 10.1038/44611
86. Pathak S, Alonso J, Schimpl M, Rafie K, Blair DE, Borodkin VS, et al. The active site of O-GlcNAc transferase imposes constraints on substrate sequence. Nat Struct Mol Biol. (2015) 22:744–50. doi: 10.1038/nsmb.3063
87. Park H, Turkalo TK, Nelson K, Folmsbee SS, Robb C, Roper B, et al. Ewing sarcoma EWS protein regulates midzone formation by recruiting Aurora B kinase to the midzone. Cell Cycle (2014) 13:2391–9. doi: 10.4161/cc.29337
88. Kreppel LK, Blomberg MA, Hart GW. Dynamic glycosylation of nuclear and cytosolic proteins. Cloning and characterization of a unique O-GlcNAc transferase with multiple tetratricopeptide repeats. J Biol Chem. (1997) 272:9308–15. doi: 10.1074/jbc.272.14.9308
89. Finn K, Lowndes NF, Grenon M. Eukaryotic DNA damage checkpoint activation in response to double-strand breaks. Cell Mol Life Sci. (2012) 69:1447–73. doi: 10.1007/s00018-011-0875-3
90. Hartwell LH, Weinert TA. Checkpoints: controls that ensure the order of cell cycle events. Science (1989) 246:629–34. doi: 10.1126/science.2683079
91. Harper JW, Elledge SJ. The DNA damage response: ten years after. Mol Cell (2007) 28:739–45. doi: 10.1016/j.molcel.2007.11.015
92. Ciccia A, Elledge SJ. The DNA damage response: making it safe to play with knives. Mol Cell (2010) 40:179–204. doi: 10.1016/j.molcel.2010.09.019
93. Li J, Xu X. DNA double-strand break repair: a tale of pathway choices. Acta Biochim Biophys Sin. (2016) 48:641–6. doi: 10.1093/abbs/gmw045
94. Furgason JM, Bahassi El M. Targeting DNA repair mechanisms in cancer. Pharmacol Ther. (2013) 137:298–308. doi: 10.1016/j.pharmthera.2012.10.009
95. Zachara NE, O'donnell N, Cheung WD, Mercer JJ, Marth JD, Hart GW. Dynamic O-GlcNAc modification of nucleocytoplasmic proteins in response to stress. A survival response of mammalian cells. J Biol Chem. (2004) 279:30133–42. doi: 10.1074/jbc.M403773200
96. Chen Q, Yu X. OGT restrains the expansion of DNA damage signaling. Nucleic Acids Res. (2016) 44:9266–78. doi: 10.1093/nar/gkw663
97. Wang P, Peng C, Liu X, Liu H, Chen Y, Zheng L, et al. OGT mediated histone H2B S112 GlcNAcylation regulates DNA damage response. J Genet Genomics (2015) 42:467–75. doi: 10.1016/j.jgg.2015.07.002
98. Hayakawa K, Hirosawa M, Tani R, Yoneda C, Tanaka S, Shiota K. H2A O-GlcNAcylation at serine 40 functions genomic protection in association with acetylated H2AZ or gammaH2AX. Epigenetics Chromatin (2017) 10:51. doi: 10.1186/s13072-017-0157-x
99. Hirosawa M, Hayakawa K, Yoneda C, Arai D, Shiota H, Suzuki T, et al. Novel O-GlcNAcylation on Ser(40) of canonical H2A isoforms specific to viviparity. Sci Rep. (2016) 6:31785. doi: 10.1038/srep31785
100. Miura Y, Sakurai Y, Endo T. O-GlcNAc modification affects the ATM-mediated DNA damage response. Biochim Biophys Acta (2012) 1820:1678–85. doi: 10.1016/j.bbagen.2012.06.013
101. Zachara NE, Molina H, Wong KY, Pandey A, Hart GW. The dynamic stress-induced “O-GlcNAc-ome” highlights functions for O-GlcNAc in regulating DNA damage/repair and other cellular pathways. Amino Acids (2011) 40:793–808. doi: 10.1007/s00726-010-0695-z
102. Lewis BA, Hanover JA. O-GlcNAc and the epigenetic regulation of gene expression. J Biol Chem. (2014) 289:34440–8. doi: 10.1074/jbc.R114.595439
103. Vardabasso C, Hasson D, Ratnakumar K, Chung CY, Duarte LF, Bernstein E. Histone variants: emerging players in cancer biology. Cell Mol Life Sci. (2014) 71:379–404. doi: 10.1007/s00018-013-1343-z
104. Pellegrini M, Celeste A, Difilippantonio S, Guo R, Wang W, Feigenbaum L, et al. Autophosphorylation at serine 1987 is dispensable for murine Atm activation >in vivo. Nature (2006) 443:222–5. doi: 10.1038/nature05112
105. Shiloh Y, Ziv Y. The ATM protein kinase: regulating the cellular response to genotoxic stress, and more. Nat Rev Mol Cell Biol. (2013) 14:197–210. doi: 10.1038/nrm3546
106. Sale JE. Translesion DNA synthesis and mutagenesis in eukaryotes. Cold Spring Harb Perspect Biol. (2013) 5:a012708. doi: 10.1101/cshperspect.a012708
Keywords: O-GlcNAc, mitosis, replication, cytokinesis, DNA damage response
Citation: Liu C and Li J (2018) O-GlcNAc: A Sweetheart of the Cell Cycle and DNA Damage Response. Front. Endocrinol. 9:415. doi: 10.3389/fendo.2018.00415
Received: 20 April 2018; Accepted: 02 July 2018;
Published: 30 July 2018.
Edited by:
Tony Lefebvre, Lille University of Science and Technology, FranceReviewed by:
Anne-Sophie Edouart, Centre National de la Recherche Scientifique (CNRS), FranceHai-Bin Ruan, University of Minnesota Twin Cities, United States
Chad Slawson, Kansas University of Medical Center Research Institute, United States
Copyright © 2018 Liu and Li. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jing Li, amluZ19saUBtYWlsLmNudS5lZHUuY24=