 Giuditta Rurale
Giuditta Rurale Luca Persani
Luca Persani Federica Marelli
Federica Marelli- 1Department of Clinical Sciences and Community Health, University of Milan, Milan, Italy
- 2Division of Endocrine and Metabolic Diseases & Laboratory of Endocrine and Metabolic Research, IRCCS Istituto Auxologico Italiano, Milan, Italy
Variations in the transcription factor Gli-similar 3 (GLIS3) gene have been associated to variable congenital endocrine defects, including both morphogenetic and functional thyroid alterations. Evidence from Glis3 knockout mice indicates a relevant role for GLIS3 in thyroid hormone biosynthesis and postnatal thyroid gland growth, with a mechanism of action downstream of the TSH/TSHR interaction. However, the pathophysiological role of this transcription factor during the embryonic thyroid development remains unexplored. In this manuscript, we will provide an overview of the current knowledge on GLIS3 function during development. As a perspective, we will present preliminary evidence in the zebrafish model in support of a potential role for this pleiotropic transcription factor in the early stages of thyroid gland development.
GLIS3 in Human Pathophysiology
The transcription factor Gli-similar 3 (GLIS3) belongs to the family of the Krüppel-like zinc finger transcription factor (1) and it is involved in several cellular processes, including proliferation, apoptosis, differentiation and development (2).
The human GLIS3 gene is located on chromosome 9p24.2 and encodes a full-length protein of 90 kD in size. GLIS3 in expressed in a tissue-specific manner, with highest levels in kidney, thyroid gland, endocrine pancreas, thymus, testis, and uterus. Lower levels of expression were also described in brain, lung, ovary, and liver (1, 3).
Several alternate transcripts of GLIS3 have been reported so far in humans. Nine out of nineteen isoforms are processed into proteins with unknown functions. The larger transcript (7.5 kb) is predominantly expressed in pancreas, thyroid, and kidney, whereas the smaller isoforms (0.8–2 kb) are mainly localized in heart, kidney, liver and skeletal muscle (4). Despite the distinct pattern of expression, so far there is no evidence of variable pathophysiological functions for these different transcripts.
The GLIS3 protein consists of a centrally located zinc finger domain (ZFD) (five tandems Cys2-His2 zinc finger motifs), a C-terminal transactivation domain (TAD) and a relatively large N-terminus that is important in the regulation of GLIS3 stability and its transcriptional activity (1, 5).
The ZFD dictates GLIS3 cytosolic or nuclear localization and is responsible for the recognition of specific DNA responsive elements, the GlisBS (Glis-binding sites) are located within the regulatory regions of target genes (1).
Once bound to the DNA, GLIS3 can repress or enhance the expression of target genes (1, 6).
Furthermore, GLIS3 stability is regulated by the binding with the “Suppressor of Fused” (SUFU), a negative regulator of Hedgehog (SHH) signaling, via a YGH motif localized into the N-terminal domain (7). SUFU not only protects Glis3 from proteasomal degradation stabilizing Glis3 protein (8), but also modulates Glis3-mediated transactivation of the promoter of target genes (9).
Based on the current knowledge, GLIS3 represents a new candidate gene for congenital hypothyroidism (CH) but its role in thyroid development and function remains largely unexplored.
Homozygous and compound heterozygous mutations in the GLIS3 gene have been associated with a rare syndrome, called NDH, characterized by neonatal diabetes (ND) and CH.
NDH patients frequently present a polycystic kidney disease, and are variably affected by additional abnormalities, such as developmental delay, hepatic fibrosis, congenital glaucoma, and facial dimorphism (1, 3, 10, 11).
The GLIS3 alterations identified in NDH patients included missense substitutions, frameshift mutations resulting in premature termination, deletions encompassing exons 5–9, 3–4, 9–11, 10–11 and larger deletions covering regions >100 kb that include exons 1–2, 1–4, or 5–9 (8).
A Chinese group recently screened GLIS3 gene in 592 CH patients and 600 controls by Next Generation Sequencing (NGS) and Multiplex Ligation-dependent Probe Amplification (MLPA) (12). They found two different variations (one deletion at chromosome 9p24.3p23, including the GLIS3 gene, and one missense variant (p.R720Q) in two CH patients with gland-in-situ (frequency of GLIS3 variations in Chinese CH: 0.3%) but not in controls. Both variations were in the heterozygous state (12).
We have recently identified rare heterozygous GLIS3 missense variants by NGS in a large cohort (18/177) of Caucasian patients with isolated CH (13). Clinically, half of the affected cases presented variable thyroid dysgenesis (athyreosis, thyroid hypoplasia, and ectopy), whereas the remaining part have in situ thyroid gland (13). Interestingly, these GLIS3 variants are associated in all 18 cases with other rare variations in genes involved in thyroid pathology, supporting a frequent oligogenic origin of CH.
Regarding the missense variations identified in NDH or CH patients no functional studies are available so far. In Figure 1B we report the alignment of the aminoacid sequences of zebrafish, mouse and human GLIS3. The residues C536, H561, and R589 associated to NDH pathology localized within the zinc-finger domain and are completely conserved across species. Concerning the CH patients (12, 13), only 2/11 missense variants, affect aminoacids (D512 and E515) belonging the first zinc-finger, but are not conserved between human and zebrafish.

Figure 1. (A) Schematic representation of the zebrafish glis3 protein (UniProt F1QB13). Yellow, Serine-Rich (SR) domain; Green, Proline-Rich (PR) domain; Light Blue, Zinc-Finger (ZF) domain; Red, Nuclear Localization Signal (NLS). (B) Alignment of the GLIS3 protein sequencing of the zebrafish (UniProt F1QB13), mouse (UniProt Q0GE24), and human (UniProt Q8NEA6). The color code of the different domains is reported above. The residues that differ between species are highlighted in gray. The missense GLIS3 variants identified in NDH (orange) and CH (green) patients are reported. (C) Aminoacid identity between zebrafish, murine and human GLIS3 proteins, performed using ClustalW2 software.
In addition, a number of genome wide association studies (GWAS) have associated several GLIS3 single nucleotide polymorphism (SNP) with elevated circulating TSH levels and low T4 (1).
Taken together, all of these data support a relevant role of GLIS3 in thyroid function and in the onset of CH, possibly contributing to the explanation of the missing hereditability of CH and the prevalent sporadic presentation of the disease (13).
Although hypothyroidism is associated with GLIS3 variants, the absence of consistent pathological features in patients makes unclear the identification of a causative mechanism (8). High TSH and low T4 characterize all of twelve NDH patients reported so far, but in presence of a wide spectrum of structural thyroid abnormalities including athyreosis, hypoplasia, perifollicular, and interstitial fibrosis, and normal thyroid anatomy. Similar phenotypic variability was previously observed in patients with mutations in NKX2.1 and PAX8 genes, involved in the specification of thyroid primordium and in the subsequent expression of genes (TG, TPO, TSHR, and NIS) responsible for the differentiation and proliferation of thyroid follicles and the synthesis of thyroid hormones (TH) (14, 15). One possible explanation for the variable CH manifestations could be attributed to the tissue-specific expression of GLIS3 transcripts of variable length. Large deletions or frameshift mutations causing profound disruptions of Glis3 structure are associated with the most severe manifestations of NDH. Instead, milder phenotypes appear correlated to missense substitutions that preserve a residual function of one or more GLIS3 transcripts (8, 15).
An additional challenge of GLIS3 defect is the high variability in the sensitivity of TSH suppression during thyroxine replacement and the consequent difficult TSH normalization (8). Several patients with elevated TSH and TG levels appear to be resistant to increasing doses of thyroxine therapy despite normalization of free T4, thus excluding alterations in the L-T4 absorption (4, 15).
Therefore, additional work is required to understand the genotype-phenotype correlations and to determine whether Gils3 acts upstream of genes involved in pathways regulating thyroid development, hormonogenesis, and the peripheral response to T4.
Glis3 Murine Models
Up to now, the murine model has been extensively used to investigate the pathological mechanism underlying the multisystem clinical presentation of NDH syndrome, and most of the researches have been focusing on pancreas development and the onset of diabetes.
The Glis3-null mice die within 1 week after birth due to the severity of neonatal diabetes (11). In mutant mice, neonatal diabetes is associated with decreased insulin levels caused by an impaired β-cell mass (11, 16), suggesting a relevant role for Glis3 in the regulation of β-cell development and endocrine function (17).
In fact, Glis3 overlaps with Pdx1 and Nkx6.1 expression in endocrine progenitors, and becomes mainly restricted to Ins+ β-cell in later stages (18). ZeRuth and co-workers demonstrated that Glis3 binds the insulin (Ins) promoter enhancing its transcription. Glis3 recruits Cbp/p300, which constitutes a scaffold for the formation of a larger transcriptional regulatory complex, including the other synergic factors Pdx1, Neurod1, and Mafa at the level of the insulin promoter (17). All of these studies indicated that GLIS3 has multiple critical regulatory functions during pancreas development and also in the adult pancreas: regulating the development of endocrine progenitors, the generations and maturation of pancreatic β-cells, regulation of insulin expression as well as in maintaining normal duct morphology (8).
As for NDH patients, the Glis3 mutant mice also developed polycystic kidney disease (19), pointing up the role of Glis3 in the maintenance of the normal renal function. Kang and co-workers demonstrated that Glis3 and its co-activator Wwtr1/TAZ are part of transcriptional regulatory networks that are critical in the function of the primary cilium (19).
As far as thyroid gland is concerned, the Refetoff and Jetten groups recently described that Glis3 is essential for TH biosynthesis and postnatal thyroid gland development in mouse, acting down-stream of TSH/TSHR system (14). In mice, GLIS3 appears to retain a relevant role in the expression of several functional proteins, such as the sodium/iodide symporter (Nis encoded by SCL5A5) and Pendrin (encoded by SLC26A4). In addition, despite a relevant role in thyroid cell proliferation, no significant thyroid developmental defects were observed in Glis3 knock-out mice. These data are partially in contrast to those observed in the NDH patients, in which thyroid dysgenesis is a common feature. However, several factors, including the type and site of the mutation, can contribute the variable expression of the thyroid phenotype (14).
For this reason, we believe that the murine models available today do not fully recapitulate the clinical manifestations of the NDH patients. A similar condition was previously experienced with the knockout of MCT8 gene in mice that fails to recapitulate the neurological manifestations of the human disease (20). This evidence made it clear that a single species would not suffice to obtain insights into the underlying pathological mechanisms. The evolutionary conservation of thyroid hormone action on neurodevelopment as well as the components regulating thyroid hormone signaling however offered the opportunity to answer different aspects of MCT8 function in brain development using different vertebrate species, such as zebrafish (21). In Mct8-deficient zebrafish embryos, it was possible to obtain the unprecedented evidence of an impaired thyroid hormone uptake at the level of the blood-brain barrier during the peri- and postnatal development (21).
Therefore, in this perspective we address the possibility of zebrafish as a viable vertebrate model to study in more details the role of GLIS3 in thyroid morphogenesis and dysfunction.
Zebrafish As a New Model to Study glis3 Functions
Zebrafish (Danio rerio) has emerged as an important and useful model system to study different human diseases. Zebrafish possesses a unique combination of features that makes it particularly well-suited for experimental and genetic analysis of vertebrate development. It has a short reproductive cycle with external fertilization and lays a large numbers of embryos per mating that can be analyzed under microscope because of their optical transparency. The availability of several genetic manipulation strategies, at costs that are definitely lower than in rodents, lead to the generation of zebrafish models for a wide variety of human diseases (22).
Like the other vertebrates, the basic unit of the zebrafish thyroid gland is the thyroid follicle, which arises from the pharyngeal endoderm (23). At 22 h post-fertilization (hpf) the zebrafish thyroid primordium starts to express the transcription factor nkx2.4, pax2a, pax8, hhex, and foxe1. As described in mice, knockdown experiments in zebrafish demonstrated that the transcription factor nkx2.4, pax2a, and hhex are required for the specification and the differentiation of thyroid primordium (24, 25). The differentiation of thyroid precursors continues up to 2 days post-fertilization (dpf) with the expression of the functional thyroid genes tg, tpo, and slc5a5. At 3 dpf the zebrafish thyroid follicles are completely developed and start to produce thyroid hormone (24).
Unlike mammals, the zebrafish thyroid is not organized as a compact gland, but is composed of single follicles localized along the ventral aorta (23). In fact, alterations in the architecture of the pharyngeal vessels are associated with severe thyroid defects, consistent with a correlation between thyroid morphology and vascular development (25). The proper amount of TH is tightly regulated by the hypothalamus-pituitary-thyroid axis, which is also preserved in zebrafish (24). As a consequence, this model has been extensively used to study the molecular mechanism underlying alteration in thyroid development (23, 25, 26) and function (27, 28). However, the mechanisms and the genes accounting for the commitment of the thyroid precursors from the endoderm are presently unknown.
The glis3 gene is evolutionary conserved across species dating back to fishes (2) and in zebrafish it is localized on the chromosome 10 and encodes a protein of 804 residues that is 88.45 kD in size. A second transcript that encodes protein of 787 residues still not characterized to date. Although the aminoacid identity between zebrafish and human is rather low (47.23%), the glis3 protein structure and the five zinc-finger motives are well-conserved also in zebrafish (Figure 1).
By in situ hybridization (ISH), we show that the zebrafish glis3 transcript is expressed since the early developmental stages with an intense signal at the pharyngeal endoderm, brain and in the glomerulus and the pronephric ducts. Of note, glis3 transcripts are not detectable when the thyroid gland development is completed at 3 dpf (Figure 2A). During embryonic development glis3 starts to be expressed during somitogenesis with a peak at 20-somite stage (19 hpf). Afterwards, the glis3 levels increase rapidly until the embryonic transition to the larval stage (Figure 2B). Consistent with clinical and experimental findings in humans and mice models, glis3 is expressed in a tissue specific manner in the adult zebrafish. It is detectable at high level in kidney, pituitary and pancreas and at lower levels in testis, ovary, brain, and pectoral fins (Figure 2C).
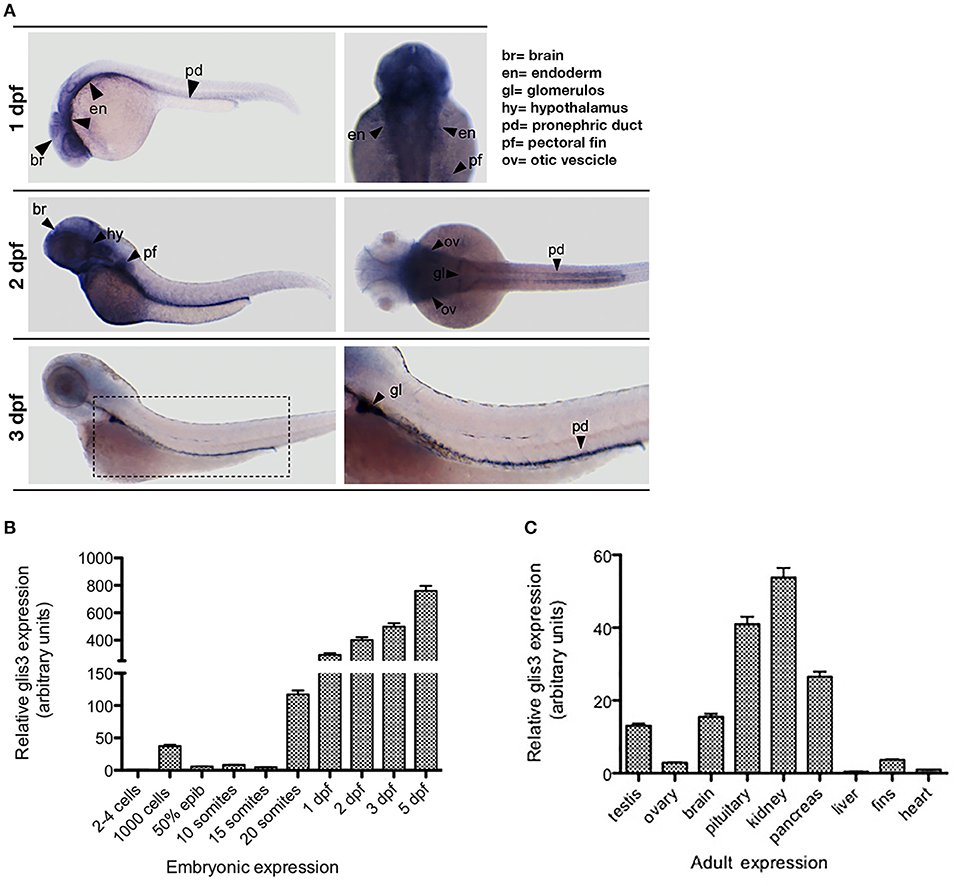
Figure 2. Ontogenetic pattern of glis3 expression in zebrafish. (A) Tissue specific expression of glis3, at different developmental stages, analyzed by ISH using glis3 antisense riboprobe followed by NBT/BCIP staining, as previously described (29). Images are representative of three experiments (30 embryos each). (B,C) qRT-PCR of glis3 relative mRNA expression during embryonic development and adult tissues. Total RNA is extracted from pools of 50 embryos/stage and from 3 adults, respectively. cDNA synthesis and qRT-PCR following standard procedures (28). Experiments are performed in triplicate and results are expressed by Mean ± SD.
The only available findings about the glis3 function in zebrafish are now-a-days related to pancreas. The glis3 knockdown embryos present Type 2 Diabetes (T2D) and a β-cell mass deficiency (30), confirming findings in mice (11).
Both Glis3 and Wwtr1 are known to contribute to renal function (19). Interestingly, wwtr1 has been shown to have an important function in the antero-posterior pattering of the pronephric progenitor field (31) as well as in the differentiation of the thyroid gland (32) in zebrafish. It is therefore tempting to hypothesize a possible interaction between glis3 and wwtr1 during thyroid growth. To date, the involvement of glis3 in the development and function of the thyroid gland and of the hypothalamic-pituitary-thyroid axis in zebrafish is presently unknown. The expression of glis3 at the pharyngeal endoderm before the appearance of nkx2.4 and pax2a expression in the thyroid primordium suggests a possible role for glis3 during the specification of the thyroid precursors, and this hypothesis is currently under investigation in our Lab.
Conclusion
To date, it is known that GLIS3 is a pleiotropic master regulator of several pathways involved in the development and function of a broad range of tissues and organs. In particular, GLIS3 is involved in the specification of several endocrine glands. Since the endocrine pancreas and thyroid arise from the same embryonic sheet, it is reasonable to think that GLIS3 could act at both sites with similar mechanisms. However, based on current findings it is possible to envisage a dual role for GLIS3 during the thyroid development and regulating thyroid function in the postnatal period.
The bewildering variability of the clinical manifestations of NDH and CH associated with GLIS3 variants cannot be explained at present. Hence, there is need to improve our understanding of the pathophysiology underlying GLIS3 action.
The establishment of a zebrafish model may constitute a valid alternative to mice in the definition of the early effects of glis3 deficiency and, eventually, provide interesting insights on the molecular mechanisms involved in thyroid development and CH pathogenesis.
Ethics Statement
The protocol for zebrafish studies was approved by the Ethic Committee of Istituto Auxologico Italiano (study 05C102_2011).
Author Contributions
GR performed the zebrafish experiments and prepared the draft of the manuscript. FM supervised the zebrafish study and contributed the draft preparation. LP conceived the study, obtained research funds and finalized the manuscript. All authors approved the final version of the manuscript.
Funding
This work was partially supported by the grant 05C102_2011 of the Ricerca Corrente of the Istituto Auxologico Italiano (Milan, Italy) and by the funds of the Ph. D. program in Experimental Medicine at the University of Milan (Milan, Italy).
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1. Lichti-Kaiser K, ZeRuth G, Jetten AM. Transcription factor gli-similar 3 (Glis3): implications for the development of congenital hypothyroidism. J Endocrinol Diabetes Obes. (2014) 2:1024.
2. Kim YS, Nakanishi G, Lewandoski M, Jetten AM. GLIS3, a novel member of the GLIS subfamily of Kruppel-like zinc finger proteins with repressor and activation functions. Nucleic Acids Res. (2003) 31:5513–25. doi: 10.1093/nar/gkg776
3. Senée V, Chelala C, Duchatelet S, Feng D, Blanc H, Cossec JC, et al. Mutations in GLIS3 are responsible for a rare syndrome with neonatal diabetes mellitus and congenital hypothyroidism. Nat Genet. (2006) 38:682–7. doi: 10.1038/ng1802
4. Dimitri P, Habeb AM, Gurbuz F, Millward A, Wallis S, Moussa K, et al. Expanding the clinical spectrum associated with GLIS3 mutations. J Clin Endocrinol Metab. (2015) 100:E1362–9. doi: 10.1210/jc.2015-1827
5. ZeRuth GT, Williams JG, Cole YC, Jetten AM. HECT E3 ubiquitin ligase itch functions as a novel negative regulator of gli-similar 3 (Glis3) transcriptional activity. PLoS ONE (2015) 10:e0131303. doi: 10.1371/journal.pone.0131303
6. Yang Y, Chang BH, Samson SL, Li MV, Chan L. The kruppel-like zinc finger protein Glis3 directly and indirectly activates insulin gene transcription. Nucleic Acids Res. (2009) 37:2529–38. doi: 10.1093/nar/gkp122
7. ZeRuth GT, Yang XP, Jetten AM. Modulation of the transactivation function and stability of Kruppel-like zinc finger protein Gli-similar 3 (Glis3) by suppressor of fused. J Biol Chem. (2011) 286:22077–89. doi: 10.1074/jbc.M111.224964
8. Jetten AM. GLIS1-3 transcription factors: critical roles in the regulation of multiple physiological processes and diseases. Cell Mol Life Sci. (2018). 75:3473–94. doi: 10.1007/s00018-018-2841-9
9. Lichti-Kaiser K, ZeRuth G, Kang HS, Vasanth S, Jetten AM. Gli-similar proteins: their mechanisms of action, physiological functions, and roles in disease. Vitam Horm. (2012) 88:141–71. doi: 10.1016/B978-0-12-394622-5.00007-9
10. Dimitri P, Warner JT, Minton JA, Patch AM, Ellard S, Hattersley AT, et al. Novel GLIS3 mutations demonstrate an extended multisystem phenotype. Eur J Endocrinol. (2011) 164:437–43. doi: 10.1530/EJE-10-0893
11. Kang HS, Kim YS, ZeRuth G, Beak JY, Gerrish K, Kilic G, et al. Transcription factor Glis3, a novel critical player in the regulation of pancreatic beta-cell development and insulin gene expression. Mol Cell Biol. (2009) 29:6366–79. doi: 10.1128/MCB.01259-09
12. Fu C, Luo S, Long X, Li Y, She S, Hu X, et al. Mutation screening of the GLIS3 gene in a cohort of 592 Chinese patients with congenital hypothyroidism. Clin Chim Acta (2018) 476:38–43. doi: 10.1016/j.cca.2017.11.011
13. de Filippis T, Gelmini G, Paraboschi E, Vigone MC, Di Frenna M, Marelli F, et al. A frequent oligogenic involvement in congenital hypothyroidism. Hum Mol Genet. (2017) 26:2507–14. doi: 10.1093/hmg/ddx145
14. Kang HS, Kumar D, Liao G, Lichti-Kaiser K, Gerrish K, Liao XH, et al. GLIS3 is indispensable for TSH/TSHR-dependent thyroid hormone biosynthesis and follicular cell proliferation. J Clin Invest. (2017) 127:4326–37. doi: 10.1172/JCI94417
15. Dimitri P, The role of GLIS3 in thyroid disease as part of a multisystem disorder. Best Pract Res Clin Endocrinol Metab. (2017) 31:175–82. doi: 10.1016/j.beem.2017.04.007
16. Watanabe N, Hiramatsu K, Miyamoto R, Yasuda K, Suzuki N, Oshima N, et al. A murine model of neonatal diabetes mellitus in Glis3-deficient mice. FEBS Lett. (2009) 583:2108–13. doi: 10.1016/j.febslet.2009.05.039
17. ZeRuth GT, Takeda Y, Jetten AM. The Kruppel-like protein Gli-similar 3 (Glis3) functions as a key regulator of insulin transcription. Mol Endocrinol. (2013) 27:1692–705. doi: 10.1210/me.2013-1117
18. Kang HS, Takeda Y, Jeon K, Jetten AM. The spatiotemporal pattern of Glis3 expression indicates a regulatory function in bipotent and endocrine progenitors during early pancreatic development and in beta, PP and ductal cells. PLoS ONE (2016) 11:e0157138. doi: 10.1371/journal.pone.0157138
19. Kang HS, Beak JY, Kim YS, Herbert R, Jetten AM. Glis3 is associated with primary cilia and Wwtr1/TAZ and implicated in polycystic kidney disease. Mol Cell Biol. (2009) 29:2556–69. doi: 10.1128/MCB.01620-08
20. Heuer H, Visser TJ. The pathophysiological consequences of thyroid hormone transporter deficiencies: insights from mouse models. Biochim Biophys Acta (2013) 1830:3974–8. doi: 10.1016/j.bbagen.2012.04.009
21. Vancamp P, Darras VM. From zebrafish to human: a comparative approach to elucidate the role of the thyroid hormone transporter MCT8 during brain development. Gen Comp Endocrinol. (2018) 265:219–29. doi: 10.1016/j.ygcen.2017.11.023
22. Lieschke GJ, Currie PD. Animal models of human disease: zebrafish swim into view. Nat Rev Genet. (2007) 8:353–67. doi: 10.1038/nrg2091
23. Wendl T, Lun K, Mione M, Favor J, Brand M, Wilson SW, et al. Pax2.1 is required for the development of thyroid follicles in zebrafish. Development (2002) 129:3751–60.
24. Marelli F, Persani L. How zebrafish research has helped in understanding thyroid diseases. F1000Res (2017) 6:2137. doi: 10.12688/f1000research.12142.1
25. Porazzi P, Calebiro D, Benato F, Tiso N, Persani L. Thyroid gland development and function in the zebrafish model. Mol Cell Endocrinol. (2009) 312:14–23. doi: 10.1016/j.mce.2009.05.011
26. Porazzi P, Marelli F, Benato F, de Filippis T, Calebiro D, Argenton F, et al. Disruptions of global and JAGGED1-mediated notch signaling affect thyroid morphogenesis in the zebrafish. Endocrinology (2012) 153:5645–58. doi: 10.1210/en.2011-1888
27. Opitz R, Maquet E, Zoenen M, Dadhich R, Costagliola S. TSH receptor function is required for normal thyroid differentiation in zebrafish. Mol Endocrinol. (2011) 25:1579–99. doi: 10.1210/me.2011-0046
28. Marelli F, Carra S, Rurale G, Cotelli F, Persani L. In vivo functional consequences of human THRA variants expressed in the zebrafish. Thyroid (2017) 27:279–91. doi: 10.1089/thy.2016.0373
29. Thisse C, Thisse B. High-resolution in situ hybridization to whole-mount zebrafish embryos. Nat Protoc. (2008) 3:59–69. doi: 10.1038/nprot.2007.514
30. O'Hare EA, Yerges-Armstrong LM, Perry JA, Shuldiner AR, Zaghloul NA. Assignment of functional relevance to genes at type 2 diabetes-associated loci through investigation of beta-cell mass deficits. Mol Endocrinol. (2016) 30:429–45. doi: 10.1210/me.2015-1243
31. Zhang J, Yuan S, Vasilyev A, Amin Arnaout M. The transcriptional coactivator Taz regulates proximodistal patterning of the pronephric tubule in zebrafish. Mech Dev. (2015) 138(Pt 3):328–35. doi: 10.1016/j.mod.2015.08.001
Keywords: GLIS3, sonic hedgehog, thyroid, congenital hypothyroidism, zebrafish
Citation: Rurale G, Persani L and Marelli F (2018) GLIS3 and Thyroid: A Pleiotropic Candidate Gene for Congenital Hypothyroidism. Front. Endocrinol. 9:730. doi: 10.3389/fendo.2018.00730
Received: 28 June 2018; Accepted: 16 November 2018;
Published: 29 November 2018.
Edited by:
Noriyuki Koibuchi, Gunma University, JapanReviewed by:
Salvatore Benvenga, Università degli Studi di Messina, ItalyJosé C. Moreno, Universität Zürich, Switzerland
Copyright © 2018 Rurale, Persani and Marelli. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Luca Persani, bHVjYS5wZXJzYW5pQHVuaW1pLml0