 Matthias Lambert
Matthias Lambert Bruno Bastide
Bruno Bastide Caroline Cieniewski-Bernard
Caroline Cieniewski-Bernard- Equipe Activité Physique, Muscle, Santé, Unité de Recherche Pluridisciplinaire Sport, Santé, Société (EA7369-URePSSS), Faculté des Sciences et Technologies, Université de Lille, Lille, France
Skeletal muscle represents around 40% of whole body mass. The principal function of skeletal muscle is the conversion of chemical energy toward mechanic energy to ensure the development of force, provide movement and locomotion, and maintain posture. This crucial energy dependence is maintained by the faculty of the skeletal muscle for being a central place as a “reservoir” of amino acids and carbohydrates in the whole body. A fundamental post-translational modification, named O-GlcNAcylation, depends, inter alia, on these nutrients; it consists to the transfer or the removal of a unique monosaccharide (N-acetyl-D-glucosamine) to a serine or threonine hydroxyl group of nucleocytoplasmic and mitochondrial proteins in a dynamic process by the O-GlcNAc Transferase (OGT) and the O-GlcNAcase (OGA), respectively. O-GlcNAcylation has been shown to be strongly involved in crucial intracellular mechanisms through the modulation of signaling pathways, gene expression, or cytoskeletal functions in various organs and tissues, such as the brain, liver, kidney or pancreas, and linked to the etiology of associated diseases. In recent years, several studies were also focused on the role of O-GlcNAcylation in the physiology and the physiopathology of skeletal muscle. These studies were mostly interested in O-GlcNAcylation during muscle exercise or muscle-wasting conditions. Major findings pointed out a different “O-GlcNAc signature” depending on muscle type metabolism at resting, wasting and exercise conditions, as well as depending on acute or long-term exhausting exercise protocol. First insights showed some differential OGT/OGA expression and/or activity associated with some differential stress cellular responses through Reactive Oxygen Species and/or Heat-Shock Proteins. Robust data displayed that these O-GlcNAc changes could lead to (i) a differential modulation of the carbohydrates metabolism, since the majority of enzymes are known to be O-GlcNAcylated, and to (ii) a differential modulation of the protein synthesis/degradation balance since O-GlcNAcylation regulates some key signaling pathways such as Akt/GSK3β, Akt/mTOR, Myogenin/Atrogin-1, Myogenin/Mef2D, Mrf4 and PGC-1α in the skeletal muscle. Finally, such involvement of O-GlcNAcylation in some metabolic processes of the skeletal muscle might be linked to some associated diseases such as type 2 diabetes or neuromuscular diseases showing a critical increase of the global O-GlcNAcylation level.
Introduction
Just over thirty years ago, the O-linked N-acetyl-β-D-glucosaminylation, termed O-GlcNAcylation, was discovered inside the mouse lymphocyte cells by Torres and Hart (1). From this discovery, about 1,400 studies were focused on this field among hundreds of other known post-translational modifications. Nowadays, scientific community shows a growing interest since half of these previous studies was published in the last 5 years, and provides more and more relevant data to better characterize the impact of O-GlcNAcylation on cellular processes. It is ubiquitous from virus to plantae and metazoan, and to date around 4000 O-GlcNAc-modified proteins have been identified (2). O-GlcNAcylation seems to be an important molecular process in biology, especially since ubiquitous OGT and OGA knockout mice experiments revealed that O-GlcNAcylation balance is crucial for embryonic stem cell viability and embryonic development (3, 4); recent data also supported the essential role of O-GlcNAcylation in adult life since inducible global knockout of OGT dramatically increased mice mortality (5).
O-GlcNAcylation is an atypical, reversible and dynamic glycosylation. Unlike the N-and O-glycans, the O-GlcNAcylation consists of the transfer of a unique monosaccharide which is not elongated, the N-acetyl-D-glucosamine, on a plethora of nucleocytoplasmic (6) and mitochondrial proteins (7). The O-GlcNAc modification is mediated by a couple of antagonist enzymes; the OGT (uridine diphospho-N-acetylglucosamine: peptide beta-N-acetyl-glucosaminyl-transferase) transfers the monosaccharide from the UDP-GlcNAc donor to a serine or threonine hydroxyl group of a protein through a beta linkage (8), while OGA (beta-N-acetylglucosaminidase) hydrolyses the O-GlcNAc moieties from O-GlcNAcylated proteins (9). Very recent data have shown that the moiety can also be added to proteins intended for the extracellular compartment, through a distinct and structurally unrelated OGT (called EGF-OGT) which works in an OGT-independent manner (10–12). Besides its reversibility, O-GlcNAcylation is also highly dynamic. Indeed, the GlcNAc moieties can be added and removed several times along the protein lifetime, and the turn-over is shorter than the protein backbone's one. Moreover, this O-GlcNAc dynamic process could reply to many environmental conditions and physiological signals such as nutriment availability, especially from its UDP-GlcNAc donor, the last product of the Hexosamine Biosynthesis Pathway (13, 14). Finally, O-GlcNAcylation can also interplay with certain other post-translational modifications such as phosphorylation and ubiquitination [for review, see (15–17)].
Nowadays, there is evidence that some fundamental protein functions are modulated through the O-GlcNAcylation, including protein-protein interactions (18, 19), protein stability (20, 21), protein activity (22), or protein localization (23). Akin to phosphorylation, the O-GlcNAcylation is involved in almost all if not all intracellular processes (15, 24, 25) and different data demonstrated that its dysregulation can play a crucial role in the etiology of several diseases including type II diabetes (26, 27), cancer (28, 29), neurodegenerative disorders (30, 31), X-linked intellectual disability (32), neuromuscular (33), or cardiovascular diseases (34, 35), and linked to aging (36).
However, recent data showed that O-GlcNAcylation is also involved in different cellular processes of the skeletal muscle, and its potential role in many disorders related to skeletal muscle defects is still undervalued. This present review discusses the involvement of O-GlcNAcylation in skeletal muscle metabolism (in particular glucose metabolism), the impact of exercise on O-GlcNAcylation, and finally, the potential role of this post-translational modification in skeletal muscle in a context of disease such as type 2 diabetes and neuromuscular disorders.
Relationship Between O-GlcNAcylation and Metabolism in Skeletal Muscle
In the human body, the skeletal muscle is an essential tissue that converts chemical energy into mechanical energy, i.e., contraction, to generate force and ensure some fundamental functions of the body such as movement production, posture control and thermoregulation (37). The skeletal muscle represents 40% of the total human body weight, contains 50 to 75% of all body proteins and accounts for 30 to 50% of whole-body protein turnover (37). It is a huge reservoir of nutrients (e.g., glycogen and amino acids), a great producer/consumer of energy and accounts for 30% of the resting metabolic rate in adult human. For instance, from basal state to fully state of contraction, the skeletal muscle can 300-fold increase its energy consumption within a few milliseconds (38). Interestingly, O-GlcNAcylation is known to be a cell nutrient sensor and the “O-GlcNAc signature” depends on the biological state of cells (39). Within the unitary contractile apparatus, named sarcomere, and more generally in the overall skeletal muscle cell, diverse proteins have been identified to be O-GlcNAcylated since 2004 (18, 40–43); the nature of these proteins is diversified including contractile, structural, cytoskeletal, metabolic, chaperones, mitochondrial proteins or proteins involved in signaling pathways. Thus, akin to phosphorylation (37, 44, 45), O-GlcNAcylation could play a significant role, still undervalued, in the skeletal muscle physiology.
Exercise-Mediated O-Glcnacylation Changes in the Skeletal Muscle
Contraction is the main function of the skeletal muscle to provide force generation and ensure movement production and posture control from baseline to exercise conditions. Skeletal muscle is able to develop several adaptations with exercise by changes in contractile protein isoforms, protein turnover, metabolism, mitochondrial functions, intracellular signaling or transcriptional response [for review, see (46–48)]. As described, O-GlcNAcylation highly depends on metabolism through the Hexosamine Biosynthesis Pathway and the UDP-GlcNAc donor. Glucose metabolism has a key role in this process since about 2–5% of the glucose enters the HBP, while the remaining glucose goes to glycogen storage or glycolysis (13, 49). In addition, a study carried out on skeletal muscle, showed that HBP also depends on fatty acids, glutamate or nucleic acids (50). Among other processes, muscle contraction directly impacts glucose homeostasis. For example, exercise increases skeletal muscle glucose uptake through an insulin-dependent/GLUT4 transport pathway (51, 52) or through the activation of CamKII by calcium released upon muscle contraction (53). Thus, we can assume that O-GlcNAcylation could be modulated through muscle-training and induces several effects on muscle functions. However, although many studies reported the importance of exercise in the modulation of cardiac O-GlcNAcylation (54–59), to date there is a very few studies interesting in the skeletal muscle.
It's long recognized that metabolic flexibility concept occurs in skeletal muscle, meaning for example its ability to increase energy supply and provide sufficient and adequate “fuel” for muscle working (48). In this context, it has been recently shown that exercise could also modulate the global O-GlcNAcylation level in rat skeletal muscle (60–62). Two different types of exercise training were applied on rat: a single acute exercise run bout to fatigue and an exhausting 6-weeks interval training program, both on treadmill. Consecutively to the acute exercise, the global level of O-GlcNAcylation was not changed in the slow-twitch oxidative soleus nor in the fast-twitch glycolytic EDL as well (62) (Table 1). In contrast, a long-term training program on rat led to an increase of the global O-GlcNAcylation level in the total extract in soleus and EDL as well, whereas its level was not altered in the myofilament fraction (62). In this study, no alteration of the OGT and OGA expression was observed in both exercise protocols, suggesting a potential regulation at the activity level following the long-term exercise. However, in another study, the same authors mentioned a decreased OGA expression in the total extract of the soleus, and not in the myofilament fraction as well as in both extracts in the EDL after a similar long-term training program on rat, suggesting a differential OGA turnover which might explain, partly at least, changes in the O-GlcNAc rate between EDL and soleus (61).
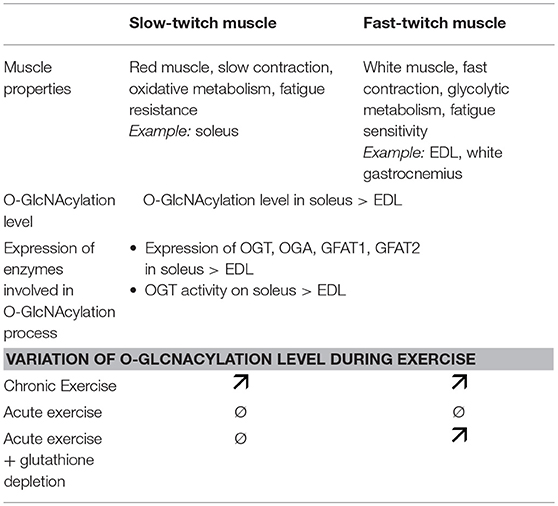
Table 1. Difference of O-GlcNAcylation process and exercise effect on slow-twitch and fast-twitch muscles. The major muscle properties of both muscle types are also indicated.
Otherwise, these O-GlcNAc adaptations following skeletal muscle activity seemed to be fully different according to the exercise protocol as well as the skeletal muscle fiber type. This could be first related to the metabolic flexibility and glucose utilization known to be different and differentially regulated depending on exercise protocols and muscle fiber types (46, 48). Little is known so far how exercise can modulate the glucose flux in the Hexosamine Biosynthesis Pathway. A study demonstrated that hindlimb skeletal muscle UDP-HexNAc concentration increased after a single swimming protocol in ad libitum-fed but not in fasted rats. In parallel, muscle glycogen content decreased and the GFAT activity was not altered in these conditions (63). Thus, it would be also interesting to determine how the glucose metabolism could operate a distinct modulation of O-GlcNAcylation in skeletal muscles depending on different exercise protocols since (i) the metabolism is different from fast and slow-twitch muscle, (ii) the O-GlcNAcylation is highly regulated through glucose metabolism and (iii) the glucose uptake through GLUT4 is highly modulated in skeletal muscle during exercise (52). These future directions could bring new insights in the involvement of the O-GlcNAcylation in the modulation of the beneficial effects of exercise in skeletal muscle, as a game changer to develop new strategies that counteract some muscular disorders or metabolic disorders such as obesity or diabetes mellitus.
Moreover, the O-GlcNAc adaptations post-exercise could also be directly linked to a differential modulation of the O-GlcNAc pattern and the O-GlcNAc processing enzymes between both muscle fiber types seen at resting conditions. Indeed, the O-GlcNAcylation level is higher in the slow soleus muscle compared with the fast EDL muscle (62, 64, 65); in parallel, the expression level of OGT, OGA, GFAT1, and GFAT2 is higher in soleus than in EDL (62), as well as the activity of OGT (64) (Table 1).
Finally, it is well known that the metabolism and stress response between the fast-twitch glycolytic fibers and the slow-twitch oxidative fibers are different (46). Thus, this concept could support the differential modulation of O-GlcNAcylation inducing variable consequences on cellular functions during basal and exercise conditions. During muscle activity, reactive oxygen species (ROS) are produced in skeletal muscle (66) and the O-GlcNAcylation has been shown to be involved in the modulation of oxidative stress through different signaling pathways including KEAP1/NRF2, FOXO, NFκB, and p53 (67–69). A recent study compared the O-GlcNAc pattern and O-GlcNAc processing enzymes expression between a single Diethyl Maleate (DEM) intraperitoneal injection in order to deplete glutathione, meaning oxidative stress in rat, and a single acute exercise on treadmill (60). Interestingly, in the fast-twitch white gastrocnemius, the global level of O-GlcNAcylation increased after acute exercise as well as glutathione depletion. On the contrary, in the slow-twitch soleus, no significant variation of the global O-GlcNAc level was observed (Table 1). These data suggest that the differential oxidant/antioxidant balance between slow and fast-twitch muscle could also be at the origin of the differential modulation of the O-GlcNAcylation level observed in the two muscle types. However, this complex interrelationship between cellular redox state and O-GlcNAcylation seems not to be the only mechanism involved in the O-GlcNAc regulation during exercise since the OGT and GFAT mRNA expressions were different between acute exercise and glutathione depletion in both muscles (60).
O-GlcNAcylation Could Mediate the Skeletal Muscle Glucose Metabolism
Recent studies demonstrated that energy metabolism, insulin sensitivity and exercise-induced glucose uptake depends on O-GlcNAcylation (65, 70). Indeed, the muscle specific knockout of OGT led to the increase of glucose uptake in skeletal muscle in basal conditions (65) as well as consequently to exercise (70). Thus, the specific inhibition of O-GlcNAcylation in skeletal muscle also led to facilitation of glucose utilization in skeletal muscle, leading to greater exercise-induced glucose disposal, involving AMPK (70). Interestingly, the enhancement of glucose uptake was correlated to an increase of glycolytic enzymes activities, suggesting that mice have greater reliance of carbohydrates for energy production (65).
It is worth to note that almost all enzymes of glycolytic pathway such as phosphofructokinase (PFK), fructose bisphosphate aldolase (FBPA), triose phosphate isomerase (TPI), glyceraldehyde-3-phosphate dehydrogenase (GAPDH), beta-enolase (BE), and pyruvate kinase (PK) are O-GlcNAcylated (40, 71–76) [and for review, see (77, 78)] (Figure 1). Thus, O-GlcNAcylation may regulate the expression and/or activity of glycolytic enzymes and might consequently be involved in the regulation of glucose metabolism in skeletal muscle. To support this important role of O-GlcNAcylation as nutrient-sensor, it was first demonstrated that O-GlcNAcylation is involved in the regulation of phosphofructokinase 1 and pyruvate kinase M2 activity (71, 73). Indeed, induction of O-GlcNAcylation at serine 529 of PFK1 inhibited PFK1 oligomerization and activity, and reduced glycolytic flux as well (71). Moreover, knockdown of OGT led to an increased PKM2 activity (73). In this previous study, the resulting decrease of O-GlcNAc PKM2 level was associated to a decreased PKM2 expression, and to a decrease of PKM2 serine phosphorylation (73). Conversely, the increase of PKM2 O-GlcNAcylation by the use of Thiamet-G, a potent OGA inhibitor, led to upregulation of PKM2 expression and a decreased PKM2 activity (73). More recently, pharmacological inhibition of OGA and knockdown of OGT were associated to a respective increase and decrease of GK expression, which is the major regulator of the glucose input into cell and therefore the major regulator of glucose metabolism (79). Thus, O-GlcNAcylation is a key regulator of enzymes of glycolysis; interestingly, two downstream enzymes of PK, lactate dehydrogenase (LD) and pyruvate dehydrogenase (PDH) are also modified by O-GlcNAc moieties (74), suggesting that O-GlcNAcylation may also be an important regulator of the utilization of the glycolysis end-product, i.e., pyruvate, through the anaerobic pathway (lactate dehydrogenase) or the aerobic pathway (TCA cycle; Figure 1). Since almost all enzymes of TCA cycle are described to be O-GlcNAc modified [aconitate hydratase (A), isocitrate dehydrogenase (IDH), ketoglutarate dehydrogenase (KGD), succinyl-CoA ligase (SL), succinate dehydrogenase (SDH) and malate dehydrogenase (MDH), so as several subunits of respiratory chain complexes (40, 80, 81)], O-GlcNAcylation might play an important role in the ATP production as well (Figure 1). However, to date, neither literature mention a potential O-GlcNAcylation of citrate synthase (CS) and fumarate hydratase (FH). In the same way, the creatine shuttle, permitting the communication between ATP site consumption (i.e., myofibrillar ATPases) and mitochondria (82), could be therefore modulated by O-GlcNAcylation since creatine kinase is itself O-GlcNAcylated (40).
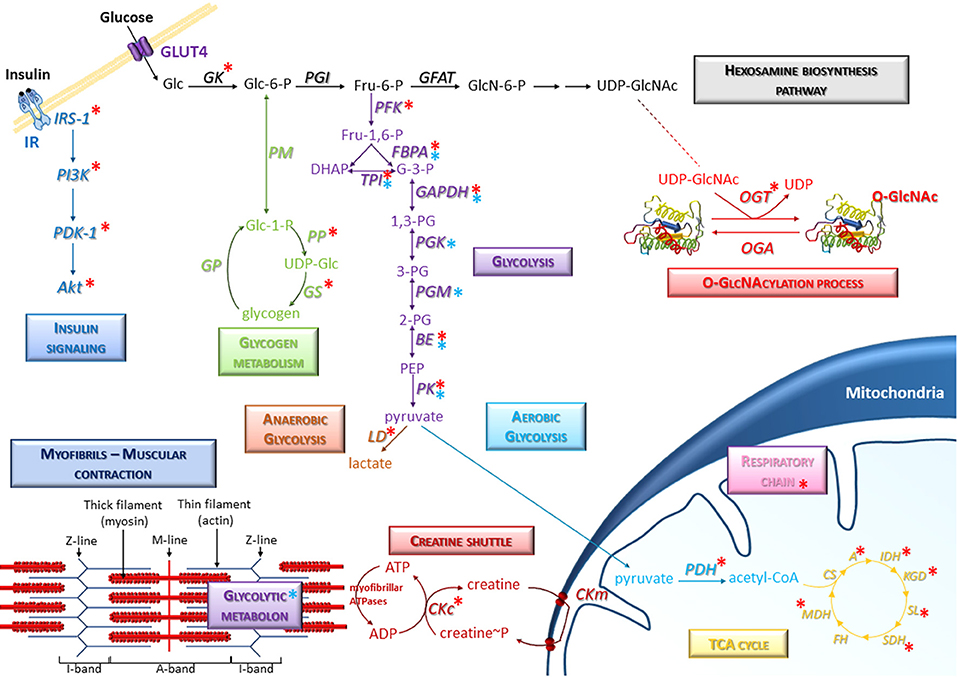
Figure 1. Representative scheme of the presence of O-GlcNAcylation on glucose metabolism in skeletal muscle. Several signaling and metabolic pathways are indicated, in particular the hexosamine biosynthesis pathway, glycolysis (anaerobic and aerobic glycolysis), glycogen metabolism, insulin signaling, and TCA cycle. Specific molecular components of skeletal muscle such as myofibrils and creatine shuttle are also represented. Red asterisks correspond to O-GlcNAcylated proteins; blue asterisks correspond to enzymes including in protein-protein complexes (such as the glycolytic metabolon) which could be potentially modulated consecutively to O-GlcNAcylation changes in skeletal muscle cells. A, Aconitase; BE, Beta-enolase; CKc, Creatine kinase cytoplasmic; CKm, Creatine kinase mitochondrial; CS, Citrate synthase; FBPA, Fructose-bisphosphate aldolase; FH, Fumarate hydratase; GAPDH, Glyceraldehyde-3-phosphate dehydrogenase; GFAT, Glutamine,fructose-6-phosphate aminotransferase; GK, Glucokinase; GP, Glycogen phosphorylase; GS, Glycogen synthase; IDH, Isocitrate dehydrogenase; KGD, Ketoglutarate dehydrogenase; LD, Lactate dehydrogenase; MDH, Malate dehydrogenase; OGA, O-GlcNAcase; OGT, O-GlcNAc transferase; PDH, Pyruvate Dehydrogenase; PFK, Phosphofructokinase; PGI, Phosphoglucose isomerase; PGK, Phosphoglycerate kinase; PGM, Phosphoglycerate mutase; PK, Pyruvate kinase; PM, phosphoglucomutase; PP, UDP-glucose pyrophosphorylase; SDH, Succinate Dehydrogenase; SL, Succinyl-CoA ligase; TPI, Triose-phosphate isomerase.
Many data suggest a close association between the myofibrils and the enzymes involved in the metabolism. Indeed, the fructose-bisphosphate aldolase (FBPA), enzyme of glycolysis and neo-glucogenesis, is known to be localized to the Z-line of the sarcomere in association with α-actinin within a multiprotein complex termed metabolon (83, 84). In the same way, the interaction between phosphoglucoisomerase (PGM), phosphofructokinase (PFK), glyceraldehyde-3-phosphate dehydrogenase (GAPDH), pyruvate kinase (PK) and aldolase (FBPA) also occurs with the thin filament (85, 86). These specific interactions between glycolytic enzyme complexes (termed glycolytic metabolon) and the contractile apparatus may ensure a very efficient and dynamic localized production of ATP for myosin ATPase and actomyosin interactions resulting in force development. Indeed, it was recently demonstrated that the global modulation of O-GlcNAcylation level in C2C12 skeletal muscle cells differentiated into myotubes led to the modulation of protein-protein interactions in multiprotein complexes; while this study focused on structural proteins, the proteomic data suggested that the glycolytic metabolon could be modulated by O-GlcNAcylation changes as well (Figure 1) (18). Indeed, several glycolytic enzymes (indicated by blue asterisks on Figure 1) were identified in protein-protein complexes which were modulated after the global O-GlcNAcylation changes, suggesting that the glycolytic metabolon could be potentially modulated consecutively to O-GlcNAcylation variations.
In addition, O-GlcNAcylation is also involved in the modulation of insulin pathway through the modulation of signaling proteins such as IRS-1, PI3K, PDK1, or Akt. O-GlcNAcylation of these upstream components of insulin signaling pathway occurs after the recruitment of OGT to the membrane, leading to the attenuation of insulin sensitivity (87–89) [for review, see (15, 90)]. Interestingly, the glycogen metabolism could be also modulated by O-GlcNAcylation through the regulation of glycogen synthase, O-GlcNAcylation acting as inhibitory mechanism of this enzyme (15, 91, 92); in addition, the UDP-glucose pyrophosphorylase (PP), which generates UDP-Glc, is also O-GlcNAcylated (75), suggesting that O-GlcNAcylation may be a regulator of glycogen synthesis. Thus, the O-GlcNAcylation, which depends itself of the glucose level through the Hexosamine Biosynthesis Pathway, could act as a nutritional sensor to regulate the glycolytic flow through the modification of glycolytic enzymes, the regulation of protein expression, the modulation of their phosphorylation level and/or the modulation of the metabolon.
Taken together, all these data were gained from different tissues or cell lines, and the precise role of O-GlcNAcylation on the regulation of glucose metabolism in the skeletal muscle remains to be clearly elucidated. In this context, it would also be wise to investigate the exact role of O-GlcNAcylation, not only in the regulation of enzymes expression and/or activities, but also in the modulation of these metabolon since OGT and OGA are also enriched in the Z-line and the I-band of the sarcomere (93). All together, these data strongly argue in favor of a key role of the O-GlcNAcylation process in the regulation of energy metabolism of skeletal muscle, in particular the utilization of glucose as “fuel” to provide energy to ensure muscle contraction.
O-GlcNAcylation and Skeletal Muscle Dysfunctions
O-GlcNAcylation Is Associated to Muscular Atrophy
Muscle atrophy arises from a defect of the balance between protein synthesis and degradation (94, 95). Both intracellular mechanisms maintain protein homeostasis and could be potentially modulated by O-GlcNAcylation (17, 96, 97), but the role of O-GlcNAcylation in the regulation of protein synthesis and degradation is not really investigated in the skeletal muscle. However, this knowledge could be crucial since muscle atrophy is often associated with impairment of contractile and structural functions, metabolism process, and changes of phenotype fiber.
O-GlcNAcylation and skeletal muscle atrophy were firstly associated subsequently to a 14- or 28-days hindlimb unloading (HU) experiments in rat (40, 64, 93). One of the most relevant data was an opposite modulation of the global O-GlcNAc level between the slow-twitch soleus and the fast-twitch EDL (64). A decrease of the global O-GlcNAcylation level was observed in the atrophied rat soleus, while in contrast it was not altered in the non-atrophied rat EDL subsequently to the 14- and 28-days hindlimb unloading. Moreover, OGT activity was also opposite in both muscles by decreasing in soleus and increasing in EDL; however, it has been shown that OGA activity increased in both muscles (64). This first report suggested that O-GlcNAcylation could be related to the muscle atrophy and plasticity processes. Interestingly, the authors demonstrated in parallel that heat-shock proteins expression was also altered. Indeed, in the rat EDL, the HSP70 expression increased, contrary to the atrophied rat soleus after a 14-days HU (64). This heat-shock protein is known to be O-GlcNAcylated, to have lectin properties (98–100), and to have the ability to increase stress tolerance while decreasing protein degradation (101, 102). Thus, in rat EDL, the increase of global O-GlcNAc level, as well as HSP70 expression, could contribute to an improvement of stress tolerance, as suggested in cardiomyocytes (103), and prevent muscle atrophy (64, 104). Interestingly, the expression of another heat-shock protein, the alphaB-crystallin which is also known to be O-GlcNAcylated (105), was decreased in the atrophied soleus and could therefore be involved in the plasticity processes (106).
More recently, different studies focused on the molecular pathways involved in skeletal muscle atrophy, including the relationship between O-GlcNAcylation, signaling pathways and muscular atrophy in skeletal muscle cells model (Figure 2). By using Thiamet-G, a potent inhibitor of the O-GlcNAcase, the C2C12 cells showed both a global increase of the O-GlcNAcylation and a modulation of some catabolic and anabolic pathways leading to atrophy (107). First, this study reported a significant decrease of Akt and GSK3β phosphorylation, as well as an increase of myostatin expression, which could lead to an inhibition of some anabolic pathways. Secondly, an increase of Atrogin-1 expression was reported and could lead to an improvement of some catabolic pathways (Figure 2). In this way, myostatin is a negative modulator of skeletal muscle growth and inhibits some protein synthesis pathways such as Akt/mTOR; it also promotes degradation of many sarcomeric proteins and seems to be dependent of Atrogin-1 (108). Interestingly, this report of molecular events following OGA deficiency in C2C12 could partly explain muscle wasting induction through glucocorticoid in stress conditions (109). Indeed, in C2C12 cells treated with dexamethasone, the OGA activity was decreased such as its expression (a similar molecular event was described when cells were treated with the Thiamet-G); in parallel, Murf-1 expression, leading to atrophy, increased (107) (Figure 2). It was suggested that dexamethasone could repress OGA gene via binding onto Glucocorticoid Response Element (107, 110); another mechanism could also involve OGT, known to be a cofactor of glucocorticoid receptors promoting transrepression (111). This is a relevant new insight regarding the use of glucocorticoids as the only available therapeutic treatment to maintain essential muscle functions in some muscular dystrophies (112). However, O-GlcNAc-mediated molecular mechanisms could be partly different between glucocorticoid-induced atrophy and disuse atrophy since the O-GlcNAc pattern is different, and the expression of O-GlcNAc processing enzymes is not changed (64).
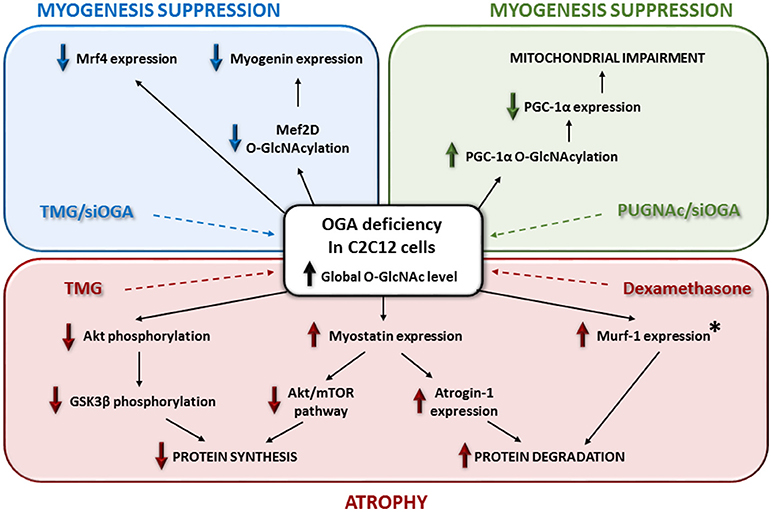
Figure 2. Relationships between O-GlcNAcylation, signaling pathways and muscular atrophy in skeletal muscle cells model. This figure depicts the impact of OGA deficiency on myogenesis and skeletal muscle atrophy. The asterisk means that Murf-1 overexpression occurs with the use of dexamethasone and not with the Thiamet-G in C2C12 cells.
In parallel, in C2C12 cells, the increase of the global O-GlcNAcylation level by the use of Thiamet-G or another OGA inhibitor such as PUGNAc or another strategy such as OGA knockdown, seemed suppress the myogenic differentiation of the muscle cells (113–115). Indeed, the terminal differentiation stage of C2C12 is altered through a decrease of mrf4, myogenin (113), and myoD expression (115) (Figure 2). Interestingly, a decreased O-GlcNAcylation of Mef2D, a transcriptional activator of myogenin, suppressed its recruitment to the myogenic factor promoter (114). Moreover, in the case of a global decrease of the O-GlcNAcylation level, the specific O-GlcNAc rate of PGC-1α led to its degradation and suppressed the mitochondrial biogenesis and myogenesis in C2C12 (115) (Figure 2). By the way, through OGA manipulation in C2C12 cells, O-GlcNAcylation seemed to be a negative regulator of the myogenesis. This conclusion is reinforced by the overexpression of an inactive OGA variant and the increase of O-GlcNAcylation in a rat model inducing skeletal muscle atrophy (116). In contrast, a skeletal muscle specific OGT knockout in mice, leading to a global decrease of O-GlcNAcylation in the tissue, did not induce muscle hypertrophy (65). Indeed, tibialis anterior, EDL and soleus of these mice showed a normal morphology and mass. Interestingly, mice exhibited reduced fat mass (65). Muscle phenotype from global OGT or OGA knockout in mice is difficult to determine since it has been shown a severe perinatal lethality, and to date there is no available data about an inducible OGA knockout or a skeletal muscle specific OGA knockout model. Along O-GlcNAcylation, all these recent studies showed complex pathways, but not fully resolved, leading to skeletal muscle atrophy.
Skeletal Muscle O-GlcNAcylation in Physiopathological Context
In various organs or tissues (e.g., heart, brain, pancreas, and kidney), O-GlcNAcylation was previously described to be both cell protective from an acute variation and deleterious from chronic and sustained variations especially through the impairment of glucose utilization or the glucose toxicity paradigm that may lead to the progression of several diseases [for review, see (31, 34, 36, 68, 117, 118)]. To date, one of the most defined examples is the involvement of O-GlcNAcylation in the progression of diabetes, characterized by hyperglycemia as the result of body's inability to correctly process blood glucose. Subsequently, all insulin sensitive tissues present hyper-O-GlcNAcylation and many complications. It appeared that a single nucleotide polymorphism in MGEA5, encoding OGA, is associated with type 2 diabetes in a mexican american population (119). Moreover, Goto-Kakizaki rats, which develop type 2 diabetes mellitus in early life, express an inactive 90 kDa isoform of OGA (120). Finally, a conditional OGA knockout in mice led to a low blood glucose concentration, a decreased insulin sensitivity, and to a perinatal death (121). However, the involvement of O-GlcNAcylation in the onset or in the progression of diabetes is still in debate (118) since a pharmacological inhibition of OGA in adipocytes did not cause insulin resistance or disruption of the glucohomeostasis (122).
Interestingly, among insulin sensitive tissues, the skeletal muscles are responsible for about 75% of insulin-stimulated uptake in the whole human body. In skeletal muscle, a global increase of O-GlcNAcylation induced insulin resistance (123), whereas insulin infusion led to an increase of the HBP flux and the O-GlcNAc content (124). It has been shown that muscular overexpression of GFAT in transgenic mice led to muscle insulin resistance (125, 126). In a same way, upregulation of GFAT expression and activity was described in skeletal muscle of diabetic patients (127). Moreover, overexpression of GFAT in mice is associated to a decrease of GLUT4 translocation to the sarcolemma (128), whereas transgenic mice overexpressing GLUT4 did not show alteration of GFAT expression or activity unlike the overexpression of GLUT1 (129), although both of these mice models showed an increase of glucose uptake and O-GlcNAcylation in muscle (130). Thus, the role of HBP in insulin resistance seems to be complex and still not resolved (131). Recent data provided that TRIB3 may be a novel link between HBP and insulin resistance in skeletal muscle (132).
Experiments with mice overexpressing OGT displayed muscle insulin resistance as well as hyperleptinemia (133). Pharmacological inhibition of OGA (123) or OGA knockdown (116) in skeletal muscle cells also induced insulin resistance. In parallel, after induction of insulin in liver, PIP3 recruited OGT from the nucleus to the membrane and caused perturbations of insulin signaling (88). Taken together, these data suggest that O-GlcNAcylation could be a link between insulin resistance and muscle impairment, since O-GlcNAcylation is involved in skeletal muscle contractility, sarcomere structuration, myogenesis, and diabetic patients often display “diabetic myopathy” (134). A significant volume of literature suggested O-GlcNAcylation linking diabetes and cardiovascular complications [for review, see (27, 35)]. In cardiac muscle from mice developing insulin resistance, mitochondrial dysfunction and changes in contractile properties were associated to an increase of O-GlcNAcylation (58). Indeed, it has been shown a correlation between increase of O-GlcNAcylation and decrease of calcium sensitivity in the cardiac tissue (135), as well as in the skeletal muscle tissue (41, 42). Interestingly, it has been shown that adenoviral transfer of OGA (136) or injection of a bacterial homologue of OGA (137), reversed the excessive O-GlcNAc content and the cardiac contractile dysfunctions. In this study, many myofibrillar proteins exhibited changes of their O-GlcNAcylation level, but the modulation of contractile properties could be also explained by a modulation of Ca2+ handling (138). Indeed, within an insulin resistance context in cardiac tissue, Serca2a expression was changed (58, 136), and an alteration of O-GlcNAc levels significantly affected Ca2+ handling, Serca2a (139) and STM1 functions (140). This role of O-GlcNAcylation should be considered also in skeletal muscle since dysfunction of contractibility as well as the Ca2+ handling were also measured in skeletal muscle of rat models of diabetes (141), via impaired Serca and GLUT4 (142).
Interestingly, OGT and OGA are highly concentrated to the sarcomere (93). Moreover, in cardiac tissue of STZ-diabetic mice, OGA, and OGT mislocalization in the sarcomere was associated to activities alteration as well as changes in the OGA interactions with actin, tropomyosin and MLC1 (137). In parallel, OGT was also mislocalized in mitochondria, the interaction between OGT and complex IV being decreased while the OGA activity decreased (143). O-GlcNAc processing enzymes distribution in the sarcomere of skeletal muscle as well as other compartments, such as mitochondria should be also investigated to better understand the involvement of O-GlcNAcylation in diabetic context. Indeed, different data support a relocalization of O-GlcNAc in atrophy or exercise muscle activity context (61, 62, 93). Recently, in C2C12 cells, an OGA knockdown induced insulin resistance and a decrease of the mitochondrial biogenesis with a decreased PGC1α expression (115).
In another context, it has been shown that the global O-GlcNAc level in skeletal muscle is increased in some human neuromuscular diseases (33). Especially, compared to normal muscle fibers, the O-GlcNAc signal seemed to be relocalized from the sarcolemma to the cytoplasm and nuclei in regenerative muscle fibers of muscular dystrophies, myositis and rhabdomyolysis. A strong O-GlcNAc content signal was also displayed in vacuolated fibers in sporadic inclusion body myositis, and distal myopathies with rimmed vacuoles, as well as in neurogenic muscular dystrophy. This O-GlcNAc raise could be associated with the stress response, since HSP70 expression is increased in the cytoplasmic compartment of these neuromuscular diseases. Moreover, a mutation in the GNE gene is known to cause distal myopathies with rimmed vacuoles. Interestingly, UDP-GlcNAc is the substrate of the GNE enzyme, and an impairment of GNE expression/activity could altered the UDP-GlcNAc content and so the O-GlcNAcylation mediated by OGT (33).
Conclusion and Perspectives
In the past ten years, more and more studies concerned O-GlcNAcylation in the skeletal muscle physiology and physiopathology. This glycosylation is mostly related to glucose metabolism, and skeletal muscle is one of the largest consumers of glucose; in addition, numerous studies showed that skeletal muscle is essential for glucose homeostasis and insulin sensitivity. Moreover, muscle plasticity allows skeletal fibers adaptations to physiological conditions in terms of contractile but also metabolism properties. Consequently, the glucose utilization will change depending on resting, wasting or exercise, as well as the fiber type composition. Interestingly, the global O-GlcNAcylation pattern in skeletal muscle changed, depending on these different conditions and fiber types. O-GlcNAcylation may be both a cause or a consequence of the modulation of the glucose utilization, in a virtuous or deleterious cycle, since most of the enzymes from the carbohydrate metabolism are known to be O-GlcNAcylated. This concept clearly raises the O-GlcNAcylation as a potential key regulator of the skeletal muscle glucose metabolism. O-GlcNAcylation has been also shown to regulate some key signaling pathways, as well cellular stress response, involved in the maintenance of the protein synthesis/degradation balance in the skeletal muscle. From these novel insights, a new paradigm is emerging considering the O-GlcNAcylation as a key factor involved in skeletal muscle physiopathology such as atrophy or insulin resistance, and more generally in neuromuscular diseases. However, O-GlcNAc involvements as a cause or consequence of skeletal muscle impairments are currently in debate. After all, it is now clear that O-GlcNAcylation is getting many involvements in the skeletal muscle physiopathology and would be confirmed in the future by larger studies of interest.
Indeed, with the exponential development of mass-spectrometry and innovative enrichment techniques, the identification of O-GlcNAc sites which are modulated subsequently to any stimuli/condition/disease will be clearly the challenge of tomorrow. In fact, O-GlcNAcylation regulates protein activity, protein localization, protein-protein interactions, and can interplay with phosphorylation or ubiquitination. This strategy will lead to a deeper understanding of the precise mechanisms by which O-GlcNAcylation can regulate skeletal muscle metabolism. Secondly, O-GlcNAc processing enzymes behavior should be more precisely investigated, since their localizations and/or interactions, and as consequence, the pattern of O-GlcNAcylation, seemed to be changed through different stimuli in skeletal muscle fibers, especially around the myofilaments. Exercise is one of these stimuli inducing complex O-GlcNAc variations, depending on the muscle phenotype, but also the kind of exercise. Importantly, due to the enhancement of glucose utilization during exercise, the O-GlcNAcylation process in skeletal muscle could be considered as a potential target to alleviate metabolic disorders. Finally, O-GlcNAcylation should be investigated in some precise muscular dystrophies or congenital myopathies, since glucose utilization is often impaired, the sarcomere could be disorganized, the mitochondria biogenesis altered, the nuclei delocalized, or the muscle plasticity changed. It will be worth knowing if O-GlcNAcylation could contribute or alleviate neuromuscular disorders or being considered as a marker of these diseases.
Author Contributions
All authors listed have made a substantial, direct and intellectual contribution to the work, and approved it for publication.
Funding
This work was supported by grant from the French National Research Agency (ANR, Agence Nationale de la Recherche, Young Researchers Program, n°11JSV8 006 01). ML is a recipient from the French Ministry for Research and Tertiary Education.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1. Torres CR, Hart GW. Topography and polypeptide distribution of terminal N-acetylglucosamine residues on the surfaces of intact lymphocytes. Evidence for O-linked GlcNAc. J Biol Chem. (1984) 259:3308–17.
2. Ma J, Hart GW. O-GlcNAc profiling: from proteins to proteomes. Clin Proteomics (2014) 11:8. doi: 10.1186/1559-0275-11-8
3. Shafi R, Iyer SP, Ellies LG, O'Donnell N, Marek KW, Chui D, et al. The O-GlcNAc transferase gene resides on the X chromosome and is essential for embryonic stem cell viability and mouse ontogeny. Proc Natl Acad Sci USA (2000) 97:5735–9. doi: 10.1073/pnas.100471497
4. Yang YR, Song M, Lee H, Jeon Y, Choi E-J, Jang H-J, et al. O-GlcNAcase is essential for embryonic development and maintenance of genomic stability. Aging Cell (2012) 11:439–48. doi: 10.1111/j.1474-9726.2012.00801.x
5. Ida S, Morino K, Sekine O, Ohashi N, Kume S, Chano T, et al. Diverse metabolic effects of O-GlcNAcylation in the pancreas but limited effects in insulin-sensitive organs in mice. Diabetologia (2017) 60:1761–9. doi: 10.1007/s00125-017-4327-y
6. Hart GW, Housley MP, Slawson C. Cycling of O-linked β-N-acetylglucosamine on nucleocytoplasmic proteins. Nature (2007) 446:1017–22. doi: 10.1038/nature05815
7. Gu Y, Ande SR, Mishra S. Altered O-GlcNAc modification and phosphorylation of mitochondrial proteins in myoblast cells exposed to high glucose. Arch Biochem Biophys. (2011) 505:98–104. doi: 10.1016/j.abb.2010.09.024
8. Haltiwanger R. S., Holt G. D., Hart G. W. Enzymatic addition of O-GlcNAc to Nuclear and Cytoplasmic Proteins. J Biol Chem. (1990) 265:2563–8.
9. Dong DL, Hart GW. Purification and characterization of an O-GlcNAc selective N-acetyl-beta-D-glucosaminidase from rat spleen cytosol. J Biol Chem. (1994) 269:19321–30.
10. Nagnan-Le Meillour P, Vercoutter-Edouart A-S, Hilliou F, Le Danvic C, Levy F. Proteomic analysis of Pig (Sus scrofa) olfactory soluble proteome reveals O-Linked-N-acetylglucosaminylation of secreted odorant-binding proteins. Front Endocrinol. (2014) 5:202. doi: 10.3389/fendo.2014.00202
11. Ogawa M, Sawaguchi S, Furukawa K, Okajima T. N-acetylglucosamine modification in the lumen of the endoplasmic reticulum. Biochim Biophys Acta (2015) 1850:1319–24. doi: 10.1016/j.bbagen.2015.03.003
12. Varshney S, Stanley P. EOGT and O -GlcNAc on secreted and membrane proteins. Biochem Soc Trans. (2017) 45:401–8. doi: 10.1042/BST20160165
13. Hanover J a, Krause MW, Love DC. The hexosamine signaling pathway: O-GlcNAc cycling in feast or famine. Biochim Biophys Acta (2010) 1800:80–95. doi: 10.1016/j.bbagen.2009.07.017
14. Hart GW, Akimoto Y. The O-GlcNAc Modification. In: Varki A, Cummings RD, Esko JD, Freeze HH, Stanley P, Bertozzi CR, Hart GW, Etzler ME, editors. Essentials of Glycobiology, 2nd edition. Cold Spring Harbor, NY: Cold Spring Harbor Laboratory Press (2009). Chapter 18. (2009).
15. Hart GW, Slawson C, Ramirez-Correa G, Lagerlof O. Cross talk between O-GlcNAcylation and phosphorylation: roles in signaling, transcription, and chronic disease. Annu Rev Biochem. (2011) 80:825–58. doi: 10.1146/annurev-biochem-060608-102511
16. van der Laarse SAM, Leney AC, Heck AJR. Crosstalk between phosphorylation and O-GlcNAcylation: friend or foe. FEBS J. (2018) 285:3152–67. doi: 10.1111/febs.14491
17. Ruan H-B, Nie Y, Yang X. Regulation of protein degradation by O-GlcNAcylation: crosstalk with ubiquitination. Mol Cell Proteomics (2013) 12:3489–97. doi: 10.1074/mcp.R113.029751
18. Lambert M, Richard E, Duban-Deweer S, Krzewinski F, Deracinois B, Dupont E, et al. O-GlcNAcylation is a key modulator of skeletal muscle sarcomeric morphometry associated to modulation of protein–protein interactions. Biochim Biophys Acta Gen Subj. (2016) 1860:2017–30. doi: 10.1016/j.bbagen.2016.06.011
19. Tarbet HJ, Toleman CA, Boyce M. A sweet embrace: control of protein–protein interactions by O-Linked β- N -Acetylglucosamine. Biochemistry (2018) 57:13–21. doi: 10.1021/acs.biochem.7b00871
20. Chu C-S, Lo P-W, Yeh Y-H, Hsu P-H, Peng S-H, Teng Y-C, et al. O-GlcNAcylation regulates EZH2 protein stability and function. Proc Natl Acad Sci. (2014) 111:1355–60. doi: 10.1073/pnas.1323226111
21. Li Y, Wang L, Liu J, Zhang P, An M, Han C, et al. O-GlcNAcylation modulates Bmi-1 protein stability and potential oncogenic function in prostate cancer. Oncogene (2017) 36:6293–305. doi: 10.1038/onc.2017.223
22. Cividini F, Scott BT, Dai A, Han W, Suarez J, Diaz-Juarez J, et al. O -GlcNAcylation of 8-Oxoguanine DNA Glycosylase (Ogg1) Impairs Oxidative Mitochondrial DNA Lesion Repair in Diabetic Hearts. J Biol Chem. (2016) 291:26515–28. doi: 10.1074/jbc.M116.754481
23. Ha JR, Hao L, Venkateswaran G, Huang YH, Garcia E, Persad S. β-Catenin is O-GlcNAc glycosylated at Serine 23: Implications for β-catenin's subcellular localization and transactivator function. Exp Cell Res. (2014) 321:153–66. doi: 10.1016/j.yexcr.2013.11.021
24. Hardivillé S, Hart GW. Nutrient regulation of signaling, transcription, and cell physiology by O- GlcNAcylation. Cell Metab. (2014) 20:208–13. doi: 10.1016/j.cmet.2014.07.014
25. Bond MR, Hanover J a. A little sugar goes a long way: the cell biology of O-GlcNAc. J Cell Biol. (2015) 208:869–80. doi: 10.1083/jcb.201501101
26. Ma J, Hart GW. Protein O-GlcNAcylation in diabetes and diabetic complications. Expert Rev Proteom. (2013) 10:365–80. doi: 10.1586/14789450.2013.820536
27. Peterson SB, Hart GW. New insights: A role for O-GlcNAcylation in diabetic complications. Crit Rev Biochem Mol Biol. (2016) 9238:1–12. doi: 10.3109/10409238.2015.1135102
28. Fardini Y, Dehennaut V, Lefebvre T, Issad T. O-GlcNAcylation: A new cancer hallmark? Front Endocrinol. (2013) 4:99. doi: 10.3389/fendo.2013.00099
29. de Queiroz RM, Carvalho Ã, Dias WB. O-GlcNAcylation: the sweet side of the cancer. Front Oncol. (2014) 4:132. doi: 10.3389/fonc.2014.00132
30. Zhu Y, Shan X, Yuzwa SA, Vocadlo DJ. The emerging link between O -GlcNAc and Alzheimer Disease. J Biol Chem. (2014) 289:34472–81. doi: 10.1074/jbc.R114.601351
31. Gong C-X, Liu F, Iqbal K. O-GlcNAcylation: a regulator of tau pathology and neurodegeneration. Alzheimer's Dement (2016) 12:1078–89. doi: 10.1016/j.jalz.2016.02.011
32. Vaidyanathan K, Niranjan T, Selvan N, Teo CF, May M, Patel S, et al. Identification and characterization of a missense mutation in the O -linked β- N -acetylglucosamine ( O -GlcNAc) transferase gene that segregates with X-linked intellectual disability. J Biol Chem. (2017) 292:8948–63. doi: 10.1074/jbc.M116.771030
33. Nakamura S, Nakano S, Nishii M, Kaneko S, Kusaka H. Localization of O-GlcNAc-modified proteins in neuromuscular diseases. Med Mol Morphol. (2012) 45:86–90. doi: 10.1007/s00795-011-0542-7
34. Dassanayaka S, Jones SP. O-GlcNAc and the cardiovascular system. Pharmacol Ther. (2014) 142:62–71. doi: 10.1016/j.pharmthera.2013.11.005
35. Wright JN, Collins HE, Wende AR, Chatham JC. O-GlcNAcylation and cardiovascular disease. Biochem Soc Trans. (2017) 45:545–53. doi: 10.1042/BST20160164
36. Banerjee PS, Lagerlöf O, Hart GW. Roles of O-GlcNAc in chronic diseases of aging. Mol Aspects Med. (2016) 51:1–15. doi: 10.1016/j.mam.2016.05.005
37. Frontera WR, Ochala J. Skeletal muscle: a brief review of structure and function. Calcif Tissue Int. (2015) 96:183–95. doi: 10.1007/s00223-014-9915-y
38. Westerblad H, Bruton JD, Katz A. Skeletal muscle: energy metabolism, fiber types, fatigue and adaptability. Exp Cell Res. (2010) 316:3093–9. doi: 10.1016/j.yexcr.2010.05.019
39. Hart GW. O-GlcNAcylation: Nutrient Sensor that Regulates Cell Physiology. In: Taniguchi N, Endo T, Hart G, Seeberger P, Wong CH, editors. Glycoscience: Biology and Medicine. Springer: Tokyo (2015) p. 1193–9. doi: 10.1007/978-4-431-54841-6_82
40. Cieniewski-Bernard C, Bastide B, Lefebvre T, Lemoine J, Mounier Y, Michalski J-C. Identification of O-linked N-acetylglucosamine proteins in rat skeletal muscle using two-dimensional gel electrophoresis and mass spectrometry. Mol Cell Proteomics (2004) 3:577–85. doi: 10.1074/mcp.M400024-MCP200
41. Cieniewski-Bernard C, Montel V, Berthoin S, Bastide B. Increasing O-GlcNAcylation level on organ culture of soleus modulates the calcium activation parameters of muscle fibers. PLoS ONE (2012) 7:e48218. doi: 10.1371/journal.pone.0048218
42. Hedou J, Cieniewski-Bernard C, Leroy Y, Michalski JC, Mounier Y, Bastide B. O-linked N-acetylglucosaminylation is involved in the Ca2+ activation properties of rat skeletal muscle. J Biol Chem. (2007) 282:10360–9. doi: 10.1074/jbc.M606787200
43. Hédou J, Bastide B, Page A, Michalski JC, Morelle W. Mapping of O-linked beta-N-acetylglucosamine modification sites in key contractile proteins of rat skeletal muscle. Proteomics (2009) 9:2139–48. doi: 10.1002/pmic.200800617
44. Kooij V, Stienen GJM, van der Velden J. The role of protein kinase C-mediated phosphorylation of sarcomeric proteins in the heart-detrimental or beneficial? Biophys Rev. (2011) 3:107–17. doi: 10.1007/s12551-011-0050-y
45. Mounier R, Théret M, Lantier L, Foretz M, Viollet B. Expanding roles for AMPK in skeletal muscle plasticity. Trends Endocrinol Metab. (2015) 26:275–86. doi: 10.1016/j.tem.2015.02.009
46. Egan B, Zierath JR. Exercise metabolism and the molecular regulation of skeletal muscle adaptation. Cell Metab. (2013) 17:162–84. doi: 10.1016/j.cmet.2012.12.012
47. Smiles WJ, Hawley JA, Camera DM. Effects of skeletal muscle energy availability on protein turnover responses to exercise. J Exp Biol. (2016) 219:214–25. doi: 10.1242/jeb.125104
48. Goodpaster BH, Sparks LM. Metabolic flexibility in health and disease. Cell Metab. (2017) 25:1027–36. doi: 10.1016/j.cmet.2017.04.015
49. Marshall S, Bacote V, Traxinger RR. Discovery of a metabolic pathway mediating glucose-induced desensitization of the glucose transport system. Role of hexosamine biosynthesis in the induction of insulin resistance. J Biol Chem. (1991) 266:4706–12.
50. Hawkins M, Barzilai N, Liu R, Hu M, Chen W, Rossetti L. Role of the glucosamine pathway in fat-induced insulin resistance. J Clin Invest. (1997) 99:2173–82. doi: 10.1172/JCI119390
51. Lee AD, Hansen PA, Holloszy JO. Wortmannin inhibits insulin-stimulated but not contraction-stimulated glucose transport activity in skeletal muscle. FEBS Lett. (1995) 361:51–4.
52. Richter EA, Hargreaves M. Exercise, GLUT4, and skeletal muscle glucose uptake. Physiol Rev. (2013) 93:993–1017. doi: 10.1152/physrev.00038.2012
53. Ojuka EO, Goyaram V, Smith JAH. The role of CaMKII in regulating GLUT4 expression in skeletal muscle. Am J Physiol Metab. (2012) 303:E322–31. doi: 10.1152/ajpendo.00091.2012
54. Myslicki JP, Belke DD, Shearer J. Role of O-GlcNAcylation in nutritional sensing, insulin resistance and in mediating the benefits of exercise. Appl Physiol Nutr Metab. (2014) 39:1205–13. doi: 10.1139/apnm-2014-0122
55. Belke DD. Swim-exercised mice show a decreased level of protein O-GlcNAcylation and expression of O-GlcNAc transferase in heart. J Appl Physiol. (2011) 111:157–62. doi: 10.1152/japplphysiol.00147.2011
56. Medford HM, Porter K, Marsh SA. Immediate effects of a single exercise bout on protein O-GlcNAcylation and chromatin regulation of cardiac hypertrophy. Am J Physiol Heart Circ Physiol. (2013) 305:H114–23. doi: 10.1152/ajpheart.00135.2013
57. Bennett CE, Johnsen VL, Shearer J, Belke DD. Exercise training mitigates aberrant cardiac protein O-GlcNAcylation in streptozotocin-induced diabetic mice. Life Sci. (2013) 92:657–63. doi: 10.1016/j.lfs.2012.09.007
58. Johnsen VL, Belke DD, Hughey CC, Hittel DS, Hepple RT, Koch LG, et al. Enhanced cardiac protein glycosylation (O-GlcNAc) of selected mitochondrial proteins in rats artificially selected for low running capacity. Physiol Genomics (2013) 45:17–25. doi: 10.1152/physiolgenomics.00111.2012
59. Cox EJ, Marsh SA. Exercise and diabetes have opposite effects on the assembly and O-GlcNAc modification of the mSin3A/HDAC1/2 complex in the heart. Cardiovasc Diabetol. (2013) 12:101. doi: 10.1186/1475-2840-12-101
60. Peternelj TT, Marsh SA, Strobel NA, Matsumoto A, Briskey D, Dalbo VJ, et al. Glutathione depletion and acute exercise increase O-GlcNAc protein modification in rat skeletal muscle. Mol Cell Biochem. (2014) 400:265–75. doi: 10.1007/s11010-014-2283-0
61. Hortemo KH, Aronsen JM, Lunde IG, Sjaastad I, Lunde PK, Sejersted OM. Exhausting treadmill running causes dephosphorylation of sMLC2 and reduced level of myofilament MLCK2 in slow twitch rat soleus muscle. Physiol Rep. (2015) 3:e12285. doi: 10.14814/phy2.12285
62. Halvorsen Hortemo K, Lunde K, Anonsen JH, Kvaløy H, Munkvik M, Rehn TA, et al. Exercise training increases protein O-GlcNAcylation in rat skeletal muscle. Physiol Rep. (2016) 4:e12896 doi: 10.14814/phy2.12896
63. Nelson BA, Robinson KA, Koning JS, Buse MG. Effects of exercise and feeding on the hexosamine biosynthetic pathway in rat skeletal muscle. Am J Physiol. (1997) 272:E848–55. doi: 10.1152/ajpendo.1997.272.5.E848
64. Cieniewski-Bernard C, Mounier Y, Michalski JC, Bastide B. O-GlcNAc level variations are associated with the development of skeletal muscle atrophy. J Appl Physiol. (2006) 100:1499–505. doi: 10.1152/japplphysiol.00865.2005
65. Shi H, Munk A, Nielsen TS, Daughtry MR, Larsson L, Li S, et al. Skeletal muscle O-GlcNAc transferase is important for muscle energy homeostasis and whole-body insulin sensitivity. Mol Metab. (2018) 11:160–77. doi: 10.1016/j.molmet.2018.02.010
66. Davies KJ, Quintanilha AT, Brooks GA, Packer L. Free radicals and tissue damage produced by exercise. Biochem Biophys Res Commun. (1982) 107:1198–205.
67. Groves JA, Lee A, Yildirir G, Zachara NE. Dynamic O-GlcNAcylation and its roles in the cellular stress response and homeostasis. Cell Stress Chaperones (2013) 18:535–58. doi: 10.1007/s12192-013-0426-y
68. Lima VV, Spitler K, Choi H, Webb RC, Tostes RC, Varki A, et al. O-GlcNAcylation and oxidation of proteins: is signalling in the cardiovascular system becoming sweeter? Clin Sci. (2012) 123:473–86. doi: 10.1042/CS20110638
69. Chen P-H, Chi J-T, Boyce M. Functional crosstalk among oxidative stress and O-GlcNAc signaling pathways. Glycobiology (2018) 28:556–64. doi: 10.1093/glycob/cwy027
70. Murata K, Morino K, Ida S, Ohashi N, Lemecha M, Park S-Y, et al. Lack of O-GlcNAcylation enhances exercise-dependent glucose utilization potentially through AMP-activated protein kinase activation in skeletal muscle. Biochem Biophys Res Commun. (2018) 495:2098–104. doi: 10.1016/j.bbrc.2017.12.081
71. Yi W, Clark PM, Mason DE, Keenan MC, Hill C, Goddard WA, et al. Phosphofructokinase 1 glycosylation regulates cell growth and metabolism. Science (2012) 337:975–80. doi: 10.1126/science.1222278
72. Champattanachai V, Netsirisawan P, Chaiyawat P, Phueaouan T, Charoenwattanasatien R, Chokchaichamnankit D, et al. Proteomic analysis and abrogated expression of O -GlcNAcylated proteins associated with primary breast cancer. Proteomics (2013) 13:2088–99. doi: 10.1002/pmic.201200126
73. Chaiyawat P, Chokchaichamnankit D, Lirdprapamongkol K, Srisomsap C, Svasti J, Champattanachai V. Alteration of O-GlcNAcylation affects serine phosphorylation and regulates gene expression and activity of pyruvate kinase M2 in colorectal cancer cells. Oncol Rep (2015) 34:1933–42. doi: 10.3892/or.2015.4178
74. Dehennaut V, Slomianny MC, Page A, Vercoutter-Edouart A-S, Jessus C, Michalski J-C, et al. Identification of Structural and Functional O -Linked N -Acetylglucosamine-bearing Proteins in Xenopus laevis Oocyte. Mol Cell Proteomics (2008) 7:2229–45. doi: 10.1074/mcp.M700494-MCP200
75. Wells L, Vosseller K, Cole RN, Cronshaw JM, Matunis MJ, Hart GW. Mapping sites of O-GlcNAc modification using affinity tags for serine and threonine post-translational modifications. Mol Cell Proteomics (2002) 1:791–804. doi: 10.1074/mcp.M200048-MCP200
76. Clark PM, Dweck JF, Mason DE, Hart CR, Buck SB, Peters EC, et al. Direct in-gel fluorescence detection and cellular imaging of O -GlcNAc-modified proteins. J Am Chem Soc. (2008) 130:11576–7. doi: 10.1021/ja8030467
77. Bacigalupa ZA, Bhadiadra CH, Reginato MJ. O-GlcNAcylation: key regulator of glycolytic pathways. J Bioenerg Biomembr. (2018) 50:189–98. doi: 10.1007/s10863-018-9742-3
78. Sharma NS, Saluja AK, Banerjee S. “Nutrient-sensing” and self-renewal: O-GlcNAc in a new role. J Bioenerg Biomembr. (2018) 50:205–11. doi: 10.1007/s10863-017-9735-7
79. Baldini SF, Steenackers A, Olivier-Van Stichelen S, Mir A-M, Mortuaire M, Lefebvre T, et al. Glucokinase expression is regulated by glucose through O-GlcNAc glycosylation. Biochem Biophys Res Commun. (2016) 478:942–8. doi: 10.1016/j.bbrc.2016.08.056
80. Ma J, Banerjee P, Whelan SA, Liu T, Wei A-C, Ramirez-Correa G, et al. Comparative Proteomics Reveals Dysregulated Mitochondrial O-GlcNAcylation in Diabetic Hearts. J Proteome Res. (2016) 15:2254–64. doi: 10.1021/acs.jproteome.6b00250
81. Cao W, Cao J, Huang J, Yao J, Yan G, Xu H, Yang P. Discovery and confirmation of O-GlcNAcylated proteins in rat liver mitochondria by combination of mass spectrometry and immunological methods. PLoS ONE (2013) 8:e76399. doi: 10.1371/journal.pone.0076399
82. Barclay CJ. Energy demand and supply in human skeletal muscle. J Muscle Res Cell Motil. (2017) 38:143–55. doi: 10.1007/s10974-017-9467-7
83. Rakus D, Mamczur P, Gizak A, Dus D, Dzugaj A. Colocalization of muscle FBPase and muscle aldolase on both sides of the Z-line. Biochem Biophys Res Commun. (2003) 311:294–9. doi: 10.1016/j.bbrc.2003.09.209
84. Mamczur P, Rakus D, Gizak A, Dus D, Dzugaj A. The effect of calcium ions on subcellular localization of aldolase-FBPase complex in skeletal muscle. FEBS Lett. (2005) 579:1607–12. doi: 10.1016/j.febslet.2005.01.071
85. Clarke FM, Masters CJ. Interactions between muscle proteins and glycolytic enzymes. Int J Biochem. (1976) 7:359–65. doi: 10.1016/0020-711X(76)90058-6
86. Méjean C, Pons F, Benyamin Y, Roustan C. Antigenic probes locate binding sites for the glycolytic enzymes glyceraldehyde-3-phosphate dehydrogenase, aldolase and phosphofructokinase on the actin monomer in microfilaments. Biochem J. (1989) 264:671–7.
87. Klein AL, Berkaw MN, Buse MG, Ball LE. O -Linked N -Acetylglucosamine modification of insulin receptor substrate-1 occurs in close proximity to multiple SH2 domain binding motifs. Mol Cell Proteomics (2009) 8:2733–45. doi: 10.1074/mcp.M900207-MCP200
88. Yang X, Ongusaha PP, Miles PD, Havstad JC, Zhang F, So WV, et al. Phosphoinositide signalling links O-GlcNAc transferase to insulin resistance. Nature (2008) 451:964–9. doi: 10.1038/nature06668
89. Whelan SA, Lane MD, Hart GW. Regulation of the O-linked beta-N-acetylglucosamine transferase by insulin signaling. J Biol Chem. (2008) 283:21411–7. doi: 10.1074/jbc.M800677200
90. Yang X, Qian K. Protein O-GlcNAcylation: emerging mechanisms and functions. Nat Rev Mol Cell Biol. (2017) 18:452–65. doi: 10.1038/nrm.2017.22
91. Parker GJ, Lund KC, Taylor RP, McClain DA. Insulin resistance of glycogen synthase mediated by o-linked N-acetylglucosamine. J Biol Chem. (2003) 278:10022–7. doi: 10.1074/jbc.M207787200
92. Parker G, Taylor R, Jones D, McClain D. Hyperglycemia and inhibition of glycogen synthase in streptozotocin-treated mice: role of O-linked N-acetylglucosamine. J Biol Chem. (2004) 279:20636–42. doi: 10.1074/jbc.M312139200
93. Cieniewski-Bernard C, Dupont E, Richard E, Bastide B. Phospho-GlcNAc modulation of slow MLC2 during soleus atrophy through a multienzymatic and sarcomeric complex. Pflügers Arch Eur J Physiol. (2014) 466:2139–51. doi: 10.1007/s00424-014-1453-y
94. Bonaldo P, Sandri M, Allen DL, Unterman TG, Amirouche A, Durieux AC, et al. Cellular and molecular mechanisms of muscle atrophy. Dis Model Mech. (2013) 6:25–39. doi: 10.1242/dmm.010389
95. Schiaffino S, Dyar KA, Ciciliot S, Blaauw B, Sandri M. Mechanisms regulating skeletal muscle growth and atrophy. FEBS J. (2013) 280:4294–314. doi: 10.1111/febs.12253
96. Zhang F, Su K, Yang X, Bowe DB, Paterson AJ, Kudlow JE. O-GlcNAc modification is an endogenous inhibitor of the proteasome. Cell (2003) 115:715–25. doi: 10.1016/S0092-8674(03)00974-7
97. Özcan S, Andrali SS, Cantrell JEL. Modulation of transcription factor function by O-GlcNAc modification. Biochim Biophys Acta Gene Regul Mech. (2010) 1799:353–64. doi: 10.1016/j.bbagrm.2010.02.005
98. Walgren JLE, Vincent TS, Schey KL, Buse MG. High glucose and insulin promote O-GlcNAc modification of proteins, including alpha-tubulin. Am J Physiol Endocrinol Metab. (2003) 284:E424–34. doi: 10.1152/ajpendo.00382.2002
99. Guinez C, Lemoine J, Michalski J-C, Lefebvre T. 70-kDa-heat shock protein presents an adjustable lectinic activity towards O-linked N-acetylglucosamine. Biochem Biophys Res Commun. (2004) 319:21–26. doi: 10.1016/j.bbrc.2004.04.144
100. Guinez C, Losfeld M-E, Cacan R, Michalski J-C, Lefebvre T. Modulation of HSP70 GlcNAc-directed lectin activity by glucose availability and utilization. Glycobiology (2006) 16:22–8. doi: 10.1093/glycob/cwj041
101. Zachara NE, O'Donnell N, Cheung WD, Mercer JJ, Marth JD, Hart GW. Dynamic O-GlcNAc modification of nucleocytoplasmic proteins in response to stress. A survival response of mammalian cells. J Biol Chem. (2004) 279:30133–42. doi: 10.1074/jbc.M403773200
102. Guinez C, Mir A-M, Leroy Y, Cacan R, Michalski J-C, Lefebvre T. Hsp70-GlcNAc-binding activity is released by stress, proteasome inhibition, and protein misfolding. Biochem Biophys Res Commun. (2007) 361:414–20. doi: 10.1016/j.bbrc.2007.07.020
103. Gong J, Jing L. Glutamine induces heat shock protein 70 expression via O-GlcNAc modification and subsequent increased expression and transcriptional activity of heat shock factor-1. Minerva Anestesiol (2011) 77:488–95.
104. Naito H, Powers SK, Demirel HA, Sugiura T, Dodd SL, Aoki J. Heat stress attenuates skeletal muscle atrophy in hindlimb-unweighted rats. J Appl Physiol. (2000) 88:359–63. doi: 10.1152/jappl.2000.88.1.359
105. Roquemore EP, Chevrier MR, Cotter RJ, Hart GW. Dynamic O-GlcNAcylation of the small heat shock protein alpha B-crystallin. Biochemistry (1996) 35:3578–86. doi: 10.1021/bi951918j
106. Atomi Y, Yamada S, Hong YM. Dynamic expression of ALPHA.B-crystallin in skeletal muscle effects of unweighting, passive stretch and denervation. Proc Japan Acad Ser B Phys Biol Sci. (1990) 66:203–8. doi: 10.2183/pjab.66.203
107. Massaccesi L, Goi G, Tringali C, Barassi A, Venerando B, Papini N. Dexamethasone-induced skeletal muscle atrophy increases O-GlcNAcylation in C2C12 Cells. J Cell Biochem. (2016) 117:1833–42. doi: 10.1002/jcb.25483
108. Lokireddy S, McFarlane C, Ge X, Zhang H, Sze SK, Sharma M, et al. Myostatin induces degradation of sarcomeric proteins through a Smad3 signaling mechanism during skeletal muscle wasting. Mol Endocrinol. (2011) 25:1936–49. doi: 10.1210/me.2011-1124
109. Braun TP, Marks DL. The regulation of muscle mass by endogenous glucocorticoids. Front Physiol. (2015) 6:12. doi: 10.3389/fphys.2015.00012
110. Ratman D, Vanden Berghe W, Dejager L, Libert C, Tavernier J, Beck IM, et al. How glucocorticoid receptors modulate the activity of other transcription factors: a scope beyond tethering. Mol Cell Endocrinol. (2013) 380:41–54. doi: 10.1016/j.mce.2012.12.014
111. Li M-D, Ruan H-B, Singh JP, Zhao L, Zhao T, Azarhoush S, Wu J, Evans RM, Yang X. O-GlcNAc transferase is involved in glucocorticoid receptor-mediated transrepression. J Biol Chem. (2012) 287:12904–12. doi: 10.1074/jbc.M111.303792
112. Wong BL, Rybalsky I, Shellenbarger KC, Tian C, McMahon MA, Rutter MM, et al. Long-term outcome of interdisciplinary management of patients with duchenne muscular dystrophy receiving daily glucocorticoid treatment. J Pediatr. (2017) 182:296–303.e1. doi: 10.1016/j.jpeds.2016.11.078
113. Ogawa M, Mizofuchi H, Kobayashi Y, Tsuzuki G, Yamamoto M, Wada S, et al. Terminal differentiation program of skeletal myogenesis is negatively regulated by O-GlcNAc glycosylation. Biochim Biophys Acta - Gen Subj. (2012) 1820:24–32. doi: 10.1016/j.bbagen.2011.10.011
114. Ogawa M, Sakakibara Y, Kamemura K. Requirement of decreased O-GlcNAc glycosylation of Mef2D for its recruitment to the myogenin promoter. Biochem Biophys Res Commun. (2013) 433:558–562. doi: 10.1016/j.bbrc.2013.03.033
115. Wang X, Feng Z, Wang X, Yang L, Han S, Cao K, et al. O-GlcNAcase deficiency suppresses skeletal myogenesis and insulin sensitivity in mice through the modulation of mitochondrial homeostasis. Diabetologia (2016) 59:1287–96. doi: 10.1007/s00125-016-3919-2
116. Huang P, Ho SR, Wang K, Roessler BC, Zhang F, Hu Y, et al. Muscle-specific overexpression of NCOATGK, splice variant of O-GlcNAcase, induces skeletal muscle atrophy. Am J Physiol Cell Physiol. (2011) 300:C456–65. doi: 10.1152/ajpcell.00124.2010
117. Nagel AK, Ball LE. Intracellular protein O-GlcNAc modification integrates nutrient status with transcriptional and metabolic regulation. Adv Cancer Res. 126 : 137–66. doi: 10.1016/bs.acr.2014.12.003
118. Qin CX, Sleaby R, Davidoff AJ, Bell JR, De Blasio MJ, Delbridge LM, et al. Insights into the role of maladaptive hexosamine biosynthesis and O-GlcNAcylation in development of diabetic cardiac complications. Pharmacol Res. (2017) 116:45–56. doi: 10.1016/j.phrs.2016.12.016
119. Lehman DM, Fu D-J, Freeman AB, Hunt KJ, Leach RJ, Johnson-Pais T, et al. A single nucleotide polymorphism in MGEA5 encoding O-GlcNAc-selective N-acetyl-beta-D glucosaminidase is associated with type 2 diabetes in Mexican Americans. Diabetes (2005) 54:1214–21. doi: 10.2337/diabetes.54.4.1214
120. Toleman C, Paterson AJ, Whisenhunt TR, Kudlow JE. Characterization of the Histone Acetyltransferase (HAT) domain of a bifunctional protein with activable O-GlcNAcase and HAT activities. J Biol Chem. (2004) 279:53665–73. doi: 10.1074/jbc.M410406200
121. Keembiyehetty C, Love DC, Harwood KR, Gavrilova O, Comly ME, Hanover JA. Conditional knock-out reveals a requirement for O-linked N-Acetylglucosaminase (O-GlcNAcase) in metabolic homeostasis. J Biol Chem. (2015) 290:7097–113. doi: 10.1074/jbc.M114.617779
122. MacAuley MS, He Y, Gloster TM, Stubbs KA, Davies GJ, Vocadlo DJ. Inhibition of O-GlcNAcase using a potent and cell-permeable inhibitor does not induce insulin resistance in 3T3-L1 adipocytes. Chem Biol. (2010) 17:937–48. doi: 10.1016/j.chembiol.2010.07.006
123. Arias EB, Kim J, Cartee GD. Prolonged incubation in PUGNAc results in increased protein O-Linked glycosylation and insulin resistance in rat skeletal muscle. Diabetes (2004) 53:921–30. doi: 10.2337/diabetes.53.4.921
124. Yki-Järvinen H, Virkamäki A, Daniels MC, McClain D, Gottschalk WK. Insulin and glucosamine infusions increase O-linked N-acetyl-glucosamine in skeletal muscle proteins in vivo. Metabolism (1998) 47:449–55. doi: 10.1016/S0026-0495(98)90058-0
125. Hebert LF, Daniels MC, Zhou J, Crook ED, Turner RL, Simmons ST, et al. Overexpression of glutamine:fructose-6-phosphate amidotransferase in transgenic mice leads to insulin resistance. J Clin Invest. (1996) 98:930–6. doi: 10.1172/JCI118876
126. Hazel M, Cooksey RC, Jones D, Parker G, Neidigh JL, Witherbee B, et al. Activation of the hexosamine signaling pathway in adipose tissue results in decreased serum adiponectin and skeletal muscle insulin resistance. Endocrinology (2004) 145:2118–28. doi: 10.1210/en.2003-0812
127. Yki-Järvinen H, Daniels MC, Virkamäki A, Mäkimattila S, DeFronzo RA, McClain D. Increased glutamine:fructose-6-phosphate amidotransferase activity in skeletal muscle of patients with NIDDM. Diabetes (1996) 45:302–7.
128. Cooksey RC, Hebert LF, Zhu J-H, Wofford P, Garvey WT, McClain DA. Mechanism of hexosamine-induced insulin resistance in transgenic mice overexpressing glutamine:fructose-6-phosphate amidotransferase: decreased glucose transporter GLUT4 translocation and reversal by treatment with thiazolidinedione. Endocrinology (1999) 140:1151–7. doi: 10.1210/endo.140.3.6563
129. Buse MG, Robinson KA, Marshall BA, Mueckler M. Differential effects of GLUT1 or GLUT4 overexpression on hexosamine biosynthesis by muscles of transgenic mice. J Biol Chem. (1996) 271:23197–202.
130. Buse MG, Robinson KA, Marshall BA, Hresko RC, Mueckler MM. Enhanced O -GlcNAc protein modification is associated with insulin resistance in GLUT1-overexpressing muscles. Am J Physiol Endocrinol Metab. (2002) 283:E241–50. doi: 10.1152/ajpendo.00060.2002
131. Buse MG. Hexosamines, insulin resistance, and the complications of diabetes: current status. Am J Physiol Endocrinol Metab. (2006) 290:E1–8. doi: 10.1152/ajpendo.00329.2005
132. Zhang W, Liu J, Tian L, Liu Q, Fu Y, Garvey WT. TRIB3 mediates glucose-induced insulin resistance via a mechanism that requires the hexosamine biosynthetic pathway. Diabetes (2013) 62:4192–200. doi: 10.2337/db13-0312
133. McClain DA, Lubas WA, Cooksey RC, Hazel M, Parker GJ, Love DC, et al. Altered glycan-dependent signaling induces insulin resistance and hyperleptinemia. Proc Natl Acad Sci USA (2002) 99:10695–9. doi: 10.1073/pnas.152346899
134. D'Souza DM, Al-Sajee D, Hawke TJ. Diabetic myopathy: impact of diabetes mellitus on skeletal muscle progenitor cells. Front Physiol. (2013) 4:379. doi: 10.3389/fphys.2013.00379
135. Ramirez-Correa GA, Jin W, Wang Z, Zhong X, Gao WD, Dias WB, et al. O-linked GlcNAc modification of cardiac myofilament proteins: a novel regulator of myocardial contractile function. Circ Res. (2008) 103:1354–8. doi: 10.1161/CIRCRESAHA.108.184978
136. Hu Y, Belke D, Suarez J, Swanson E, Clark R, Hoshijima M, Dillmann WH. Adenovirus-mediated overexpression of O-GlcNAcase improves contractile function in the diabetic heart. Circ Res. (2005) 96:1006–13. doi: 10.1161/01.RES.0000165478.06813.58
137. Ramirez-Correa GA, MA J, Slawson C, Zeidan Q, Lugo-Fagundo NS, Xu M, et al. Removal of abnormal myofilament O-GlcNAcylation restores Ca2+ sensitivity in diabetic cardiac muscle. Diabetes (2015) 64:3573–87. doi: 10.2337/db14-1107
138. Wende AR. Unsticking the broken diabetic heart: O- GlcNAcylation and calcium sensitivity. Diabetes (2015) 64:3339–41. doi: 10.2337/dbi15-0001
139. Suarez J, Hu Y, Makino A, Fricovsky E, Wang H, Dillmann WH. Alterations in mitochondrial function and cytosolic calcium induced by hyperglycemia are restored by mitochondrial transcription factor A in cardiomyocytes. AJP Cell Physiol. (2008) 295:C1561–8. doi: 10.1152/ajpcell.00076.2008
140. Zhu-Mauldin X, Marsh SA, Zou L, Marchase RB, Chatham JC. Modification of STIM1 by O-linked N-acetylglucosamine (O-GlcNAc) attenuates store-operated calcium entry in neonatal cardiomyocytes. J Biol Chem. (2012) 287:39094–106. doi: 10.1074/jbc.M112.383778
141. Eshima H, Tanaka Y, Sonobe T, Inagaki T, Nakajima T, Poole DC, et al. In vivo imaging of intracellular Ca2+ after muscle contractions and direct Ca2+ injection in rat skeletal muscle in diabetes. AJP Regul Integr Comp Physiol. (2013) 305:R610–8. doi: 10.1152/ajpregu.00023.2013
142. Safwat Y, Yassin N, Gamal El Din M, Kassem L. Modulation of Skeletal muscle performance and SERCA by exercise and adiponectin gene therapy in insulin-resistant rat. DNA Cell Biol. (2013) 32:378–85. doi: 10.1089/dna.2012.1919
Keywords: O-GlcNAcylation, slow-twitch muscle, fast-twitch muscle, glucose metabolism, exercise, skeletal muscle atrophy
Citation: Lambert M, Bastide B and Cieniewski-Bernard C (2018) Involvement of O-GlcNAcylation in the Skeletal Muscle Physiology and Physiopathology: Focus on Muscle Metabolism. Front. Endocrinol. 9:578. doi: 10.3389/fendo.2018.00578
Received: 21 June 2018; Accepted: 11 September 2018;
Published: 16 October 2018.
Edited by:
Tony Lefebvre, Lille University of Science and Technology, FranceReviewed by:
Anna Krzeslak, University of Łódz, PolandYobana Perez-Cervera, Benito Juárez Autonomous University of Oaxaca, Mexico
Abderrahman Maftah, University of Limoges, France
Copyright © 2018 Lambert, Bastide and Cieniewski-Bernard. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Caroline Cieniewski-Bernard, Y2Fyb2xpbmUuY2llbmlld3NraS1iZXJuYXJkQHVuaXYtbGlsbGUuZnI=
†Present Address: Matthias Lambert, Division of Genetics and Genomics, Boston Children's Hospital, Boston, MA, United States; Department of Pediatrics, Harvard Medical School, Boston, MA, United States