 Ramiro Alsina
Ramiro Alsina Milagros Trotta
Milagros Trotta Viviana Florencia Bumaschny
Viviana Florencia Bumaschny- 1Universidad de Buenos Aires, CONICET, Instituto de Fisiología y Biofísica “Bernardo Houssay” (IFIBIO), Grupo de Neurociencia de Sistemas, Buenos Aires, Argentina
- 2Universidad de Buenos Aires, Facultad de Medicina, Departamento de Ciencias Fisiológicas, Buenos Aires, Argentina
The arcuate nucleus of the hypothalamus is a key regulator of energy balance and glucose homeostasis. In particular, arcuate proopiomelanocortin (POMC) neurons inhibit food intake, stimulate energy expenditure and increase glucose tolerance. The interruption of insulin or glucose signaling in POMC neurons leads to glucose intolerance without changing energy homeostasis. Although it was previously shown that POMC neurons are necessary for normal glucose homeostasis, the participation of POMC neuropeptide, by mechanisms independent of energy balance, remains to be demonstrated. To study the role of POMC in the regulation of glucose homeostasis, we performed glucose and insulin tolerance tests in non-obese mice lacking hypothalamic POMC expression. We found that POMC deficiency leads to glucose intolerance and insulin resistance in female mice before the onset of obesity or hyperphagia. Conversely, POMC deficiency does not impair glucose homeostasis in non-obese male mice. Interestingly, females completely normalize both glucose and insulin tolerance after genetic POMC restoration. Next, to further study sex dimorphism of POMC neurons regarding glucose homeostasis, we measured glucose-elicited changes in C-FOS by performing immunofluorescence in brain slices of POMC-EGFP mice. Remarkably, we found that glucose-induced C-FOS expression in POMC neurons is more than 3-fold higher in female than in male mice. Altogether, our results reveal a key role of arcuate POMC in the regulation of glucose homeostasis in females. Since POMC reactivation completely reverses the diabetogenic phenotype, arcuate POMC could be a potential target for diabetes therapy.
Introduction
Diabetes mellitus is a condition affecting 422 million people all over the world (1). Type 2 diabetes is the predominant type, and it mainly results from excess body weight and physical inactivity (1). In obese patients and mice, type 2 diabetes greatly improves or even reverts after mild body weight loss (2–4). It is accepted that the improvement in glucose homeostasis is a consequence of fat mass reduction, especially in the liver. However, since the hypothalamus is a key regulator of both glucose and energy homeostasis, hypothalamic mechanisms may also be implicated in the anti-diabetic consequences of losing weight (5, 6).
Within the arcuate nucleus of the hypothalamus, proopiomelanocortin (POMC) neurons sense the energy status of the organism integrating peripheral signals such as leptin, glucose and insulin, among others (7). In turn, POMC neurons coordinate responses to maintain energy balance and glucose homeostasis. Since melanocyte-stimulating hormones (α- and β-MSH) derived from POMC peptide are anorexigenic, POMC deficient patients and mice are extremely obese (8, 9).
Regarding the role of POMC neurons in glucose homeostasis, some studies using electrophysiology on brain slices showed that POMC neuron activity can be modulated by changes in extracellular glucose levels (10–12). Moreover, deleting components of the insulin, leptin or glucose signaling cascades in POMC neurons impairs glucose tolerance and insulin sensitivity in genetically engineered mice (10, 12, 13). In addition, glucose induces both Pomc mRNA expression as well as α-MSH release from hypothalamic neurons (12, 14). Although the role of POMC neurons in glucose homeostasis has been well established, it remains to be elucidated if hypothalamic POMC peptide is directly involved, especially considering that these neurons may also co-secrete glutamate, gamma-aminobutyric acid and Cocaine and Amphetamine Regulated Transcript (15).
We have previously shown that hypothalamic POMC deficiency leads both to obesity and type 2 diabetes (2). Interestingly, restoration of POMC expression in extremely obese POMC-deficient mice induces partial body weight loss but complete normalization of glycemia (2). These results suggest that the reestablishment of glucose homeostasis may not only be a consequence of losing weight but also of Pomc restoration itself, which would imply a direct protective role of POMC against type 2 diabetes. In the present work, in order to test the role of POMC in glucose homeostasis independently of its role in body weight maintenance, we determine glucose tolerance and insulin sensitivity in non-obese POMC-deficient mice.
Materials and Methods
Animal Care
Mice were kept under standard laboratory conditions, with controlled photoperiod (lights on from 7 a.m. to 7 p.m.), tap water and standard lab chow available ad libitum. Mice were weaned at P21. All procedures were approved by the Institutional Animal Care and Use Committee of the School of Medicine, University of Buenos Aires.
Mouse Lines
arcPomc−/− [Figure 1A; (2)], POMC-EGFP (16) and Cre:ERT [B6.Cg-Tg(cre/Esr1)5Amc/J] (17) mice were kindly provided by Marcelo Rubinstein, INGEBI-CONICET and bred as previously described (2).
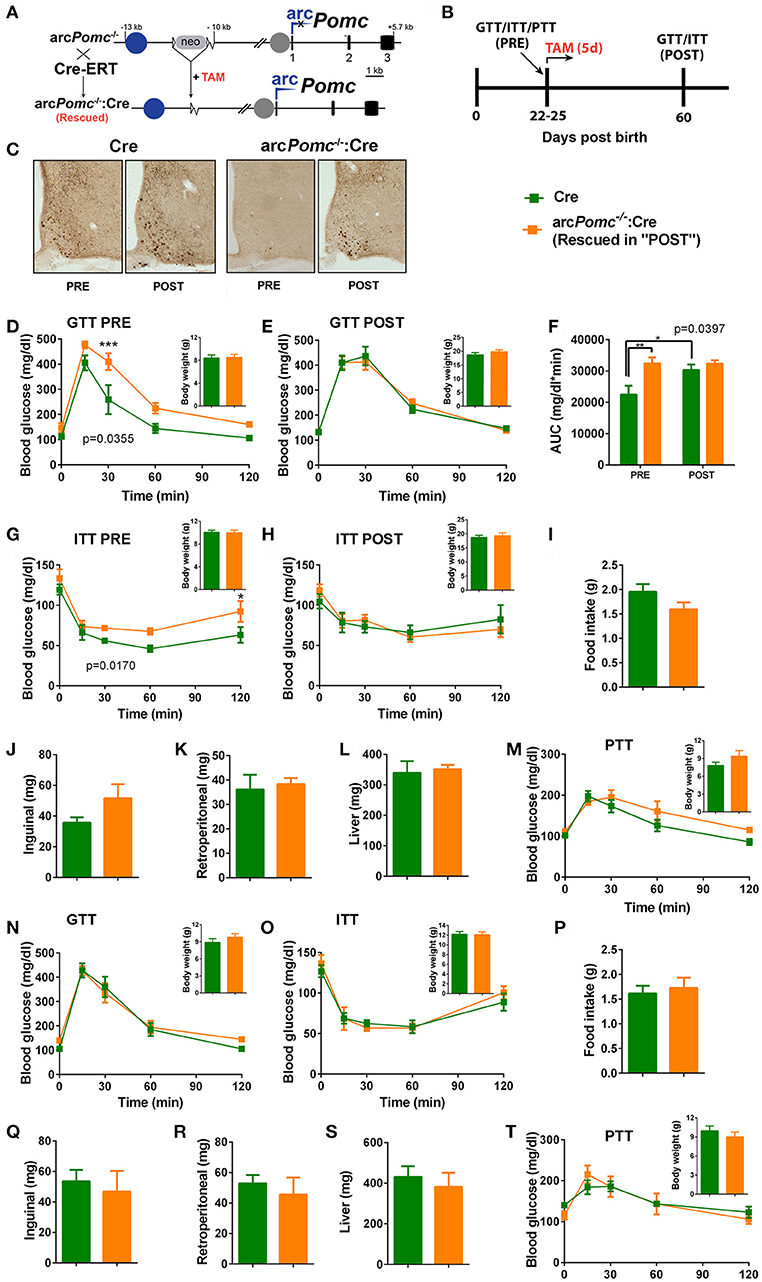
Figure 1. Pomc regulation of glucose homeostasis in a mouse model of reversible hypothalamic Pomc deficiency. (A) A reversible arcuate Pomc deficient mouse line (arcPomc−/−) was intercrossed with Cre-ERT line to restore Pomc expression. arcPomc−/−contains an insertion of a neomycin resistance cassette (neo), flanked by loxP sites (triangles), interrupting Pomc neuronal enhancer activity (Blue circle: nPE1 enhancer; gap after neo: Deleted nPE2 enhancer). Gray circle: intact Pomc pituitary promoter. arc: arcuate Pomc transcription. Black rectangles: Pomc exons. Pomc expression can be restored by treating arcPomc−/−:Cre mice with tamoxifen (TAM). (B) General experimental design. Mice are weaned at P21 and subjected either to Glucose, Insulin or Pyruvate Tolerance Tests (GTT, ITT, or PTT) at P22-P25 (PRE). Mice are treated with TAM for five consecutive days immediately after the first GTT or ITT. At P60, GTT or ITT was repeated (POST). (C) Examples of brain coronal sections subjected to immunohistochemistry for POMC in non-treated mice (PRE) and mice treated with TAM (POST). Note that arcPomc−/−:Cre mice only show POMC immune-positive neurons after POMC restoration by TAM treatment. Sections correspond to P60-P80 mice. (D-E) GTTs and body weights (insets) of females before (PRE) or after (POST) POMC restoration. p: genotype effect of RMA. **p < 0.01 (Bonferroni). n = 7–8. (F) Area under the curves (AUC) of (D,E). p: genotype effect of RMA. *p < 0.05; **p < 0.01 (Bonferroni). (G,H) ITTs and body weights of females before (PRE) or after (POST) POMC restoration. p: genotype effect of RMA. *p < 0.05 (Bonferroni). n = 11. (I) Daily food intake of female mice measured from P21 to P25. T-test, p > 0.05. n = 8–11. (J–L) Weights of retroperitoneal and inguinal fat pads, and livers of female mice at P25. T-test, p > 0.05. n = 6. (M) PTTs and body weights of females before POMC restoration. RMA: genotype and genotype x time effects, p > 0.05. n = 5–6. (N,O) GTT and ITT of weanling male mice, respectively. RMA, genotype and genotype x time effect: p > 0.05. Insets: body weights. n = 6–8 (GTT) and n = 7 (ITT). (P) Daily food intake of male mice from P21 to P25. n = 10–12. T-test, p > 0.05. (Q–S) Weights of retroperitoneal and inguinal fat pads, and livers of male mice at P25. T-test, p>0.05. n = 5–6. (T) PTTs and body weights of males before POMC restoration. RMA: genotype and genotype x time effects, p > 0.05. n = 5–6. In all graphs, error bars correspond to ±SEM.
Glucose, Insulin and Pyruvate Tolerance Tests (GTT, ITT and PTT)
Three cohorts of postnatal day 22–25 (P22-25) mice were subjected to GTT, ITT, or PTT (Figure 1B). To avoid excessive weight loss in weanling mice, GTTs and PTTs were performed following 5-h fasting (8 a.m. to 1 p.m.), as suggested (18), and received an i.p injection of glucose (2 g/kg; Sigma) or sodium pyruvate (2 g/kg; Anedra). For ITTs mice were fasted for 2 h (8 a.m. to 10 a.m.) and i.p injected with human insulin (Humulin R; 1 U/kg; Lilly). Blood samples from the tail tip were taken to measure glucose with a One Touch® glucometer (LifeScan, Johnson & Johnson), before and after injections, as described previously (19). Immediately after GTT or ITT, mice were treated with tamoxifen and re-tested at P60.
CRE Induction by Tamoxifen Administration
Mice were injected i.p. with 50 mg/kg/day tamoxifen (Sigma) during five consecutive days, with a solution of 5 mg tamoxifen/ml of semsame oil (Sigma) prepared as described (20).
Food Intake and Fat Determination
Another cohort of individually housed arcPomc−/−:Cre and Cre mice was used for food intake assessment from P21 to P25. After that, mice were euthanized by cervical dislocation and unilateral subcutaneous (inguinal) and visceral (retroperitoneal) fat pads, as well as livers, were dissected and weighed.
Immunohistochemistry
Mice were anesthetized with 5% chloral hydrate, perfused with 4% paraformaldehyde and brains were cut into 35 μm coronal sections with a frozen microtome (Leica). Hypothalamic POMC reactivation was confirmed by immunohistochemistry (Figure 1C) using a rabbit polyclonal anti rat-ACTH antibody (1:1,000, A.F. Parlow, National Hormone and Peptide Program, Harbor-UCLA Medical Center) and developed with diaminobenzidine (Vector Labs) as previously described (2). For C-FOS detection, P22-P25 POMC-EGFP mice were fasted from 8 a.m. to 1 p.m., and then i.p injected either with glucose (2 g/kg) or saline. 90-120 min later brains were processed as stated above, immunostained for C-FOS (rabbit anti C-FOS, Merck, 1:1,000) and developed with anti-rabbit-Cy3 antisera (Jackson ImmunoResearch, 1:500). Double immunohistochemistry: (1) mouse monoclonal IgG2a anti C-FOS (Santa Cruz Biotechnologies, 1:4,000) followed by anti-mouse IgG2a-Alexa Fluor 555 antisera (ThermoFisher, 1:1,000); (2) rabbit anti ER alpha antibody (Millipore, 1:10,000) followed by biotinylated anti-rabbit antisera (Vector Labs, 1:200) and developed by streptavidin-Alexa Fluor 647 (Jackson ImmunoResearch, 1:15,000). No staining was performed to visualize POMC neurons since POMC-EGFP mice express EGFP in this neuronal population. Micrographs were taken with an AxioImager M2 motorized fluorescent microscope with Apotome2 structured illumination (Zeiss). Positive neurons were counted using Image J software (21).
Statistical Analysis
All data are presented as the mean ± SEM and were analyzed by Student's unpaired two-tailed t-test, one or two way ANOVA (OWA, TWA) or repeated measures ANOVA (RMA), using GraphPad Prism version 6.00 for Windows (GraphPAd Software, La Jolla California, United States). Post hoc Bonferroni's test was used when necessary. P < 0.05 was considered significant. After RMA, significant Genotype Effect was only taken into account when Interaction (Genotype x Time) was not significant. The total area under the curve (AUC) was calculated using the trapezoidal rule.
Results
With the aim of studying the role of POMC in glucose homeostasis, we used an arcuate specific Pomc knockout mouse model (arcPomc−/−) (2). These mice bare a floxed neomycine-resistance gene (Neo) immediately downstream of the hypothalamic Pomc neuronal enhancer module (Figure 1A). Neo cassette prevents arcuate Pomc expression while preserving transcription in the nucleus of the solitary tract and the pituitary gland, avoiding corticosterone insufficiency (2).
Arcuate Pomc deficiency leads to obesity the fifth week after birth, which predisposes to Type 2 Diabetes in adult mice (2). Thus, in order to dissect the role of POMC in the regulation of glucose homeostasis independently of obesity mechanisms, we performed GTTs in arcPomc−/− mice immediately after weaning, while animals still have normal body weights. Interestingly, we found that despite no significant differences in basal glycemia, female arcPomc−/− are less tolerant to a glucose overload than WT littermates [RMA, genotype effect: F(1, 9) = 5.992, p = 0.0369, n = 5–6 per group].
To further confirm that hypothalamic POMC prevents glucose intolerance, we restored eutopic POMC expression by crossing arcPomc−/− mice with a tamoxifen inducible Cre mouse line (Figure 1A). We have previously shown that POMC recovery at P25 completely prevents hyperphagia and obesity in arcPomc−/−:Cre mice (2). Like arcPomc−/−, arcPomc−/−:Cre female mice showed decreased glucose tolerance despite normal body weight (Figure 1D). However, glucose tolerance is completely normalized in arcPomc−/−:Cre after POMC restoration, which further suggests a protective role of POMC against diabetes (Figures 1E,F).
In order to address if glucose tolerance impairment triggered by POMC deficiency is caused by decreased insulin sensitivity, another cohort of arcPomc−/−:Cre and Cre female mice were subjected to an ITT. Interestingly, non-obese arcPomc−/−:Cre females show decreased insulin sensitivity that was normalized by POMC restoration (Figures 1G,H). Glucose intolerance and insulin resistance of arcPomc−/−:Cre females before POMC restoration are not caused by increased inguinal, retroperitoneal or liver fat stores, nor food intake, because no significant differences were found when compared to Cre control mice (Figures 1I–L). Altogether, these results suggest that POMC prevents glucose intolerance by improving insulin sensitivity through mechanisms not related to energy balance regulation or fat storage.
Although it has been previously shown that hypothalamic α-MSH enhances insulin inhibition of hepatic gluconeogenesis in rats (22), we found no significant differences in PTTs between female arcPomc−/−:Cre and Cre littermates (Figures 1M).
Contrary to females, non-obese POMC deficient male mice showed normal glucose tolerance and insulin sensitivity, while exhibiting normal food intake, inguinal and retroperitoneal adiposity, liver weights and pyruvate tolerance (Figures 1N–T). To further characterize if POMC neurons also show sexual dimorphism in terms of response to glucose overload, we measured C-FOS immunoreactivity in POMC neurons of POMC-EGFP mice injected either with glucose or saline (Figure 2A). Surprisingly, glucose elicited greater C-FOS expression in arcuate POMC neurons of females than males (Figure 2B). Notably, while C-FOS expression in POMC neurons of glucose treated females was 71.8 ± 5.5% higher than that of saline treated controls, males showed a difference of only 29.5 ± 5.7% (t-test: p < 0.01). Since a subpopulation of POMC neurons express estrogen receptor alpha (ESR1), which was postulated to control glucose homeostasis (23), we studied C-FOS induction by glucose overload specifically in POMC-ESR1 neurons (Figure 2C). Interestingly, we found that, despite the percentage of POMC neurons expressing ESR1 is similar in males and females (38.9 ± 1.5% and 42.8 ± 1.7%, respectively), glucose significantly increased C-FOS expression only in POMC-ESR1 neurons of female mice (Figure 2D).
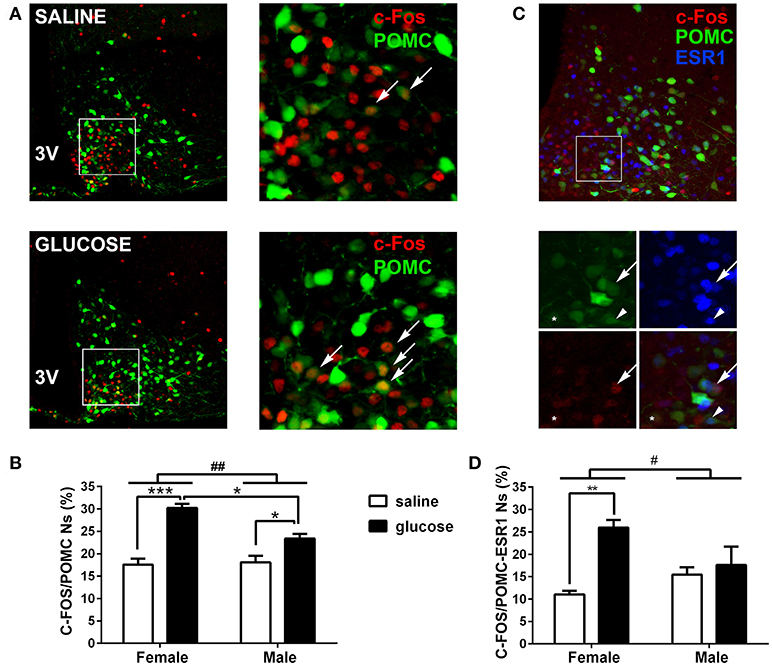
Figure 2. Induction of C-FOS expression by peripheral glucose overload in POMC neurons. (A) Representative coronal brain sections of POMC-EGFP female mice injected at P22-25 with saline (top) or glucose (bottom), subjected to anti-C-FOS immunofluorescence. Right panels show magnified images of squared areas depicted in left panels. Green: EGFP expression of POMC neurons. Red: C-FOS immunopositive neurons. Arrows indicate examples of POMC neurons positive for C-FOS. (B) Percentage of POMC neurons expressing C-FOS in male and female mice treated either with saline or glucose. POMC neurons per hemi-section: 58.45 ± 10.7; sections per animal: 7 ± 1.7 (mean ± SD). n = 4–5. (C) Representative coronal brain section of POMC-EGFP female mice injected at P22-25 with glucose (top), subjected to double C-FOS and ERS1 immunofluorescence. Bottom panels show magnified images of squared area depicted in top panel. Green: EGFP expression of POMC neurons. Red: C-FOS immunopositive neurons. Blue: ERS1 immunopositive neurons. Arrows: POMC/C-FOS/ESR1 neuron; *: POMC/C-FOS. Arrowhead: POMC/ESR1 neuron. (D) Percentage of POMC/ESR1 neurons expressing C-FOS in male and female mice treated either with saline or glucose. POMC neurons per hemi-section: 50.6 ± 14.1; sections per animal: 6.1 ± 0.9 (mean ± SD). n = −3–4. Error bars correspond to ±SEM. #p < 0.05 and ##p < 0.01 (TWA, sex x treatment effect); *p < 0.05, **p < 0.01, and ***p < 0.001 (Bonferroni).
Discussion
In the present work, we studied the regulation of glucose homeostasis by using a reversible POMC knockout mouse model with a unique feature, in which glucose homeostasis and insulin sensitivity can be tested before and after POMC restoration, providing strong evidence for an association between hypothalamic POMC expression and regulation of glucose homeostasis. Our results show that POMC deficiency impairs insulin sensitivity and glucose tolerance in non-obese juvenile female mice. These findings are in line with previous studies showing that the disruption of glucose signaling in POMC neurons impairs glucose homeostasis (10, 12, 13). It was also previously shown that POMC deficiency leads to decrease insulin sensitivity in obese and food restricted mice, both with elevated body fat composition (24). In addition, here we found that POMC deficiency leads to insulin resistance and glucose intolerance before the onset of obesity in ad libitum-fed female mice characterized by normal food intake, body weight and inguinal, retroperitoneal and liver fat stors. Therefore, the protective role of POMC in glucose homeostasis is presumably achieved by mechanisms independent of those involved in energy balance control. One of the major findings of our study is that POMC recovery completely restores insulin sensitivity and glucose tolerance, which emphasizes the importance of POMC in the regulation of glucose homeostasis. This regulation may be mediated by α-MSH since it was shown that insulin sensitivity is impaired in α-MSH receptor knockout mice through mechanisms involving the sympathetic neural system (25, 26). Regardless the pathway involved, since pyruvate tolerance was unaltered in our model of POMC deficiency, impaired glucose uptake rather than liver glucose production might be responsible for insulin intolerance.
Sexual dimorphism is an interesting feature of POMC neurons concerning the regulation of glucose homeostasis. Here, we found that POMC deletion in hypothalamic neurons leads to glucose intolerance and insulin resistance only in females. Furthermore, glucose-elicited POMC neuron activity is greater in female than in males and the subpopulation of POMC-ESR1 neurons response to glucose only in female mice. However, since glucose was injected systemically, our experiments do not distinguish between a direct or indirect action of glucose in POMC neurons. In either case, we speculate that sexual dimorphism found in our study is a consequence of estradiol (E2) facilitation of POMC activity since it was previously shown that estradiol prevents insulin resistance in POMC neurons of diet induced obese female but not male mice (27). Furthermore, it was demonstrated that E2 increases excitatory inputs, C-FOS protein and Pomc mRNA expression in POMC neurons of wild-type females (28, 29). Interestingly, female mice lacking estrogen receptor alpha only in POMC neurons develop insulin resistance and glucose intolerance (23). We hypothesize the existence of a protective pathway linking glucose signaling, ESR1, hypothalamic POMC and glucose homeostasis which is specific to females. Remarkably, women have lower prevalence of diabetes than men despite having higher prevalence of obesity (30). On this regard, our results shed light on the mechanisms underlying gender differences in diabetes pathophysiology.
In summary, we show for the first time a protective role of hypothalamic POMC peptide against glucose intolerance by mechanisms that are independent of POMC role in energy balance. Finally, since Pomc reactivation completely reverses the diabetogenic phenotype, arcuate POMC might be a potential target for diabetes therapy, particularly type 2 diabetes.
Author Contributions
VB designed research. RA, MT, and VB performed research. VB contributed new reagents, analytic tools. RA, MT, and VB analyzed data. VB wrote the paper and RA, MT revised it. All authors have approved the final article.
Funding
This work was supported by Fundación Florencio Fiorini, Agencia Nacional de Promoción Científica y Tecnológica (PICT2014-2000), Consejo Nacional de Investigaciones Científicas y Técnicas (PIP 2014-487), and Universidad de Buenos Aires, Argentina.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We thank Marcelo Rubinstein, Juan Belforte, Jessica Tollkuhn, Mario Perelló, Lorena Rela, and Gustavo Murer for valuable mice and reagents; Jesica Unger, Verónica Risso and Analía López Díaz, for technical assistance; Camila Zold, for editing the manuscript; and Johnson&Johnson Medical S.A., Argentina, for donating glucometer strips.
References
1. WHO (2016). Global Report on Diabetes-World Health organization. Geneva. Available online at: http://apps.who.int/iris/bitstream/10665/204871/1/9789241565257_eng.pdf?ua=1
2. Bumaschny VF, Yamashita M, Casas-Cordero R, Otero-Corchon V, de Souza FS, Rubinstein M, et al. Obesity-programmed mice are rescued by early genetic intervention. J Clin Invest. (2012) 122:4203–12. doi: 10.1172/JCI62543
3. Huo L, Gamber K, Greeley S, Silva J, Huntoon N, Leng XH, et al. Leptin-Dependent control of glucose balance and locomotor activity by POMC neurons. Cell Metab. (2009) 9:537–47. doi: 10.1016/j.cmet.2009.05.003
4. Knowler WC, Barrett-Connor E, Fowler SE, Hamman RF, Lachin JM, Walker EA, et al. Reduction in the incidence of type 2 diabetes with lifestyle intervention or metformin. N Engl J Med. (2002) 346:393–403. doi: 10.1056/NEJMoa012512
5. Routh VH, Hao L, Santiago AM, Sheng Z, Zhou C. Hypothalamic glucose sensing: making ends meet. Front Syst Neurosci. (2014) 8:236. doi: 10.3389/fnsys.2014.00236
6. Schwartz MW, Seeley RJ, Tschop MH, Woods SC, Morton GJ, Myers MG, et al. Cooperation between brain and islet in glucose homeostasis and diabetes. Nature (2013) 503:59–66. doi: 10.1038/nature12709
7. Xu Y, Elmquist JK, Fukuda M. Central nervous control of energy and glucose balance: focus on the central melanocortin system. Ann N Y Acad Sci. (2011) 1243:1–14. doi: 10.1111/j.1749-6632.2011.06248.x
8. Krude H, Biebermann H, Luck W, Horn R, Brabant G, Gruters A. Severe early-onset obesity, adrenal insufficiency and red hair pigmentation caused by POMC mutations in humans. Nat Genet. (1998) 19:155–7. doi: 10.1038/509
9. Yaswen L, Diehl N, Brennan MB, Hochgeschwender U. Obesity in the mouse model of pro-opiomelanocortin deficiency responds to peripheral melanocortin. Nat Med (1999) 5:1066–70. doi: 10.1038/12506
10. Claret M, Smith MA, Batterham RL, Selman C, Choudhury AI, Fryer LG, et al. AMPK is essential for energy homeostasis regulation and glucose sensing by POMC and AgRP neurons. J Clin Invest. (2007) 117 2325–36. doi: 10.1172/JCI31516
11. Ibrahim N, Bosch MA, Smart JL, Qiu J, Rubinstein M, Rønnekleiv OK, et al. Hypothalamic proopiomelanocortin neurons are glucose responsive and express KATP channels. Endocrinology (2003) 144:1331–40. doi: 10.1210/en.2002-221033
12. Parton LE, Ye CP, Coppari R, Enriori PJ, Choi B, Zhang CY, et al. Glucose sensing by POMC neurons regulates glucose homeostasis and is impaired in obesity. Nature (2007) 449:228–32. doi: 10.1038/nature06098
13. Hill JW, Elias CF, Fukuda M, Williams KW, Berglund ED, Holland WL, et al. Direct insulin and leptin action on pro-opiomelanocortin neurons is required for normal glucose homeostasis and fertility. Cell Metab. (2010) 11:286–97. doi: 10.1016/j.cmet.2010.03.002
14. Bady I, Marty N, Dallaporta M, Emery M, Gyger J, Tarussio D, et al. Evidence from glut2-null mice that glucose is a critical physiological regulator of feeding. Diabetes (2006) 55:988–95. doi: 10.2337/diabetes.55.04.06.db05-1386
15. Mercer AJ, Hentges ST, Meshul CK, Low MJ. Unraveling the central proopiomelanocortin neural circuits. Front Neurosci. (2013) 7:19. doi: 10.3389/fnins.2013.00019
16. Cowley MA, Smart JL, Rubinstein M, Cerdan MG, Diano S, Horvath TL, et al. Leptin activates anorexigenic POMC neurons through a neural network in the arcuate nucleus. Nature (2001) 411:480–4. doi: 10.1038/35078085
17. Hayashi S, McMahon AP. Efficient recombination in diverse tissues by a tamoxifen-inducible form of Cre: a tool for temporally regulated gene activation/inactivation in the mouse. Dev Biol. (2002) 244:305–18. doi: 10.1006/dbio.2002.0597
18. Ayala JE, Bracy DP, McGuinness OP, Wasserman DH. Considerations in the design of hyperinsulinemic-euglycemic clamps in the conscious mouse. Diabetes (2006) 55:390–7. doi: 10.2337/diabetes.55.02.06.db05-0686
19. Perez Millan MI, Luque GM, Ramirez MC, Noain D, Ornstein AM, Rubinstein et al. Selective disruption of dopamine D2 receptors in pituitary lactotropes increases body weight and adiposity in female mice. Endocrinology (2014) 155:829–39. doi: 10.1210/en.2013-1707
20. Metzger D, Chambon P. Site- and time-specific gene targeting in the mouse. Methods (2001) 24:71–80. doi: 10.1006/meth.2001.1159
21. Abràmoff MD, Magalhães PJ, Ram SJ. Image Processing With ImageJ. 4th Edn. Biophotonics International, Vol. 11 (2004).
22. Obici S, Feng Z, Tan J, Liu L, Karkanias G, Rossetti L. Central melanocortin receptors regulate insulin action. J Clin.Invest. (2001) 108:1079–85. doi: 10.1172/JCI12954
23. Zhu L, Xu P, Cao X, Yang Y, Hinton AO, Xia Y, et al. The ERα-PI3K cascade in proopiomelanocortin progenitor neurons regulates feeding and glucose balance in female mice. Endocrinology (2015) 156:4474–91. doi: 10.1210/en.2015-1660
24. Chhabra KH, Adams JM, Fagel B, Lam DD, Qi N, Rubinstein M, et al. Hypothalamic POMC deficiency improves glucose tolerance despite insulin resistance by increasing Glycosuria. Diabetes (2016) 65:660–72. doi: 10.2337/db15-0804
25. Fan W, Dinulescu DM, Butler AA, Zhou J, Marks DL, Cone RD. The central melanocortin system can directly regulate serum insulin levels. Endocrinology (2000) 141:3072–9. doi: 10.1210/en.141.9.3072
26. Sohn JW, Harris LE, Berglund ED, Liu T, Vong L, Lowell BB, et al. Melanocortin 4 receptors reciprocally regulate sympathetic and parasympathetic preganglionic neurons. Cell (2013) 152:612–9. doi: 10.1016/j.cell.2012.12.022
27. Qiu J, Bosch MA, Meza C, Navarro UV, Nestor CC, Wagner EJ. Estradiol protects proopiomelanocortin neurons against insulin resistance. Endocrinology (2018) 159:647–64. doi: 10.1210/en.2017-00793
28. Gao Q, Mezei G, Nie Y, Rao Y, Choi CS, Bechmann I, et al. Anorectic estrogen mimics leptin's effect on the rewiring of melanocortin cells and Stat3 signaling in obese animals. Nat Med. (2007) 13:89–94. doi: 10.1038/nm1525
29. Priest CA, Roberts JL. Estrogen and tamoxifen differentially regulate beta-endorphin and cFos expression and neuronal colocalization in the arcuate nucleus of the rat. Neuroendocrinology (2000) 72:293–305. doi: 10.1159/000054598
Keywords: proopiomelanocortin, glucose tolerance, insulin sensitivity, diabetes, hypothalamus, Esr1
Citation: Alsina R, Trotta M and Bumaschny VF (2018) Hypothalamic Proopiomelanocortin Is Necessary for Normal Glucose Homeostasis in Female Mice. Front. Endocrinol. 9:554. doi: 10.3389/fendo.2018.00554
Received: 28 March 2018; Accepted: 30 August 2018;
Published: 19 September 2018.
Edited by:
Toru Hosoi, Hiroshima University, JapanReviewed by:
Guojun Shi, University of Michigan, United StatesMiguel López, Universidade de Santiago de Compostela, Spain
Abhiram Sahu, University of Pittsburgh, United States
Copyright © 2018 Alsina, Trotta and Bumaschny. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Viviana Florencia Bumaschny, dmJ1bWFzY2hueUBmbWVkLnViYS5hcg==
† These authors have contributed equally to this work